

Abstract
Cancer cells have long been recognized to exhibit unique bioenergetic requirements. The apoptolidin family of glycomacrolides are distinguished by their selective cytotoxicity towards oncogene transformed cells, yet their molecular mechanism remains uncertain. We used photoaffinity analogs of the apoptolidins to identify the F1 subcomplex of mitochondrial ATP synthase as the target of apoptolidin A. CryoEM of apoptolidin and ammocidin-ATP synthase complexes revealed a novel shared mode of inhibition that was confirmed by deep mutational scanning of the binding interface to reveal resistance mutations which were confirmed using CRISPR-Cas9. Ammocidin A was found to suppress leukemia progression in vivo at doses that were tolerated with minimal toxicity. The combination of cellular, structural, mutagenesis, and in vivo evidence define the mechanism of action of apoptolidin family glycomacrolides and establish a path to address OXPHOS-dependent cancers.
Introduction
The development of effective cancer therapeutics is constrained by the need to identify targets and drugs that selectively eliminate cancer cells with limited toxicity to healthy cells. One approach to addressing this challenge is to identify functional dependencies that are unique to cancer; even if an ‘essential’ target is present in non-cancerous cells, the unique demands of cancer cells create vulnerabilities that can be leveraged therapeutically1. Alterations in bioenergetics have long been recognized as a hallmark of cancer2–4, with Warburg’s observation that some cancer cells convert glucose to lactate in the presence of adequate oxygen. Although Warburg metabolism predominates in many cancers, there is increasing recognition that some cancers and subsets of cancer cells are dependent on mitochondrial oxidative phosphorylation (OXPHOS) to serve both bioenergetic and biosynthetic needs5,6. Notably, OXPHOS dependence appears to be a recurrent vulnerability of cancer stem cells (CSCs) and leukemia stem cells (LSCs), which are often responsible for treatment failure7–11. Despite these vulnerabilities, outside of a small group of genetically defined tumor subsets12, therapeutics designed for metabolic targets in cancer have largely fallen short of their great promise13.
The apoptolidin family of glycosylated macrolides (glycomacrolides) was discovered in a series of phenotypic screens for compounds that induce apoptosis selectively in oncogene-transformed cells. Apoptolidin A, produced by Nocardiopsis sp., was discovered to selectively induce apoptosis in E1A transformed rat fibroblasts14, while ammocidin A, a structurally related glycomacrolide produced by Saccharothrix sp., was discovered in a screen for compounds that selectively induce apoptosis in HRas-G12V transformed BaF3 cells15. When evaluated in the NCI-60 screening program, apoptolidin A was noted to be among the top 0.1% most cell line selective cytotoxic agents tested at that time, and early studies of its mechanism of action suggested that it targets ATP synthase, a mitochondrial membrane protein complex involved in oxidative phosphorylation16. These findings prompted multiple groups to develop new analogs via fermentation, semi-synthesis, and total synthesis. Structure-activity relationship (SAR) studies of these analogs supported a role for OXPHOS inhibition at ATP synthase in the mechanism of apoptolidin family compounds, but subtle differences in the bioactivity of apoptolidins compared to other classical ATP synthase inhibitors such as oligomycin suggested a distinct biochemical target17,18. Given the selectivity of apoptolidin family compounds ex vivo and emerging evidence of the importance of OXPHOS in cancer, identification of the apoptolidin target could reveal new targetable dependencies in cancer metabolism. In this study, we applied photoaffinity probes of apoptolidin family macrolides and electron cryomicroscopy (cryoEM) to identify their specific target and binding site on the F1, rather than FO, subcomplex of ATP synthase. Resistance mutations uncovered by deep mutational scanning eliminated both binding and cytotoxicity, confirming the mechanistic role for ATP synthase in their selective cytotoxicity. Ammocidin A was able to suppress leukemia development in mouse xenografts with minimal toxicity, demonstrating that ATP synthase can be effectively targeted in OXPHOS dependent cancers.
Results
Glycomacrolides target the F1 subcomplex of ATP synthase
The apoptolidin family of glycomacrolides, including apoptolidin A [Apop A, (1)], apoptolidin H [Apop H, (2)], and ammocidin A [Ammo A, (3)] all incorporate a 20-membered macrolide conjugated to three deoxy sugar units and are produced by taxonomically related actinobacteria. While superficially structurally similar, there are multiple differences in the functionality of the macrolide cores, decorating sugars, and in the positioning of glycosyl units. In order to identify the target of this family, we synthesized photoaffinity probe analogs of apoptolidin family macrolides (Fig. 1a). The C9 2-deoxyglucose was selected as the site of modification as previous studies had demonstrated that it could tolerate modification with retention of nanomolar cytotoxicity19. Apoptolidin A PA [Apop A-PA (4)] incorporates a diazirine, which provides for in situ photoreactive crosslinking when exposed to UV-A light, as well as an alkyne, which allows for post-treatment ligation of fluorophores or affinity enrichment tags using click chemistry20. The MV-4–11 acute myeloid leukemia cell line was selected as a primary model system for target identification based on its high sensitivity to apoptolidin family compounds. The activity of Apop A-PA was comparable to Apop A with regards to both cytotoxicity (Fig. 1b,c) and suppression of S6 [Ser235/236] phosphorylation (pS6 – Extended Data Fig. 1a), a marker of activation of the AMPK pathway which has been implicated in the mechanism of action of apoptolidin18. Consistent with previous studies using fluorescent live cell apoptolidin probes19, Apop A-PA exhibits mitochondrial localization when applied to adherent apoptolidin sensitive H292 cells and probed with rhodamine azide after fixation (Fig. 1d).
Fig. 1 |. Photoaffinity probes of apoptolidin family glycomacrolides retain cellular activity.

a, Structures of natural products and probes used in this study with key differences between apoptolidins and ammocidin highlighted; b, Concentration response curves for apoptolidin family glycomacrolides and c, probe compounds in MV-4–11 cells at 72 h by MTT assay showing retention of activity (P = 0.005, two-sided ratio test, n = 3 replicates per dose); d, Confocal microscopy of H292 cells treated with 200 nM Apop A-PA for 1 h, photocrosslinked, fixed, and conjugated with rhodamine azide, showing mitochondrial localization of Apop A-PA.
To identify apoptolidin family binding proteins, MV-4–11 cells were treated with DMSO, Apop A-PA alone, or Apop A-PA combined with various potential competitors (Fig. 2a). Proteins were photocrosslinked prior to lysis and then conjugated via azide-alkyne Huisgen cycloaddition to a trifunctional TAMRA-azide-desthiobiotin reagent, which facilitated both gel-based fluorescent profiling and subsequent affinity enrichment. Gel-based profiling of Apop A-PA adducts (Fig. 2b) revealed concentration-dependent labelling of a ~50 kDa target with an apparent EC50 of ~10 nM. Adduction of the 50 kDa target was eliminated by competition with excess Apop A, as well as Ammo A, suggestive of a shared putative binding site for the two glycomacrolides. Apop H, lacking the C-27 disaccharide and known to have reduced cytotoxicity relative to Apop A21, exhibited decreased competitive displacement of Apop A-PA. Furthermore, oligomycin (Oligm) did not exhibit any competition, suggesting that the 50 kDa target engaged by Apop A, Apop H, and Ammo A was not the target of Oligm, as previously proposed22. To confirm similar adduction specificity across the family, we synthesized photoaffinity probes Apoptolidin H-PA [Apop H-PA, (5)] and ammocidin A-PA [Ammo A-PA, (6)], which also retain cytotoxicity similar to the parent compounds (Extended Data Fig. 1b,c). Gel-based profiling of adducted proteins demonstrated the same 50 kDa band (Extended Data Fig. 1d,e). Similar binding patterns were observed in K562 cells (Fig. 2c), as well as HRas-G12V transformed BaF3s (Extended Data Fig. 1f) and H292 lung cancer cells (Extended Data Fig. 1g), comparable to those used in previous studies on apoptolidin family glycomacrolides15,23. The trienoate and C21 hemiketal functional groups of Apop A are potential electrophiles that could allow apoptolidin to act as a covalent inhibitor. However, irreversible binding to the 50 kDa target was dependent on both the diazirine and exposure to UV light (Extended Data Fig. 1h), suggesting that covalent modification of the target does not play a role in target engagement in naturally occurring apoptolidin family macrolides.
Fig. 2 |. Identification of F1 subcomplex of ATP synthase as the target of apoptolidin A.

a, Schematic of affinity enrichment workflow and competition experiments used to distinguish specific and non-specific binding partners – (T = [specific] target, N = non-specific binder, U = unbound protein); b, Gel-based profiling of Apop A-PA adducts in MV-4–11 cell or c, K562 cells; d, Volcano plot of identified proteins from TMT multiplexed quantitative proteomics of 1 μM Apop A-PA + vehicle (probe) or 20 μM Apop A (competition) treated MV-4–11 cells in duplicate, adjusted p-values calculated using Limma; e, Illustration of the mitochondrial ATP synthase; f, Immunoblot of MV-4–11 cells treated with Apop A-PA [1 μM] ± Apop A [20 μM] showing specific enrichment of ATP5B, non-specific enrichment of VDAC and TIMM17B, and no enrichment of ATP5G1; g, Inhibition of ATP synthesis after 16 h treatment with Ammo A, Apop A, or Oligm measured in MV-4–11 leukemia cells expressing the ATP:ADP reporter Perceval HR.
To identify the putative targets of Apop A in the MV-4–11 proteome, affinity enrichment was performed after in situ photocrosslinking of Apop A-PA with or without excess Apop A. Following lysis and cycloaddition to TAMRA-azide-desthiobiotin, samples were affinity enriched using streptavidin resin and labeled for multiplexed mass spectrometry quantification using isobaric tandem mass tags (TMT) (Extended Data Fig. 2a,b). Quantitative proteomics on the enriched tagged samples revealed a single, highly enriched protein relative to the competition control: ATP5B, the β subunit of ATP synthase (Fig. 2d).
