

ABSTRACT
Albumin is the most abundant protein in blood plasma and acts as a carrier for many circulating molecules. Hypoalbuminaemia, mostly caused by either renal or liver disease or malnutrition, can perturb vascular homeostasis and is involved in the development of multiple diseases. Here we review four functions of albumin and the consequences of hypoalbuminaemia on vascular homeostasis. (i) Albumin is the main determinant of plasma colloid osmotic pressure. Hypoalbuminaemia was therefore thought to be the main mechanism for oedema in nephrotic syndrome (NS), however, experimental studies showed that intrarenal mechanisms rather than hypoalbuminaemia determine formation and, in particular, maintenance of oedema. (ii) Albumin functions as an interface between lysophosphatidylcholine (LPC) and circulating factors (lipoproteins and erythrocytes) and the endothelium. Consequently, hypoalbuminaemia results in higher LPC levels in lipoproteins and erythrocyte membrane, thereby increasing atherosclerotic properties of low-density lipoprotein and blood viscosity, respectively. Furthermore, albumin dose-dependently restores LPC-induced inhibition of vasodilation. (iii) Hypoalbuminaemia impacts on vascular nitric oxide (NO) signalling by directly increasing NO production in endothelial cells, leading to reduced NO sensitivity of vascular smooth muscle cells. (iv) Lastly, albumin binds free fatty acids (FFAs). FFAs can induce vascular smooth muscle cell apoptosis, uncouple endothelial NO synthase and decrease endothelium-dependent vasodilation. Unbound FFAs can increase the formation of reactive oxygen species by mitochondrial uncoupling in multiple cell types and induce hypertriglyceridemia in NS. In conclusion, albumin acts as an interface in the circulation and hypoalbuminaemia impairs multiple aspects of vascular function that may underlie the association of hypoalbuminaemia with adverse outcomes. However, hypoalbuminaemia is not a key to oedema in NS. These insights have therapeutic implications.
Keywords: albumin, free fatty acids, lysophosphatidylcholine, nephrotic syndrome, oxidative stress
INTRODUCTION
Albumin is the most abundant protein in the human circulation. Albumin has a turnover time in humans of ∼25 days being produced at a rate of 10.5 g/day and being cleared renally (∼6%), gastrointestinally (∼10%) and catabolized (∼84%) [1]. Changes in serum albumin can be the consequence of reduced production (hepatic disease, reduced protein intake or due to a negative acute-phase response in inflammation [2]) or increased loss of albumin [nephrotic syndrome (NS), protein-losing enteropathy or catabolism during severe disease]. Albumin is synthesized in the liver, in hepatocytes it is translated from a single gene as preproalbumin [3]. The preproalbumin protein is transported to the endoplasmic reticulum where the N-terminal prepropeptide is cleaved by a serine protease. Afterwards, it is transported to the Golgi system before it is secreted into the circulation as a simple protein containing only amino acids without additives or prosthetic groups [1, 4]. Therefore it is one of the few plasma proteins without a carbohydrate additive, making albumin a non-glycoprotein. More recently, it has been suggested that albumin can undergo post-translational modification under specific situations, modifying its interaction with other molecules such as exogenous drugs [3]. One example of such a modification is glycation of albumin, which is decreased in NS but increased in diabetes mellitus [5].
Albumin consists of eight and one-half double loops formed by disulphide bonds involving adjacent half-cysteine residues, resulting in a molecular mass of 65–66 kDa. Albumin consists of 35 cysteine amino acids that form Cys-Cys pairs, except for Cys-34 having a sulfhydryl additive. It can be further divided into three homologous helical domains, numbered I, II and III, formed by two longer loops separated by a shorter loop [4]. This structure is well-preserved across species, having the same distribution of half-cysteines. Although the three domains are homologous, there ligand-binding functions are different. Within each domain the first two loops (loops 1–2, 4–5 and 7–8) are grouped together as subdomains IA, IIA, IIIA, respectively [4]. Additionally, loop 3, 6 and 9 are grouped as IB, IIB, IIIB, responsible for the three-dimensional structure and function of albumin. The main function of serum albumin is to bind and transport small molecules in the circulation. Due to its three-domain design, it has multiple binding sites and thus is capable of binding a wide variety of molecules. Hydrophobic organic anions of 100–600 Da, including long-chain free fatty acids (FFAs), haematin and bilirubin, bind the strongest to albumin. Smaller molecules such as tryptophan, ascorbate and 25-hydroxyvitamin D3 are bound highly specifically but less strongly to albumin [4, 6]. For an extensive overview of the different molecules that can be bound to albumin, we refer to Peters et al. [4]. Through the binding of all these compounds, albumin can function as a depot, extending the available quantities beyond their solubility. In addition, albumin can bind circulating toxins, rendering them harmless until they can be cleared by the liver and/or kidneys. Importantly, several drugs are bound by albumin and thus the loss of circulating albumin can disrupt normal pharmacokinetics, thereby affecting half-life and plasma unbound drug concentration and ultimately altering normal metabolism and the function of drugs [7].
ALBUMIN, NS AND OEDEMA
In NS, defects in glomerular barrier function result in leakage of proteins into the pre-urine, surpassing the protein reuptake capacity of the proximal tubule epithelial cells [8]. This cascade can result in heavy proteinuria (>3.5 g/day) with subsequent hypoalbuminaemia, which, together with sodium retention, water retention and generalized oedema, is one of the hallmarks of NS [9]. Historically, hypoalbuminaemia was thought to be the main mechanism of oedema formation associated with NS (Figure 1). This so-called underfill hypothesis was based on the findings of Frank Starling, who formulated Starling's Principle of how plasma and tissue colloid osmotic and hydrostatic pressure are responsible for maintaining plasma–interstitial fluid equilibrium [10] (see Figure 1 for the full Starling's Principle). Interstitial albumin concentrations vary between tissues [11, 12]. It is dependent on plasma concentration of albumin [12], together with the local transcapillary escape rate and the passage of albumin from the interstitial to intravascular compartment via the lymph or diffusion [13]. Interestingly, the transcapillary escape rate has been shown to be increased in common diseases inducing vascular leakage, such as diabetes [14]. Since albumin is one of the strongest determinants (∼50%) of the plasma colloid osmotic pressure, it was hypothesized that the low plasma colloid osmotic pressure resulted in fluid flux into the interstitium, thus inducing generalized oedema and a hypovolemic state [13]. The kidneys try to prevent volume depletion by activation of the renin–angiotensin–aldosterone system (RAAS) and subsequently increase sodium and water retention, inducing a vicious circle [13].
FIGURE 1:

Paradigm shift of the pathophysiological mechanisms of oedema formation in NS. Historically Starling's Principle was thought to be the main mechanism, but due to observations from clinical and experimental studies this was rejected. More recent studies show a role for urinary protein-activated plasmin promoting ENaC activation in the cortical collecting duct.
J v, trans-endothelial filtration volume; Lp, hydraulic conductivity of the membrane; S, surface area; P, hydrostatic pressure; π, oncotic pressure; ACE, angiotensin-converting enzyme; uPA(R), urokinase (receptor).
Multiple observations from clinical and experimental studies investigating NS shifted this paradigm. The hypothesis implied that patients with NS are hypovolaemic, but data from clinical studies demonstrated that only a small portion actually are hypovolaemic and that the majority of patients are euvolaemic or hypervolaemic [15–18]. Plasma oncotic pressure was reduced in children with NS, while hypovolemic symptoms were only present in ∼27% of the children with NS [19]. Hypovolaemia was accompanied with an activation of the RAAS; however, in a large cohort of NS patients, RAAS activation was not associated with blood volume [20]. Additionally, increased plasma renin activity was only increased in NS patients with hypovolemic symptoms [19]. It is suggested that minimal change disease represents a unique NS phenotype with increased neurohumoral activity, independent of blood volume [21]. In children with NS due to minimal change disease, sodium retention occurs before hypoproteinaemia and while renal blood flow is high [22]. After steroid-induced remission of NS is achieved, natriuresis is promoted while plasma protein content and blood volume are still low [23]. In contrast to all this evidence against the hypovolaemia hypothesis, there is one study showing that fractional sodium excretion is only reduced in NS patients with symptoms of hypovolaemia [19].
