

Abstract
Radiation is a common anticancer therapy for prostate cancer, which transforms tumor-associated normal fibroblasts to myofibroblasts, resulting in fibrosis. Oxidative stress caused by radiation-mediated mitochondrial damage is one of the major contributors to fibrosis. As diabetics are oxidatively stressed, radiation-mediated reactive oxygen species cause severe treatment failure, treatment-related side effects, and significantly reduced survival for diabetic prostate cancer patients as compared to non-diabetic prostate cancer patients. Hyperglycemia and enhanced mitochondrial damage significantly contribute to oxidative damage and disease progression after radiation therapy among diabetic prostate cancer patients. Therefore, reduction of mitochondrial damage in normal prostate fibroblasts after radiation should improve the overall clinical state of diabetic prostate cancer patients. We previously reported that MnTE-2-PyP, a manganese porphyrin, reduces oxidative damage in irradiated hyperglycemic prostate fibroblasts by scavenging superoxide and activating NRF2. In the current study, we have investigated the potential role of MnTE-2-PyP to protect mitochondrial health in irradiated hyperglycemic prostate fibroblasts. This study revealed that hyperglycemia and radiation increased mitochondrial ROS via blocking the mitochondrial electron transport chain, altered mitochondrial dynamics, and reduced mitochondrial biogenesis. Increased mitochondrial damage preceeded an increase in myofibroblast differentiation. MnTE-2-PyP reduced myofibroblast differentiation, improved mitochondrial health by releasing the block on the mitochondrial electron transport chain, enhanced ATP production efficiency, and restored mitochondrial dynamics and metabolism in the irradiated-hyperglycemic prostate fibroblasts. Therefore, we are proposing that one of the mechanisms that MnTE-2-PyP protects prostate fibroblasts from irradiation and hyperglycemia-mediated damage is by protecting the mitochondrial health in diabetic prostate cancer patients.
Keywords: Radiation, Diabetes, Manganese porphyrin, Mitochondria, ROS, Fibroblast metabolism
Graphical abstract
A. In non-stressed conditions, PGC1α and NRF2 transcribe mitochondrial protein to supply new mitochondria in the cell. Oxidative phosphorylation (OXPHOS) produces ATP and physiological levels of ROS through the electron transport chain. Normal metabolic function is carried out by non-stressed mitochondria and there is no fibrosis.B. In hyperglycemia and radiation exposure, PGC1α and NRF2 levels are reduced, resulting in less mitochondrial biogenesis. Mitochondrial OXPHOS complexes are also inhibited, resulting in mitochondria consuming increased amount of oxygen to meet ATP demands and producing increased amount of ROS. PGAM5 was not released into the cytosol, and NRF2 was not localized to outer membrane of mitochondria; therefore, mitophagy was reduced. This accumulation of damaged mitochondria and ROS in the cells results in pathologic metabolism, increased lipid oxidation, and fibrosis.C. In MnTE-2-PyP treated, irradiated and hyperglycemic cells, PGC1α and NRF2 levels are maintained, resulting in increased mitochondrial biogenesis. Mitochondrial OXPHOS complexes are increased, which reduces ROS levels. Mitochondria consume less oxygen and produce higher amount of ATP. PGAM5 is released into the cytosol, and NRF2 localizes to the mitochondrial outer membrane, resulting in mitophagy. The elimination of damaged mitochondria and reduction of ROS, results in normal metabolism, reduced lipid peroxidation, and inhibition of fibrosis.

Highlights
-
•
MnTE-2-PyP protects mitochondria from radiation and hyperglycemia-induced stress.
-
•
MnTE-2-PyP reduced mitochondrial ROS by restoring the levels of OXPHOS complexes.
-
•
MnTE-2-PyP increased the number of healthy mitochondria and enhanced ATP production efficiency.
-
•
Mitochondrial protection by MnTE-2-PyP inhibits myofibroblast differentiation.
-
•
MnTE-2-PyP treatment partly restores radiation-mediated metabolic changes.
1. Introduction
Radiation therapy (RT) is a common anticancer therapy for prostate cancer (PCa), but it also damages normal tissues surrounding the tumor. RT-mediated normal tissue damage not only causes severe side effects but also significantly contributes to enhanced radioresistant metastatic tumor growth and therapy resistance. More than 15% of all cancer patients are diabetic, which enhances the overall mortality rate and tumor recurrence by 42% and 21% respectively as compared to the non-diabetic cancer patients [1,2]. Specifically, diabetic prostate cancer (PCa) patients have increased therapy resistance, metastasis, and RT-mediated tissue damage, which results in 30% decreased survival in diabetic PCa patients as compared to non-diabetic PCa patients [3,4].
We and others have previously shown that radiation increases prostate tissue fibrosis [5,6], which is a significant cause for therapy resistance [7]. In the irradiated tumor microenvironment, fibroblasts are the major regulators of post-RT fibrosis. Radiation exposure in diabetic PCa patients increases reactive oxygen species (ROS) [[8], [9], [10]], which can result in mitochondrial damage. Damaged mitochondria enhance cellular oxidative stress, which starts a vicious cycle of ROS-mediated tissue damage after RT. Therefore, in a diabetic irradiated environment, protection against mitochondrial damage in prostate fibroblasts may protect normal tissue after RT, maintain healthy metabolism, and increase the survival of the diabetic PCa patients.
Radiation increases ROS-induced normal tissue damage via fibrosis, inflammation, and loss of antioxidant defenses and mitochondrial function. It is well reported that MnTE-2-PyP (T2E), a manganese porphyrin, scavenges ROS and acts as a potent radioprotector for normal cells during and after radiation. We have reported that T2E reduces normal tissue fibrosis, inflammation, and antioxidant damage via inhibition of NOX4-TGFβ-mediated pro-fibrotic signaling [11], inhibition of the NFκB-p50- mediated proinflammatory response and upregulation of the NRF2-mediated antioxidant response [12]. Previously, we have reported that T2E protects irradiated hyperglycemic prostate fibroblasts by increasing antioxidant defenses elicited by NRF2 [12], a master transcriptional regulator of antioxidant signaling. T2E increased total NRF2 levels and nuclear localization of NRF2 both in hyperglycemic and normoglycemic conditions after radiation [12]. Although DNA binding of NRF2 was significantly higher after T2E treatment in normoglycemia, it was not in hyperglycemic conditions. Rather, T2E increased AP1/NRF2-mediated secondary antioxidant response in hyperglycemic conditions by increasing DNA binding of AP1 [12]. T2E increased cytoprotective effects against radiation both in normoglycemic and hyperglycemic conditions. Therefore, we hypothesized that DNA binding of NRF2 was not the only mechanism for T2E-mediated cytoprotection in hyperglycemia. One of the major cytoprotective roles of NRF2, besides transcriptional regulation of cytoprotective genes, is maintaining mitochondrial health. NRF2 is well reported as a vital regulator for ROS mediated mitochondrial damage protection [13,14]. NRF2 localization to the outer mitochondrial membrane was previously reported [15,16]. PGC1α, an important regulatory molecule for mitochondrial biogenesis [17], also protects cells through NRF2 activation [18]. Therefore, to determine the potential of T2E as a mitochondrial protector, we investigated mitochondrial health, function, and cellular metabolism in irradiated hyperglycemic prostate fibroblasts in the presence or absence of T2E in our current study.
To mimic a diabetic irradiated environment, we have irradiated human prostate fibroblasts in a hyperglycemic environment in the presence or absence of T2E. This study revealed that in both radiated and non-radiated conditions, T2E increased mitochondrial NRF2 levels and total expression levels of PGC1α irrespective of glycemic levels. However, when NRF2 was knocked down, T2E was still able to protect mitochondrial health, which suggests that NRF2 is not directly involved in T2E-mediated mitochondrial protection. T2E increased mitochondrial ATP synthase levels, total mitochondrial number, and healthy mitochondrial number in the irradiated hyperglycemic cells. Mitochondrial efficiency, as measured by ATP production, was increased after T2E treatment. In addition, there was a large increase in mitochondrial ROS in irradiated and hyperglycemic fibroblasts, which was reduced significantly by T2E treatment. These results indicate that T2E protects from radiation and hyperglycemia-mediated mitochondrial damage. This study also revealed that mitochondrial damage preceeds the transformation of fibroblasts to myofibroblasts, which was prevented by T2E, in an irradiated, hyperglycemic environment. Thus, another way T2E inhibits fibrosis is by protecting mitochondrial health of fibroblasts. Finally, we have demonstrated the alterations of the metabolites, especially lipid changes, due to mitochondrial damage in irradiated and hyperglycemic fibroblasts. Specifically, radiation and hyperglycemia increased lipid oxidation, which was reduced to normal levels by T2E treatment. This is the first study to report that T2E restores healthy mitochondrial function and metabolic profiles in irradiated and hyperglycemic fibroblasts.
2. Materials and methods
2.1. Cell culture
P3158 are normal human prostate fibroblasts that were immortalized by pBABE-hygro-hTERT plasmid (Addgene, plasmid cat #1773) and kindly gifted by Dr. J Tyson McDonald. RPMI-1640 (Hyclone, cat #: SH30027.01) media, supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin were used to culture P3158 cells. Cells cultured in this media (containing 11.1 mM or 200 mg/dL glucose) are referred to as control or normo-glycemic condition in this study. To mimic hyperglycemia in diabetics, an extra 20 mM or 360 mg/dL glucose were added in the media, where the final glucose concentration was 560 (200 + 360) mg/dL. This condition is referred to as high glucose (HG) condition or hyperglycemia in this study. We have reported that the addition of 20 mM glucose was enough to induce hyperglycemia-associated oxidative stress without affecting osmotic pressure in P3158 cells [12]. In the presence or absence of 20 mM glucose (for HG) and/or 30 μM MnTE-2-PyP (referred as T2E), P3158 cells were plated. After 24 h, half of the cells were irradiated with 3 Gy of X-ray using the Rad Source RS-2000 irradiator (referred as RAD). Our previous study showed that 3 Gy of radiation on the human prostate fibroblasts (P3158) increased ROS and decreases NRF2 activity, which was recovered by MnTE-2-PyP in normoglycemic and hyperglycemic conditions [12]. Therefore, to investigate MnTE-2-PyP-mediated mitochondrial protection in irradiated and hyperglycemic prostate fibroblasts, we have selected 3 Gy of radiation. Cells were then incubated in 37 °C with 5% CO2. All the assays were executed on the fifth day after radiation exposure, if not otherwise mentioned. There were eight groups for each study: 1. Control (CON), 2. HG, 3. T2E, 4. T2E + HG (TG), 5. RAD, 6. RAD + HG (RG), 7. RAD + T2E (RT) and 8. RAD + HG + T2E (RGT), if not mentioned otherwise.
2.2. Western blot
Whole cell, cytosolic, and mitochondrial protein extracts were prepared by plating 2.0 × 106 cells/T75 flask. Cells were treated with HG, T2E and RAD as mentioned above. Five days post-radiation, whole cell extracts, cytosolic and mitochondrial extracts were prepared. Cell pellets were lysed by cell lysis buffer [120 mM NaCl, 50 mM Tris- HCl, 5 mM EDTA, 1% NP-40 and complete protease inhibitor cocktail tablets (Roche, cat # 11697498001; 1 tablet/50 ml)] to prepare the whole cell extract. Mitochondrial and cytosolic extracts were prepared by following the manufacturer's instructions for the Mitochondria Isolation Kit for Mammalian Cells (Thermo Scientific, reference # 89874). Total protein concentrations were measured by Bradford reagent (Bio Rad, cat # 500-0006). Samples were electrophoresed on Bolt 4–12% Bis-Tris Plus gels (Thermo Fisher Scientific, cat # NW04120BOX) and transferred to nitrocellulose membranes (Life Technologies, cat # IB23002). Nonfat milk (5%) in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.5% Tween 20) was used to block the membrane for 2 h before incubation of the membrane with primary antibodies overnight. Membranes were then washed with TBST and incubated with horseradish peroxidase (HRP) conjugated secondary antibodies (1:10,000 dilution) for 1–2 h at room temperature. Blots were then developed with an ECL detection system (Thermo Fisher Scientific) and exposed to film after washing with TBST. ImageJ was used to analyze densitometry of the scanned images from the films. Ponceau staining of total proteins were performed as a loading control for each blot. In some cases, TOMM20 was used as a mitochondrial protein loading control. We have used the following primary antibodies in this study: α-SMA (Abcam, cat # ab5694, 1:3000 dilution), NRF2 (Abcam, cat # ab62352, 1:1000 dilution), PGC1α (Abcam, cat # ab54481, 1:1000 dilution), TOMM20 (Abcam, cat # ab56783, 1:1000 dilution), OXPHOS complexes cocktail (Abcam, cat # ab110413, 1:1000 dilution), DRP1 (Cell Signaling Technology, cat # 8570, 1:1000 dilution), MFN1 (Cell Signaling Technology, cat # 14739, 1:1000 dilution), MFN2 (Cell Signaling Technology, cat # 11925, 1:1000 dilution), OPA1 (Cell Signaling Technology, cat # 80471, 1:1000 dilution), PGAM5 (Abcam, cat # ab244218, 1:1000 dilution), and PINK1 (Cell Signaling Technology, cat # 6946, 1:1000 dilution). Secondary goat anti-rabbit antibody (Invitrogen, reference # A24537) and secondary goat anti-mouse antibody (Invitrogen, reference # A24524) were used with 1: 10,000 dilutions.
2.3. Immunofluorescence staining
Cells were treated with HG, T2E and RAD and were incubated for five days post-radiation. Cells were then fixed in formalin for 10 min after washing with PBS once. Then cells were washed thrice with PBS and permeabilized with 0.5% Triton X in PBS for 8 min. Cells were blocked with 5% goat serum in PBS for 1 h after washing thrice with PBS. Then the primary antibodies were applied for 2 h at room temperature followed by washing with PBS. A fluorescence tagged secondary antibody was applied for 1 h at room temperature in the dark. Finally, cells were washed with PBS, mounted with ProLong Gold antifade reagent with DAPI (Invitrogen, cat #P36931), coverslipped, and imaged using a Leica DM4000 B LED microscope.
Mitochondria were stained with 150 nM of Mitotracker Red CMXRos (Invitrogen, Molecular probes, Cat #M7512) in serum free media for 20 min at 37 °C in 5% CO2, in the dark, followed by washing with Hank's balanced salt solution (HBSS) buffer three times. For staining with another antibody along with Mitotracker Red CMXRos, the above-mentioned steps for fixation, permeabilization, blocking and immunostaining were followed after Mitotracker staining.
Antibodies used for these experiments are as follows: ATP synthase Beta (Thermo Fisher Scientific, cat # A-21351, 1:250 dilution), NRF2 (Abcam, cat # ab62352, 1:250 dilution), α-SMA (Abcam, cat # ab5694, 1:500 dilution), Alexa fluor 488 goat anti-rabbit fluorescence secondary antibody (Life technology, reference # A11008, 1:500 dilution) and Alexa fluor 524 goat anti-rabbit fluorescence secondary antibody (Life technology, reference # A11008, 1:500 dilution).
2.4. Mitochondrial ROS measurement
After treatment with HG, T2E and RAD, cells were incubated for five days after radiation, as indicated in the previous section. Then cells were trypsinized and washed with HBSS (with Ca2+, Mg2+) buffer. Cells were then incubated in the dark with 5 μM of MitoSOX Red (Invitrogen, Molecular probes, cat #M36008, excitation/emission wavelength 510/580 nm) for 10 min at 37 °C in 5% CO2. After washing with HBSS (with Ca2+, Mg2+) buffer, cells were subjected to flow cytometry using a LSRII Green 532 Flow Cytometer (BD Biosciences, San Jose, CA) using the 488 excitation laser and a 575/26 with 550 long pass emission filter.