ATP synthase is a ~600 kDa multiprotein complex responsible for converting the electrochemical proton gradient generated by the electron transport chain into chemical energy by the synthesis of ATP from ADP and phosphate. ATP synthase is composed of two subcomplexes: the membrane-embedded FO region, which contains the proton channel and is the target of oligomycin, and the soluble F1 region, which contains the catalytic sites (Fig. 2e). The F1 subcomplex is composed of five distinct subunits with the stoichiometry α3β3γδε and is largely conserved across the kingdoms of life24. The three catalytic sites are located at the interfaces of the α and β subunits with the catalytic residues localized to the β subunit, which is encoded by the gene ATP5F1B in humans. The three catalytic αβ sites are found in different conformations designated by their nucleotide content as αDPβDP (ADP-bound), αTPβTP (ATP-bound), αEβE (empty). During ATP synthesis, proton translocation through the FO region induces rotation of the central rotor complex that drives ATP synthesis in the F1 region25. The enzyme can also catalyze ATP hydrolysis (ATPase), which causes the rotor to reverse direction and pump protons from the matrix to the intermembrane space.
Several non-specific adducts were enriched in the competition conditions compared to vehicle (Extended Data Fig. 2c,d). These include the mitochondrial membrane proteins VDAC and TIMM17B, as well as other membrane proteins, many of which are documented non-specific targets of alkyl diazirines26. Immunoblotting confirmed the specific interaction of Apop A-PA with the β subunit of ATP synthase and nonspecific interactions with VDAC, and no interaction with ATP5G1, the c subunit in the FO region that is target of Oligm (Fig. 2f). Several attempts were made to identify the adduction site of Apop A-PA on the β subunit using shotgun proteomics, but these were unsuccessful, likely due to the large size of the theoretical adduct. β subunit binding by Apop A-PA was also confirmed by direct in-gel digestion of Coomassie dye stained bands after affinity enrichment (Extended Data Fig. 2e). Binding specificity to the β subunit was also confirmed in HEK-293 landing pad (HEK293-LP) cells expressing transgenic ATP5F1B with a C-terminal FLAG-tag (Extended Data Fig. 2f).
ATP synthase had been proposed as a target of apoptolidin family glycomacrolides based on similar cytotoxicity profiles to oligomycin in the NCI-60 screening program16. Due to their common general classification as macrolides, apoptolidin and oligomycin were proposed to share a SAR model and both bind to the FO region of ATP synthase27. More recent studies questioned whether the ATP synthase FO region is truly the mechanistic target of apoptolidin due to the discrepancy between in vitro (low μM) and ex vivo (low nM) activity17 and different effects on cell signaling18. These data demonstrate that apoptolidin and oligomycin act via distinct mechanisms at different binding sites on ATP synthase. Of note, apoptolidin and ammocidin represent the first known bacterial compounds to target the F1 ATP synthase, as all previously described F1 inhibitors are of fungal origin28, while bacterial ATP synthase inhibitors such as oligomycin target the FO region29.
To evaluate whether apoptolidin was acting as an inhibitor of ATP synthase at the cytotoxic concentration, ATP synthesis was evaluated in MV-4–11 cells expressing the intracellular ADP/ATP reporter, Perceval HR30. Treatment with Apop A, Ammo A, or Oligm A elicited an increase in ADP/ATP ratio at concentrations comparable with their cytotoxicity (Fig. 2g). In vitro, Apop A and Ammo A exhibited similar low nano-molar inhibition of ATPase activity by purified Saccharomyces cerevisiae ATP synthase (Extended Data Fig. 3a). Apop A-PA adduction was effectively displaced by equal or greater concentrations of Apop A (Extended Data Fig. 3b), suggesting that the parent compound and probe have similar binding affinities for ATP5, in line with their comparable cytotoxicity in cell-based assays. Competition experiments with known F1 binding inhibitors revealed uncompetitive binding with aurovertin and a lack of binding to the efrapeptin-bound conformation (Extended Data Fig. 3c). Kinetics studies in isolated mouse liver mitochondria and in yeast F1 subcomplexes under non-saturating conditions revealed mixed inhibition (Extended Data Fig. 3d,e) consistent with an allosteric binding site distal to the catalytic sites at the interfaces of the α and β subunits.
Surprisingly, pre-treatment of cells with the uncoupling agents carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) or 2,4-dinitrophenol (DNP) also eliminated adduction of Apop A-PA to the β subunit (Extended Data Fig. 3f). This observation suggested that either transport of Apop A-PA to the mitochondrial matrix was membrane potential dependent and/or the addition of uncouplers eliminated the apoptolidin binding site. Under uncoupled conditions, the decrease in mitochondrial matrix pH causes a conformational change in the inhibitory factor (ATPIF1, IF1) that allows it to bind to the F1 subcomplex and inhibit the ATPase activity of ATP synthase31. To test if IF1 binding was interfering with apoptolidin binding, CRISPR/Cas9 was used to knock out ATPIF1 in K562 leukemia cells (Extended Data Fig. 3g). In this setting, FCCP eliminated binding of Apop A-PA to the β subunit in the parental line, but not in ATP5IF1-KO cells (Extended Data Fig. 3h), suggesting that IF1 prevents binding of apoptolidin to ATP synthase.
CryoEM reveals glycomacrolide bindings site on ATP synthase
The novel mode of inhibition of glycomacrolides revealed by photoaffinity labeling and enzymatic assays prompted a search for their binding site by structure studies of S. cerevisiae ATP synthase. CryoEM and image analysis yielded 3D maps of the ATP synthase bound to Ammo A and Apop A at 3.1 and 3.4 Å resolutions, respectively, allowing construction of atomic models for the complexes. In order to investigate conformational changes induced in ATP synthase upon inhibitor binding, we also determined a structure of the yeast ATP synthase without either inhibitor under otherwise identical conditions, and refined the F1 region of the map to 4.2 Å resolution (Extended Data Fig. 4a–i, Supplementary Table 2).
Density corresponding to the inhibitor is found in the catalytic F1 region of ATP synthase (Fig. 3a, pink density, Fig. 3b, green density). Despite the presence of three-fold pseudosymmetry in the F1 region, which provides three nearly equivalent potential glycomacrolide binding sites, only a single inhibitor molecule was found to bind at the αDPβDP interface. This binding site is formed mainly by subunits αDP, βDP, and γ, although neighboring βTP and αE subunits provide additional protein-inhibitor contacts (Fig. 3c). Despite the functional differences in the aglycone, attachment site, and sequence of glycosylation between Apop A and Ammo A, both molecules bind to ATP synthase in a strikingly similar manner (Fig. 3a,b). For clarity, this discussion is focused on the higher-resolution ammocidin structure. The core macrolide is buried deeply inside the binding pocket and interacts with multiple hydrophobic residues of nearby subunits, while the head and tail sugar groups form fewer interactions with the protein (Fig. 3c,d). At current resolution, it is possible to build both ammocidin and apoptolidin models into the cryoEM density each with two different orientations, differing by rotation of the macrolide ring by ~180° (Extended Data Fig. 4j,k). The first of these conformations was identified as the most likely fit due to better agreement with the experimental density and a slightly more negative free energy of binding from molecular docking (−11.7 vs −9.9 kcal/mol for ammocidin and −11.8 vs −10.8 kcal/mol for apoptolidin). The structure indicates that introducing photoaffinity probes at the C2’ position of the C9 deoxysugar of apoptolidin, and the C3’ position of ammocidin (Fig. 1a) is unlikely to interfere with drug binding, supporting the reliability of the photoaffinity labeling experiments described above. βTP-D386 is located close to the C9 sugar and represents a likely site of photoaffinity adduct formation26. Together, these observations indicate that the high-affinity binding of ammocidin and apoptolidin to ATP synthase are achieved through extensive interactions between the core macrolides and their hydrophobic binding site in the enzyme (Fig. 3d). Comparison of the inhibitor-free and inhibitor-bound structures shows that inhibitor binding forces subunit βDP to move outward to adopt a more “open” conformation (Fig. 3e, red arrows and Movie 1), while the conformation of the non-catalytic αDP subunit remains largely unchanged except for the residues in direct contact with the inhibitor. This “open” conformation of βDP is incompatible with nucleotide binding and explains why the only natural substrate found in the inhibitor-bound structures is a phosphate ion at the βTP subunit (Fig. 3f). In addition to making βDP incapable of binding substrates, the inhibitors likely also block the rotation of ATP synthase by interacting strongly with subunits αDP, βDP, and γ, preventing the conformational changes needed for ATP hydrolysis and thereby inhibiting the rotary catalytic mechanism of the enzyme24.
Fig. 3 |. Structure of glycomacrolide bound yeast ATP synthase bound reveals a novel mode of inhibition.

a, Atomic model of the F1 region of yeast ATP synthase bound to ammocidin, cryoEM density of ammocidin is shown in pink; b, Close-up view of the ammocidin (top) and apoptolidin (bottom) binding site; c, Ammocidin binding site residues; d, Ammocidin binding pocket is largely formed with hydrophobic residues (yellow surface); e, Ammocidin binding induces conformational change of βDP subunit (red arrows) and forces it to adopt a more “open” conformation; f, Cross section through the F1 region shows that βDP does not bind to nucleotide (green circles) when bound to ammocidin.