There are several oedema-preventing mechanisms that help maintain plasma and interstitial fluid equilibrium [13]. Mobilization of interstitial protein storage into the circulation is the main mechanism in hypoalbuminaemia-induced reduction in plasma oncotic pressure [13]. Only when the interstitial oncotic pressure cannot be further reduced will oedema occur. Interestingly, it has been shown that NS patients have oedema at higher levels of plasma oncotic pressure [13]. Joles et al. [24] demonstrated that plasma oncotic pressure due to hypoproteinaemia must be below ∼8 mmHg for sodium to be actively retained, which is lower than in a significant portion of the NS patients [20]. In patients with NS, transcapillary oncotic gradient is maintained both at diagnosis and during (complete or partial) recovery [25]. This was also observed in Nagase analbuminaemic rats, which have a decrease in plasma colloid osmotic pressure but a maintained capillary-interstitial oncotic gradient due to a parallel decrease in interstitial colloid osmotic pressure [26]. Similar findings have been observed in a different rat model of hypoproteinaemia induced by starvation [27]. Rapid induction of hypoproteinaemia has been achieved by repeated plasmapheresis in dogs. In this model of hypoproteinaemia, a compensatory decrease in tissue oncotic pressure has been demonstrated and even an increased fluid recovery from haemorrhage [28]. Interestingly, studies conducted in this model have also demonstrated that moderate hypoproteinaemia does increase extra-cellular volume fraction, but does not alter blood volume or neurohumoral activation [24, 29, 30]. Strikingly, sodium balance was achieved when protein levels were still low at the onset of recovery of plasmapheresis [24, 30]. In line with these findings, in a unilateral nephrotic rat model, only the diseased kidney retains sodium [31]. Ultimately 43% of patients with congenital analbuminaemia do not experience oedema formation and 38% only experience mild ankle oedema [32]. Altogether, these findings suggest that generalized oedema in NS is not merely due to hypoalbuminaemia, as volume regulation even in the absence of albumin is mostly maintained, but that an intrarenal mechanism is responsible, the so-called overflow hypothesis.
In the overflow hypothesis for oedema formation in NS, the proteolytic activation of epithelial sodium channels (ENaCs) has been suggested to be the driving factor of sodium and subsequent fluid retention (Figure 1) [33]. Activation of ENaCs is the result of cleavage of the γ-subunit by urinary plasmin, a serine protease [34, 35]. ENaC activation by cleavage of the γ-subunit by proteases has been suggested to be a physiological process [33]. However, glomerular defects in NS result in increased protein and proteases in the urine. In the urinary tract, plasminogen is predominantly converted to biologically active plasmin by urokinase-type plasminogen activator (uPA) [36]. Indeed, in the urine of NS patients, plasmin and plasminogen levels were increased [35, 37]. In a large cohort of NS patients, the urinary plasminogen–plasmin:creatinine ratio was an independent predictor of oedema formation, and a decrease after treatment was associated with oedema remission [38]. Furthermore, urine of patients with NS induced an increase in sodium current in vitro, which could be inhibited by amiloride and anti-plasmin [35]. In experimental NS, the serine protease inhibitor aprotinin prevented proteolytic activation of ENaCs [39]. This notion is further supported by findings in several patient case reports wherein inhibition of ENaCs has been shown to resolve resistant oedema in NS [40–42]. However, there are some indications that uPA-produced plasmin is not the sole contributor to NS-associated sodium retention [33]. Bohnert et al. [43] demonstrated that uPA does promote plasminogen–plasmin conversion but that it is not essential for oedema formation in experimental NS. This is in line with findings in other studies that demonstrate an inhibitory effect on sodium retention in experimental NS by uPA inhibition [44]. In conclusion, although the specific mechanisms are not completely unravelled, there seems to be a role for proteolytic activation of ENaCs in NS.
ALBUMIN AND LYSOPHOSPHATIDYLCHOLINE (LPC)
LPC is a phospholipid that is present as a minor component of the cell membrane and is present in circulating plasma. In contrast, phosphatidylcholines (PCs) are the major phospholipid component of cell membranes as well as lipoproteins. LPC is produced by the hydrolysis of PCs by phospholipase A2 (PLA2), removing one of the fatty acid groups on the sn-2 position (Figure 2) or as a product by the lecithin cholesterol acyltransferase reaction (LCAT). In the circulation, LPC has a short half-life due to rapid degradation, which prevents impairment of a variety of vascular functions. Albumin is one of the most important transporters; normally 80% of LPC in the circulation is bound to albumin [45, 46]. A single molecule of albumin can bind up to five LPC molecules, suggesting multiple binding sites [47]. Albumin acts as an interface that binds LPC and transports it back to different tissues, but mainly the liver. In the liver, LPC can be formed back to PCs by lysophosphatidylcholine acyltransferase (LPCAT) in the presence of acyl-CoA. Subsequently, PCs are incorporated in the surface of very-low-density lipoproteins (VLDLs) and high-density lipoproteins (HDLs) and so released back into the circulation (the so-called Lands’ cycle).
FIGURE 2:
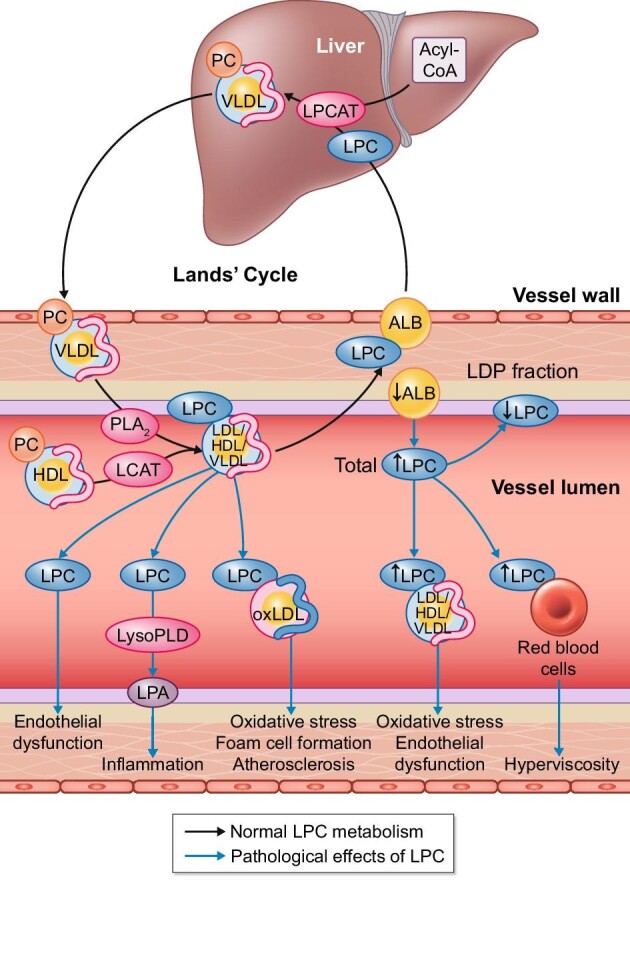
Normal and pathophysiological LPC metabolism and the role of albumin.
The Lands’ cycle is responsible for rapid alterations in phosplipid composition of lipoproteins. These phospholipids are mainly PC. Both PLA2 and LCAT are capable of hydrolysis of PC to LPC in the circulation. LPC is mostly bound to albumin (ALB), rendering its detrimental effects ineffective. Albumin-bound LPC is transported to the liver and LPCAT catalyses the conversion back to PC. The accumulation of non-albumin-bound LPC has multiple detrimental effects on vascular function.
LDP, lipoprotein-deficient plasma; LysoPLD, lysophospholipase D; LPA, lysophosphatidic acid; oxLDL, oxidized low-density lipoprotein.