2.5. Mitochondrial oxygen consumption rate determination
Cells (0.3 × 106) were plated in a T25 flask and treated with HG, T2E and RAD as mentioned before. Twenty-four hours after radiation, cells were trypsinized and reseeded on XFe96 seahorse assay cell culture plate at a density of 0.3 × 10 -5 cells/well. The total media volume was 200 μl/well. HG and T2E were added in proper concentrations after reseeding. After 48 h of reseeding, cells were washed and replaced with fresh Mitostress assay medium. After 1 h incubation in a non-CO2 incubator, mitochondrial oxygen consumption rate (OCR) was measured using the XFe96 analyzer (Seahorse Biosciences, Santa Clara, CA, USA). Oligomycin, carbonyl cyanide-4-phenyl hydrazone (FCCP), and Rotenone/Antimycin were used to measure basal, ATP linked, maximal, non-mitochondrial OCR and spare capacity. OCR data was normalized to total protein content of the cells, measured by Bradford reagent.
2.6. Measurement of ATP levels
P3158 cells were treated with HG, RAD and T2E as previously described. After 5 days of radiation treatment, cells were trypsinized and counted. Sterile deionized water (1 ml), pre-heated to 135 °C, was added to 2.0 × 105 cells, and incubated in boiling water for 3 min. The cell suspension was then centrifuged at 12,000 g for 7 min at 4 °C. The supernatant (20 μl) was added to 180 μl of the ATP measurement reaction buffer, prepared by following the manufacture's protocol for ATP determination kit (Invitrogen Molecular probes, cat # A22066). Luminescence was measured to determine relative levels of ATP in experimental groups by using a Tecan Infinite M200 Pro plate reader. Deionized water (20 μl) was used to determine the background luminescence. Three technical replicates were used to read the luminescence for each sample for every biological replicate.
2.7. Measurement of total and healthy mitochondria levels
Cells were treated with HG, T2E and RAD as described above. After 5 days post-radiation, cells were trypsinized and washed with PBS once. Cells were treated with 100 nM of both Mitotracker Red CMXRos (Invitrogen, Molecular probes, cat #M7512) and Mitotracker Green (Invitrogen, Molecular probes, cat #M7514) in serum free media for 20 min at 37 °C in 5% CO2 in the dark, followed by washing with HBSS buffer three times. Cells were subjected to flow cytometry using a LSRII Green 532 Flow Cytometer (BD Biosciences, San Jose, CA) using the Y/G 605/15 laser for Mitotracker Red CMXRos (MTR) and blue 530/30 laser for Mitotracker Green (MTG). We have used carbonyl cyanide m-chlorophenylhydrazone (CCCP) as a positive control for altering mitochondrial membrane potential. We treated P3158 cells with 10 μM CCCP for 1 h before staining the cells with Mitotracker green and red.
2.7.1. siRNA treatment
P3158 cells (75,000 cells/well) were seeded in a 6 well plate and transfected with 25 nM of control siRNA (Ambion, cat # 4390843) or NRF2 siRNA (Ambion, cat# 4392420). After 24 h, cells were treated with HG (20 mM) and T2E (30 μM) and incubated for another 24 h followed by irradiation (3 Gy). Cells were harvested and whole cell lysates were prepared for western blot analysis 4 days post-radiation.
2.8. Metabolite extraction protocol
P3158 cells (1 × 106) were plated in 100 mm dishes and treated with HG, T2E and RAD, as described above. Six biological replicates for each experimental condition were collected 5 days post-radiation for the metabolomics study. The cells were washed with 1 ml of Nanopure water followed by pipet mixing and slow speed centrifugation. The cells were then centrifuged at 5000 g for 10 min at 4 °C and the cell wash was discarded. Quality control (QC) samples were prepared by combining equal amounts from each biological replicate. The same extraction procedure was applied to the QC and biological replicate samples. Cells were resuspended in 1 ml of 80% methanol and submitted to mechanical lysis with zirconia beads in a FastPrep® homogenizer (15 s at 1200 rpm repeated three times). The samples were placed in an ice bath for 30 s between cycles. The lysed sample was then centrifuged at 20,000 g for 20 min at 4 °C. The supernatant was collected and 1 ml of 50% methanol and 50% Nanopure water (v:v) was added to each cell pellet and vortexed for 10 s. The sample was centrifuged, the supernatant was collected and then combined with the previous supernatant. Each fibroblast cell sample was split 60:40 to prepare an NMR and mass spectrometry (MS) sample, respectively. Each sample was transferred to a rotary evaporator and then lyophilized to dryness.
After extraction of the aqueous metabolites, 4 ml of 2:1:1 chloroform: methanol: water mixture was added to each cell pellet in a glass vial. The pellet was then vortexed for 30 s and centrifuged at 20,000 g for 20 min at 4 °C and the organic layer was collected. The process was repeated a total of three times and the organic layers were combined. Each sample was lyophilized to dryness on a rotary evaporator. The organic metabolome extraction was only used to prepare a MS sample.
2.9. Preparation of metabolomics samples for nuclear magnetic resonance (NMR) and mass spectrometry (MS) analysis
After lyophilization, each dried aqueous cell extraction allocated for NMR analysis was resuspended in 0.3 ml of a 50 mM potassium phosphate buffer at pH 7.2 (uncorrected) in “100%” D2O containing 50 μM of 3-(trimethylsilyl) propionic-D4 (TMSP-D4) as a chemical shift reference. The sample was then transferred to a 3 mm NMR tube for data collection. After lyophilization, each dried aqueous cell extract allocated for MS analysis was resuspended in 100 μl of Nanopure water containing 0.1% of formic acid. After lyophilization, each dried organic cell extract was resuspended in 100 μl of isopropanol containing 0.1% formic acid.
2.10. One-dimensional (1D) 1H NMR data acquisition
1D 1H NMR spectra were collected on a Bruker Avance III-HD 700 MHz spectrometer equipped with a quadruple resonance QCI-P cryoprobe (1H, 13C, 15N, 31P) with z-axis gradients. A Bruker SampleJet sample changer with IconNMR and auto tune and match (ATM) were used to automate the NMR data collection. The 1D 1H NMR data were collected at 298K with 32K data points, a spectrum width of 11 ppm, 64 scans and 4 dummy scans. The spectra were collected using excitation sculpting [19] to remove the solvent and maintain a flat baseline.
2.11. Liquid chromatography-mass spectrometry (LC-MS) data acquisition
LC-MS metabolomics was performed on a Waters Acquity ultra performance liquid chromatography (UPLC) system coupled to a Xevo G2-XS Q-TOF (Waters MS Technologies, Manchester, UK) equipped with an electrospray ionization (ESI) source operating in positive ionization mode. The metabolites were separated with an HSS T3 column (Waters, 1.0 mm × 50 mm, 1.8 μm) with a 31-min linear gradient from 0.1% to 85% mobile phase B. Mobile phase A was 0.1% formic acid in water and mobile phase B was 0.1% formic acid in acetonitrile. The column and autosampler temperatures were set to 40 °C and 5 °C, respectively. The flow rate was set to 95 μl/min. The ionization source condition was set as follows: capillary voltage of 3.2 kV, sampling cone voltage of 40 V, and source offset voltage of 80 V. The source temperature was set to 120 °C and the desolvation temperature was set to 500 °C. The cone and desolvation gas flows were set to 50 and 800 L/h, respectively. Data acquisition was obtained in the MSE mode, which simultaneously records exact mass precursor ion and fragment ion information. MSE was performed with a low collision energy of 4 eV and a high collision energy ramped from 15 to 50 eV. The data was collected using an m/z range of 50 to 1200 with a scan time of 0.05 s. The data were acquired using an independent reference lock mass via the LockSpray interface to ensure accuracy and reproducibility during the MS analysis. Leucine Enkephalin was used as the reference compound ([M+H]+ = 556.2771).
LC-MS lipidomics was performed on a Waters Acquity UPLC system coupled to a Xevo G2-XS Q-TOF (Waters MS Technologies, Manchester, UK) equipped with an ESI source operating in either the positive or negative ionization mode. The lipids were separated on a C18 column (1.0 × 50 mm, 1.7 μm, Waters) with a 20-min linear gradient from 40% to 99% mobile phase B. Mobile phase A was an acetonitrile and water mixture (60:40, v/v) containing 10 mM ammonium formate and 0.1% formic acid. Mobile phase B was an isopropanol and acetonitrile mixture (90:10, v/v) containing 10 mM ammonium formate and 0.1% formic acid. The column and autosampler temperatures were set to 40 °C and 5 °C, respectively. The flow rate was set to 50 μl/min. The ionization source condition was set as follows: capillary voltage of 3.2 kV, sampling cone voltage of 40 V, and a source offset voltage of 80 V. The source and desolvation temperatures were set to 120 °C and 500 °C, respectively. The cone and desolvation gas flows were set to 50 and 800 L/h, respectively. MSE was performed with a low collision energy of 4 eV, and the high collision energy was ramped from 15 to 40 eV. The data was collected using an m/z range of 100–2000, a scan time of 0.05 s, and an independent reference lock mass via the LockSpray interface to ensure accuracy and reproducibility during the MS analysis. Leucine Enkephalin was used as the reference compound ([M+H]+ = 556.2771, [M − H]-=554.2615). The EquiSplash® Lipidomics® mass spectrometry standards (Avanti polar lipids, Inc.) were used as a quality-control standard before collecting the experimental lipidomics samples.
2.12. NMR data processing and statistical analysis
The 1D 1H NMR spectra were processed with our MVAPACK metabolomics toolkit (http://bionmr.unl.edu/mvapack.php) to generate a data matrix [20]. The spectra were processed with a 1.0 Hz exponential apodization function, a single round of zero-filling, and a Fourier transformation. Spectra was referenced and aligned to TMSP-D4 [20]. Solvent signals and noise regions were excluded, and the spectra were either aligned using the Icoshift [20] peak alignment algorithm or binned using an adaptive intelligent binning algorithm [20]. The 1D 1H NMR data matrix was pareto scaled and normalized with the median in order to generate a normal gaussian distribution using Metaboanalyst 5.0. The orthogonal projection to latent structures discriminant analysis (OPLS-DA) models was validated using permutation testing with n = 1000, R2, Q2, p-values were generated using Metaboanalyst 5.0 or MVAPACK. Discriminatory features were identified using Chenomx Suite 8.0 and Human Metabolome database (HMDB). A 1H chemical shift uncertainty of 0.08 ppm was used to match experimental chemical shifts.
2.13. LC-MS data processing and statistical analysis
The LC-MS datasets were imported into the Progenesis® QI metabolomics software (version 2.4, Nonlinear Dynamics, Newcastle, UK). The chromatographic peak alignment and peak picking were performed in an automatic manner with QC runs as references. Features were automatically deconvoluted for isotopes and adducts; and then associated with a retention time and an m/z value. For the positive ionization mode, adducts were assigned to either an [M+H]+, [M+Na]+, [M + NH4]+, [M–H2O + H]+ or [M+K]+. For the negative ionization mode, [M–H2O–H]-, [M − H]-, or [M + FA-H]- adducts were selected. Features were selected based on coefficients of variation (CVs) relative to QC samples where features with CVs over 30% were eliminated. Features not detected in at least 50% of the samples were also omitted. Signal imputation was performed using the k-nearest neighbor (knn) method [21]. All features underwent a log2 transformation.
LC-MS metabolic features were normalized with cyclic loess, and pareto scaled using R-packages ‘affy’, ‘limma’, and ‘Stats’ in R (4.0.3) [[22], [23], [24], [25]]. Principal component analysis (PCA) and orthogonal projections to latent structures discriminant analysis (OPLS-DA) models were produced. Using Metaboanalyst and MVAPACK, Loading plots, variable importance in projection (VIP) features and S-plots were generated from the OPLS-DA model; and were employed to visualize the relative importance of the differential variables and to acquire a list of peak indices. Metabolites were considered differential when presenting a discriminant p-corr value, VIP>1.0, a minimum fold change ≥1.0 and/or an ANOVA p-value ≤0.05. As previously described, the qualified filtered variables were submitted to putative metabolite annotation using MetaScope with theoretical fragment search against HMDB using Progenesis QI software [26].
2.14. Enrichment pathway and network analysis
After statistical analysis and compound identification, the statistically significant features from both the LC-MS and NMR data sets were subjected to enrichment pathway using Metaboanalyst 5.0 (https://www.metaboanalyst.ca/). Significantly perturbed metabolite pathways were identified using the Pathway Analysis module by selecting all the metabolite names identified by the NMR and MS metabolomics data analysis.
2.15. Statistical analyses
Statistical analyses of NMR-based and LC-MS-based metabolomics data is described as mentioned in respective sections. For all the other experiments, statistical analyses were executed using GraphPad Prism 6 Software version 6.0.5. Mean and standard deviation from three or more independent experiments were plotted unless otherwise indicated. Unpaired two-tailed t-test between two groups and one way ANOVA followed by post hoc Tukey's test were used to determine statistically significant differences between experimental groups and for multiple comparisons.
3. Results
3.1. MnTE-2-PyP protects against mitochondrial damage
NRF2 is a protector of mitochondrial health [13,14]. Other than acting as a transcription factor to express mitochondrial proteins, there are reports showing localization of NRF2 to the outer membrane of mitochondria [15,16]. We have previously reported that T2E upregulates NRF2 in fibroblasts [12]. Thus, we studied mitochondrial localization of NRF2 in the context of hyperglycemia and radiation. In the hyperglycemic conditions, T2E treatment significantly increased mitochondrial localization of NRF2 both in non-radiated and radiated groups as compared to the HG and RG groups. In the normoglycemic condition, T2E caused a trend of increased localization of NRF2 to the mitochondria, though the increase did not reach statistical significance (Fig. 1A). To validate this finding, we have studied NRF2 by immunofluorescence staining, where the mitochondria are labeled with Mitotracker Red dye. Co-localization of NRF2 and Mitotracker dye was evident in the TG and RGT groups (Fig. 1B). We have also observed colocalization of TOMM20 and NRF2 in a high-resolution image in the RGT treated cells (Fig. 1B). Purity of mitochondrial protein extraction was measured by the presence of TOMM20 protein in the mitochondrial extract but not in the cytosolic extract (Supplementary Fig. 1A).
Fig. 1.