Deep mutational scanning of glycomacrolide binding site
Having determined that the F1 region of ATP synthase is a target of Apop A and Ammo A, we next sought to determine whether binding to ATP synthase was essential for the cytotoxicity of apoptolidin by identifying one or more resistance mutations in the complex that prevent binding and cytotoxicity. Genomic analysis and multiple sequence alignment of ATP synthase genes from the apoptolidin producer Nocardiopsis sp. FU-40, revealed a substitution of phenylalanine at position ATP5B-L394 (Extended Data Fig. 5a) that we hypothesized would cause a clash that prevents binding. Introduction of ATP5B-L394F into MV-4–11 or K562 cell lines by CRISPR/Cas9 homology-directed repair (HDR)32 revealed decreased sensitivity to Ammo A, but unexpectedly increased sensitivity to Apop A compared to the parental line (Extended Data Fig. 5b–e). Comparing the binding poses of Apop A to Ammo A, ATP5B-L394 interacts most closely with the cyclic hemiketal that exhibits large differences in binding pose to account for the differing attachments of the disaccharide at C24 vs C27 (Extended Data Fig. 5f). Although this substitution does not provide cross resistance against both macrolides, the diverging effects on sensitivity support the binding mode identified by cryoEM of the yeast enzyme.
In order to identify a mutation that confers complete resistance to both apoptolidin and ammocidin, a deep mutational scanning approach was taken, using the Bxb1 “landing pad” system33 to conduct a pooled screen of all possible missense mutations in the binding site. Mutagenesis targets were identified by selecting all residues within 4.5 Å of apoptolidin or ammocidin in the atomic models, identifying 21 residues across the α (5), β (8), and γ (8) subunits (Extended Data Fig. 6a), and a mutant library containing all 420 point mutants of the three genes was generated using nicking mutagenesis34 (Extended Data Fig. 6b,c).
After transfection of the mutant library and selection for cells that had undergone successful recombination, each pool of mutant-expressing cells was exposed to varying concentrations of Apop A or Ammo A (Fig. 4a). Immunofluorescence confirmed that the transgenic ATP synthase subunits were properly expressed and trafficked to the mitochondria (Extended Data Fig. 6d). Cells were cultured in galactose-containing media to enforce OXPHOS dependence and select against loss of function mutants. After two rounds of selection, resistant mutants were expanded, and the integrated variants were amplified by PCR and subjected to deep sequencing. Allelic enrichment was consistent across biological replicates and no enrichment was seen in cells passaged without inhibitors (Extended Data Fig. 6e). Comparison of the variant abundance in the plasmid library to the variant abundance in successfully integrated cells allows for inference of variant fitness in the absence of any treatment. Notably, ATP5B (β subunit) I393, L394, and E398, which all make close contact with ammocidin in the atomic model of the inhibited ATP synthase, were enriched for wildtype alleles, suggesting that mutations at these positions result in a loss of fitness (Extended Data Fig. 6f).
Fig. 4 |. Deep mutational scanning and CRISPR/Cas9 directed editing of binding site residues reveals mutations which confer resistance to ammocidin and apoptolidin.
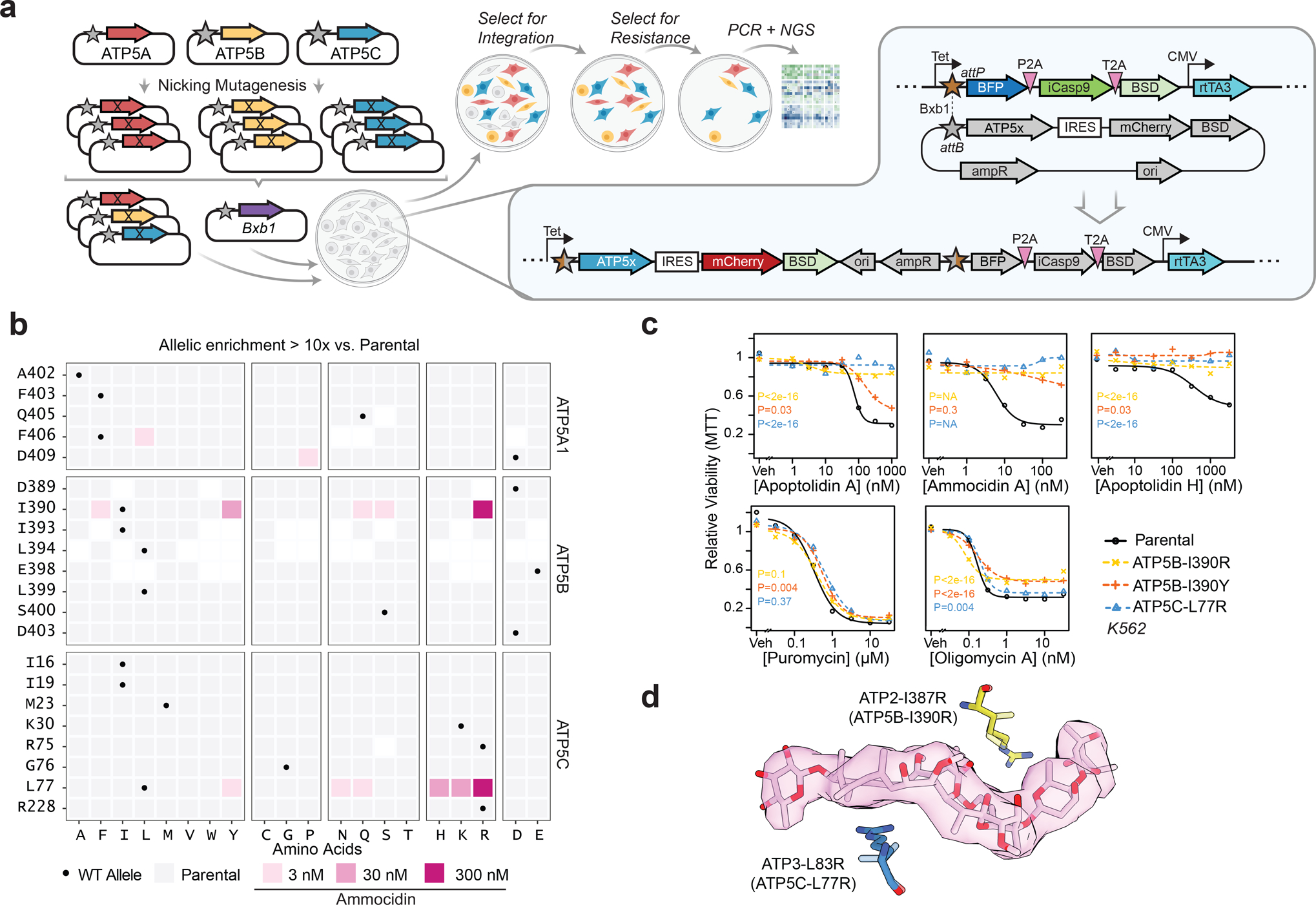
a, Illustration of the Bxb1 landing pad system consisting of an attP containing landing pad integrated at a single site in the genome and an attB transfer plasmid containing the variant gene of interest (ATP5x), colored genes are expressed in the presence of doxycycline; b, Tile plot of allelic enrichment after selection, filled based on the highest dose at which >10-fold enrichment was observed; c, Comparison of drug sensitivity between K562 Parental and K562 CRISPR/Cas9 knock-in resistant mutants demonstrating cross resistance to glycomacrolides with retained sensitivity to Oligm and puromycin (n = 3 per dose, P values computed for lower-bound of fitted log-logistic model, two-sided); d, Modeling of L83R and I387R mutations in the ammocidin cryoEM model (~ human residues in parenthesis).
Comparing variant abundance between the parental cell line and those treated with Ammo A revealed two key mutational hot-spots: ATP5B-I390 and ATP5C-L77. Mutations at these positions conferred varying degrees of resistance to both apoptolidin and ammocidin (Fig. 4b, Extended Data Fig. 7), with positively charged residues exhibiting enrichment upon exposure to Ammo A or Apop A (Extended Data Fig. 6g,h). Substituting these mutations in the atomic model suggests that introduction of arginine at these positions likely alters binding site electrostatics and disrupts the hydrophobic interactions necessary for binding (Fig.4d). Resistance was confirmed by expressing each mutation independently in 239LP cells (Extended Data Fig. 8a–c), as well as with CRISPR/Cas9 genome editing (Extended Data Fig. 8d–i) of the native alleles in K562 (Fig. 4c) and MV-4–11 leukemia cell lines (Extended Data Fig. 8j). Consistent with the results of the deep mutational scanning experiment, introduction of the ATP5B-I390R or ATP5C-L77R mutations conferred complete resistance to Apop A and Ammo A and eliminated binding of Apop A-PA as assessed by gel-based profiling (Extended Data Fig. 8k), while the ATP5B-I390Y variant raised the EC50s by 3 to 10-fold (Fig. 4c). None of the mutations tested notably affected sensitivity to Oligm or puromycin, reflecting retention of a functional electron transport chain. These data establish that ATP synthase is the sole mechanistic target of apoptolidin family glycomacrolides; binding to the F1 subcomplex of ATP synthase is necessary for the selective cytotoxicity of the family.
The binding modes of apoptolidin family macrolides revealed by cryoEM and deep mutational scanning studies also help contextualize the previous studies on the SAR of apoptolidin, which were notable in that no single modification was able to eliminate its activity17,23. This observation is consistent with its binding site, which is dominated by hydrophobic interactions each of which contributes a fractional amount to its nanomolar affinity. The structure also provides clues to the role of the disaccharide that is key for its activity: although loss of the disaccharide on Apop H reduces its activity by >10-fold, it still retains sub-micromolar activity ex vivo. Importantly, this residual activity is eliminated by introduction of the apoptolidin resistance mutations ATP5B-I390R and ATP5C-L77R (Fig. 4c), suggestive of a shared binding mode of the macrolide core.