Hypoalbuminaemia can therefore limit the LPC return capacity to the liver, which can be detrimental to vascular homeostasis. Indeed, the total concentration of LPC in the blood increases in hypoalbuminaemia due to proteinuria [46]. However, LPC concentrations in lipoprotein-deficient plasma were decreased, showing that there was a shift of LPC binding to VLDL, intermediate-density lipoprotein and LDL, independent of the lipidaemic status of the patient [46]. Importantly, this phenomenon is observed not only in severe hypoalbuminaemia, but also in subnormal albumin (29 g/L) [46]. Furthermore, LPC has been shown to be increased about five times in oxidized LDL and about seven times in oxidized lipoprotein A [48]. This shift prolongs the half-life of LPC in the circulation and thereby impairs vascular function. For instance, it is well known that oxidized LDL plays a pivotal role in atherosclerosis development through multiple pathogenic mechanisms, and it is thought that this could be attributed in part to LPC [49]. Inhibition of endothelial cell migration is often observed in atherosclerosis in the presence of oxidized LDL [50] or LPC specifically [51, 52]. Increased oxidative stress is a known mediator of atherosclerosis and, in particular, oxidative stress due to LPC has been shown to induce endothelial cell apoptosis. Interestingly, adding albumin might reduce cytotoxicity in a dose-dependent manner in cultured Jurkat T cells treated with 20 µmol of LPC [47]. Furthermore, LPC is associated with endothelial dysfunction, which is both associated with atherosclerosis development and with the loss of endothelial-dependent vasodilation [48, 49, 53]. Loss of endothelial-dependent, but not endothelial-independent, vasodilation has been demonstrated in aortic rings incubated with LPC [54]. In Nagase analbuminaemic rats, renal LPC concentrations and renal blood flow were normal, but intrarenal infusion of LPC induced more pronounced vasoconstriction and doubled the LPC concentration, while it had no effect in control rats [55]. Notably, in isolated rat aortic rings [54] and in vivo in Nagase analbuminaemic rats [55], albumin restored LPC-induced vasoconstrictor effects. This demonstrates that LPC can induce vascular dysfunction by impairing multiple vascular cell types and that albumin is able to prevent or restore these impairments.
Besides the shift of LPC binding to lipoproteins in hypoalbuminaemia, experiments in Nagase analbuminaemic and nephrotic rats have shown that LPC increasingly binds to red blood cells in hypoalbuminaemia [56]. Even though hypoalbuminaemia reduces plasma viscosity, binding of LPC to red blood cells resulted in reduced red blood cell deformability and consequently in increased whole blood viscosity [56]. Moreover, this phenomenon was still present when red blood cells were suspended in serum, excluding fibrinogen as the culprit. Albumin supplementation or suspending red cells from analbuminaemic rats in normal rat plasma reduced binding of LPC from red blood cell membranes, increased LPC plasma levels and restored blood viscosity to normal [56]. Furthermore, LPC might also bind to various classes of leucocytes and thereby modulate inflammatory responses. However, whether LPC induces a pro- or anti-inflammatory response seems to be cell specific. For example, exogenous LPC promotes the expression of adhesion molecules in endothelial cells, induces the release of TNF-α and interleukins 1β and 6 from adipocytes, activates macrophages and B cells and increases interferon-γ release from peripheral leucocytes [45]. In contrast, the effects of neutrophil activation [57] as well as eosinophil activation and migration are inhibited by LPC, suggesting an anti-inflammatory effect of LPC in specific conditions such as allergic reactions [58].
ALBUMIN AND NITRIC OXIDE SIGNALLING
We previously described the detrimental effect of hypoalbuminaemia and subsequent increased circulating LPC on endothelium-dependent vasodilation. However, it has been suggested that albumin also plays a role in regulating endothelium-dependent vasodilation directly. Nitric oxide (NO) is considered one of the most important endothelium-dependent vasodilator mechanisms in most vascular beds also has multiple paracrine effects [59]. NO can be produced from the substrate L-arginine by three different isoforms of the NO synthase protein [59]. The isoform, endothelial nitric oxide synthase (eNOS), is considered the most important isoform contributing to vasomotor control. Endothelium-derived NO diffuses to vascular smooth muscle cells (VSMCs) surrounding the endothelial cells and stimulates soluble guanylate cyclase (sGC), catalysing the conversion of guanosine triphosphate to cyclic guanosine monophosphate (cGMP). Subsequent activation of cGMP-dependent protein kinases results in a reduction in intracellular Ca2+, hyperpolarization and thus vasodilation [59]. NO is a highly reactive molecule and previous research has shown that NO plays an important role in redox reactions [59–61]. The main molecules that can react with NO are metals, thiols, oxygen (O2) and superoxide (O2–) [62], particularly the reaction with O2– resulting in peroxynitrite (ONOO–) and O2 resulting in NO2 (nitrogen dioxide), both considered powerful reactive nitrogen species [62]. Storage of gaseous NO is tricky due to its reactive nature, therefore it can be stored in different forms. Storage of NO as the metabolites NO2– (nitrite) and NO3– (nitrate) in the circulation is most common [63]. Additionally, red blood cells are known to be able to store, as an S-nitroso (SNO) adduct or as iron nitrosyl to haemoglobin, and release NO in the case of hypoxia or increased metabolic demand [63, 64]. A similar phenomenon is observed in albumin, as the single free cysteine, Cys-34, is particularly capable of binding NO as an SNO adduct [60, 61, 64]. This makes NO more stable and makes albumin-SNO a circulating reservoir for NO [61, 65]. Indeed, the concentration of unbound NO in the circulation is about 3 nM, while 7 µM of S-nitrosothiols are present, of which 96% are protein-bound, of which 82% is bound to albumin [61].
Besides the evidenced capacity of albumin to bind NO and form albumin-SNO, it has been shown that albumin-SNO has a physiological role in vascular homeostasis and can influence vasomotor tone. In rabbits, inhibition of eNOS resulted in an increase in mean arterial pressure associated with an inverse decrease in albumin-SNO [61]. In mongrel dogs, an intravenous injection of SNO–bovine serum albumin (BSA) markedly reduced mean arterial pressure, produced epicardial vasodilation, increased coronary blood flow and inhibited platelet adhesion in a dose-dependent manner [66]. Although it was less potent than nitroglycerin and S-nitroso-cysteine, it has a longer duration, reflecting an increased half-life [66]. Another study nicely demonstrated that this effect is endothelium independent, as endothelium-denuded aortic arteries showed dose-dependent vasodilation to SNO-BSA [60].
With these findings on the physiological function of albumin-SNO, the question arises if changes in albumin can also alter NO production or signalling. In end-stage renal disease (ESRD) patients on haemodialysis, lower plasma albumin was observed and was associated with impaired vascular function [67]. From clinical studies performed in NS patients, it is known that peripheral vascular function is impaired [68–70]. This is mainly due to the blunting of endothelium-dependent vasodilation or a loss of NO-mediated vasodilation specifically, while endothelium-independent vasodilation is preserved [68–70]. It is unclear if this is mainly the effect of NS-associated dyslipidaemia or hypoalbuminaemia [68, 69]. In Nagase analbuminaemic rats, infusion with an NO donor produced an similar reduction in mean arterial pressure compared with healthy rats. However, the recovery time of the Nagase analbuminaemic rats was significantly reduced, suggesting the lack of ‘long-lived’ NO-producing prolonged vasodilation [65]. These alterations were associated with a 300% increase in plasma S-nitrosothiols 30 min after NO-donor administration in healthy rats, while no changes in Nagase analbuminaemic rats were observed [65]. Bevers et al. [71] demonstrated that NO metabolites were increased in the plasma of Nagase analbuminaemic rats. Interestingly, aortic eNOS protein content was similar between Nagase analbuminaemic and healthy rats. However, they went on to show that hypoalbuminaemia increased NO production (measured by two independent methods: 4,5-diaminofluorescein diacetate and electron paramagnetic resonance) as well as eNOS activity of cultured endothelial (bEnd.3) cells in vitro [71, 72]. Moreover, this corresponded with a concomitant increase in eNOS-produced NO metabolites (Griess colorimetry), representing ‘short-life’ NO [71]. This suggests that albumin binding of NO, presumably at the endothelial surface, can limit diffusion of NO to VSMCs. Hypoalbuminaemia results in an increased flux of NO to the vascular smooth muscle, presumably necessitating downregulation of guanylate cyclase [73].