MnTE-2-PyP protected from radiation and hyperglycemia-induced mitochondrial damage in normal prostate fibroblasts. Human prostate fibroblast cells were treated with 20 mM glucose (HG) and 30 μM MnTE-2-PyP (T2E) followed by 3 Gy of X-rays (RAD). 5 days after radiation, cells were harvested. A. Mitochondrial and whole cell proteins were extracted and immunoblotted for mitochondrial NRF2, total PGC1α and TOMM20. Quantification of 3 independent western blots is shown below. B. Mitochondrial localization of NRF2. Left: Representative images (20X) of human prostate fibroblasts stained with Mitotracker Red (red) and NRF2 (green). Scale bar = 100 μm. Right: Upper two images represent Mitotracker Red (red) and NRF2 (green) in 40X magnification. RGT group but not the control group showed co-localization of Mitotracker Red and NRF2 in yellow color (areas indicated by white arrows). Scale bar = 50 μm. Right lower panel represents a confocal image (63X) of colocalization between (white signals, analyzed by Image J) TOMM20 (green) and NRF2 (red) in the RGT group. Scale bar = 25 μm. C. Representative images of mitochondrial ATP synthase protein expression. Mean fluorescent intensity of ATP synthase staining per cell is shown below. Scale bar = 100 μm. All figures are representative of at least 3 independent experiments. CON = control, HG = high glucose, T2E = 30 μM MnTE-2-PyP treated, TG = T2E + HG treated, RAD = 3 Gy X-ray treated, RG = RAD + HG treated, RT = RAD + T2E treated and RGT = RAD + HG + T2E treated.(*), ($), (#) and (@) denote a significant difference (p ≤ 0.5) as compared to control, HG, RAD and RAD + HG group respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
PGC1α, an important regulator of mitochondrial biogenesis, maintains mitochondrial number and health via a mutual collaboration with NRF2 [27]. Therefore, we investigated PGC1α levels in our study. T2E increased the total expression of PGC1α in all conditions (control, HG, RAD and RG groups). Although the increase in PGC1α expression was statistically significant only in TG and RT groups as compared to HG only and RAD only groups (Fig. 1A). This data indicates that there is a possibility of increased mitochondrial biogenesis in T2E treated conditions.
Thus, it was necessary to study overall mitochondrial levels in our study. We measured total levels of TOMM20, an outer membrane protein of the mitochondria. TOMM20 levels were significantly lower in HG, RAD and RG groups as compared to the control. T2E increased TOMM20 levels in TG and RGT as compared to HG and RG group respectively (Fig. 1A).
After measuring TOMM20, we also measured the levels of a vital functional protein, mitochondrial ATP synthase, which is an important component of the mitochondrial electron transport chain, by immunofluorescence staining (Fig. 1C). Our study revealed that ATP synthase expression per cell was significantly lower in RAD and RG groups as compared to controls. ATP synthase expression was significantly lower in RG group as compared to HG only group as well. T2E treatment significantly increased ATP synthase expression as compared to control, HG, RAD and RG groups (Fig. 1C). In addition, we have also co-stained the cells with Mitotracker red to visualize mitochondrial health. Mitotracker intensity was also increased in T2E treated cells (Supplementary Fig. 1B).
Therefore, our data demonstrate that in the hyperglycemic and irradiated condition, mitochondrial localization of NRF2, and expression of PGC1α, TOMM20, and mitochondrial ATP synthase were increased by T2E treatment in prostate fibroblast cells.
3.2. MnTE-2-PyP reduces mitochondrial ROS
Damaged mitochondria produce an excess amount of ROS, which is a major source of hyperglycemia and radiation-mediated oxidative damage. Therefore, we measured mitochondrial ROS by MitoSox dye per cell (Fig. 2A). As mitochondrial ATP synthase levels correspond to the number of intact mitochondria, the amount of mitochondrial ROS was also normalized by average mitochondrial ATP synthase levels per cell (Fig. 2B). We found that RAD and RG significantly increased mitochondrial ROS as compared to controls. T2E treatment significantly decreased mitochondrial ROS levels in the radiated groups (Fig. 2B).
Fig. 2.

MnTE-2-PyP reduced radiation and hyperglycemia-induced mitochondrial ROS levels in normal prostate fibroblasts. Human prostate fibroblast cells were treated with 20 mM glucose (HG) and 30 μM MnTE-2-PyP (T2E) followed by 3 Gy of X-rays (RAD). 5 days after radiation, cells were harvested. A. Mitochondrial ROS was measured using MitoSox Red via flow cytometry. B. Mitochondrial ROS levels were normalized by average levels of ATP synthase per cell. C. Representative western blot images of levels of oxidative phosphorylation (OXPHOS) protein complexes in whole cell extract. D. Quantification of OXPHOS complex-I (C–I). E. Quantification of OXPHOS complex-II (C-II). F. Quantification of OXPHOS complex-III (C-III). G. Quantification of OXPHOS complex-IV (C-IV). H. Quantification of OXPHOS complex-V (C–V). All figures are representative of at least 3 independent experiments. CON = control, HG = high glucose, T2E = 30 μM MnTE-2-PyP treated, TG = T2E + HG treated, RAD = 3 Gy X-ray treated, RG = RAD + HG treated, RT = RAD + T2E treated and RGT = RAD + HG + T2E treated.
(*), ($), (#) and (@) denote a significant difference (p ≤ 0.5) as compared to control, HG, RAD and RAD + HG group respectively.
Oxidative phosphorylation (OXPHOS) is a major source of ROS production in mitochondria. Therefore, we measured the levels of OXPHOS proteins to investigate the source of mitochondrial ROS in the irradiated, hyperglycemic environment (Fig. 2C). HG, RAD and RG downregulated the total levels of OXPHOS complex-I, II, III and IV (Fig. 2D, 2E, 2F and 2G). This indicates both hyperglycemia and radiation may reduce electron transport chain activity by reducing OXPHOS protein complexes. The total levels of OXPHOS complex I, II and III were restored in T2E, TG and RT groups as compared to the control, HG and RAD groups respectively (Fig. 2D, 2E and 2F). However, after RG treatment, T2E only restored complex III levels (Fig. 2F) but had no effect on complex I and II (Fig. 2D and 2E). The levels of complex IV were not restored by T2E in any experimental group. Instead, T2E treatment downregulated complex IV levels as compared to their PBS treated counterparts (Fig. 2G). Although total levels of complex V were not affected by HG and/or RAD treatment, levels of complex V expression were significantly enhanced in TG, RT and RGT as compared to HG, RAD and RG groups respectively (Fig. 2H). Due to changes in mitochondrial mass in HG, RAD, RG and T2E treatments, we have further normalized the OXPHOS complex protein levels by TOMM20 protein levels (Supplementary Fig. 2). Most complexes followed similar patterns when normalized to either ponceau or TOMM20. However, complex IV and V levels were significantly upregulated in RAD and RG groups when normalized to TOMM20 but did not change when normalized to ponceau.
These data indicate that blockage of the electron transport chain is likely the major source of mitochondrial ROS production in the hyperglycemic and irradiated conditions. Blocked electrons flow back and produce superoxide via coenzyme Q after reacting with molecular oxygen. Overall downregulation of complex I, II, III and increase in the levels of complex IV per cell increased ROS in RAD and RG groups. T2E reduces mitochondrial ROS production by eliminating the blockage of the electron transport chain and reopening the electron flow via restoring the levels of OXPHOS complexes.
To find out the role of NRF2 in T2E-mediated mitochondrial protection, we have downregulated NRF2 in control and RG treated cells by siRNA treatment in the presence or absence of T2E (Supplementary Figs. 3A and 3B) and measured OXPHOS complex V and complex III, as these proteins were restored by T2E in RG treated samples (Supplementary Fig. 3C, 3D and 3E). In the control group, complex V was significantly downregulated in the absence of NRF2 but in RG or T2E treated groups complex V was not altered significantly (Supplementary Figs. 3C and 3D). The T2E mediated restoration of complex III was also not significantly affected by NRF2 knockdown (Supplementary Figs. 3C and 3E). Therefore, NRF2 is not solely responsible for T2E mediated mitochondrial OXPHOS protein protection.
3.3. MnTE-2-PyP increases mitochondrial efficiency
Hyperglycemia and radiation decreased mitochondrial health and T2E restored it. Therefore, it was expected that mitochondrial function of the irradiated and hyperglycemia treated fibroblasts would be reduced, which would be restored by T2E. To study the activity of mitochondria, we measured the mitochondrial oxygen consumption rate (OCR). By using Oligomycin, FCCP, Rotenone, and Antimycin A we have measured mitochondrial OCR during basal respiration (Fig. 3A), ATP production (Fig. 3B), maximal respiration (Fig. 3C), spare respiratory capacity (Fig. 3D), proton leak (Fig. 3E), and non-mitochondrial oxygen consumption (Fig. 3F). In non-radiated hyperglycemic cells (HG), OCR was downregulated during all the events and T2E treatment significantly increased OCR as compared to control and HG only treatment. We can summarize from this data that overall mitochondrial activity was down regulated in HG treatment and T2E treatment significantly increased overall mitochondrial OCR, which was evident from the time dependent OCR curve as well (Supplementary Fig. 1C).
Fig. 3.

MnTE-2-PyP increased mitochondrial ATP synthesis efficiency after radiation and hyperglycemia stress in normal prostate fibroblasts. Human prostate fibroblast cells were treated with 20 mM glucose (HG) and 30 μM MnTE-2-PyP (T2E) followed by 3 Gy of X-rays (RAD). Cells were subjected to Seahorse bio-analyzer to measure mitochondrial oxygen consumption rate (OCR). A. Basal respiration, B. ATP linked OCR, C. Maximal respiration, D. Spare respiratory capacity, E. Proton leak linked OCR and F. Non-mitochondrial OCR were measured. G. Total levels of ATP were measured in human prostate fibroblasts after 5 days of radiation. H. ATP levels were normalized by average value of ATP linked OCR (Fig. 3B). Levels of ATP produced using per unit of ATP linked OCR were measured. All graphs are representative of at least 3 independent experiments.
CON = control, HG = high glucose, T2E = 30 μM MnTE-2-PyP treated, TG = T2E + HG treated, RAD = 3 Gy X-ray treated, RG = RAD + HG treated, RT = RAD + T2E treated and RGT = RAD + HG + T2E treated.
(*), ($), (#) and (@) denote a significant difference (p ≤ 0.5) as compared to control, HG, RAD and RAD + HG group respectively.
Surprisingly, both normoglycemic and hyperglycemic irradiated groups showed significantly higher levels of OCR for basal, ATP linked, maximal and spare respiratory capacity as compared to control. T2E treatment reduced the OCR to the control levels. This data was not expected as higher OCR denotes higher mitochondrial activity. However, complex V, ATP synthase, was enhanced when normalized to TOMM20. Therefore, to determine if the enhanced OCR could be used for enhanced ATP production per cell in RAD and RG groups, we measured ATP levels in each condition as mitochondrial ATP production is the final endpoint for the mitochondrial metabolic activity. We measured total levels of ATP in equal number of cells and normalized the total ATP levels by the ATP linked OCR. This data revealed mitochondrial efficiency i.e., the amount of ATP produced per unit of oxygen consumption [28]. T2E treated cells in the irradiated group produced higher amounts of ATP (Fig. 3G) using lower amounts of oxygen (Fig. 3B) as compared to RAD and RG groups. Therefore, T2E significantly increases mitochondrial efficiency both in normoglycemic and hyperglycemic conditions after radiation as compared to the PBS treated irradiated cells (Fig. 3H). Mitochondrial efficiency was not altered significantly in other experimental groups.
OCR linked proton leakage was significantly increased in the RAD group as compared to the controls and T2E treatment restored proton leakage back to control levels. In the RG group, the OCR linked proton leak was not altered (Fig. 3E). Non-mitochondrial OCR was significantly higher in all the irradiated groups as compared to controls and T2E treatment did not reduce the non-mitochondrial OCR to the control levels (Fig. 3F).
3.4. MnTE-2-PyP maintains mitochondrial homeostasis
Balance in mitochondrial fission, fusion, mitophagy, and biogenesis maintains mitochondrial homeostasis in the healthy cellular environment. Blockage or over representation of any of these processes reduces the number of healthier mitochondria and increases accumulation of damaged mitochondria in the cells. Therefore, a disproportionate increase or decrease in any of these processes contributes to increased mitochondrial ROS and cellular damage in stressed conditions.
Therefore, we measured levels of mitochondrial fission and fusion proteins in mitochondrial protein extracts via western blots. We measured the levels of dynamin-related protein 1 (DRP1) as a representative of a mitochondrial fission (Fig. 4A) protein and mitofusin-1 (MFN1), mitofusin-2 (MFN2), and optic atrophy 1 (OPA1) as markers of mitochondrial fusion proteins. Our data suggests that in the non-radiated groups there was no significant change in mitochondrial fission or fusion processes as the levels of fission protein, DRP1, and the fusion proteins, MFN1 and OPA1, were not altered (Fig. 4A). However, levels of MFN2, a mitochondrial fusion protein, were significantly reduced in the TG group as compared to the HG group (Fig. 4B). In the normoglycemic-irradiated group mitochondrial levels of DRP1 were significantly reduced and MFN1 and OPA1 levels were increased significantly as compared to controls (Fig. 4A). T2E restored the levels of DRP1, MFN1 and OPA1 to control levels (Fig. 4A). We may conclude from this data that in a normoglycemic-irradiated condition, where mitochondrial ROS and mitochondrial damage were significantly high, mitochondrial fission was also blocked and T2E restored it to the control levels. It is reported that the long form of OPA1 helps in mitochondrial fusion and the short form preserves mitochondrial bioenergetics [29,30]; the cooperation between the two forms of OPA1 is necessary for complete mitochondrial fusion [31]. In our study, we have separately quantified the long and short form of OPA1 and normalized the data with the total OPA1 expression. However, we did not find significant up or downregulation of any of the specific forms of OPA1 (data not shown). In irradiated, hyperglycemic cells, mitochondrial fusion proteins were not significantly altered.
Fig. 4.

MnTE-2-PyP maintained mitochondrial homeostasis after radiation and hyperglycemia stress in normal prostate fibroblasts. Human prostate fibroblast cells were treated with 20 mM glucose (HG) and 30 μM MnTE-2-PyP (T2E) followed by 3 Gy of X-rays (RAD). 5 days after radiation, cells were harvested. Mitochondrial and cytosolic proteins were extracted. A. Representative western blots for DRP1 (mitochondrial fission protein), MFN1, MFN2 and OPA1 (mitochondrial fusion proteins) were measured and quantified. B. Quantification of PGAM5 in the cytosolic protein extract were measured. C. Cells were harvested at 30 h and 5 days post radiation. PINK1 protein levels were quantified in mitochondrial protein extracts. All images and graphs are representative of at least 3 independent experiments. CON = control, HG = high glucose, T2E = 30 μM MnTE-2-PyP treated, TG = T2E + HG treated, RAD = 3 Gy X-ray treated, RG = RAD + HG treated, RT = RAD + T2E treated and RGT = RAD + HG + T2E treated.
(*), ($), (#) and (@) denote a significant difference (p ≤ 0.5) as compared to control, HG, RAD and RAD + HG group respectively.
Data from these experiments suggest that in a normoglycemic condition, radiation significantly decreases mitochondrial fission and increases mitochondrial fusion, which was restored by T2E. However, under hyperglycemia, mitochondrial fission and fusion were not altered by radiation significantly.
Blockage of mitochondrial fission can block mitophagy and increase accumulation of damaged mitochondria inside the cells. It is reported that mitochondrial stress can be sensed by PGAM5, a mitochondrial inner membrane protein [32]. Under stress, PGAM5 relocates to the cytosol after being cleaved from the inner mitochondrial membrane. Thus, cytosolic PGAM5 serves as a signal for increasing mitophagy to eliminate damaged mitochondria from cells [32,33] as well as to increase mitochondrial biogenesis [34] to maintain healthy mitochondrial levels in the cells. PGAM5 senses mitochondrial damage and localizes to the cytosolic side of the mitochondrial outer membrane, where PGAM5 binds to NRF2. NRF2 localizes to the outer mitochondrial membrane via Keap1. This PGAM5-NRF2 complex acts as a signal for mitochondrial biogenesis and mitophagy to protect mitochondrial health [16,33,34]. Therefore, we measured the levels of cytosolic PGAM5. As expected, T2E significantly increased cytosolic levels of PGAM5 (Fig. 4B) without affecting the levels of total mitochondrial PGAM5 (Supplementary Fig. 1D).