Ammocidin A inhibits leukemia growth in vivo
Although apoptolidin and ammocidin are distinguished by their ability to induce apoptosis in transformed cells while sparing healthy cells, ATP synthase, the target of apoptolidin, is ubiquitously expressed and considered ‘essential’ in non-cancerous cells. Though the ATP synthase inhibitor Oligm is widely used as a tool compound in cellular assays, its lipophilicity and narrow therapeutic index render it poorly suited for in vivo applications35. These studies on older ATP synthase inhibitors prompted us to question whether inhibition of ATP synthase by apoptolidin family glycomacrolides could be leveraged therapeutically and whether a therapeutic index between cancerous cells and healthy cells exists. Ammo A was selected as the lead compound for in vivo studies due to its serum stability, higher potency, and the known proclivity of Apop A to isomerize in vivo to isoapoptolidin A via ester migration of C2036. Preliminary pharmacokinetic studies confirmed that ammocidin injected intraperitoneally was bioavailable in blood (Extended Data Fig. 9a) and dose escalation studies established ammocidin 0.1 mg/kg/day as the maximum tolerated dose, with minimal detectable toxicity over 14 days (Extended Data Fig. 9b.)
Based on the efficacy observed for ammocidin in vitro, we tested the compound as a monotherapy in an in vivo model. Antileukemic efficacy was evaluated in a systemic murine NSGS xenograft model37 using MV-4–11 human leukemia cells. Engrafted mice were treated with vehicle, 0.1 mg/kg or 0.03 mg/kg Ammo A dosed daily, five days on, two days off, for two weeks (Fig. 5a). Weekly chimerism analyses were conducted, and the percentage of MV-4–11 cells were quantified via flow cytometric analysis of murine peripheral blood using anti-human CD45 (hCD45) and anti-human CD33 (hCD33) monoclonal antibodies. At day 28, vehicle mice became moribund, and all mice were sacrificed. Blood, bone marrow, and splenic tissues were harvested for chimerism analysis37. Treatment with ammocidin resulted in dramatically decreased bone marrow leukemia burden in mice treated with 0.1 mg/kg ammocidin (Fig. 5b). A trend towards leukemia suppression was noted in the spleen, however, this effect was not statistically significant. Leukemia burden was also assessed by immunohistochemistry for hCD45 in bone marrow, which similarly revealed dose-dependent leukemia suppression in Ammo A treated mice (Fig. 5c). The degree of efficacy seen for Ammo A monotherapy in this cell derived xenograft model is consistent with recent studies that suggest that leukemia cells, and in particular, therapy resistant leukemia stem cells (LSCs), are exquisitely sensitive to OXPHOS inhibition7,38–42.
Fig. 5 |. Ammocidin A inhibits leukemia growth in vivo.
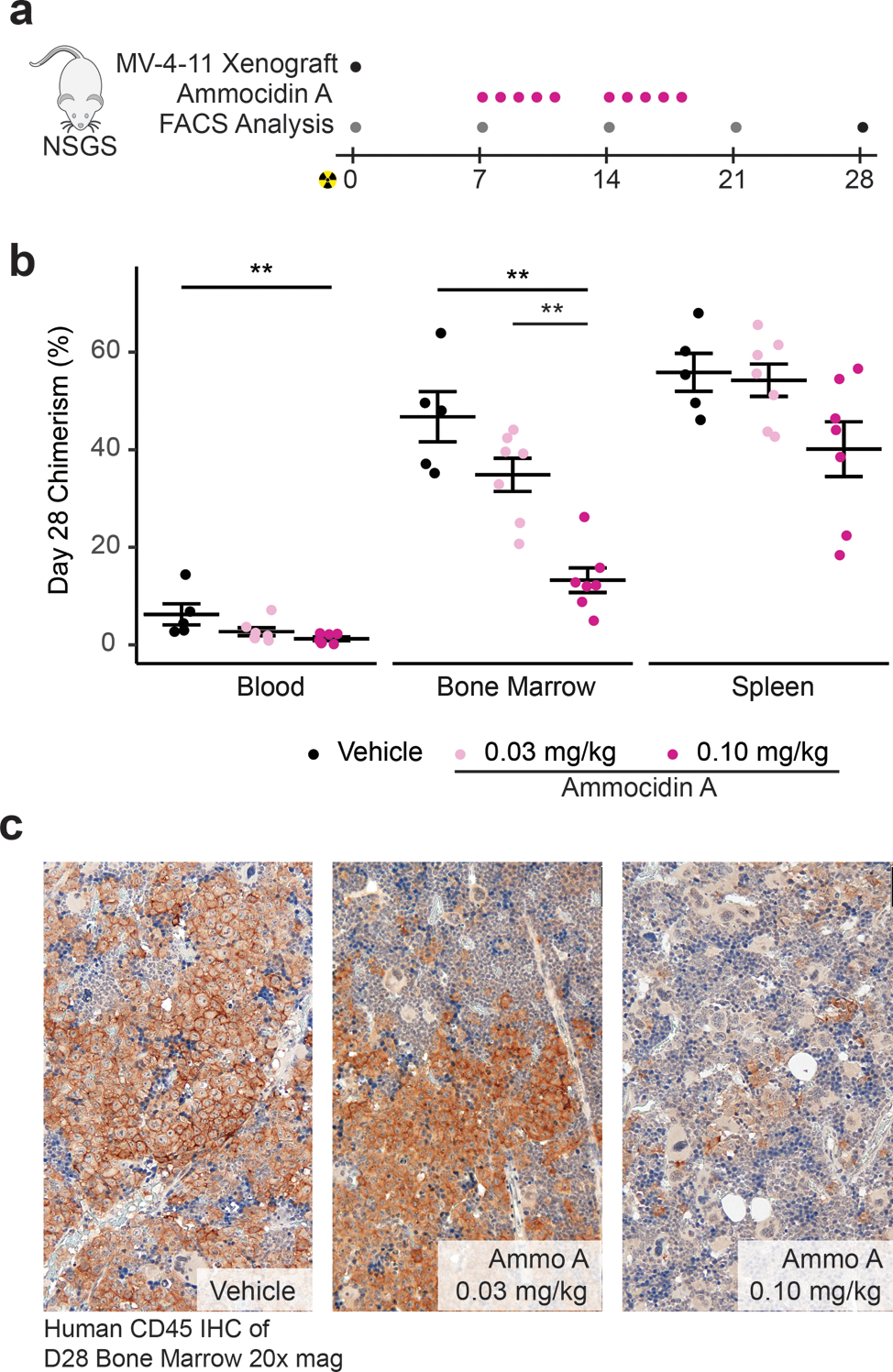
a, Experimental scheme for xenograft experiments—grey dots indicate assessment of human MV-4–11 chimerism in blood only, pink dots on days of ammocidin dosing, and black dot for terminal analysis on day 28 (e.g. assessment of chimerism in the blood, marrow, and spleen); b, Assessment of human MV-4–11 chimerism in the blood, bone marrow, and spleen at day 28; bars represent mean ± S.E.M for each group. Statistical differences between treatment groups were determined using a two-sided Wilcox test with Holm-Bonferroni correction for multiple comparisons (n = 5 – 7 mice per group, ** = P < 0.01, exact P values provided in source data); c, Immunohistochemistry against human CD45 in day 28 marrow showing dose dependent decrease in leukemia burden.
Discussion
Alterations in bioenergetics, including OXPHOS dependency, have long been recognized as a hallmark of cancer, however, efforts to target this vulnerability have been hampered by a dearth of metabolic inhibitors with sufficient selectivity and acceptable toxicity profiles43. This unmet need is especially acute as many of the cancers that appear to be most vulnerable to OXPHOS inhibition are those for which few therapeutic options exist, including acute myeloid leukemia and glioblastoma. Previous efforts to target mitochondrial bioenergetics in leukemia include inhibitors of mitochondrial complex I38,44, complex III6, and the mitochondrial translation machinery responsible for synthesizing the mitochondrially encoded components of the electron transport chain45. In addition to efforts to target the electron transport chain directly, recent studies have suggested that venetoclax, an inhibitor of BCL-2 recently approved for multiple hematolymphoid malignancies, targets LSCs by inhibiting OXPHOS9,10,40. There is also evidence that OXPHOS inhibitors can be combined synergistically to increase the efficacy of venetoclax therapy45,46, suggesting a possible route for efficacious combination therapies with apoptolidin family glycomacrolides.
Recent studies suggest that the respiratory complexes are under varying degrees of selective pressure in tumor evolution47, which suggests some respiratory complexes may be better therapeutic targets than others. Despite the abundant literature supporting OXPHOS as a target in cancer, it is still somewhat surprising that ammocidin possesses a favorable therapeutic index in vivo. While an organism may not tolerate genetic ablation of ATP synthase, transient inhibition with a small molecule may provide an opportunity for selectivity by sparing healthy cells that can temporarily meet their bioenergetic needs via alternative pathways. Alternatively, ammocidin may possess unique pharmacokinetic and pharmacodynamic properties that contribute to its selective cytotoxicity and provide a path towards further optimization. Though further study on the mechanism and scope of this therapeutic index is warranted, this work demonstrates that apoptolidin family glycomacrolides are unique among all OXPHOS inhibitors currently in clinical development or any previously described ATP synthase inhibitors.
Chemical probes, particularly compounds derived from natural products discovered in phenotypic screens, have contributed much to our understanding about the functional dependencies of cancer cells. Though tool compounds may modulate pathways and phenotypes in desirable ways, to be considered a chemical probe a compound must act selectively through a defined mechanism of action48. In order for a compound to be considered a therapeutic candidate, it must be bioavailable, capable of modulating its target in vivo, and possess an acceptable therapeutic index48. Though ATP synthase was proposed as the target of apoptolidin based its cytotoxicity profile in the NCI-60 screen16, a lack of target validation precluded its use as a chemical probe and apoptolidin’s reduced stability precluded its development as a therapeutic agent. This work defines the mechanism of action of the entire class of apoptolidin glycomacrolides including Apop A and Ammo A, combining evidence from cellular target engagement, structural biology, and mutagenesis of the binding site. The in vivo studies using Ammo A represent the first application of this compound class in vivo and demonstrate that ammocidin family compounds are bioavailable, well-tolerated, and efficacious at suppressing leukemia growth in human xenografts. Combined with the structural insights and past work on the SAR of apoptolidin family compounds, this work establishes a path towards the therapeutic development of apoptolidin family glycomacrolides to address OXPHOS dependency in cancer and to study the reliance of cellular OXPHOS in human physiology and disease.