Indeed, endothelium-independent vasodilation to exogenous NO-donor sodium nitroprusside was markedly blunted in aortic rings of Nagase analbuminaemic rats versus normal control rats (median effective dose increased by nearly two orders of magnitude). Thus even in isolation, the downregulation of endothelium-independent vasodilation was maintained. Endothelium-dependent vasodilation to acetylcholine was slightly, but significantly, stronger in Nagase analbuminaemic rats compared with healthy rats, but only at the high doses [71]. Overall, these demonstrate that hypoalbuminaemia results in a shift from ‘long-life’ NO to ‘short-life’ NO, resulting in an increase in NO production with subsequent NO-mediated desensitization as a negative feedback mechanism to maintain vascular homeostasis (Figure 3).
FIGURE 3:
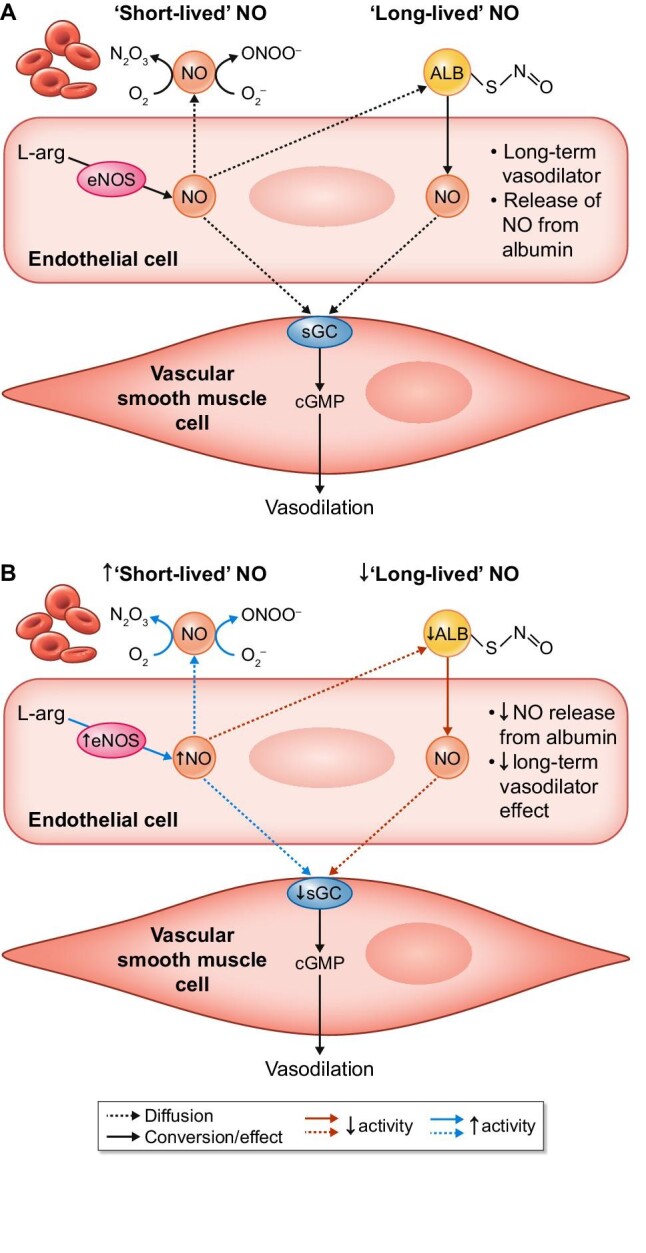
The role of albumin and hypoalbuminaemia in vascular NO signalling.
NO is a potent vasodilator produced by eNOS. It induces vasodilation by stimulating soluble guanylyl cyclase (sGC) that increases cGMP concentrations in VSMCs. NO in the circulation is short-lived, as it is highly reactive. Circulating albumin (ALB) can bind NO as an S-nitrosothiol (-S-N=O), thereby prolonging its half-life. This figure shows the role of NO and albumin in (A) normoalbuminaemia and (B) hypoalbuminaemia.
N2O3, dinitrogen trioxide; O2, oxygen; ONOO−, peroxynitrite; O2–, superoxide radical; L-arg, L-arginine.
ALBUMIN AND FFAS
The inhibitory effects on albumin of NO production and synthesis by endothelial cells described above were actually a serendipitous finding. The primary aim of that study was to expose bEnd.3 cells to the FFAs oleate and palmitate to test the hypothesis that fatty acid inhibits endothelial NO production. However, we could only do this by adding these fatty acids to an albumin solution. Thus we also compared fatty acid–free albumin in the medium to albumin-free medium and discovered a marked depression of pNOroduction even without fatty acids [71, 72]. Normally, FFAs are bound to albumin in a 0.7:1 FFA albumin molar ratio in the circulation, and this ratio can increase to up to 8:1 in diseases such as diabetes mellitus [74] and NS [75]. Other studies also showed the pro-oxidative effect of oleate albumin complexes on endothelial cells and demonstrated a reduction in eNOS function; however, they did not investigate the sole effect of albumin or oleate [76]. Besides the effect on NO production by endothelial cells, albumin-bound palmitate was capable of inducing apoptosis in cultured VSMCs due to increased oxidative stress compared with albumin alone [77]. Interestingly, albumin-bound oleate did not induce this increase in oxidative stress and apoptosis, and even attenuated palmitate-induced oxidative stress and apoptosis [77]. Additionally, FFAs have a lipotoxic effect on pancreatic β-cells, contributing to impaired insulin production in obese patients [78, 79]. Moreover, albumin polymorphisms reduced β-cell cytotoxicity by increasing affinity of palmitate and oleate binding [78].
For a long time it was unsure how proteinuria resulted in hypertriglyceridemia, as observed in NS [80]. Experimental studies conducted in rats demonstrated that both hypoalbuminaemia and proteinuria contribute separately to the development of hypertriglyceridemia in NS [81, 82]. It was postulated that hypertriglyceridemia in NS was not the result of increased production, but rather decreased clearance [80, 82, 83]. An elaborate study from Clement et al [84]. went on to demonstrate that circulating angiopoietin-like 4 (ANGPTL4) plays a pivotal role in the development of hypertriglyceridemia, proteinuria, low circulating albumin and renal and systemic feedback mechanisms (Figure 4). Briefly, urinary loss of albumin results in hypoalbuminaemia and increased FFA content bound to albumin. Subsequently, the increased FFA:albumin ratio in plasma and tissue stimulates upregulation of ANGPTL4 in skeletal muscle, heart and adipose tissue [84, 85]. NGPTL4 inhibits endothelial-bound lipoprotein lipase activity, which inhibits FFA generation from triglycerides and thus induces hypertriglyceridemia. This comprises the local feedback loop, as reduced FFA results in a decrease in the FFA:albumin ratio [84, 85]. The systemic feedback loop reduces proteinuria by binding of ANGPTL4 to αvβ5 integrins that are present at the basement membrane of glomerular endothelial cells. These integrins bind to different extracellular matrix proteins, e.g. vitronectin, and regulate vascular barrier integrity [86], subsequently lowering the leakage of protein into the urine [84, 85]. Albumin that is filtered by the glomeruli is reabsorbed by the proximal tubule epithelial cells, as discussed above [87–89]. Normally albumin is taken up in the proximal tubules in the kidney by receptor-mediated endocytosis, preventing urinary loss of albumin [87–89]. It should be noted, however, that this hypothesis might not yet be fully understood, as some studies show ANGPTL4 to be unaltered in NS and proteinuria [90]. It has been shown that excess albumin in the epithelial tubular cells activates protein kinase C, which subsequently increases NAD(P)H oxidase activity and thus reactive oxygen species production [91]. However, it has been suggested that this is not solely the effect of albumin, but also the effect of albumin-bound FFAs [92, 93]. Indeed, albumin-bound FFAs are capable of inducing higher levels of oxidative stress in proximal tubules than free albumin, by mitochondrial production of superoxide and downregulation of antioxidants [92, 94]. In a similar study, albumin had a proliferative effect on proximal tubular cells, while albumin–linoleate complexes were tubulotoxic [93]. Furthermore, oleate-bound albumin exerted a fibrogenic effect through protein kinase C and fibronectin signalling [93]. This study demonstrates that albumin-bound FFAs are capable of influencing and aggravating the detrimental effects of albumin on proximal tubular cells.