Since we observed increased levels of cytosolic PGAM5, mitochondrial NRF2 and PGC1α in T2E treated samples, we hypothesized that T2E may increase mitophagy to eliminate damaged mitochondria and thus reduce the source of oxidative stress from the cells. The accumulation of PINK1 to the mitochondrial membrane is the first step in mitophagy. We measured PINK1 protein levels in the mitochondrial protein extract after 30 h and 5 days post-radiation. Thirty hours post-radiation, T2E significantly increased PINK1 accumulation in the mitochondria of the irradiated-hyperglycemic condition (Fig. 4C). This data suggests that in the irradiated hyperglycemic condition, T2E may increase the mitophagy process. However, as the number of damaged mitochondria were decreased in T2E treated cells after 5 days of radiation, T2E-mediated PINK1 accumulation in the mitochondria was also reduced after 5 days (Fig. 4C). This data suggests that T2E reduced the number of damaged mitochondria in irradiated and non-irradiated cells. Cytosolic PINK1 levels were not altered (Supplementary Fig. 1E).
Therefore, these experiments suggest that overall levels of healthy mitochondria will be restored by T2E treatment via mitochondrial biogenesis and increased mitophagy in damaged cells after hyperglycemia and radiation stress.
3.5. MnTE-2-PyP increases the number of mitochondria and improves mitochondrial health
To investigate the total number and overall health of mitochondria in cells, we stained cellular mitochondria with Mitotracker Green (MTG) and Mitotracker CMXRos Red (MTR), which stains all the mitochondria and mitochondria with intact membrane potential, respectively. As a positive control for mitochondrial damage, we have treated cells with CCCP and detected a decreased MTR intensity levels in CCCP treated cells as compared to the control (Supplementary Fig. 4A). According to our previous study, cell death varies from 10% −20% (highest in RG group) in all the experimental conditions [12]. To avoid any possible presence of dead cells in the current flow cytometry analysis, we only collected attached cells. Additionally, we have analyzed larger and more granular cell population only (P1), selected by forward (FSC-A) and side scatter (SSC-A) area plot (Supplementary Fig. 4B). There were no differences in the total percentage of cells in the P1 population among the different groups (Supplementary Fig. 4C).
We then plotted P1 cell population by MTG intensity on the X-axis and MTR intensity on the Y-axis (Supplementary Fig. 4D). There was no overlap of MTG and MTR staining during detection (Supplementary Fig. 4D, top panel). We have denoted 4 quadrants of the P1 cell population distribution as Q1, Q2, Q3 and Q4. Q2 represents the healthy mitochondria containing cells, as Mitotracker intensities were the highest in Q2. Conversely, Q3 represents the damaged mitochondria containing cells, as Mitotracker intensities were lowest in Q3. Whereas Q1 and Q4 can be considered as semi-damaged cell populations, where either number (MTG, in Q1) or the membrane potential (MTR, in Q4) of cellular mitochondria were compromised. We have measured the percentage of cells and average intensities of MTG and MTR in all these quadrants.
RAD and RG groups showed increased number of cells and reduced average Mitotracker intensities in Q1 as compared to the control. In T2E, RT and RGT groups, cell numbers were decreased and the average MTG and MTR intensities were increased as compared to their PBS treated counterpart (Fig. 5, panel A). This indicates T2E increases mitochondrial number and health in this semi-damaged cell population. Most of the cells were in Q2. The percentage of cells and the average MTR intensity in Q2 were not altered significantly in any of the experimental groups but average MTG intensity was increased in all the T2E treated groups (Fig. 5, panel B). This suggests T2E increased total mitochondrial numbers in this cell population. The Q2 population had two different sub-populations (represented in Supplementary Fig. 4D). We have separately analyzed the percentage of cells and average Mitotracker intensities in those two populations. The major sub-population of Q2 showed a similar trend as compared to the whole Q2 population (data not shown). RAD and RG treatment increased damaged cell numbers in Q3, which were reduced to control levels by T2E treatment. MTG and MTR intensities were decreased in RG group. T2E alone and RGT groups increased the average MTG and MTR intensities in Q3 population (Fig. 5, panel C). This data suggests T2E not only decreased the damaged cell population also increased the mitochondrial health and number in this population. All irradiated groups showed increased cell number and decreased Mitotracker intensities in Q4 (Fig. 5, panel D).
Fig. 5.

MnTE-2-PyP increased the number of healthy mitochondria after radiation and hyperglycemia stress in normal prostate fibroblasts. Human prostate fibroblast cells were treated with 20 mM glucose (HG) and 30 μM MnTE-2-PyP (T2E) followed by 3 Gy of X-rays (RAD). 5 days after radiation, cells were harvested, stained with Mitotracker Green (MTG) and Mitotracker Red (MTR) and subjected to flow cytometry. A. Left: Fold change of percentage of cells in quadrant 1 (Q1). Middle: Average intensity of MTG staining in Q1. Intensity of MTG staining in Q1 was normalized by percentage of cells in Q1. Right: Average intensity of MTR staining in Q1. Intensity of MTR staining in Q1 was normalized by percentage of cells in Q1. B. Left: Fold change of percentage of cells in quadrant 2 (Q2). Middle: Average intensity of MTG staining in Q2. Intensity of MTG staining in Q2 was normalized by percentage of cells in Q2. Right: Average intensity of MTR staining in Q2. Intensity of MTR staining in Q2 was normalized by percentage of cells in Q2. C. Left: Fold change of percentage of cells in quadrant 3 (Q3). Middle: Average intensity of MTG staining in Q3. Intensity of MTG staining in Q3 was normalized by percentage of cells in Q3. Right: Average intensity of MTR staining in Q3. Intensity of MTR staining in Q3 was normalized by percentage of cells in Q3. D. Left: Fold change of percentage of cells in quadrant 4 (Q4). Middle: Average intensity of MTG staining in Q4. Intensity of MTG staining in Q4 was normalized by percentage of cells in Q4. Right: Average intensity of MTR staining in Q4. Intensity of MTR staining in Q4 was normalized by percentage of cells in Q4. All data is representative of at least 3 independent experiments. CON = control, HG = high glucose, T2E = 30 μM MnTE-2-PyP treated, TG = T2E + HG treated, RAD = 3 Gy X-ray treated, RG = RAD + HG treated, RT = RAD + T2E treated and RGT = RAD + HG + T2E treated. (*) and (@) denote a significant difference (p ≤ 0.5) as compared to control and RAD + HG group respectively.
3.6. MnTE-2-PyP mediated mitochondrial protection inhibits myofibroblast differentiation
Under stressed conditions, such as hyperglycemia and radiation, fibroblasts are prone to differentiate to myofibroblasts, which results in fibrosis. Mitochondrial damage and myofibroblast differentiation are a cyclic process. Mitochondrial damage in a stressed condition leads to an increase in ROS and thereby promotes myofibroblast differentiation [35,36]. The combination of radiation and hyperglycemia exerts greater stress on the fibroblasts as compared to radiation or hyperglycemia alone. Therefore, we wanted to determine the initiating factor for this vicious cyclic process in hyperglycemia and radiation. We measured the mitochondrial ATP synthase (red) and α-SMA stress fibers (green) in prostate fibroblasts after radiation and hyperglycemia treatment in the presence or absence of T2E consecutively for 5 days post-radiation. Mitochondrial ATP synthase is a measure of mitochondrial health and α-SMA stress fibers indicate myofibroblast differentiation. Representative images (Fig. 6A) show that after 5 days of radiation, the RG group, but not the RGT group, differentiated to myofibroblasts. Mitochondrial ATP synthase levels were higher in the RGT group as compared to the RG group (Fig. 6A). We quantified stress fiber formation and ATP synthase intensity levels over 5 days post-radiation and determined that from day 3 post-radiation, levels of ATP synthase started to continuously increase in the RGT group. In the RG group, ATP synthase levels never significantly increased over time (Fig. 6B). α-SMA stress fiber formation started increasing in the RG group from day 4 post-radiation. In the RGT group, α-SMA stress fiber insignificantly increased in day 2 but from day 3 onward α-SMA staining intensity declined (Fig. 6D). We conclude that there is a significant difference between the slopes of the lines, measured by linear regression analysis of the means of the intensity values of ATP synthase expression (Fig. 6C) and percentage of stress fiber containing cells (Fig. 6E) between the RG and RGT groups (p value = 0.008 and p value < 0.0001 respectively). Therefore, we conclude that protection of mitochondria by T2E preceeds inhibition of α-SMA stress fiber formation.
Fig. 6.

MnTE-2-PyP-mediated mitochondrial protection is prior to inhibition of myofibroblast differentiation after radiation and hyperglycemia stress in normal prostate fibroblasts. Human prostate fibroblasts were treated with 20 mM glucose (HG) and 30 μM MnTE-2-PyP (T2E) followed by 3 Gy of X-rays (RAD). 1–5 days post-radiation, cells were subjected to immunofluorescence assay with mitochondrial ATP synthase (red) and α-SMA (green) antibody. A. Representative image of human prostate fibroblasts stained with mitochondrial ATP synthase (red), α-SMA (green) antibody and DAPI (blue), 5 days after radiation. Scale bar = 100 μm. B. Intensity of mitochondrial ATP synthase per cell over time was quantified by Image J. C. Differences between the slopes of the lines were measured by linear regression analysis of the means of the intensity values using GraphPad Prism. D. Percentage of α-SMA stress fiber containing cells over time was quantified by Image J. E. Differences between the slopes of the lines were measured by linear regression analysis of the means of the intensity values using GraphPad Prism. All data is representative of at least 3 independent experiments. (*) denotes a significant difference (p ≤ 0.5) as compared to RAD + HG group. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.7. Metabolic profiles of fibroblasts exposed to radiation and hyperglycemia stress
NMR and MS were used to identify altered metabolites and lipids in HG, RAD, and RG groups. Metabolomics and lipidomics techniques were also employed to assess if T2E treatment reversed the altered metabolome or lipidome or induced a counterbalance effect by increasing cytoprotective metabolites or lipids to maintain an overall healthy profile after exposure to radiation and hyperglycemic stress. To maximize the coverage of the metabolome and lipidome, we utilized a combination of 1D 1H NMR metabolomics, LC-MS metabolomics, and LC-MS lipidomics. A schematic outline of the metabolomics and lipidomics workflow is shown in Supplementary Fig. 5. NMR metabolomics emphasized polar or aqueously soluble metabolites, while LC-MS metabolomics, which by using a C18 column to separate aqueous compounds from the cell lysate, primarily detected semi-polar metabolites. Conversely, the LC-MS lipidomics method observed a diverse class of lipids from the organic extract of the cell lysate. The detected lipids range from semi non-polar lipids like lyso-phospholipids to non-polar lipids like cholesteryl esters. The two data-independent acquisition platforms, NMR and LC-MS, and the two different OMIC techniques, lipidomics and metabolomics, were able to differentiate between the HG, RAD, RG, and the T2E treated groups. Specifically, each experimental group displayed unique metabolome and lipidome profiles as evident from both a multivariate and univariate statistical analysis.
3.8. Overall alteration of metabolic and lipid profiles in human prostate fibroblasts are not overlapped after HG, RAD, RG and T2E treatment
An unsupervised principal component analysis (PCA) was used to measure the global differences of the metabolic profiles in each stressed condition (HG/RAD/RG) in the presence or absence of T2E as compared to the control using the NMR and LC-MS datasets. The LC-MS spectral data consisted of total of 1531 features (Supplementary Fig. 6A, 6B and 6C), while the 1D 1H NMR spectral data contained a total of 119 features (Supplementary Fig. 6D, 6E and 6F), for metabolomics. Most of the PCA models exhibited statistically relevant separations between all sets of groups (Supplementary Table 1).
To further explore the impact of radiation and hyperglycemia stress on fibroblasts, we also characterized changes to the lipidome. Importantly, the exact same samples used for metabolomics were also used to analyze the lipidomes by using LC-MS spectral data. The PCA models for the lipidome were generated from the three comparative groups for each comparison set using RPLC-MS in the positive ionization mode with 223 lipid features (Supplementary Fig. 7A, 7B and 7C) and in the negative ionization mode with 550 lipid features (Supplementary Fig. 7D, 7E and 7F). Both the PCA and OPLS-DA lipidomics models showed statistically valid separation between each set of groups (Supplementary Tables 1 and 2). Average cross-permutation number was 1000 for OPLS-DA analysis.
To identify significantly different metabolites in three types of oxidative stresses (HG, RAD and RG) as compared to control, we have analyzed the pairwise comparisons (HG vs control, RAD vs Control and RG vs Control) using supervised orthogonal projections to latent structures discriminant analysis (OPLS-DA). Specifically, the OPLS model was used to derive variable importance in projection (VIP) features. Similarly, the pair-wised OPLS-DA models showed distinct group separations. The OPLS models were statistically relevant based on average R2 and Q2 values of 0.996 ± 0.0003 and 0.9 ± 0.1, respectively. The OPLS model was also validated using a cross-permutation test (n = 1000) with a resulting average p value of 0.008 ± 0.008 (Supplementary Table 2). Separate PCA and OPLS models were created for each NMR and LC-MS dataset.
Overall, the PCA and OPLS models for the metabolomics and lipidomics datasets demonstrated that radiation and hyperglycemia stress induced significant perturbations in the global cellular lipidomes metabolism relative to controls. Also, T2E treatment did not fully restore the global metabolism to controls, but, instead, induced a distinct metabolome and lipidome. The NMR and LC-MS features with VIP scores >1.0 from the OPLS models, p values < 0.01 from a univariate Student's t-test, and a fold change >1.0 were selected for further detailed individual and pathway analysis.
In the B and D parts of Fig. 7, Fig. 8, Fig. 9, we have represented those selective metabolites, which are involved significantly in the altered pathways and are also significantly altered by T2E.
Fig. 7.

MnTE-2-PyP-mediated restoration of altered metabolic and lipid profile in hyperglycemic human prostate fibroblasts. Human prostate fibroblast cells were treated with 20 mM glucose (HG) and 30 μM MnTE-2-PyP (T2E). After 5 days, cells were harvested and subjected to metabolomics and lipidomics analyses A. Enriched pathways, analyzed from altered metabolites through HG treatment, were analyzed by MetaboAnalyst and are displayed with fold enrichment. B. Representative metabolite changes from the CON, HG, and HG + T2E (TG) treatment data sets. (*) Denotes p-values < 0.05 and VIP >1.C. Enriched pathways, analyzed from altered lipids through HG treatment, were analyzed by MetaboAnalyst and are displayed with fold enrichment. D. Representative lipid changes from the CON, HG, and HG + T2E (TG) treatment data sets. (*) Denotes p-values < 0.05 and VIP >1. CON = control, HG = high glucose and TG = T2E + HG treated cells. (*) and ($) denote a significant difference (p ≤ 0.5) as compared to control and HG group respectively. All data is representative of at least 5 independent experiments.
Fig. 8.