Materials and Methods
Cell Culture.
MV-4–11, HEK-293T, and H-292 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). K562 cells were kindly provided by Dr. Gregor Neuert respectively and were validated by STR profiling at the University of Arizona genetics core. BaF3 cells were a kind gift of Dr. Christine Lovly, Vanderbilt University Medical Center. HEK-293-LP cells were a kind gift of Kenneth Matreyek, Case Western Reserve University. Cell lines were tested for mycoplasma contamination using the ATCC Universal Mycoplasma detection kit before use. MV-4–11 and K562 cells were maintained with Gibco IMDM Glutamax media supplemented with 10% FBS and 1% Pen/Strep. H292 cells were maintained with RPMI 1640 supplemented with 10% FBS and 1% Pen/Strep. HEK-293 cells were cultured in DMEM media, 10% FBS, 1% Pen/Strep. Cells grown with galactose media were grown in glucose free DMEM media supplemented with 10% FBS, 1% Pen/Strep, and 10 mM galactose. Cells were maintained at a density of 5 ×104- 1 × 106, except where single cells were isolated to generate clonal populations.
Molecular Biology.
All plasmids were prepared using commercial MiniPrep kits (Qiagen, Hilden, Germany). All enzymes were obtained from New England Biolabs and used per the manufacturer’s recommendations. PCR reactions were carried out using Q5 High-Fidelity Polymerase (New England Biolabs, Ipswich, MA) per the manufacturer’s protocols. Primers were ordered from Genosys (Sigma-Aldirch, Burlington, MA) or Integrated DNA Technologies (Coralville, IA). Except where noted, genomic DNA was extracted using QuikExtract DNA reagent (Lucigen, Middleton, WI). Except where noted, non-viral plasmids were maintained in competent DH5-alpha E. coli, prepared by the Vanderbilt Molecular Cell Biology Resource (MCBR), lentiviral and retroviral plasmids were maintained in Stbl2 E. coli (ThermoFisher, Waltham, MA), and cloned plasmids were initially transformed into competent cells included with kits. Gibson assemblies were carried out using the NEB Gibson Assembly Cloning kit. Site Directed Mutagenesis was carried out using the QuikChange Lightning II cloning kit (Agilent, Santa Clara, CA). PCR cloning was carried out using the NEB PCR cloning kit. All Sanger sequencing was completed by Genewiz, Inc (South Plainfield, NJ).
Immunoblotting.
Cell lysates were prepared using RIPA buffer (see lysis and click chemistry methods below) and run on SDS Tris-Glycine gels (Precast 4 – 20% Bio-Rad, or hand-casted 20%). Gels were transferred to low-background PVDF membranes using the BioRad TransBlot Turbo system and blocked with 5% milk powder in TBS + 0.1% Tween 20 (TBST) for 1 hr at room temperature. Primary antibodies were diluted at 1:1000 unless otherwise specified in 5% BSA in TBST + 0.1% sodium azide and incubated overnight at 4 °C. Membranes were washed for 5 min x 5 with TBST, and appropriate Starbright Blue 700 secondary antibody (Bio-Rad, Hercules, CA) for 1 hr at room temperature, followed by 5 × 5 min washes with TBST. Membranes were imaged using the ChemiDoc MP imaging system. Antibodies used for immunoblotting were: ATP5B (Proteintech, 17247–1AP), VCAC1 (CST #4661), TIMM17B (Proteintech 11062–1-AP), FLAG® (Sigma Aldrich, M2), ATP5G1 (Abcam, EPR13908), Actin (BioRad, 12004163).
Photoaffinity Labeling.
~5 – 30M cells per condition were washed twice with OptiMEM media and resuspended in 1 mL of OptiMEM media in a 12 well plate. Each well was treated with vehicle (DMSO) or competitor as indicated for 1 hr at 37 °C followed by the addition of vehicle or photoaffinity probe for 1 hr at 37 °C (maximum DMSO concentration of 0.5% v/v). The samples where then irradiated with 365 nm light for 15 min on ice using a Stratalinker 2400 (Agilent). The samples were then washed twice with 1 mL of 1x PBS and then lysed as described below or flash frozen at −80 °C for later processing.
Lysis and Click Reaction.
Cell pellets were lysed in 100 μL of radioimmunoprecipitation assay buffer (RIPA: 150 mM NaCl, 50 mM Tris pH 7.5, 1 % Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS). After lysis, the samples were centrifuged for 1 min at 28,000 g and the lysates transferred to a microcentrifuge tube. Protein concentration was determined using the BCA assay (Pierce) and each sample was adjusted to a concentration of 1 mg/mL. Each 100 – 1,000 μg click reaction was carried out at the following final concentrations: 100 μM azide (as indicated: Azide Fluor 545 – Sigma Aldrich OR TAMRA-Azide-Desthiobiotin – Broadpharm), 800 μM CuSO4, 1.6 mM BTTAA ligand (Click Chemistry Tools), and 5 mM sodium ascorbate. The azide was added directly to the cell lysate while the CuSO4, BTTA, and sodium ascorbate (fresh) were prepared as a master mix and added to each tube. The samples were vortexed briefly and incubated at 37 °C for 1 hr. The proteins were precipitated using a methanol-chloroform precipitation using a 3:1:4 ratio of methanol, chloroform, and water. The pellet was washed once with 3:1 MeOH/CHCl3, and once with methanol and dried briefly at room temp, taking care to avoid over drying the pellet. Samples used for in-gel analysis were resuspended in 1 x loading buffer + BME (Bio-rad), heated for 15 min at 37 °C followed by sonication for 15 min to aid solubilization. The samples were boiled for 5 min at 95 °C, centrifuged at 20,800 g x 1 min, and 10 μg of protein was loaded into each lane. Samples used for affinity purification were processed as described below.
Affinity Enrichment.
Protein pellets functionalized with the TAMRA-Azide-Desthiobiotin (Click Chemistry Tools, Scottsdale, AZ) were precipitated by methanol/chloroform as described above and resuspended in 500 μL of 6M Urea + 25 mM ammonium bicarbonate, with 140 μL of 10% SDS was added to aid resolubilization. 6 mL of 1 x PBS was added to each sample to lower the urea concentration to < 0.5 M. 50 μL of high-capacity streptavidin beads (Pierce, Waltham, MA) were washed three times with PBS, added to each sample, and incubated for 2 hrs at room temperature on a rotator. Each sample was then loaded onto a 0.8 mL spin column (Pierce) in 500 μL portions using a vacuum manifold. The beads were then washed with 4 × 500 μL volumes of 1% SDS in PBS, 4 volumes of 4M Urea in PBS, 4 volumes of 1M sodium chloride, and 1 volume of 1% SDS in PBS. The bead slurry was then resuspended in 100 μL of 50 mM biotin, 1% SDS, in PBS (pH 7.2), incubated at 37 °C for 10 minutes, agitated for 10 minutes, and then collected into a fresh 1.5 mL Eppendorf tube via centrifugation at 5200 g x 3 min. The biotin elution was repeated once with a second 100 μL volume of 50 mM biotin, 1% SDS in PBS. The two biotin elution fractions were combined and precipitated with methanol/chloroform as described above and resuspended in either 1 x loading buffer (if running a gel and silver staining) or in 0.1% RapiGest SF (Waters Corp, Milford, MA) in 0.1 M HEPES pH 8.0 if proceeding to MudPIT proteomics. Silver staining was carried out using the Pierce Silver Stain kit, per the manufacturers protocols.
TMT-Multiplexed Proteomics Sample Preparation.
TMT samples were prepared as follows: RapiGest resuspended samples (in 50 μL) were reduced by the addition of 0.5 μL 0.5 M TCEP (final concentration, 5 mM) for 30 min at room temperature and cysteines alkylated using 1 μL 0.5 M iodoacetamide (final concentration, 10 mM) for 30 min at room temperature protected from light. Samples were digested by addition of 0.25 μg of sequencing grade trypsin dissolved in 50 mM acetic acid at 0.5 μg/mL (Promega) overnight at 37 °C with vigorous shaking. Sample volume was adjusted to 60 μL with H2O and TMT labeling was carried out using the TMT sixplex isobaric label (Thermo Scientific) using 100 μg of reagent dissolved in 100% acetonitrile per sample and incubated at room temperature for 1 hr. The labeling reaction was quenched with 4 μL of 10% w/v of ammonium bicarbonate (final concentration of 0.4% w/v) at room temperature for 1 hr. Samples were then acidified with formic acid (final conc 5% v/v) and concentrated in vacuo to ~1/6th of its original volume to remove the acetonitrile and sample volume was adjusted to 600 μL with 0.1% formic acid. Samples were then heated at 42 °C for 30 min to precipitate the RapiGest surfactant and insoluble components were removed by centrifugation at 28,000 g x 30 min. Digested, TMT-labeled samples were stored at −80 °C until analysis.
In-gel Digest Proteomics Sample Preparation.
Gel bands were excised and washed three times with 25 mM ammonium bicarbonate in 1:1 acetonitrile/water, and then dried using a vacuum centrifuge. Proteins were reduced with the addition of 10 mM DTT in 25 mM ammonium bicarbonate in 10% acetonitrile (v/v), and heating at 56 °C for 1 hr. Next, proteins were alkylated with 55 mM iodoacetamide at room temperature for 45 minutes. Gel pieces were then rinsed with 25 mM ammonium bicarbonate is aqueous solution, followed by 25 mM ammonium bicarbonate in 1:1 acetonitrile/water. Rinses were repeated and gels pieces were dried using a vacuum centrifuge. Enough trypsin (12.5 ng/μL in 25 mM ammonium bicarbonate in aqueous solution) was added to cover the gel pieces and samples were incubated on ice for 30 min. Excess trypsin solution was removed and 25 mM ammonium bicarbonate in aqueous solution was added. Samples were then incubated at 37 °C for 8 hrs. After digestion, three volumes of water were added to the gel samples and they were vortexed for 10 min, sonicated for 5 min, and the resulting supernatant was collected. Gel pieces then had 45% water/50% acetonitrile/5% formic acid (v/v/v) added and were vortexed for 10 min, sonicated for 5 min, and the resulting supernatant was collected. The extraction with 45% water/50% acetonitrile/5% formic acid (v/v/v) was repeated and supernatant was again collected. Samples were concentrated using a vacuum centrifuge and analyzed.