FIGURE 4:

The role of albumin (ALB) in free FFA metabolism in vascular and tubular homeostasis.
ALB is capable of binding FFAs and hypoalbuminaemia increases the FFA:ALB ratio. Increased circulating FFAs bound to ALB uncouples endothelial mitochondria that produce superoxide radicals (O2–) and in turn uncouple eNOS, further inducing vascular dysfunction. Peripheral tissues increase their uptake of FFAs and subsequently release ANGPTL4, which in a local feedback loop reduces production of FFAs from triglycerides (TG) by lipoprotein lipase (LPL). In a systemic feedback, loop proteinuria is reduced by ANGPTL4 binding to αvβ5 integrins at the surface of the glomerular endothelial cell. ALB can induce oxidative stress and a fibrogenic response in proximal tubule epithelial cells, which seems to be aggravated when FFAs are bound to ALB, resulting in tubulotoxicity.
BH4, tetrahydrobiopterin; Rac1, Ras-related C3 botulinum toxin substrate 1; PKC, protein kinase C; NOX, NADPH-oxidase; SOD, superoxide dismutase.
ALBUMIN IN THE CLINICAL SETTING
Reduced albumin plasma levels are a strong predictor of morbidity and mortality regardless of the implicated disease [7]. Specifically, the association of hypoalbuminaemia and worse outcome in ESRD has long been established and is mostly due to cardiovascular complications [95, 96]. Interestingly, this phenomenon is independently associated to both protein malnutrition and hypoalbuminaemia, but only hypoalbuminaemia is associated to the prevalence of vascular disease in patients on dialysis [97]. In this study, a 1 g/L decrease in albumin was associated with a 10% increase in mortality. This adds to the hypothesis that hypoalbuminaemia is not merely due to malnutrition in patients with ESRD. Other mechanisms linking hypoalbuminaemia and worse outcome in ESRD, such as inflammation, have been suggested and investigated over the years. Indeed C-reactive protein has been shown to be a strong predictor of death in haemodialysis patients and the predictive value of albumin was dependent on C-reactive protein [98]. Inflammation reduces the synthesis of albumin by inhibiting its transcription. This is mediated by interleukin-6/1β and tissue necrosis factor-α [7, 96]. The hypothesis of hypoalbuminaemia as an acute phase protein that is reduced in response to inflammation is strengthened by the fact that hypoalbuminaemia is also commonly present in severely ill patients such as those in the intensive care unit. In a meta-analysis, hypoalbuminaemia has proven to be a strong and dose-dependent predictor of poor outcome in such patients [99].
Hypoalbuminaemia in various diseases might be an epiphenomenon and therefore albumin might be a suitable marker but not a therapeutic target. Indeed, multiple studies have aimed to investigate the supplementation of albumin in the clinical setting. Theoretically, albumin 4.5% is about four times as effective in volume expansion as crystalloids [100]. However, in hypovolaemia and hypoalbuminaemia, human albumin is not superior in reducing mortality as compared with the much cheaper alternatives such as other colloid and crystalloid solutions [101]. It has been hypothesized that in a highly selected subgroup of patients, human albumin supplementation might be indicated. For example, a subgroup analysis of the Saline versus Albumin Fluid Evaluation trial [102] and a meta-analysis [103] demonstrated that albumin infusion tended to improve outcome in sepsis and septic shock patients. In contrast, a more recent meta-analysis [104] extracting new information from a large randomized controlled trial [105] showed no benefit of adding albumin supplementation of crystalloids in severe sepsis. Therefore albumin supplementation is recommended only in hypovolaemia as a second choice when treatment with crystalloids does not suffice [106]. This is also in line with the recommendation of the European Rare Kidney Disease Reference Network–European Society for Pediatric Nephrology Working Group to supplement albumin in infants with congenital NS, mainly to increase intravascular volume and not serum albumin concentration per se [107]. Intradialytic hypotension can result in organ hypoperfusion and therefore must be treated. In those patients, albumin infusion for volume expansion has been investigated in small trials and showed a beneficial haemodynamic effect with hyperoncotic but not iso-oncotic albumin solutions [108]. In patients on chronic haemodialysis treatment, hypoalbuminaemia is common partly due to loss of albumin by current dialysis techniques [109]. Whereas smaller membrane pores prevent albumin loss, they also limit the removal of middle molecules to the dialysate [109]. Medium cut-off membranes were developed to increase middle molecule clearance, but might cause more albumin loss compared with high-flux membranes [109, 110]. Larger trials comparing these two dialysis modalities are needed to further evaluate this. The therapeutic value of albumin supplementation in NS patients overall remains a knowledge gap, mainly due to the lack of well-performed randomized controlled trials [111], but small trials do not show any benefit [112].
CONCLUSION
This review provides an overview of the role of albumin in physiological and pathophysiological conditions associated with hypoalbuminaemia. Indeed, as we have presented above, albumin plays a pivotal role in binding of multiple small and large molecules in the circulation. Although hypoalbuminaemia is most pronounced in NS, it can also be the consequence of inflammation, underfeeding (catabolism) and hepatic diseases. Moreover, a reduction of albumin can aggravate multiple common cardiovascular diseases and therefore should be considered and evaluated in patients at risk for developing cardiovascular disease. Additionally, it has proven to be an excellent prognostic marker in severe illness [113, 114]. However, increasing albumin levels by albumin infusion has not been proven beneficial in all critically ill intensive care patients [113, 115] or in hypotensive patients on kidney replacement therapy [115] but possibly has a beneficial effect in limited subgroups [113]. Mechanistic studies further investigating the interaction between hypoalbuminaemia and inflammation might help us understand the lack of improved prognosis [2]. The beneficial effect of albumin infusion for the treatment of oedema in NS patients remains controversial [112], warranting better studies [111]. The detrimental effects of hypoalbuminaemia can be easily overlooked in both (preclinical) research and clinical practice. Therefore we feel that albumin as a marker and effector of multiple pro-inflammatory and pro-oxidative processes should be included in the analysis of studies performed within the field of nephrology, cardiovascular and inflammatory disease. We can conclude that albumin is definitely not a mere sponge but functions as an important interface between tissues and circulating factors in order to maintain homeostasis in multiple systems and tissues.
Contributor Information
Jens van de Wouw, Division of Experimental Cardiology, Department of Cardiology, Erasmus MC, University Medical Center, Rotterdam, The Netherlands.
Jaap A Joles, Department of Nephrology and Hypertension, University Medical Center, Utrecht, the Netherlands.
FUNDING
This work was supported by a grant from the Netherlands CardioVascular Research Initiative, an initiative with support of the Dutch Heart Foundation [CVON2014-11 (RECONNECT)] and from the Dutch CardioVascular Alliance, an initiative with support of the Dutch Heart Foundation (Grant 2020B008; RECONNEXT).
CONFLICT OF INTEREST STATEMENT
The authors declare that there are no conflicts of interests regarding the publication of this article.