MnTE-2-PyP-mediated restoration of altered metabolic and lipid profile in radiated human prostate fibroblasts. Human prostate fibroblast cells were treated with 30 μM MnTE-2-PyP (T2E) followed by 3 Gy of X-rays (RAD). 5 days after radiation, cells were harvested, and subjected to metabolomics and lipidomics analyses. A. Enriched pathways, analyzed from altered metabolites through RAD treatment, were analyzed by MetaboAnalyst and are displayed with fold enrichment. B. Representative metabolite changes from the CON, RAD, and RAD + T2E (RT) treatment data sets. (*) Denotes p-values < 0.05 and VIP >1. C. Enriched pathways, analyzed from altered lipids through RAD treatment, were analyzed by MetaboAnalyst and are displayed with fold enrichment. D. Representative lipid changes from the CON, RAD, and RAD + T2E (RT) treatment data sets. (*) Denotes p-values < 0.05 and VIP >1. CON = control, RAD = 3 Gy X-ray treated and RT = RAD + T2E treated cells. (*) and (#) denote a significant difference (p ≤ 0.5) as compared to control and RAD group respectively. All data is representative of at least 5 independent experiments.
Fig. 9.

MnTE-2-PyP-mediated restoration of altered metabolic and lipid profile in radiated-hyperglycemic human prostate fibroblasts. Human prostate fibroblast cells were treated with 20 mM glucose (HG) and 30 μM MnTE-2-PyP (T2E) followed by 3 Gy of X-rays (RAD). 5 days after radiation, cells were harvested and subjected to metabolomics and lipidomics analyses. A. Enriched pathways, analyzed from altered metabolites through RAD + HG (RG) treatment, were analyzed by MetaboAnalyst and are displayed with fold enrichment. B. Representative metabolite changes from the CON, RG, and RAD + HG + T2E (RGT) treatment data sets. (*) Denotes p-values < 0.05 and VIP >1. C. Enriched pathways, analyzed from altered lipids through RAD + HG (RG) treatment, were analyzed by MetaboAnalyst and are displayed with fold enrichment. D. Representative lipid changes from the CON, RG, and RAD + HG + T2E (RGT) treatment data sets. (*) Denotes p-values < 0.05 and VIP >1.
CON = control, RG = RAD + HG treated and RGT = RAD + HG + T2E treated cells. (*) and (@) denote a significant difference (p ≤ 0.5) as compared to control and RAD + HG group respectively. All data is representative of at least 5 independent experiments.
3.9. High-glucose induced metabolic and lipidomic alterations and T2E-mediated restoration of metabolites and lipids
A total of 17 metabolites from the NMR and LC-MS datasets were altered due to HG treatment (Supplementary Table 3), which, not surprisingly, were shown to contribute to the pathways including Warburg effect, gluconeogenesis, and lactose degradation according to the top three altered pathways on the metabolites sets enrichment analysis (Fig. 7A). The effect of T2E on HG (HG + T2E) was evaluated by comparing the relative abundance of these metabolites. Our data revealed that levels of LysoPC, LysoPE, lactate and oleoyl tyrosine were significantly reduced in HG treated cells as compared to the control and T2E treatment restored the levels of these metabolites (Fig. 7B, Supplementary Table 3). The lyso-glycerophospholipids, LysoPC and LysoPE, and lactate are involved in gluconeogenesis and Warburg effect pathways. Oleoyl tyrosine, an N-acylamide, is involved in lipid signaling [37,38].
Furthermore, we looked at the lipid levels with the same pairwise comparison, control versus HG treatment. Confirming that the high-glucose alters overall lipidomic changes with statistically significant OPLS-DA model (Supplementary Table 2). A total of 37 lipids from the LC-MS lipidomics datasets were also altered due to HG (Supplementary Table 4). This result suggests high glucose disrupts the lipidome to a larger extent than the metabolome. Notably, some of the key metabolites identified above from the metabolomics dataset are properly classified as lipids. A pathway analysis identified oxidized glycerophospholipids, sterol conjugates, and glycerophosphoserine as the top 3 (p value < 0.05) lipid pathways altered by HG treatment (Fig. 7C, Supplementary Table 4). PHHdiA-PG and PHODA-PS, which are representative lipids of the oxidized glycerophospholipids pathway, and increased in HG, were decreased by T2E to the control levels. Notably, another oxidized phospholipid, PKdiA-PS, was decreased in HG and remained lower in TG. Overall, these results suggest that T2E reduced the oxidization of phospholipids, even to levels lower than control. Interestingly, sphingosines (such as, 3-ketosphingosine and glucosyl sphingosine) and phospholipids (such as, phosphatidyl-serine) were also significantly increased in HG and returned to control levels following a T2E treatment. Conversely, sterol conjugates, another lipid pathway altered by high glucose, did not return to control levels by T2E (Fig. 7D, Supplementary Table 4).
3.10. Radiation induced metabolic and lipidomic alterations and T2E-mediated restoration of metabolites and lipids
A total of 17 metabolites from the NMR and LC-MS metabolomics datasets were altered due to radiation exposure (Supplementary Table 5). A pathway enrichment analysis identified ketone body metabolism, butyrate metabolism, and mitochondrial electron transport chain, among others, as the top metabolic pathways altered by exposure to radiation (Fig. 8A). Notably, these pathways were also among the top 7 dysregulated pathways due to HG (Fig. 7A). Radiation exposure also resulted in a modest increase in TCA cycle metabolites (such as, succinic acid and acetic acid), which are relevant to electron transport chain metabolism. These TCA cycle metabolites were reduced to control levels after T2E treatment (Fig. 8B, Supplementary Table 5). Similarly, furan fatty acids (such as., dimethyl-pentyl-furanundecanoic acid) and pyridinoline were increased following exposure to radiation and reduced closer to the control levels in the T2E treated irradiated cells. Furan fatty acids (i.e., Dimethyl-pentyl-furanundecanoic acid) are antioxidants that inhibit lipid peroxidation [[39], [40], [41]]. Pyridinoline cross-linking is associated with fibrosis [42]. Wu et al. noted that antioxidants are able to reduce the elevated levels of pyridinoline cross-links [43]. Thus, these data supports that T2E is acting as an antioxidant in fibroblast cells when exposed to radiation, as has been previously reported [11,12].
We next evaluated lipidomics from control, RAD and RT treatment to see if there was any normalization effect of the drug on irradiated cells. OPLS-DA model confirmed that the radiation alters total lipid levels significantly (Supplementary Table 2). A total of 52 lipids from the LC-MS lipidomics datasets were also altered due to exposure to radiation (Supplementary Table 6). Again, this result suggests the lipidome is more sensitive than the metabolome to radiation. A pathway analysis identified glycerophosphoserines, oxidized glycerophospholipids, and fatty aldehydes, among others, as highly altered (p value < 0.05) lipid pathways (Fig. 8C). The relative concentrations of a few representative glycerophospholipids (Fig. 8D and Supplementary Table 6), such as, phosphoserines, phosphoethanolamine, phosphoinositols were either increased or decreased by radiation exposure but return to control levels in the RT group. PHODA-PS is an oxidized phospholipid that was increased by radiation treatment (Fig. 8D), was also increased by high glucose (Fig. 7D), presumably due to an oxidative environment. T2E reduced the levels of PHODA-PS in both conditions to levels lower than control. There was also a modest but statistically significant increase in fatty aldehydes, which are derived from lipid peroxidation [44], due to radiation treatment. Fatty aldehydes were reversed back to control levels by T2E (RT group) treatment.
3.11. RAD + HG treatment induced metabolic and lipidomic alterations in the healthy fibroblast cells and T2E-mediated restoration of metabolites and lipids
In total, 14 metabolites from the NMR and LC-MS metabolomics datasets were altered due to an irradiated and hyperglycemic environment (Supplementary Table 7). An enrichment pathway analysis identified glucose-alanine, lactose degradation, and glutamate metabolism, among others, as the top metabolic pathways (p value < 0.05) altered under an irradiated, hyperglycemic condition (Fig. 9A). Glucose, methylxanthine, TCA cycle metabolites (i.e., succinic acid and acetic acid), glycolipids (i.e., megastigmene-diol-apiosyl-glucoside and lyso-phosphatidic acid) were all increased in an irradiated, hyperglycemic environment and returned towards normal levels with a T2E treatment (Fig. 9B, Supplementary Table 7).
OPLS-DA scores plot also showed significant alteration of lipid levels in RG treated cells (Supplementary Table 2). Alteration in 32 lipids were identified due to RG treatment (Supplementary Table 8). A pathway enrichment analysis identified that glycerophosphoserine, oxidized glycerophospholipids, and sterol esters, among others, are the most altered (p value < 0.05) lipid pathways due to an irradiated and hyperglycemic environment (Fig. 9C). Representatives of glycerophospholipids are shown in Fig. 9D. Phosphoserines were decreased in the RG group and returned toward original levels after T2E treatment. PKOOA-PE, an oxidized phospholipid, increased in the RG treatment and T2E treatment reduced the levels of PKOOA-PE; whereas the levels of PHODA-PS did not change with the T2E treatment. Also, steroid conjugates, such as 4-Hydroxyandrostenedione glucuronide returned to its normal levels in the presence of T2E treatment (Supplementary Table 8).
In summary of the results of our study, hyperglycemia and radiation damage mitochondrial health and thereby increase mitochondrial ROS, which alters metabolism and increases lipid oxidation. T2E restores mitochondrial health, reduces mitochondrial ROS and oxidized lipid levels, and restores many metabolites and lipids to control levels.
4. Discussion
Mitochondrial damage produces more ROS in the diabetic irradiated environment, which starts a vicious cycle of oxidative stress, pathogenic metabolic changes, and cellular damage [[8], [9], [10]]. Therefore, we speculate that protection of mitochondria would be a beneficial approach to mitigate the feedforward cycle of oxidative stress in an irradiated, diabetic environment. In a diabetic environment, different compounds, such as, resveratrol [45] and melatonin [46] have been reported as mitochondrial protectors. We have previously published that T2E acts as a radioprotector [11,12,47]. In a diabetic environment, T2E decreases mitochondrial ROS [48]. However, the mechanism of mitochondrial ROS reduction and whether T2E protects mitochondrial health during radiation and hyperglycemia has not been studied. The goal of this current study was to determine whether T2E can protect mitochondrial health and improve metabolic health during radiation and hyperglycemia stress. T2E increased the levels of TOMM20, a mitochondrial membrane transport protein, PGC1α, the major regulator of mitochondrial biogenesis via coordinated action with NRF2 [27] and mitochondrial ATP synthase. This data suggest that T2E protects mitochondria from radiation and hyperglycemia mediated damage. In our previous study, we have reported that T2E increases expression and transcriptional activity of NRF2 [12]. Although NRF2 protects mitochondrial health [13,49,50], mitochondrial localization of NRF2 is not as well studied [15,51]. Therefore, we investigated the mitochondrial localization of NRF2 in this study and found increased localization of NRF2 to the mitochondria after T2E treatment. We have previously reported that T2E oxidizes Keap1 via H2O2, which stabilizes NRF2 levels [5]. Other reports also showed that a sublethal dose of H2O2 promotes NRF2 localization to the outer mitochondrial membrane [15]. T2E also increases PGAM5 levels in the cytosolic side of the outer mitochondrial membrane, which makes a complex with Keap1, inhibits NRF2 degradation, and promotes localization to the cytosolic side of mitochondrial outer membrane [16,32,34,51]. Therefore, we may speculate that generation of H2O2 and increased levels of cytosolic PGAM5 by T2E treatment are responsible for NRF2 stabilization on the mitochondrial membrane. However, NRF2 knockdown in T2E treated cells did not fully reverse T2E mediated mitochondrial OXPHOS complex protein protection. Therefore, other members besides NRF2 play a role in T2E-mediated mitochondrial protection. It should be noted that we did not investigate the role of NRF2 in other endpoints of T2E-mediated mitochondrial protection. Therefore, NRF2 could be playing a role in other endpoints, such as mitophagy, mitochondrial dynamics, which were not investigated. In the future, we plan to determine the potential role of NRF2 in these other T2E mediated mitochondrial protection endpoints.
Mitochondrial ROS is a major indicator of overall mitochondrial health, which was significantly increased in hyperglycemic and irradiated cells. T2E significantly reduced mitochondrial ROS levels across all conditions. T2E can enter the mitochondria [52] and directly scavenge ROS by its antioxidant potential. Previously, we have reported that in a normoglycemic condition, the activity of MnSOD, a major superoxide scavenger in mitochondria, was enhanced by T2E due to altering the acetylation status of MnSOD [5]. In addition to these T2E-mediated ROS scavenging mechanisms in the mitochondria, another way to reduce mitochondrial ROS levels is to block the production of mitochondrial ROS. The source of mitochondrial ROS is the electron transport chain via the OXPHOS complexes. In the HG, RAD and RG groups, complex I, II and III levels were significantly reduced, and OCR was increased in the RAD and RG groups. Therefore, there was abundant molecular oxygen to receive free electrons and to produce superoxide in the absence of electron acceptors, such as, complex I, II and III, which results in increased ROS production. Complex I and II levels were restored to control levels by T2E treatment after HG and RAD treatment but not after RG treatment. T2E rescued the complex III levels in the RG condition. It is specifically reported that the blockage of complex III in diabetes produces mitochondrial ROS [53,54]. Electrons released from the electron donors, NADH and FADH2, are transported to OXPHOS complex I and II respectively. Via coenzyme Q, electrons from both the complexes are then transferred to complex III, cytochrome-C, complex IV, and finally to molecular oxygen. Therefore, complex III acts as a hub to accept all the electrons to continue the electron transport process and ATP production. This data suggests that during radiation and hyperglycemia, electron flow was blocked, which pushed the free electrons to produce more superoxide by binding with molecular oxygen. T2E released the blockage and maintained normal electron flow and reduced mitochondrial ROS production.
Due to changes in mitochondrial mass in HG, RAD, RG and T2E treatment, we further normalized the levels of OXPHOS complexes by TOMM20. Interestingly, after normalization by TOMM20, complex-IV was expressed at the highest levels in the RG group followed by the RAD group (Supplementary Fig. 2D). When normalized to ponceau, this group had low levels of complex IV expression. These two groups also showed increased OCR (Fig. 3B). Although the reduction of complex IV is considered a ROS producing process [55], complex IV is a rate limiting step for the respiratory chain via an ATP-mediated allosteric inhibition [56,57]. Inhibition of ATP synthesis and simultaneously consumption of higher amounts of oxygen leads to ROS production in the mitochondria. Therefore, increased expression of complex-IV per mitochondria in RAD and RG groups could be responsible for increased ROS production in RAD and RG groups, which was significantly reduced by T2E, resulting in increased ATP production, which may act as a feedback inhibitor of complex IV [56].
The levels of ATP synthase (complex V) were also upregulated by T2E treatment (Fig. 1, Fig. 2H). In fact, T2E increased ATP production more efficiently as compared to normoglycemic-irradiated and hyperglycemic-irradiated cells (Fig. 3G and H). After normalization to TOMM20 levels, Complex-V expression was increased in the RAD and RG groups (Supplementary Fig. 2E); however, we did not observe higher levels of overall ATP levels in these groups (Fig. 3G and H). Therefore, we hypothesize that increases in the ATP synthase subunit (complex V) in the RAD and RG groups, is a complementary phenomenon by the damaged cells to meet the ATP demands in stressed conditions. Therefore, we speculate that reduced mitochondrial efficiency (Fig. 3H), blockage of complex-I, II, III and increase in complex-IV are responsible factors for increased mitochondrial ROS in irradiated and hyperglycemic conditions, which were restored by T2E.