Liquid Chromatography – Tandem Mass Spectrometry.
MudPIT microcolumns were prepared and peptide samples were directly loaded onto the columns using a high-pressure chamber. Samples were then desalted for 30 min with buffer A (95% water, 4.9% acetonitrile, 0.1% formic acid v/v/v). LC-MS/MS analysis was performed using a Q-Exactive HF (Thermo Fisher) mass spectrometer equipped with an Ultimate-3000 RSLCnano system (Thermo Fisher). MudPIT experiments were performed with 10 μL sequential injections of 0, 10, 30, 60, and 100% buffer C (500mM ammonium acetate in buffer A), followed by a final injection of 90% buffer C with 10% buffer B (99.9% acetonitrile, 0.1% formic acid v/v) and each step followed by a 130 minute gradient from 5% to 80% B with a flow rate of 300 nL/min on a 20 cm fused silica microcapillary column (ID 100 μm) ending with a laser-pulled tip filled with Aqua C18, 3 μm, 125 Å resin (Phenomenex). Electrospray ionization (ESI) was performed directly from the analytical column by applying a voltage of 2.0 V with an inlet capillary temperature of 275 °C. Data-dependent acquisition of mass spectra was carried out by performing a full scan from 300–1800 m/z with a resolution of 60,000. The top 15 peaks for each full scan were fragmented by HCD using normalized collision energy of 38, 0.7 m/z isolation window, 120 ms maximum injection time, at a resolution of 15,000 scanned from 100 to 1800 m/z and dynamic exclusion set to 60 s. Peptide identification and TMT-based protein quantification was carried out using Proteome Discoverer 2.3. MS/MS spectra were extracted from Thermo Xcalibur .raw file format and searched using SEQUEST against a Uniprot human proteome database (released 03/2014 and containing 20337 entries). The database was curated to remove redundant protein and splice-isoforms and supplemented with common biological MS contaminants. Searches were carried out using a decoy database of reversed peptide sequences and the following parameters: 10 ppm peptide precursor tolerance, 0.02 Da fragment mass tolerance, minimum peptide length of 6 amino acids, trypsin cleavage with a maximum of two missed cleavages, dynamic methionine modification of 15.9949 Da (oxidation), static cysteine modification of 57.0215 Da (carbamidomethylation), and static N-terminal and lysine modifications of 229.1629 Da (TMT sixplex).
Statistical Analysis of TMT Proteomics.
DataProtein matches and TMT quantification were processed using the DEP pipeline 49. Briefly, raw TMT quantifications were normalized using the vsn package. Differential enrichment was determined using limma. Enriched proteins were selected based on an adjusted p-value of 0.05 and log2 fold change of 2 (See Supplementary Table 1). Plots were generated using ggplot2.
MTT Viability Assay.
The MTT assay protocol was adapted from Deguire et al.19 Briefly, a 100 μL of suspension cells at 100,000 per mL were added to wells of a microtiter plate precoated with 0.5 μL of test compound at 200x in DMSO and incubated for 48 – 72 hrs (as noted). MTT reagent was dissolved in fresh media at 1 mg/mL and 100 μL was added to each well to achieve a final concentration of 0.5 mg/mL and incubated for 2 hrs at 37 °C. Cells were centrifuged at 800 g x 5 min and decanted. MTT crystals were redissolved in 100 μL of DMSO, allowed to incubate for 5 min at RT, and read at 560 nM using a SpectraMax plus 384 plate reader (Molecular Devices, San Jose, CA). Absorbance values were normalized by background subtraction (wells without cells) and normalized such that vehicle treated cells had a viability of 1.0. Concentration response curves were fit using the DRC R package with a four-parameter log-logistic function. Statistical testing of differences between concentration response curves was carried out using the EDcomp function with default parameters for comparison of IC50s and the paramcomp function for comparison of other parameters.
Sulforhodamine B (SRB) Proliferation Assay.
HEK-293 or H292 cells were treated as above (MTT assay) in TC treated 96 well plates with 100 μL of media. After 48 – 72 hrs, cells were fixed by the gentle addition of 25 μL of cold 50% (w/v) trichloroacetic acid (TCA) and incubated at 4 °C for 1 h. Plates were then washed 4x by submersion in a basin of tap water and allowed to dry overnight. Cells were stained by the addition of 50 μL of 0.04% (w/v) SRB dissolved in 1% (v/v) acetic acid to each well and allowed to incubate at room temperature for 1 hr. The SRB solution was decanted and plates were washed with 4× 100 μL volumes of 1% (v/v) acetic acid and allowed to dry. The SRB reagent was redissolved by the addition of 10 mM Tris base (pH 10.5), allowed to incubate for 10 min, and absorbance was measured at 510 nM using a SpectraMax plus 384 plate reader (Molecular Devices, San Jose, CA). Absorbance values were normalized by background subtraction (wells without cells) and normalized such that vehicle treated cells had a viability of 1.0. IC50 curves were fit using the DRC package with a four-parameter log-logistic function.
Confocal Microscopy.
Coverslips, (22 mm diameter, 1.5H thickness) (Thorlabs) were washed with 1M HCl for 1 hrs at 55 °C, washed with DI water and 70% ethanol, and coated with poly-D-lysine (100 μg/mL in sterile water), and allowed dry. Adherent cells were plated on top of coverslips and grown till confluent. For imaging of Apop A PA localization, cells were treated with 200 μM Apop A PA for 1 h and photocrosslinked for 15 min as described under ‘photoaffinity labeling’. Mitotracker Deep Red FM (Invitrogen) was added 30 min prior to the end of the experiment at a final concentration of 100 nM. Cells were washed 2x with PBS (containing Ca++ and Mg++) and fixed with 1.6% PFA for 15 min. Apop A PA treated cells were permeabilized with ice cold methanol for 20 min at −20 °C, while cells intended for immunofluorescence were permeabilized with 1% Triton X100 for 15 min. Cells were washed with 1% BSA in PBS twice, while cells intended for antibody staining were then blocked with 5% BSA in PBS. Azide Fluor 545 labeling was carried out as described under ‘lysis and click reaction’ with the azide concentration reduced to 100 μM, and samples were washed with 1% BSA in PBS after 1 hr at 37 °C. Samples for immunofluorescence were stained for overnight with primary antibodies, washed 4x with 1% BSA in PBS, followed by 1 hr staining at room temperature with secondary antibodies (goat anti-mouse alexa 488 antibody, Invitrogen A-11008), followed by 4x washes with 1% BSA in PBS. Samples were stained briefly with DAPI (1 μg/mL) mounted on slides using ProLong Glass mountant (Invitrogen), allowed to set for 72 hrs, imaged using a Zeiss 880 Airyscan confocal microscope, and processed using Zeiss Zen software.
Yeast growth and ATP synthase purification.
ATP synthase was purified from yeast strain USY006 50 bearing 6×His tags at the N-termini of the β subunits as described previously but with the following modifications 51. Yeast was cultured in an 11 L fermenter (New Brunswick Scientific) and the mitochondria were prepared by breaking yeast cell wall by bead beating. All subsequent purification steps were performed at 4 °C. Mitochondria were washed with phosphate buffer (50 mM sodium phosphate, pH 9.0, 5 mM 6-aminocaproic acid, 5 mM benzamidine, 1 mM PMSF) for 30 min before being collected by centrifugation at 184,000 g for 30 min. Membranes were solubilized by resuspending in buffer (50 mM Tris-HCl, pH 7.4, 10% [v/v] glycerol, 1% [w/w] dodecyl-β-D-maltoside [DDM, Anatrace], 5 mM 6-aminocaproic acid, 5 mM benzamidine, 1 mM PMSF) and mixed for 1 hour. Insoluble material was removed by centrifugation at 184,000 g for 30 min and the supernatant containing solubilized protein was collected. Imidazole was added to 40 mM and NaCl to 300 mM and the sample was loaded onto a HisTrap HP 5 mL column (MilliporeSigma, Burlington, MA) equilibrated with wash buffer (50 mM Tris-HCl, pH 7.4, 10% [v/v] glycerol, 0.05% [w/w], 40 mM imidazole, 300 mM NaCl, 5 mM 6-aminocaproic acid, 5 mM benzamidine, 1 mM PMSF). The column was washed with 5 column volumes of wash buffer and ATP synthase was eluted with the wash buffer containing 300 mM imidazole before being loaded to a Superose 6 increase column (MilliporeSigma) equilibrated with buffer (20 mM Tris-HCl, pH 7.4, 10% [v/v] glycerol, 0.05% [w/w], 100 mM NaCl, 5mM MgCl2). Fractions containing ATP synthase were pooled and protein was concentrated to ~15 mg/ml prior to storage at −80 °C.
Yeast F1-ATPase Preparation.