REFERENCES
- 1. Levitt DG, Levitt MD. Human serum albumin homeostasis: a new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int J Gen Med 2016; 9: 229–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaysen GA. Biochemistry and biomarkers of inflamed patients: why look, what to assess. Clin J Am Soc Nephrol 2009; 4(Suppl 1): S56–S63 [DOI] [PubMed] [Google Scholar]
- 3. Hawkins JW, Dugaiczyk A. The human serum albumin gene: structure of a unique locus. Gene 1982; 19: 55–58 [DOI] [PubMed] [Google Scholar]
- 4. Peters T. All about Albumin: Biochemistry, Genetics, and Medical Applications. San Diego, CA: Academic Press, 2008 [Google Scholar]
- 5. Wang Z, Xing G, Zhang L. Glycated albumin level is significantly decreased in patients suffering nephrotic syndrome. Prog Mol Biol Transl Sci 2019; 162: 307–319 [DOI] [PubMed] [Google Scholar]
- 6. Varshney A, Sen P, Ahmad E et al. Ligand binding strategies of human serum albumin: how can the cargo be utilized? Chirality 2010; 22: 77–87 [DOI] [PubMed] [Google Scholar]
- 7. Gatta A, Verardo A, Bolognesi M. Hypoalbuminemia. Intern Emerg Med 2012; 7(Suppl 3): S193–S199 [DOI] [PubMed] [Google Scholar]
- 8. Garg P, Rabelink T. Glomerular proteinuria: a complex interplay between unique players. Adv Chronic Kidney Dis 2011; 18: 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaysen GA, Myers BD, Couser WG et al. Mechanisms and consequences of proteinuria. Lab Invest 1986; 54: 479–498 [PubMed] [Google Scholar]
- 10. Starling EH. On the absorption of fluids from the connective tissue spaces. J Physiol 1896; 19: 312–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gyenge CC, Tenstad O, Wiig H. In vivo determination of steric and electrostatic exclusion of albumin in rat skin and skeletal muscle. J Physiol 2003; 552: 907–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wiig H, DeCarlo M, Sibley L et al. Interstitial exclusion of albumin in rat tissues measured by a continuous infusion method. Am J Physiol 1992; 263: H1222–H1233 [DOI] [PubMed] [Google Scholar]
- 13. Joles JA, Rabelink TJ, Braam B et al. Plasma volume regulation: defences against edema formation (with special emphasis on hypoproteinemia). Am J Nephrol 1993; 13: 399–412 [DOI] [PubMed] [Google Scholar]
- 14. Broekhuizen LN, Lemkes BA, Mooij HL et al. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia 2010; 53: 2646–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Geers AB, Koomans HA, Roos JC et al. Preservation of blood volume during edema removal in nephrotic subjects. Kidney Int 1985; 28: 652–657 [DOI] [PubMed] [Google Scholar]
- 16. Koomans HA, Braam B, Geers AB et al. The importance of plasma protein for blood volume and blood pressure homeostasis. Kidney Int 1986; 30: 730–735 [DOI] [PubMed] [Google Scholar]
- 17. Vande Walle J, Donckerwolcke R, Boer P et al. Blood volume, colloid osmotic pressure and F-cell ratio in children with the nephrotic syndrome. Kidney Int 1996; 49: 1471–1477 [DOI] [PubMed] [Google Scholar]
- 18. Geers AB, Koomans HA, Boer P et al. Plasma and blood volumes in patients with the nephrotic syndrome. Nephron 1984; 38: 170–173 [DOI] [PubMed] [Google Scholar]
- 19. Vande Walle JG, Donckerwolcke RA, Koomans HA. Pathophysiology of edema formation in children with nephrotic syndrome not due to minimal change disease. J Am Soc Nephrol 1999; 10: 323–331 [DOI] [PubMed] [Google Scholar]
- 20. Geers AB, Koomans HA, Roos JC et al. Functional relationships in the nephrotic syndrome. Kidney Int 1984; 26: 324–330 [DOI] [PubMed] [Google Scholar]
- 21. Siddall EC, Radhakrishnan. The pathophysiology of edema formation in the nephrotic syndrome. Kidney Int 2012; 82: 635–642 [DOI] [PubMed] [Google Scholar]
- 22. Vande Walle JG, Donckerwolcke RAMG, van Isselt JW et al. Volume regulation in children with early relapse of minimal-change nephrosis with or without hypovolaemic symptoms. Lancet 1995; 346: 148–152 [DOI] [PubMed] [Google Scholar]
- 23. Brown EA, Markandu N, Sagnella GA et al. Sodium retention in nephrotic syndrome is due to an intrarenal defect: evidence from steroid-induced remission. Nephron 1985; 39: 290–295 [DOI] [PubMed] [Google Scholar]
- 24. Joles JA, Koomans HA, Kortlandt W et al. Hypoproteinemia and recovery from edema in dogs. Am J Physiol 1988; 254: F887–F894 [DOI] [PubMed] [Google Scholar]
- 25. Koomans HA, Kortlandt W, Geers AB et al. Lowered protein content of tissue fluid in patients with the nephrotic syndrome: observations during disease and recovery. Nephron 1985; 40: 391–395 [DOI] [PubMed] [Google Scholar]
- 26. Joles JA, Willekes-Koolschijn N, Braam B et al. Colloid osmotic pressure in young analbuminemic rats. Am J Physiol 1989; 257: F23–F28 [DOI] [PubMed] [Google Scholar]
- 27. Fiorotto M, Coward WA. Pathogenesis of oedema in protein-energy malnutrition: the significance of plasma colloid osmotic pressure. Br J Nutr 1979; 42: 21–31 [DOI] [PubMed] [Google Scholar]
- 28. Joles JA, Kortlandt W, de Mik H et al. Effect of hypoproteinemia on blood volume recovery after moderate hemorrhage in conscious splenectomized dogs. J Surg Res 1989; 47: 515–519 [DOI] [PubMed] [Google Scholar]
- 29. Manning RD, Guyton AC. Effects of hypoproteinemia on fluid volumes and arterial pressure. Am J Physiol 1983; 245: H284–H293 [DOI] [PubMed] [Google Scholar]
- 30. Manning RD Jr. Effects of hypoproteinemia on renal hemodynamics, arterial pressure, and fluid volume. Am J Physiol 1987; 252: F91–F98 [DOI] [PubMed] [Google Scholar]
- 31. Ichikawa I, Rennke HG, Hoyer JR et al. Role for intrarenal mechanisms in the impaired salt excretion of experimental nephrotic syndrome. J Clin Invest 1983; 71: 91–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Russi E, Weigand K. Analbuminemia. Klin Wochenschr 1983; 61: 541–545 [DOI] [PubMed] [Google Scholar]
- 33. Hinrichs GR, Jensen BL, Svenningsen P. Mechanisms of sodium retention in nephrotic syndrome. Curr Opin Nephrol Hypertens 2020; 29: 207–212 [DOI] [PubMed] [Google Scholar]
- 34. Passero CJ, Mueller GM, Rondon-Berrios H et al. Plasmin activates epithelial Na+ channels by cleaving the gamma subunit. J Biol Chem 2008; 283: 36586–36591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Svenningsen P, Bistrup C, Friis UG et al. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol 2009; 20: 299–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Svenningsen P, Hinrichs GR, Zachar R et al. Physiology and pathophysiology of the plasminogen system in the kidney. Pflugers Arch 2017; 469: 1415–1423 [DOI] [PubMed] [Google Scholar]
- 37. Vaziri ND, Gonzales EC, Shayestehfar B et al. Plasma levels and urinary excretion of fibrinolytic and protease inhibitory proteins in nephrotic syndrome. J Lab Clin Med 1994; 124: 118–124 [PubMed] [Google Scholar]
- 38. Chen JL, Wang L, Yao XM et al. Association of urinary plasminogen-plasmin with edema and epithelial sodium channel activation in patients with nephrotic syndrome. Am J Nephrol 2019; 50: 92–104 [DOI] [PubMed] [Google Scholar]
- 39. Bohnert BN, Menacher M, Janessa A et al. Aprotinin prevents proteolytic epithelial sodium channel (ENaC) activation and volume retention in nephrotic syndrome. Kidney Int 2018; 93: 159–172 [DOI] [PubMed] [Google Scholar]