Overall OCR (basal, ATP linked, maximal, spare respiratory capacity, proton leak and non-mitochondrial oxygen consumption) was reduced by HG and was increased by T2E (Fig. 3A–F). Levels of OXPHOS complexes (Fig. 2) and overall mitochondrial protection (Fig. 1) also followed a similar trend. Therefore, we may surmise that in moderate stress such as hyperglycemia only, the mechanism of T2E-mediated mitochondrial protection is to increase OCR to reach the ATP demand without reducing mitochondrial efficiency significantly (Fig. 3H). In the case of severe stress, such as, RAD and RG, the mitochondrial damage was higher. Therefore, to meet the ATP requirement, T2E significantly increased mitochondrial efficiency by producing increased levels of ATP while consuming less oxygen as compared to irradiated PBS treated groups (Fig. 3H). Proton leak (Fig. 3E) and non-mitochondrial (Fig. 3F) oxygen consumption were not altered in RGT group as compared to RG group. However, in the radiation only group, OCR linked to proton leak was upregulated by radiation, which was significantly reduced by T2E (Fig. 3E), while at the same time increased production of ATP (Fig. 3G) was observed. Additionally, ATP generated from glycolysis is also contributing to the total ATP levels in the cells. Therefore, to get a more accurate measure of T2E mediated mitochondrial ATP generation efficiency, in the future, we would like to investigate the glycolytic capacity and the levels and activity of uncoupling proteins (UCPs) in hyperglycemic and irradiated conditions.
Maintenance of healthy mitochondria in a stressed condition depends on mitochondrial biogenesis and elimination of damaged mitochondria via mitophagy. For an energy efficient cell, if one portion of mature tubular mitochondria is damaged and another part of it is still active, the cell will use the active parts of the mitochondria to produce energy. After being sliced out from tubular mitochondria via fission, the damaged part of the mitochondria will be eliminated from the cells via mitophagy and the remaining mitochondria will be fused with newly made smaller mitochondria to make a mature, tubular mitochondria. Therefore, increased mitophagy, mitochondrial biogenesis and fission after stress restore mitochondrial homeostasis in the cell. After sensing mitochondrial damage, PGAM5 is cleaved from the inner mitochondrial membrane and relocated to the cytosol, which then acts as a signal for the PINK1/Parkin to relocate to the mitochondrial membrane and initiate mitophagy [32,33]. Our data revealed that radiation significantly decreases mitochondrial fission and increases mitochondrial fusion, which was restored by T2E. In RAD and RG conditions, cytosolic PGAM5 levels (Fig. 4B) were decreased. Mitochondrial accumulation of PINK1 was decreased in the RG group after radiation (30 h), which was restored by T2E treatment. Therefore, in a normoglycemic-irradiated condition, blockage of mitochondrial fission and in a hyperglycemic-irradiated condition, blockage of mitophagy also contributed to the accumulation of damaged mitochondria and increased ROS in the cells.
To maintain the healthy mitochondria in cells exposed to cellular stress, cytosolic PGAM5 also signals a mitochondrial biogenesis process [34]. Cytosolic PGAM5-mediated mitochondrial biogenesis and the mitophagy process requires NRF2 localization on the outer surface of the mitochondrial outer membrane [16,51]. We had already observed that along with cytosolic levels of PGAM5 (Fig. 4B), levels of mitochondrial NRF2 and total levels of PGC1α were significantly increased by T2E treatment (Fig. 1A). Therefore, it was expected that the total number of mitochondria and mitochondrial health would be increased in T2E treated samples. We measured mitochondrial number (MTG) and mitochondrial health (MTR) (Fig. 5), which showed that not only is there an overall increase in the number and health of mitochondria in the healthy cells, T2E also improves the number and membrane potential of the mitochondria in the damaged cell population. This strengthens the potential of T2E as a mitochondrial protector not only as a preventive therapy but also as a recovery therapy after mitochondrial stress.
Oxidative stress, mitochondrial damage and fibrosis form a vicious cycle during stress-induced cellular damage. This study also revealed that the mitochondrial protection (increase in ATP synthase) by T2E is a prior event (starts from day 3 post-radiation) to myofibroblast differentiation (α-SMA stress fiber formation), which starts increasing by day 4 post-radiation in irradiated hyperglycemic cells.
Mitochondria maintain normal metabolic function in cells. Regulation of lipid metabolism is also one of the vital functions executed by mitochondria. Therefore, it was expected that T2E-mediated mitochondrial protection in the stressed conditions would lead to a healthier metabolic profile in fibroblasts, which may prevent fibroblasts from differentiation into myofibroblasts. We have observed that the metabolite and lipid profiles were different in HG, RAD and RG groups as compared to control. Notably, a larger number of lipids (32–52) compared to metabolites (14–17) were dysregulated by either high glucose or radiation exposure. Moreover, T2E treatment reversed some, but not all, of these changes.
Metabolites involved in oxidation of branched chain fatty acid, phytanic acid peroxisomal oxidation, caffeine metabolism and protein metabolic pathways, such as, ketone body metabolism, butyrate metabolism, and carnitine synthesis were altered in all stressed groups. That indicates these pathways are sensitive to any kind of stress studied here (HG, RAD and RG). We found some hyperglycemia or radiation specific metabolic alterations also. Aspartate metabolism, phospholipid biosynthesis and fatty acid biosynthesis were altered in RAD and RG groups, but not in the HG group. Therefore, these pathways were affected by radiation and not by hyperglycemia. As per expectations, glucose metabolism related pathways (such as, glycolysis, citric acid cycle and electron transport chain) were altered, especially in the hyperglycemic groups.
This study revealed a major change in lipids metabolism, especially, oxidation of lipids. We observed similar alterations of glycerophosphoserine levels and oxidation of glycerophospholipids, sterol and steroid conjugates, glycerophosphocholines in all the stressed conditions (HG, RAD and RG) as compared to the control, which suggests that these alterations also contribute to the increased oxidative stress in all these stressed environments. T2E reduced the oxidization of phospholipids. We observed that levels of phospholipids (LysoPC (18:0), LysoPE (20:0), Oleoyl tyrosine), which are a major structural component of the cell membrane, were reduced in HG treated cells and were rescued by T2E treatment. Recently, it was reported that decreased LysoPC (18:0) is associated with decreased insulin sensitivity in diabetic patients [58]. The association of tyrosine in insulin resistance is also reported in obese children [59]. In our study, we also observed an HG-mediated decrease in N-Oleoyl tyrosine, which was rescued to control levels by T2E treatment. Therefore, improvement of insulin sensitivity by manganese porphyrin treatment in diabetic mice [60,61] can partially be explained by rescuing LysoPC (18:0) and tyrosine levels in a hyperglycemic environment. The most abundant phospholipids in the mitochondria are phosphatidylethanolamine (PE) and phosphatidylcholine (PC), with the majority of PE being produced in the mitochondria [62]. Therefore, mitochondrial damage in HG treatment and T2E-mediated mitochondrial protection explained the reduced levels of PC and PE in the HG treated cells and rescued levels in T2E + HG treated cells. Reduction of cell membrane phospholipids, such as, PE, after radiation exposure has been reported before [63]. Our study revealed radiation-mediated decrease in PE that was rescued by T2E, which suggests that T2E may restore or protect the cell membrane from radiation damage. As lipids are one of the major components for cellular and mitochondrial membranes, increased lipid oxidation by HG, RAD and RG supports the theory of cellular and mitochondrial membrane damage after stress [64]. Anabolic and catabolic changes in lipid and fatty acid metabolism also enhance the possibility of ferroptosis mediated cell death and mitochondrial membrane damage [65,66]. Glycosphingolipids are another vital component for cell and mitochondrial membranes [67], which are protected by T2E after stressed conditions. Oxidized phospholipids are formed in many inflammatory diseases and they are a product of non-enzymatic lipid peroxidation in the presence of oxidative stress [68]. Some phospholipids (such as, 3-ketosphingosine and glucosyl sphingosine) were downregulated back to their normal levels in the presence of T2E after HG treatment. It was previously reported that the sphingolipids regulate signal transduction pathways and affect cell fate (i.e., apoptosis, cell proliferation, etc.) [69,70]. This study provides a potential protective role of T2E against mitochondrial damage after radiation by restoring glycolipid and phospholipid metabolism.
In an oxidatively stressed environment, prostate fibroblasts surrounding the prostate tumor make the tumor microenvironment fibrotic, which promotes tumor growth. We observed that radiation treatment increased the levels of pyridinoline, a profibrotic metabolite [42], which were reduced by T2E. Radioresistance is a clinical problem for PCa and increased radioresistance was associated with increased levels of glucosyl sphingolipid in different cancers including prostate tumors [[71], [72], [73]]. Our study revealed that radiation significantly increased glucosyl sphingosine levels (6.29-fold) as compared to the control, which was reduced by T2E (Supplementary Table 4). We can speculate from this data that fibroblasts in an irradiated tumor microenvironment may enhance fibrosis and radioresistance via pyridinoline and glucosyl sphingosine respectively, which are reduced by T2E treatment [47,74]. The combination treatment of radiation and hyperglycemia significantly increased the levels of hydroxyandrostenedione glucuronide, a precursor of an androgen receptor agonist [75]. Therefore, increased production of this metabolite from the prostate fibroblasts in the irradiated and hyperglycemic tumor microenvironment, may increase androgen receptor signaling in the PCa cells even after androgen deprivation therapy (ADT), which may lead to ADT resistant PCa cell growth in a diabetic irradiated PCa environment. T2E significantly decreased the levels of hydroxyandrostenedione glucuronide. Therefore, we speculate that T2E treatment may sensitize the PCa cells to ADT and decrease PCa growth in a diabetic, irradiated environment. We have also observed that levels of phosphatidylserine (PS) were increased in HG and RAD treatment as compared to the control, but the combination treatment (RG) decreased PS levels. In healthy cells, PS is located predominantly in the inner part of the plasma membrane. In excess stress, such as, RG, damaged plasma membranes release PS to the extracellular environment; therefore, we observed a significant reduction in PS levels in RG group. Increased levels of PS in the extracellular vesicle serve as a biomarker and a signal for immune suppression in pathologic conditions including cancers [[76], [77], [78]]. Therefore, our data suggests a detailed future study about the localization and externalization of PS in the tumor microenvironment of diabetic irradiated PCa models in context of immune suppression in PCa. From this study, we may surmise that in an irradiated and hyperglycemic environment, damaged mitochondria of prostate fibroblast cells produce tumor promoting metabolites, which may provide survival advantages to the PCa cells via radioresistance, ADT-resistance, immune-suppression or enhancing profibrotic signaling. T2E treatment protects mitochondria and reduces the tumor promoting transformation of prostate fibroblasts that may reduce PCa progression in a diabetic environment after radiation.
This study has some limitations. Mitochondrial protection by T2E was investigated in P3158 cells only. We have previously published that P3158 cells, mouse primary prostate cells and human primary prostate fibroblasts become activated after radiation, elicit a fibrotic response and T2E activates NRF2 in all these cell types [5,6,11,12]. Therefore, mitochondrial protection and metabolic changes by T2E in P3158 will most likely be reflected in the other fibroblast types also. However, mechanisms of T2E mediated mitochondrial protection and metabolic alterations should be conducted in other cells as well.
In the future, we would like to determine the mechanisms of T2E mediated mitochondrial and metabolic changes. According to our previous and current investigations, T2E mediated mitochondrial protection is likely mediated by multiple pathways. Data from this study revealed, NRF2 is not solely responsible for T2E mediated mitochondrial protection (Supplementary Fig. 3). Previously we reported that T2E mediated inhibition of a profibrotic response (α-SMA expression) was not completely dependent on NRF2 [12]. We have also published before that T2E reduces ROS by its direct ROS scavenging capacity, reduces NOX4 expression and stabilizes NRF2 by Keap1 oxidation, which elicits an antioxidant response. Direct ROS scavenging by T2E may be one of the mechanisms. This study also showed that T2E enhanced cytosolic PGAM5, enhanced PGC1α and restored cytoprotective metabolites. The combination of all these events restores mitochondrial health and metabolism. Further extensive studies need to be conducted to determine if these factors are causal for T2E-mediated mitochondrial protection and metabolic alterations. This study also provides significant rationale for studying the role of lipid metabolic pathways, beta oxidation and alteration in membrane lipids in radiation and hyperglycemia mediated cellular damage and T2E mediated restoration in the future. Identification of deleterious metabolites and altered metabolic pathways in a irradiated and hyperglycemic environment will provide probable future targets to reduce treatment resistance and radiation-mediated complications in diabetic PCa patients.
In conclusion, this study strongly suggests that T2E is a robust and novel mitochondrial protector in an irradiated and hyperglycemic environment. This study also demonstrates the mechanism of inhibition of mitochondrial ROS by T2E by restoring OXPHOS protein levels and maintaining normal flow of the electron transport chain. This study indicates that use of T2E along with radiation in diabetic and non-diabetic PCa patients will reduce mitochondrial damage, lipid oxidation, and restore a healthier metabolic profile in the irradiated normal cells, which will provide better efficacy of RT and better quality of life after radiation treatment for these patients.
Declaration of competing interest
Dr. Oberley-Deegan is a consultant with BioMimetix Pharmaceutical, Inc. and hold equities in BioMimetix Pharmaceutical, Inc. There are no conflicts of interest for the other authors.
Acknowledgments
This project was supported by following funding.
ROD: National Institute of Health (NIH) grant: (1R01CA17888), NIH SP20 GM103480 CoBRE, NIH PG20 GM104320, Fred and Otis Globe Medical Research Foundation.