Preparation of F1-ATPase followed the same protocol as intact ATP synthase preparation until the mitochondrial were pelleted after bead beating. Protocols were derived from Mueller, 2004 50. Unless otherwise noted, all steps were done at 4 °C. Submitochondrial particles (SMPs) were prepared by resuspending mitochondria at 10 mg/mL in sonication buffer (SB - 0.25 M sucrose, 50 mM phosphate buffer, 5 mM 6-aminocaproic acid, 5 mM benzamidine, 1 mM PMSF, pH 7.5) with 1 mM EDTA and sonicated at 150W × 1 min in 5 s on 10 s off cycles, and centrifuged at 5000 g for 10 min. The supernatant containing SMPs was centrifuged at 100,000 g for 60 min at 4 °C. The pellet was washed twice with SB without EDTA, resuspended at 20 mg/mL, and warmed to room temperature. F1 ATPase was extracted from SMPs by the addition of 0.5 volumes of PBS saturated chloroform and vortexed for 30 s. ADP was added at a final concentration of 2 mM and the phases were separated by centrifugation at 3000 g for 10 min at 15 °C. The aqueous layer was collected, methanol was added to a final concentration of 10% v/v, followed by the addition of Buffer D (10% MeOH, 1.2 M NaCl, 40% [w/v] glycerol, 0.25 M sucrose, 50 mM phosphate, 5 mM 6-aminocaproic acid, 5 mM benzamidine, pH 7.5). The crude F1 preparation was loaded onto a 5mL HisTrap HP 5 mL column at 4 °C and washed with 300 mL of 97 % wash buffer (10% MeOH, 10% glycerol, 0.3M NaCl, 0.25 M sucrose, 50 mM phosphate, 5 mM 6-aminocaproic acid, 5 mM benzamidine, 1 mM PMSF, pH 7.5) + 3% elution buffer (wash buffer + 0.4 M imidazole). F1 ATPase was eluted with 100% wash buffer and the F1-containing fractions were pooled and concentrated to < 1 mL. F1 ATPase was further purified by means of gel-filtration at room temperature with a Superdex 200 increase 10/300 GL column with SDX buffer (0.25 M sucrose, 0.2 M NaCl, 50 mM Tris, 1 mM EDTA, 1 mM ATP, 0.5 mM PMSF, pH 8.0) at 0.5 mL/min, the F1 containing fractions were pooled, and precipitated by the addition of saturated ammonium sulfate (70%), and stored at 4 °C.
CryoEM Grid Preparation.
CryoEM grids of drug-free ATP synthase were prepared by removing glycerol from the sample with Zeba Spin Desalting Column (Thermo Fisher Scientific) immediately before freezing. Glycerol-free ATP synthase was then applied to home-made52 holey-gold grids that had been glow discharged in air for 2 min. Grids were blotted with modified Vitrobot Mark III (Thermo Fisher Scientific) for 26 s with ~100% humidity at 4 °C before being plunge frozen in a liquid ethane/propane mixture53. Grids of ATP synthase with ammocidin and apoptolidin were prepared the same way except that ATP synthase was incubated with either 60 μM ammocidin or 20 μM apoptolidin for 1 hour and the glycerol removal step was performed with buffers containing the same concentrations of inhibitors.
CryoEM Data collection.
CryoEM movies of yeast ATP synthase bound to ammocidin and apoptolidin were collected with a Titan Krios G3 electron microscope operated at 300 kV and equipped with a prototype Falcon 4 camera (Thermo Fisher Scientific). Automatic data collection was performed with EPU (Thermo Fisher Scientific). For the ammocidin dataset, 4345 movies each containing 30 fractions were collected at a nominal magnification of 75000×, corresponding to a calibrated pixel size of 1.03 Å. The total exposure and the camera exposure rate were ~45 e-/Å2 and 5.0 e-/pix/s, respectively. The apoptolidin dataset, consisting of 4019 movies with 29 fractions each were collected at same magnification as the ammocidin dataset. The total exposure and the camera exposure rate for this dataset were ~43 e-/Å2 and 4.8 e-/pix/s, respectively. The drug-free ATP synthase dataset was collected on a Tecnai F20 electron microscope (Thermo Fisher Scientific) operated at 200 kV and equipped with a K2 Summit camera (Gatan). A dataset consisting 190 movies each containing 30 fractions was manually collected at a nominal magnification of 25000×, corresponding to a calibrated pixel size of 1.45 Å. The total exposure and the camera exposure rate were ~36 e-/Å2 and 5.0 e-/pix/s, respectively.
Image analysis.
Image analysis was performed with cryoSPARC v2 54 except where noted. Movie frames were aligned with MotionCor2 55 using a 7×7 grid and contrast transfer function (CTF) parameter were estimated in patches. Two templates displaying a sideview and an oblique view of ATP synthase for particle picking were generated with 2D classification of manually selected particles with a box size of 320×320. Template picking generated 654,097 particle images for the ammocidin dataset, 1,189,085 particle images for the apoptolidin dataset and 38,514 particle images for the drug-free dataset. After cleaning by 2D classification, 329,297, 621,136, and 34,035 particle images remained for the corresponding datasets. Ab initio 3D classification and heterogeneous refinement identified three classes for each dataset corresponding to the three main rotational states of ATP synthase. For each dataset, the maps of the three states were aligned based on the F1 region and a masked local non-uniform refinement 56 of the region was performed. For the high-resolution ammocidin and apoptolidin datasets, individual particle defocus parameters were estimated, and a second masked refinement was performed with updated CTF parameters. Image parameters were then converted to Relion 3.0 format with the pyem package and individual particle motion was re-estimated with Bayesian Polishing. For the apoptolidin dataset, particle images were downscaled to a pixel size of 1.2875 Å with Relion 3.0. The resulting particles were imported back to cryoSPARC v2 and another round of masked non-uniform refinement of the F1 region was performed. For the ammocidin and apoptolidin datasets, individual particle defocus was re-estimated, followed by a final round of masked refinement. The final maps of the ammocidin, apoptolidin, and drug-free dataset contain 289,501, 477,847 and 34,035 particle images and reached resolutions of 3.1 Å, 3.3 Å and 4.2 Å, respectively. The ammocidin and apoptolidin maps were locally sharpened with a COSMIC2 implementation of DeepEMhancer.
Atomic model building.
To model the F1 region of the ammocidin-bound structure, individual subunits of the F1 region of a yeast ATP synthase crystal structure (2XOK) was fit rigidly into the ammocidin map with UCSF Chimera. The model was then manually adjusted in Coot and ISOLDE before being refined in Phenix. The model of the apoptolidin bound structure was built in a similar manner, with the ammocidin model as the starting structure. To demonstrate the conformational change induced upon inhibitor binding, individual subunits of the same crystal structure (2XOK) were fit into the drug-free map as rigid bodies to generate Figure 2D and Movie 1. Figures and supplementary movie were generated with UCSF Chimera and USCF ChimeraX. Molecular docking was performed with AutoDock Vina.
Isolation of Mouse Liver Mitochondria.
All steps were performed on ice. Freshly harvested mouse livers with the gallbladder removed were washed four times in mitochondrial isolation buffer (IB = 0.2 M sucrose, 10 mM Tris-MOPS, 1 mM EGTA/Tris, pH 7.4), weighed, and cut into small pieces with scissors. Liver pieces were resuspended in 2 volumes of IB, transferred to a 45 mL Potter-Elvehjem homogenizer, and homogenized with four strokes of the pestle. The homogenate was diluted with an additional 3 volumes of IB, transferred to a 50 mL conical vial, and centrifuged at 600g x 10 min at 4 °C to pellet nuclei. The supernatant was transferred to a 50 mL Nalgene Oak Ridge Centrifuge Tube and centrifuged at 7000g x 10min at 4 °C in a Sorvall RC 2-B centrifuge equipped with an SA-600 rotor to pellet mitochondria. The mitochondrial pellet was resuspended in 5 volumes of IB and centrifuged at 7000g x 10 min at 4 °C a second time to remove microsomes. The mitochondrial pellet was resuspended at 20 mg/mL as determined using a BCA assay (Pierce).
Enzyme Coupled ATP Synthesis Assay.
The respiration buffer (RB) consisted of 240 mM mannitol, 100 mM KCl, 1mM EGTA, 20 mM MgCl2, 10 mM KH2PO4, and 0.1% (w/v) fatty acid free BSA (pH 7.2). 20 mM Succinate, pH 7.2 with KOH (S3674) was used as a substrate in all experiments. The assay readout consisted of 5 mM glucose, 2.5 U/ml hexokinase (Sigma H4502), 2.5 U/ml glucose-6-phosphate dehydrogenase (Sigma G8529), and 1.6 mM NADP+(Sigma N8035). 20 μM P1,P5-di(adenosine-5) pentaphosphate (AP5A) was used to inhibit adenylate kinase which could otherwise convert 2 ADP → AMP + ATP. The assay reagents were prepared at 2 × in RB and 100 μL was dispensed into each well of UV transparent 96 well plate. 50 μL of 4x succinate was then added, followed by 25 μL of 8x mitochondria. The plate was incubated at 25 °C for 5 min to allow the mitochondria to energize. The reaction was initiated by the addition of 25 μL of 8x ADP using a multichannel pipette to yield a total volume in the well of 200 μL. Absorbance was monitored at 340 nM using a SpectraMax Plus 384 plate reader (Molecular Devices) set at 25 °C using Softmax Pro 4.3 acquisition software.
Enzyme Coupled ATPase Assay for F1 ATPase.
The assay buffer consisted of 60 mM Tris-Acetate pH 7.8; 1 mM MgCl2, 2.5 mM phosphoenolpyruvate; 1 mM KCN. The working solution was prepared at 2 × for a final concertation of 0.4 mM NADH; 3 U/mL of pyruvate kinase (PK) + 4.5 U/mL Lactate Dehydrogenase (LDH) [Sigma P0294]; ±10 μM carbonylcyanide p-trifluoromethoxyphenylhydrazone (FCCP). Unless otherwise specified, ATP was added as a 4x solution at 200 μM. Absorbance was monitored at 340 nm using a SpectraMax Plus 384 plate reader (Molecular Devices) set at 25 °C using Softmax Pro 4.3 acquisition software. 50 μL of enzyme was added to 100 μL of working solution and monitored for 2 min, 50 μL of ATP was then added for a final volume of 200 μL and absorbance was monitored over 10 minutes.
Enzyme Coupled ATPase Assay for ATP synthase.