- 40. Hinrichs GR, Mortensen LA, Jensen BL et al. Amiloride resolves resistant edema and hypertension in a patient with nephrotic syndrome; a case report. Physiol Rep 2018; 6: e13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hoorn EJ, Ellison DH. Diuretic resistance. Am J Kidney Dis 2017; 69: 136–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yamaguchi E, Yoshikawa K, Nakaya I et al. Liddle's-like syndrome associated with nephrotic syndrome secondary to membranous nephropathy: the first case report. BMC Nephrol 2018; 19: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bohnert BN, Daiminger S, Worn M et al. Urokinase-type plasminogen activator (uPA) is not essential for epithelial sodium channel (ENaC)-mediated sodium retention in experimental nephrotic syndrome. Acta Physiol (Oxf) 2019; 227: e13286. [DOI] [PubMed] [Google Scholar]
- 44. Hinrichs GR, Weyer K, Friis UG et al. Urokinase-type plasminogen activator contributes to amiloride-sensitive sodium retention in nephrotic range glomerular proteinuria in mice. Acta Physiol (Oxf) 2019; 227: e13362. [DOI] [PubMed] [Google Scholar]
- 45. Law SH, Chan ML, Marathe GK et al. An updated review of lysophosphatidylcholine metabolism in human diseases. Int J Mol Sci 2019; 20: 1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vuong TD, Stroes ES, Willekes-Koolschijn N et al. Hypoalbuminemia increases lysophosphatidylcholine in low-density lipoprotein of normocholesterolemic subjects. Kidney Int 1999; 55: 1005–1010 [DOI] [PubMed] [Google Scholar]
- 47. Kim YL, Im YJ, Ha NC et al. Albumin inhibits cytotoxic activity of lysophosphatidylcholine by direct binding. Prostaglandins Other Lipid Mediat 2007; 83: 130–138 [DOI] [PubMed] [Google Scholar]
- 48. Heermeier K, Schneider R, Heinloth A et al. Oxidative stress mediates apoptosis induced by oxidized low-density lipoprotein and oxidized lipoprotein(a). Kidney Int 1999; 56: 1310–1312 [DOI] [PubMed] [Google Scholar]
- 49. Rader DJ, Dugi KA. The endothelium and lipoproteins: insights from recent cell biology and animal studies. Semin Thromb Hemost 2000; 26: 521–528 [DOI] [PubMed] [Google Scholar]
- 50. Murugesan G, Chisolm GM, Fox PL. Oxidized low density lipoprotein inhibits the migration of aortic endothelial cells in vitro. J Cell Biol 1993; 120: 1011–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murugesan G, Fox PL. Role of lysophosphatidylcholine in the inhibition of endothelial cell motility by oxidized low density lipoprotein. J Clin Invest 1996; 97: 2736–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chaudhuri P, Colles SM, Damron DS et al. Lysophosphatidylcholine inhibits endothelial cell migration by increasing intracellular calcium and activating calpain. Arterioscler Thromb Vasc Biol 2003; 23: 218–223 [DOI] [PubMed] [Google Scholar]
- 53. Joles JA, Stroes ES, Rabelink TJ. Endothelial function in proteinuric renal disease. Kidney Int Suppl 1999; 71: S57–S61 [DOI] [PubMed] [Google Scholar]
- 54. Vuong TD, de Kimpe S, de Roos R et al. Albumin restores lysophosphatidylcholine-induced inhibition of vasodilation in rat aorta. Kidney Int 2001; 60: 1088–1096 [DOI] [PubMed] [Google Scholar]
- 55. Vuong TD, Braam B, Willekes-Koolschijn N et al. Hypoalbuminaemia enhances the renal vasoconstrictor effect of lysophosphatidylcholine. Nephrol Dial Transplant 2003; 18: 1485–1492 [DOI] [PubMed] [Google Scholar]
- 56. Joles JA, Willekes-Koolschijn N, Koomans HA. Hypoalbuminemia causes high blood viscosity by increasing red cell lysophosphatidylcholine. Kidney Int 1997; 52: 761–770 [DOI] [PubMed] [Google Scholar]
- 57. Curcic S, Holzer M, Frei R et al. Neutrophil effector responses are suppressed by secretory phospholipase A2 modified HDL. Biochim Biophys Acta 2015; 1851: 184–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Knuplez E, Curcic S, Theiler A et al. Lysophosphatidylcholines inhibit human eosinophil activation and suppress eosinophil migration in vivo. Biochim Biophys Acta Mol Cell Biol Lipids 2020; 1865: 158686. [DOI] [PubMed] [Google Scholar]
- 59. Liaudet L, Soriano FG, Szabó C. Biology of nitric oxide signaling. Crit Care Med 2000; 28; N37–N52. [DOI] [PubMed] [Google Scholar]
- 60. Stamler JS, Simon DI, Osborne JA et al. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA 1992; 89: 444–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stamler JS, Jaraki O, Osborne J et al. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci UA 1992; 89: 7674–7677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ali AA, Coulter JA, Ogle CH et al. The contribution of N2O3 to the cytotoxicity of the nitric oxide donor DETA/NO: an emerging role for S-nitrosylation. Biosci Rep 2013; 33: e00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lundberg JO, Weitzberg E, Gladwin MT. The nitrate–nitrite–nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov 2008; 7: 156–167 [DOI] [PubMed] [Google Scholar]
- 64. Stamler JS. S-nitrosothiols in the blood: roles, amounts, and methods of analysis. Circ Res 2004; 94: 414–417 [DOI] [PubMed] [Google Scholar]
- 65. Minamiyama Y, Takemura S, Inoue M. Albumin is an important vascular tonus regulator as a reservoir of nitric oxide. Biochem Biophys Res Commun 1996; 225: 112–115 [DOI] [PubMed] [Google Scholar]
- 66. Keaney JF, Simon DI, Stamler JS et al. NO forms an adduct with serum albumin that has endothelium-derived relaxing factor-like properties. J Clin Invest 1993; 91: 1582–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pannier B, Guerin AP, Marchais SJ et al. Postischemic vasodilation, endothelial activation, and cardiovascular remodeling in end-stage renal disease. Kidney Int 2000; 57: 1091–1099 [DOI] [PubMed] [Google Scholar]
- 68. Dogra GK, Watts GF, Herrmann S et al. Statin therapy improves brachial artery endothelial function in nephrotic syndrome. Kidney Int 2002; 62: 550–557 [DOI] [PubMed] [Google Scholar]
- 69. Watts GF, Herrmann S, Dogra GK et al. Vascular function of the peripheral circulation in patients with nephrosis. Kidney Int 2001; 60: 182–189 [DOI] [PubMed] [Google Scholar]
- 70. Stroes ES, Joles JA, Chang PC et al. Impaired endothelial function in patients with nephrotic range proteinuria. Kidney Int 1995; 48: 544–550 [DOI] [PubMed] [Google Scholar]
- 71. Bevers LM, van Faassen EE, Vuong TD et al. Low albumin levels increase endothelial NO production and decrease vascular NO sensitivity. Nephrol Dial Transplant 2006; 21: 3443–3449 [DOI] [PubMed] [Google Scholar]
- 72. Gremmels H, Bevers LM, Fledderus JO et al. Oleic acid increases mitochondrial reactive oxygen species production and decreases endothelial nitric oxide synthase activity in cultured endothelial cells. Eur J Pharmacol 2015; 751: 67–72 [DOI] [PubMed] [Google Scholar]
- 73. Vaziri ND, Wang XQ. cGMP-mediated negative-feedback regulation of endothelial nitric oxide synthase expression by nitric oxide. Hypertension 1999; 34: 1237–1241 [DOI] [PubMed] [Google Scholar]
- 74. Cistola DP, Small DM. Fatty acid distribution in systems modeling the normal and diabetic human circulation. A 13C nuclear magnetic resonance study. J Clin Invest 1991; 87: 1431–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Thomas ME, Schreiner GF. Contribution of proteinuria to progressive renal injury: consequences of tubular uptake of fatty acid bearing albumin. Am J Nephrol 1993; 13: 385–398 [DOI] [PubMed] [Google Scholar]
- 76. Esenabhalu VE, Schaeffer G, Graier WF. Free fatty acid overload attenuates Ca2+ signaling and NO production in endothelial cells. Antioxid Redox Signal 2003; 5: 147–153 [DOI] [PubMed] [Google Scholar]
- 77. Mattern HM, Hardin CD. Vascular metabolic dysfunction and lipotoxicity. Physiol Res 2007; 56: 149–158 [DOI] [PubMed] [Google Scholar]
- 78. Tuei VC, Ha JS, Ha CE. Effects of human serum albumin complexed with free fatty acids on cell viability and insulin secretion in the hamster pancreatic beta-cell line HIT-T15. Life Sci 2011; 88: 810–818 [DOI] [PubMed] [Google Scholar]