RP: The Nebraska Center for Integrated Biomolecular Communication (P20GM113126, NIGMS). The research was performed in facilities renovated with support from the National Institutes of Health (RR015468-01).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102301.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
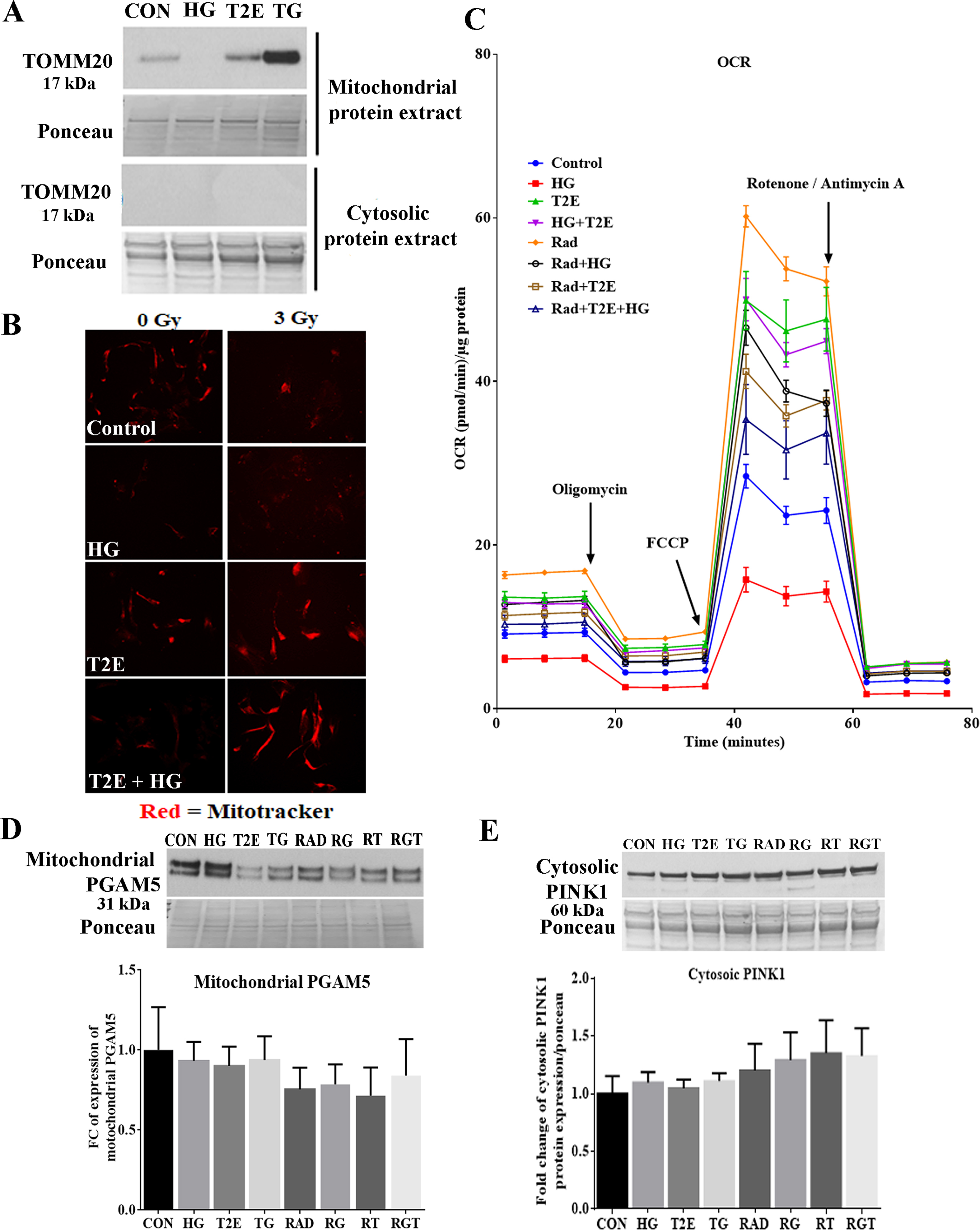{kind=link}
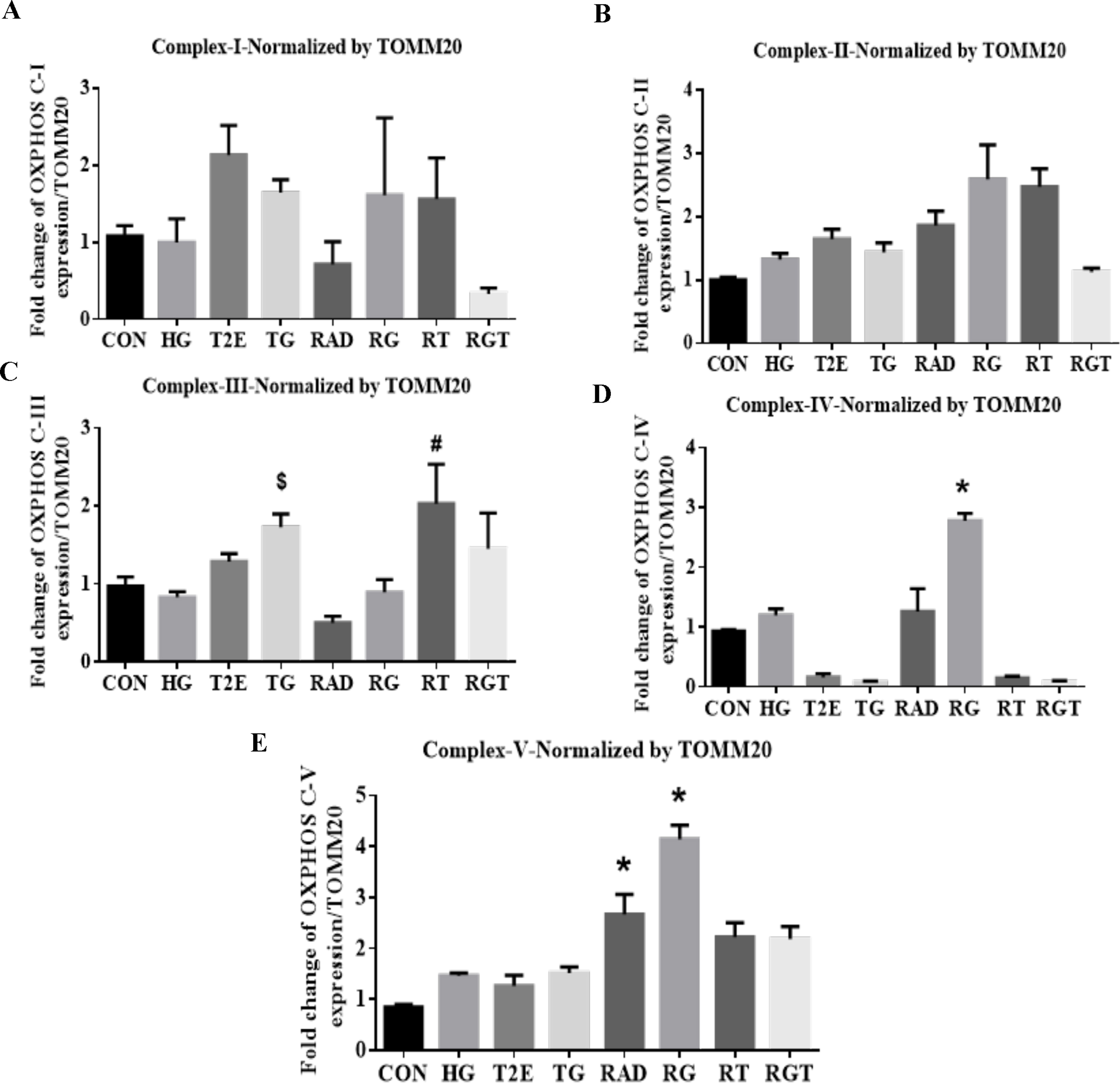{kind=link}
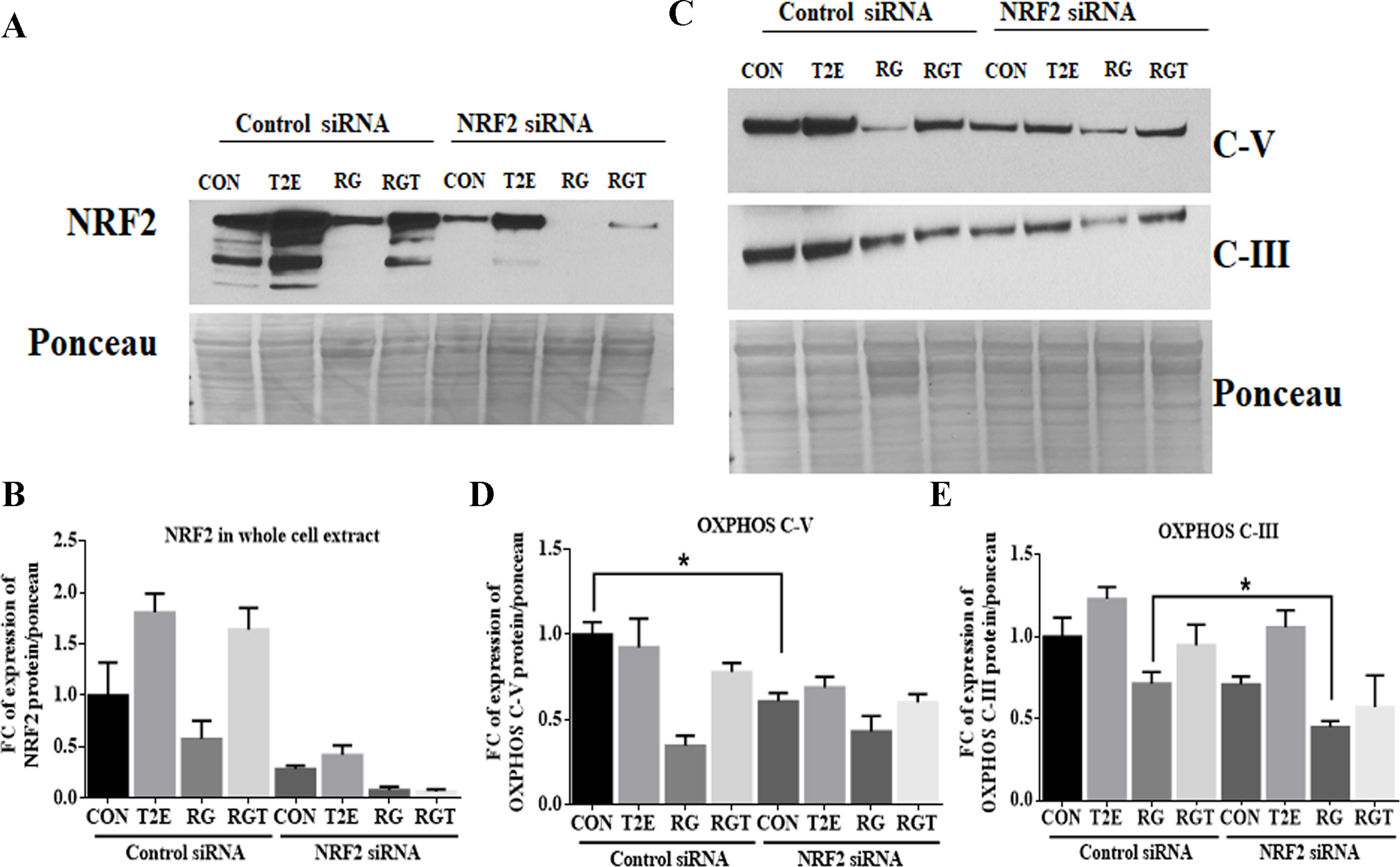{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Barone B.B., Yeh H.C., Snyder C.F., Peairs K.S., Stein K.B., Derr R.L., Wolff A.C., Brancati F.L. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: a systematic review and meta-analysis. JAMA. 2008;300(23):2754–2764. doi: 10.1001/jama.2008.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barone B.B., Yeh H.C., Snyder C.F., Peairs K.S., Stein K.B., Derr R.L., Wolff A.C., Brancati F.L. Postoperative mortality in cancer patients with preexisting diabetes: systematic review and meta-analysis. Diabetes Care. 2010;33(4):931–939. doi: 10.2337/dc09-1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D'Amico A.V., Braccioforte M.H., Moran B.J., Chen M.H. Causes of death in men with prevalent diabetes and newly diagnosed high- versus favorable-risk prostate cancer. Int. J. Radiat. Oncol. Biol. Phys. 2010;77(5):1329–1337. doi: 10.1016/j.ijrobp.2009.06.051. [DOI] [PubMed] [Google Scholar]
- 4.Lee J., Giovannucci E., Jeon J.Y. Diabetes and mortality in patients with prostate cancer: a meta-analysis. SpringerPlus. 2016;5(1):1548. doi: 10.1186/s40064-016-3233-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shrishrimal S., Chatterjee A., Kosmacek E.A., Davis P.J., McDonald J.T., Oberley-Deegan R.E. Manganese porphyrin, MnTE-2-PyP, treatment protects the prostate from radiation-induced fibrosis (RIF) by activating the NRF2 signaling pathway and enhancing SOD2 and sirtuin activity. Free Radic. Biol. Med. 2020;152:255–270. doi: 10.1016/j.freeradbiomed.2020.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shrishrimal S., Kosmacek E.A., Chatterjee A., Tyson M.J., Oberley-Deegan R.E. The SOD mimic, MnTE-2-PyP, protects from chronic fibrosis and inflammation in irradiated normal pelvic tissues. Antioxidants. 2017;6(4) doi: 10.3390/antiox6040087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Straub J.M., New J., Hamilton C.D., Lominska C., Shnayder Y., Thomas S.M. Radiation-induced fibrosis: mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 2015;141(11):1985–1994. doi: 10.1007/s00432-015-1974-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edwards J.L., Quattrini A., Lentz S.I., Figueroa-Romero C., Cerri F., Backus C., Hong Y., Feldman E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia. 2010;53(1):160–169. doi: 10.1007/s00125-009-1553-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West I.C. Radicals and oxidative stress in diabetes. Diabet. Med. 2000;17(3):171–180. doi: 10.1046/j.1464-5491.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- 10.Wolff S.P. Diabetes mellitus and free radicals. Free radicals, transition metals and oxidative stress in the aetiology of diabetes mellitus and complications. Br. Med. Bull. 1993;49(3):642–652. doi: 10.1093/oxfordjournals.bmb.a072637. [DOI] [PubMed] [Google Scholar]
- 11.Chatterjee A., Kosmacek E.A., Oberley-Deegan R.E. MnTE-2-PyP treatment, or NOX4 inhibition, protects against radiation-induced damage in mouse primary prostate fibroblasts by inhibiting the TGF-beta 1 signaling pathway. Radiat. Res. 2017;187(3):367–381. doi: 10.1667/RR14623.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chatterjee A., Kosmacek E.A., Shrishrimal S., McDonald J.T., Oberley-Deegan R.E. MnTE-2-PyP, a manganese porphyrin, reduces cytotoxicity caused by irradiation in a diabetic environment through the induction of endogenous antioxidant defenses. Redox Biol. 2020;34:101542. doi: 10.1016/j.redox.2020.101542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryoo I.G., Kwak M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018;359:24–33. doi: 10.1016/j.taap.2018.09.014. [DOI] [PubMed] [Google Scholar]
- 14.Tufekci K.U., Civi Bayin E., Genc S., Genc K. The nrf2/ARE pathway: a promising target to counteract mitochondrial dysfunction in Parkinson's disease. Parkinsons Dis. 2011;2011:314082. doi: 10.4061/2011/314082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strom J., Xu B., Tian X., Chen Q.M. Nrf2 protects mitochondrial decay by oxidative stress. Faseb. J. 2016;30(1):66–80. doi: 10.1096/fj.14-268904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Mealey G.B., Plafker K.S., Berry W.L., Janknecht R., Chan J.Y., Plafker S.M. A PGAM5-KEAP1-Nrf2 complex is required for stress-induced mitochondrial retrograde trafficking. J. Cell Sci. 2017;130(20):3467–3480. doi: 10.1242/jcs.203216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ventura-Clapier R., Garnier A., Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc. Res. 2008;79(2):208–217. doi: 10.1093/cvr/cvn098. [DOI] [PubMed] [Google Scholar]
- 18.Choi H.I., Kim H.J., Park J.S., Kim I.J., Bae E.H., Ma S.K., Kim S.W. PGC-1alpha attenuates hydrogen peroxide-induced apoptotic cell death by upregulating Nrf-2 via GSK3beta inactivation mediated by activated p38 in HK-2 Cells. Sci. Rep. 2017;7(1):4319. doi: 10.1038/s41598-017-04593-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simpson A.J., Brown S.A. Purge NMR: effective and easy solvent suppression. J. Magn. Reson. 2005;175(2):340–346. doi: 10.1016/j.jmr.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 20.Worley B., Powers R. MVAPACK: a complete data handling package for NMR metabolomics. ACS Chem. Biol. 2014;9(5):1138–1144. doi: 10.1021/cb4008937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang C., Li Y., Luo J. A novel method to detect functional microRNA regulatory modules by bicliques merging. IEEE ACM Trans. Comput. Biol. Bioinf. 2016;13(3):549–556. doi: 10.1109/TCBB.2015.2462370. [DOI] [PubMed] [Google Scholar]
- 22.Tanabe S., Mohanty R., Lindroth H., Casey C., Ballweg T., Farahbakhsh Z., Krause B., Prabhakaran V., Banks M.I., Sanders R.D. Cohort study into the neural correlates of postoperative delirium: the role of connectivity and slow-wave activity. Br. J. Anaesth. 2020;125(1):55–66. doi: 10.1016/j.bja.2020.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y.H., Dudoit S., Luu P., Lin D.M., Peng V., Ngai J., Speed T.P. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30(4):e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin L.X., Tuschl T., Singer S. An empirical evaluation of normalization methods for MicroRNA arrays in a liposarcoma study. Cancer Inf. 2013;12:83–101. doi: 10.4137/CIN.S11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crook A., De Lima Leite A., Payne T., Bhinderwala F., Woods J., Singh V.K., Powers R. Radiation exposure induces cross-species temporal metabolic changes that are mitigated in mice by amifostine. Sci. Rep. 2021;11(1):14004. doi: 10.1038/s41598-021-93401-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gureev A.P., Shaforostova E.A., Popov V.N. Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC-1alpha signaling pathways. Front. Genet. 2019;10:435. doi: 10.3389/fgene.2019.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salin K., Auer S.K., Rey B., Selman C., Metcalfe N.B. Variation in the link between oxygen consumption and ATP production, and its relevance for animal performance. Proc. Biol. Sci. 2015;282(1812):20151028. doi: 10.1098/rspb.2015.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Del Dotto V., Fogazza M., Carelli V., Rugolo M., Zanna C. Eight human OPA1 isoforms, long and short: what are they for? Biochim. Biophys. Acta Bioenerg. 2018;1859(4):263–269. doi: 10.1016/j.bbabio.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 30.Lee H., Smith S.B., Yoon Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J. Biol. Chem. 2017;292(17):7115–7130. doi: 10.1074/jbc.M116.762567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ge Y., Shi X., Boopathy S., McDonald J., Smith A.W., Chao L.H. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. Elife. 2020;9 doi: 10.7554/eLife.50973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamaguchi A., Ishikawa H., Furuoka M., Yokozeki M., Matsuda N., Tanimura S., Takeda K. Cleaved PGAM5 is released from mitochondria depending on proteasome-mediated rupture of the outer mitochondrial membrane during mitophagy. J. Biochem. 2019;165(1):19–25. doi: 10.1093/jb/mvy077. [DOI] [PubMed] [Google Scholar]
- 33.Park Y.S., Choi S.E., Koh H.C. PGAM5 regulates PINK1/Parkin-mediated mitophagy via DRP1 in CCCP-induced mitochondrial dysfunction. Toxicol. Lett. 2018;284:120–128. doi: 10.1016/j.toxlet.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 34.Bernkopf D.B., Jalal K., Bruckner M., Knaup K.X., Gentzel M., Schambony A., Behrens J. Pgam5 released from damaged mitochondria induces mitochondrial biogenesis via Wnt signaling. J. Cell Biol. 2018;217(4):1383–1394. doi: 10.1083/jcb.201708191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X., Zhang W., Cao Q., Wang Z., Zhao M., Xu L., Zhuang Q. Mitochondrial dysfunction in fibrotic diseases. Cell Death Dis. 2020;6(1):80. doi: 10.1038/s41420-020-00316-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimura T., Sasatani M., Kawai H., Kamiya K., Kobayashi J., Komatsu K., Kunugita N. Radiation-induced myofibroblasts promote tumor growth via mitochondrial ROS-activated TGFbeta signaling. Mol. Cancer Res. 2018;16(11):1676–1686. doi: 10.1158/1541-7786.MCR-18-0321. [DOI] [PubMed] [Google Scholar]
- 37.Cohen L.J., Esterhazy D., Kim S.H., Lemetre C., Aguilar R.R., Gordon E.A., Pickard A.J., Cross J.R., Emiliano A.B., Han S.M., Chu J., Vila-Farres X., Kaplitt J., Rogoz A., Calle P.Y., Hunter C., Bitok J.K., Brady S.F. Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature. 2017;549(7670):48–53. doi: 10.1038/nature23874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raboune S., Stuart J.M., Leishman E., Takacs S.M., Rhodes B., Basnet A., Jameyfield E., McHugh D., Widlanski T., Bradshaw H.B. Novel endogenous N-acyl amides activate TRPV1-4 receptors, BV-2 microglia, and are regulated in brain in an acute model of inflammation. Front. Cell. Neurosci. 2014;8:195. doi: 10.3389/fncel.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simons K., Toomre D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000;1(1):31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 40.Watson A.D. Thematic review series: systems biology approaches to metabolic and cardiovascular disorders. Lipidomics: a global approach to lipid analysis in biological systems. J. Lipid Res. 2006;47(10):2101–2111. doi: 10.1194/jlr.R600022-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Sethi J.K., Vidal-Puig A.J. Thematic review series: adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J. Lipid Res. 2007;48(6):1253–1262. doi: 10.1194/jlr.R700005-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Slot A.J., van Dura E.A., de Wit E.C., De Groot J., Huizinga T.W., Bank R.A., Zuurmond A.M. Elevated formation of pyridinoline cross-links by profibrotic cytokines is associated with enhanced lysyl hydroxylase 2b levels. Biochim. Biophys. Acta. 2005;1741(1–2):95–102. doi: 10.1016/j.bbadis.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 43.Wu H., Hung L., Leung P., Wan K. [The effects of antioxidants on pyridinoline cross linkage formation in human fibroblasts culture from hypertrophic scars] Zhonghua Yixue Zazhi. 2002;82(9):590–592. [PubMed] [Google Scholar]