- 79. Koshkin V, Wang X, Scherer PE et al. Mitochondrial functional state in clonal pancreatic beta-cells exposed to free fatty acids. J Biol Chem 2003; 278: 19709–19715 [DOI] [PubMed] [Google Scholar]
- 80. Kaysen GA, Gambertoglio J, Felts J et al. Albumin synthesis, albuminuria and hyperlipemia in nephrotic patients. Kidney Int 1987; 31: 1368–1376 [DOI] [PubMed] [Google Scholar]
- 81. Shearer GC, Stevenson FT, Atkinson DN et al. Hypoalbuminemia and proteinuria contribute separately to reduced lipoprotein catabolism in the nephrotic syndrome. Kidney Int 2001; 59: 179–189 [DOI] [PubMed] [Google Scholar]
- 82. Davies RW, Staprans I, Hutchison FN et al. Proteinuria, not altered albumin metabolism, affects hyperlipidemia in the nephrotic rat. J Clin Invest 1990; 86: 600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vaziri ND. Disorders of lipid metabolism in nephrotic syndrome: mechanisms and consequences. Kidney Int 2016; 90: 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Clement LC, Mace C, Avila-Casado C et al. Circulating angiopoietin-like 4 links proteinuria with hypertriglyceridemia in nephrotic syndrome. Nat Med 2014; 20: 37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Clement LC, Mace C, Del Nogal Avila M et al. The proteinuria-hypertriglyceridemia connection as a basis for novel therapeutics for nephrotic syndrome. Transl Res 2015; 165: 499–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. McCurley A, Alimperti S, Campos-Bilderback SB et al. Inhibition of αvβ5 integrin attenuates vascular permeability and protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 2017; 28: 1741–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Brunskill NJ. Molecular interactions between albumin and proximal tubular cells. Exp Nephrol 1998; 6: 491–495 [DOI] [PubMed] [Google Scholar]
- 88. Whaley-Connell AT, Morris EM, Rehmer N et al. Albumin activation of NAD(P)H oxidase activity is mediated via Rac1 in proximal tubule cells. Am J Nephrol 2007; 27: 15–23 [DOI] [PubMed] [Google Scholar]
- 89. Brunskill NJ. Mechanisms of albumin uptake by proximal tubular cells. Am J Kidney Dis 2001; 37: S17–S20 [DOI] [PubMed] [Google Scholar]
- 90. Cara-Fuentes G, Segarra A, Silva-Sanchez C et al. Angiopoietin-like-4 and minimal change disease. PLoS One 2017; 12: e0176198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Imai E, Nakajima H, Kaimori JY. Albumin turns on a vicious spiral of oxidative stress in renal proximal tubules. Kidney Int 2004; 66: 2085–2087 [DOI] [PubMed] [Google Scholar]
- 92. Ishola DA, Post JA, van Timmeren MM et al. Albumin-bound fatty acids induce mitochondrial oxidant stress and impair antioxidant responses in proximal tubular cells. Kidney Int 2006; 70: 724–731 [DOI] [PubMed] [Google Scholar]
- 93. Arici M, Brown J, Williams M et al. Fatty acids carried on albumin modulate proximal tubular cell fibronectin production: a role for protein kinase C. Nephrol Dial Transplant 2002; 17: 1751–1757 [DOI] [PubMed] [Google Scholar]
- 94. Long KR, Rbaibi Y, Gliozzi ML et al. Differential kidney proximal tubule cell responses to protein overload by albumin and its ligands. Am J Physiol Renal Physiol 2020; 318: F851–F859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Foley RN, Parfrey PS, Harnett JD et al. Hypoalbuminemia, cardiac morbidity, and mortality in end-stage renal disease. J Am Soc Nephrol 1996; 7: 728–736 [DOI] [PubMed] [Google Scholar]
- 96. van Gelder MK, Abrahams AC, Joles JA et al. Albumin handling in different hemodialysis modalities. Nephrol Dial Transplant 2018; 33: 906–913 [DOI] [PubMed] [Google Scholar]
- 97. Cooper BA, Penne EL, Bartlett LH et al. Protein malnutrition and hypoalbuminemia as predictors of vascular events and mortality in ESRD. Am J Kidney Dis 2004; 43: 61–66 [DOI] [PubMed] [Google Scholar]
- 98. Yeun JY, Levine RA, Mantadilok V et al. C-reactive protein predicts all-cause and cardiovascular mortality in hemodialysis patients. Am J Kidney Dis 2000; 35: 469–476 [DOI] [PubMed] [Google Scholar]
- 99. Vincent JL, Dubois MJ, Navickis RJ et al. Hypoalbuminemia in acute illness: is there a rationale for intervention? A meta-analysis of cohort studies and controlled trials. Ann Surg 2003; 237: 319–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mendez CM, McClain CJ, Marsano LS. Albumin therapy in clinical practice. Nutr Clin Pract 2005; 20: 314–320 [DOI] [PubMed] [Google Scholar]
- 101. Albumin R. Human albumin solution for resuscitation and volume expansion in critically ill patients. Cochrane Database Syst Rev 2011; 11: CD001208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. SAFE Study Investigators, Finfer S, McEvoy S et al. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37: 86–96 [DOI] [PubMed] [Google Scholar]
- 103. Xu JY, Chen QH, Xie JF et al. Comparison of the effects of albumin and crystalloid on mortality in adult patients with severe sepsis and septic shock: a meta-analysis of randomized clinical trials. Crit Care 2014; 18: 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Patel A, Laffan MA, Waheed U et al. Randomised trials of human albumin for adults with sepsis: systematic review and meta-analysis with trial sequential analysis of all-cause mortality. BMJ 2014; 349: g4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Caironi P, Tognoni G, Masson S et al. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med 2014; 370: 1412–1421 [DOI] [PubMed] [Google Scholar]
- 106. Rozga J, Piatek T, Malkowski P. Human albumin: old, new, and emerging applications. Ann Transplant 2013; 18: 205–217 [DOI] [PubMed] [Google Scholar]
- 107. Boyer O, Schaefer F, Haffner D et al. Management of congenital nephrotic syndrome: consensus recommendations of the ERKNet-ESPN working group. Nat Rev Nephrol 2021; 17: 277–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hryciw N, Joannidis M, Hiremath S et al. Intravenous albumin for mitigating hypotension and augmenting ultrafiltration during kidney replacement therapy. Clin J Am Soc Nephrol 2021; 16: 820–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kalantar-Zadeh K, Ficociello LH, Bazzanella J et al. Slipping through the pores: hypoalbuminemia and albumin loss during hemodialysis. Int J Nephrol Renovasc Dis 2021; 14: 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ward RA, Beck W, Bernardo AA et al. Hypoalbuminemia: a price worth paying for improved dialytic removal of middle-molecular-weight uremic toxins? Nephrol Dial Transplant 2019; 34: 901–907 [DOI] [PubMed] [Google Scholar]
- 111. Ho JJ, Adnan AS, Kueh YC et al. Human albumin infusion for treating oedema in people with nephrotic syndrome. Cochrane Database Syst Rev 2019; 7: CD009692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Dorhout Mees EJ. Does it make sense to administer albumin to the patient with nephrotic oedema? Nephrol Dial Transplant 1996; 11: 1224–1226 [DOI] [PubMed] [Google Scholar]
- 113. Vincent JL, Russell JA, Jacob M et al. Albumin administration in the acutely ill: what is new and where next? Crit Care 2014; 18: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hryciw N, Joannidis M, Hiremath S et al. Intravenous albumin for mitigating hypotension and augmenting ultrafiltration during kidney replacement therapy. Clin J Am Soc Nephrol 2020; 16: 820–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Soeters PB, Wolfe RR, Shenkin A. Hypoalbuminemia: pathogenesis and clinical significance. JPEN J Parenter Enteral Nutr 2019; 43: 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]