- 44.Demozay D., Rocchi S., Mas J.C., Grillo S., Pirola L., Chavey C., Van Obberghen E. Fatty aldehyde dehydrogenase: potential role in oxidative stress protection and regulation of its gene expression by insulin. J. Biol. Chem. 2004;279(8):6261–6270. doi: 10.1074/jbc.M312062200. [DOI] [PubMed] [Google Scholar]
- 45.Diao J., Wei J., Yan R., Fan G., Lin L., Chen M. Effects of resveratrol on regulation on UCP2 and cardiac function in diabetic rats. J. Physiol. Biochem. 2019;75(1):39–51. doi: 10.1007/s13105-018-0648-7. [DOI] [PubMed] [Google Scholar]
- 46.Karamitri A., Jockers R. Melatonin in type 2 diabetes mellitus and obesity. Nat. Rev. Endocrinol. 2019;15(2):105–125. doi: 10.1038/s41574-018-0130-1. [DOI] [PubMed] [Google Scholar]
- 47.Chatterjee A., Zhu Y., Tong Q., Kosmacek E.A., Lichter E.Z., Oberley-Deegan R.E. The addition of manganese porphyrins during radiation inhibits prostate cancer growth and simultaneously protects normal prostate tissue from radiation damage. Antioxidants. 2018;7(1) doi: 10.3390/antiox7010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sidlauskaite E., Gibson J.W., Megson I.L., Whitfield P.D., Tovmasyan A., Batinic-Haberle I., Murphy M.P., Moult P.R., Cobley J.N. Mitochondrial ROS cause motor deficits induced by synaptic inactivity: implications for synapse pruning. Redox Biol. 2018;16:344–351. doi: 10.1016/j.redox.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dinkova-Kostova A.T., Abramov A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015;88(Pt B):179–188. doi: 10.1016/j.freeradbiomed.2015.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holmstrom K.M., Kostov R.V., Dinkova-Kostova A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr Opin Toxicol. 2016;1:80–91. doi: 10.1016/j.cotox.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lo S.C., Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp. Cell Res. 2008;314(8):1789–1803. doi: 10.1016/j.yexcr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spasojevic I., Chen Y., Noel T.J., Yu Y., Cole M.P., Zhang L., Zhao Y., St Clair D.K., Batinic-Haberle I. Mn porphyrin-based superoxide dismutase (SOD) mimic, MnIIITE-2-PyP5+, targets mouse heart mitochondria. Free Radic. Biol. Med. 2007;42(8):1193–1200. doi: 10.1016/j.freeradbiomed.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 54.Korshunov S.S., Skulachev V.P., Starkov A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416(1):15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 55.Reichart G., Mayer J., Zehm C., Kirschstein T., Tokay T., Lange F., Baltrusch S., Tiedge M., Fuellen G., Ibrahim S., Kohling R. Mitochondrial complex IV mutation increases reactive oxygen species production and reduces lifespan in aged mice. Acta Physiol. 2019;225(4) doi: 10.1111/apha.13214. [DOI] [PubMed] [Google Scholar]
- 56.Arnold S., Kadenbach B. The intramitochondrial ATP/ADP-ratio controls cytochrome c oxidase activity allosterically. FEBS Lett. 1999;443(2):105–108. doi: 10.1016/s0014-5793(98)01694-9. [DOI] [PubMed] [Google Scholar]
- 57.Kadenbach B. Regulation of cytochrome c oxidase contributes to health and optimal life. World J. Biol. Chem. 2020;11(2):52–61. doi: 10.4331/wjbc.v11.i2.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Semba R.D., Gonzalez-Freire M., Moaddel R., Sun K., Fabbri E., Zhang P., Carlson O.D., Khadeer M., Chia C.W., Salem N., Jr., Ferrucci L. Altered plasma amino acids and lipids associated with abnormal glucose metabolism and insulin resistance in older adults. J. Clin. Endocrinol. Metab. 2018;103(9):3331–3339. doi: 10.1210/jc.2018-00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hellmuth C., Kirchberg F.F., Lass N., Harder U., Peissner W., Koletzko B., Reinehr T. Tyrosine is associated with insulin resistance in longitudinal metabolomic profiling of obese children. J. Diabetes Res. 2016;2016:2108909. doi: 10.1155/2016/2108909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brestoff J.R., Brodsky T., Sosinsky A.Z., McLoughlin R., Stansky E., Fussell L., Sheppard A., DiSanto-Rose M., Kershaw E.E., Reynolds T.H.t. Manganese [III] tetrakis [5,10,15,20]-Benzoic acid porphyrin reduces adiposity and improves insulin action in mice with pre-existing obesity. PLoS One. 2015;10(9) doi: 10.1371/journal.pone.0137388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coudriet G.M., Delmastro-Greenwood M.M., Previte D.M., Marre M.L., O'Connor E.C., Novak E.A., Vincent G., Mollen K.P., Lee S., Dong H.H., Piganelli J.D. Treatment with a catalytic superoxide dismutase (SOD) mimetic improves liver steatosis, insulin sensitivity, and inflammation in obesity-induced type 2 diabetes. Antioxidants. 2017;6(4) doi: 10.3390/antiox6040085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shiao Y.J., Lupo G., Vance J.E. Evidence that phosphatidylserine is imported into mitochondria via a mitochondria-associated membrane and that the majority of mitochondrial phosphatidylethanolamine is derived from decarboxylation of phosphatidylserine. J. Biol. Chem. 1995;270(19):11190–11198. doi: 10.1074/jbc.270.19.11190. [DOI] [PubMed] [Google Scholar]
- 63.Benderitter M., Vincent-Genod L., Pouget J.P., Voisin P. The cell membrane as a biosensor of oxidative stress induced by radiation exposure: a multiparameter investigation. Radiat. Res. 2003;159(4):471–483. doi: 10.1667/0033-7587(2003)159[0471:tcmaab]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 64.Yukawa O., Miyahara M., Shiraishi N., Nakazawa T. Radiation-induced damage to mitochondrial D-beta-hydroxybutyrate dehydrogenase and lipid peroxidation. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1985;48(1):107–115. doi: 10.1080/09553008514551111. [DOI] [PubMed] [Google Scholar]
- 65.Matsko C.M., Hunter O.C., Rabinowich H., Lotze M.T., Amoscato A.A. Mitochondrial lipid alterations during Fas- and radiation-induced apoptosis. Biochem. Biophys. Res. Commun. 2001;287(5):1112–1120. doi: 10.1006/bbrc.2001.5696. [DOI] [PubMed] [Google Scholar]
- 66.Yuan Z.H., Liu T., Wang H., Xue L.X., Wang J.J. Fatty acids metabolism: the bridge between ferroptosis and ionizing radiation. Front. Cell Dev. Biol. 2021;9:675617. doi: 10.3389/fcell.2021.675617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Annunziata I., Sano R., d'Azzo A. Mitochondria-associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis. 2018;9(3):328. doi: 10.1038/s41419-017-0025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Que X., Hung M.Y., Yeang C., Gonen A., Prohaska T.A., Sun X., Diehl C., Maatta A., Gaddis D.E., Bowden K., Pattison J., MacDonald J.G., Yla-Herttuala S., Mellon P.L., Hedrick C.C., Ley K., Miller Y.I., Glass C.K., Peterson K.L., Binder C.J., Tsimikas S., Witztum J.L. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolemic mice. Nature. 2018;558(7709):301–306. doi: 10.1038/s41586-018-0198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bassoy E.Y., Baran Y. Bioactive sphingolipids in docetaxel-induced apoptosis in human prostate cancer cells. Biomed. Pharmacother. 2012;66(2):103–110. doi: 10.1016/j.biopha.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 70.El Alwani M., Wu B.X., Obeid L.M., Hannun Y.A. Bioactive sphingolipids in the modulation of the inflammatory response. Pharmacol. Ther. 2006;112(1):171–183. doi: 10.1016/j.pharmthera.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 71.Huang X., Taeb S., Jahangiri S., Emmenegger U., Tran E., Bruce J., Mesci A., Korpela E., Vesprini D., Wong C.S., Bristow R.G., Liu F.F., Liu S.K. miRNA-95 mediates radioresistance in tumors by targeting the sphingolipid phosphatase SGPP1. Cancer Res. 2013;73(23):6972–6986. doi: 10.1158/0008-5472.CAN-13-1657. [DOI] [PubMed] [Google Scholar]
- 72.Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer. 2018;18(1):33–50. doi: 10.1038/nrc.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Snider A.J., Seeds M.C., Johnstone L., Snider J.M., Hallmark B., Dutta R., Moraga Franco C., Parks J.S., Bensen J.T., Broeckling C.D., Mohler J.L., Smith G.J., Fontham E.T.H., Lin H.K., Bresette W., Sergeant S., Chilton F.H. Identification of plasma Glycosphingolipids as potential biomarkers for prostate cancer (PCa) status. Biomolecules. 2020;10(10) doi: 10.3390/biom10101393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu Y., Kosmacek E.A., Chatterjee A., Oberley-Deegan R.E. MnTE-2-PyP suppresses prostate cancer cell growth via H2O2 production. Antioxidants. 2020;9(6) doi: 10.3390/antiox9060490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.du Toit T., Bloem L.M., Quanson J.L., Ehlers R., Serafin A.M., Swart A.C. Profiling adrenal 11beta-hydroxyandrostenedione metabolites in prostate cancer cells, tissue and plasma: UPC(2)-MS/MS quantification of 11beta-hydroxytestosterone, 11keto-testosterone and 11keto-dihydrotestosterone. J. Steroid Biochem. Mol. Biol. 2017;166:54–67. doi: 10.1016/j.jsbmb.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 76.Birge R.B., Boeltz S., Kumar S., Carlson J., Wanderley J., Calianese D., Barcinski M., Brekken R.A., Huang X., Hutchins J.T., Freimark B., Empig C., Mercer J., Schroit A.J., Schett G., Herrmann M. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23(6):962–978. doi: 10.1038/cdd.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang W., Fa H., Xiao D., Wang J. Targeting phosphatidylserine for Cancer therapy: prospects and challenges. Theranostics. 2020;10(20):9214–9229. doi: 10.7150/thno.45125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zwaal R.F., Comfurius P., Bevers E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005;62(9):971–988. doi: 10.1007/s00018-005-4527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.