To measure the ATP hydrolysis activity of ATP synthase, enzyme-coupled ATPase activity assays were performed in 96-well plates with 160 μL buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 10% [v/v] glycerol, 5 mM MgCl2, 0.2 mM NADH, 2 mM ATP, 1 mM phosphoenol pyruvate, 3.2 units pyruvate kinase, 8 units lactate dehydrogenase, 0.05% [w/v] DDM, DMSO [v/v] 2%, 2 nM ATP synthase). NADH concentration was monitored at 24 °C with a Synergy Neo2 Multi-Mode Assay Microplate Reader (BioTek) measuring absorbance at 340 nm. Inhibition with ammocidin and apoptolidin was determined by adding different concentrations of the inhibitors to assay mixture. Assays were performed in triplicates with two independently purified batches of yeast ATP synthase.
Generation of Perceval-HR MV-4–11s.
Lentiviral particles were generated using H293T maintained in high glucose DMEM supplemented with 10% FBS, without antibiotics at approximately 50 – 60% confluence on the day of transfection. The transfection mixture consistent of 1 μg FUGW-PercevalHR (Addgene #49083), 750 ng psPAX2 (Addgene #12260), and 250 ng of pMD2.G (Addgene #12259) using FuGENE 6 (Promega, Madison, WI) per manufacturers protocol. After 16 hrs the media was replaced with DMEM supplement with 10% FBS and 1% Pen/Strep and viral supernatant was harvested at 24 hrs and 48 hrs and sterile filtered using 45 μM syringe filter. Viral media was used immediately or frozen at −80 °C for later use. MV-4–11 cells were seeded into 6 well plates in 2 mL of media. 500 μL of viral supernatant was added to each well, along with 500 μL of 36 μg/mL DEAE-Dextran in DMEM + 10% FBS + 1% Pen/Strep (final DEAE-Dextran concentration 6 μg/mL.) After 48 hours, YFP positive cells were single cell sorted into 96 well plates to generate clonal reporter lines.
Generation of HRas-G12V BaF3s.
Platinum-E (Plat-E) packing cells selected for 2 days in DMEM + 10% FBS + 10 μg/mL blasticidin + 1 μg/mL puromycin in 100 mm tissue culture treated dishes. Selection media was replaced with DMEM + 10% FBS without antibiotics overnight. A transfection mix consisting of 500 μL OPTI-MEM + 30 μL FuGENE 6 was allowed to incubate for 5 min at room temperature. 10 μg of pBABE-puro (Addgene #1764) or pBabe-puro Ras V12 (Addgene #1768) was added to the transfection mix, allowed to incubate for 10 minutes, then added to Plat-E cells and incubated overnight. The media was removed and replaced with fresh DMEM + 10% FBS + 1% PenStrep. Viral supernatant was harvested after 48 hrs, sterile filtered, and added to BaF3 cells (gift of Dr. Christine Lovly). BaF3 cells were seeded at 50,000 /mL per well of a 6 well plate in RPMI 1640 + 10% FBS + 1% PenStrep, supplemented with recombinant murine IL-3 (Gibco, PMC0035) at 1 μg/mL. 1 mL of viral supernatant and polybrene (final conc. 10 μg/mL) were added to each well, and ‘spinfected’ for 1 hr at 800g, 30 °C. The cells were allowed to incubate with virus for 48 hrs, at which point the viral media was removed and replaced with RPMI 1640 + 10% FBS + 1% PenStrep + 1 μg/mL mIL-3 + 2 μg/mL Puromycin.
ATP Synthase Mutant Library Preparation.
ATP synthase ORF clones were purchased from GenScript USA Inc (Piscataway, NJ, USA) [ATP5A1, NM001001937.1; ATP5B NM_001686.4; ATP5C NM_001001973.3] were amplified by PCR and subcloned into an attB-SCN5A-IRES-mCherry-2A-BSD plasmid (gift of Dan Roden), replacing SCN5A, by Gibson assembly. Successful assembly was confirmed by Sanger sequencing using ag122 and ag123 primers (See Supplementary Table 3). Mutagenic primers were designed in three pools, one for each ORF, using the published primer design script, specifying a single codon for each of 20 amino acids without stop codons at a total of 21 positions (420 total variants), with 30 nt homology arms on both sides of the mutated codon (See Supplementary Table 3 for primer sequences) 57. Primers were obtained as three separate 50 pm oPools from Integrated DNA technologies (Coralville, IA, USA). Nicking mutagenesis was carried out exactly as described in Wrenbeck EE, 2016 34, Supplementary Protocol 1, except for reversing the use of the Nt.BbvCI and Nb.BbvCI enzymes to degrade the proper strands. Primer ag123 was used to synthesize the complementary strand. The product of each mutagenesis reaction was transformed into XL1-Blue Electroporation-Competent Cells (Agilent Technologies, Santa Clara, CA, USA) and serial dilutions were plated to determine the total number of colonies. All colonies from the 25 cm x 25 cm LB plate were collected, and plasmid was purified by MiniPrep. The three pools were combined in a 5:8:8 ATP5A1:ATP5B:ATP5C ratio to reflect the number of variants present in each pool. A ‘wildtype’ pool was generated by combing the parental plasmids in the same 5:8:8 ratio for use as a negative control.
Mutant Library Transfection and Selection.
HEK-293T landing pad cells33 were maintained in glucose free DMEM (Gibco) supplemented with 10% heat inactivated FBS (Gibco), ±1% Pen/Strep, and 10 mM Galactose. On day −1, 500,000 cells with an ‘empty’ landing pad (LP-Neg) cells were plated into 6 well plates in antibiotic free media. On day 0, the cells were transfected with 1 μg of mutant library and 100 μg of Bxb1 expressing plasmid (pCAG–NLS–HA–Bxb1; Addgene #51271, a gift from Pawel Pelczar) in triplicate. On day 1, the cells were replated into 60 mm TC dishes with antibiotic containing media and gene expression was induced with 1 μg/mL doxycycline. On day 3, cells were treated with 100 μg/mL Blasticidin S (Gibco) and 10 nM AP1903 (MedChemExpress) to select for cells which expressed the transgene and had undergone successful integration; selection was continued for 7 days. On day 10, HEK-293-integrated cells were plated into 6 well TC plates and treated with DMSO, Apop A, Ammo A at various concentrations (final DMSO concentration 0.25% v/v). Selective pressure was applied for two rounds of 4 days of exposure to each compound, followed by 3 days for recovery in glycomacrolide free media. After selection, surviving cells were allowed to expand until confluent at which point genomic DNA was extracted using the Qiagen DNeasy Blood & Tissue Kit.
Amplicon Preparation and Sequencing.
The integrated ATP synthase genes were PCR amplified using primers designed to anneal to the landing pad (TRE3G_fwd) and plasmid IRES sequence (IRES_rev) using 5% of the genomic DNA as a template and Q5 polymerase (New England Biolabs - NEB) at 100 μL scale. The PCR product was purified using the Monarch PCR & DNA clean-up kit (NEB). All library preparation and sequencing was carried out at the Vanderbilt Technologies for Advanced Genomics (VANTAGE) center. Sequencing libraries were prepared by tagmentation using the Nextera Flex library preparation kit (Illumina Inc. San Diego, CA, USA) using the manufacturers protocol. PE150 sequencing was carried out on the Novaseq 6000 platform at VATNAGE. Data are deposited in the gene expression omnibus (GEO), GSM5224467.
Analysis of Saturation Mutagenesis Experiments.
Demultiplexed reads were down-sampled to 5M paired reads using seqtk, aligned to the reference ORFs using bwa-mem with a gap opening penalty of 100, and converted to BAM format using samtools. Variant frequency was determined using gatk version 4.1.8.1. AnalyzeSaturationMutagenesis was used for each ORF. The resulting amino acid frequency tables were analyzed using R with the tidy data deposited on GEO as a supplementary file. The log10 fold change for each amino acid was calculated by subtracting the log10 transformed frequency of each treated replicate to its parental frequency.
Generation and Validation of CRISPR lines.
Isogenic mutants were generated using CRISPR-Cas9 gene editing as described in 32, using pSpCas9(BB)-2A-GFP (PX458), a gift from Feng Zhang (Addgene plasmid # 48138). Guide sequences (see supplement) were generated by annealing complementary oligonucleotides and golden-gate cloning with BbsI into PX458 and confirmed by sanger sequencing. 101–110 nt ssODN HDR templates were ordered with 50 bp of homology on either side of the cut site. HDR templates were designed to eliminate the PAM sequence to prevent re-cutting and designed to incorporate one or more synonymous mutations to add or remove restriction sites to facilitate screening of clones. 200,000 cells were electroporated using the Neon Transfection system (ThermoFisher), with 500 ng of PX458 plasmid ± 10 pmol ssODN template. Single cells were FACS sorted into U-bottom 96 well plates into conditioned media, selecting GFP+/PI- negative cells. Clones were expanded and genotyped by PCR of the targeted region, followed by restriction digest to identify edited clones. Successfully edited clones were further confirmed by PCR cloning and sanger sequencing of 6–8 colonies. ATPIF-1 K.O. lines were generated using PX458 without a donor template as described above. Individual clones were screened using intracellular flow cytometry against ATPIF1.
In vivo Murine Modeling.
All animal experiments were conducted in accordance to guidelines approved by the IACUC at Vanderbilt University Medical Center. Male NSGS [NOD-scid IL2Rgnull3Tg (hSCF/hGM-CSF/hIL3)] mice (The Jackson Laboratory), 6 – 8 weeks old were irradiated with 100 cGy microwave radiation. Twenty-four hours later, mice were transplanted with 1 × 106 MV-4–11 cells via tail vein injections in each irradiated mouse. Mice were randomized post xenograft transplantation into cages of 5. Prior to treatment, peripheral microchimerism was documented at week 1. Upon establishing microchimerism, mice were treated with either 0.1 mg/kg or 0.03 mg/kg ammocidin in saline or saline vehicle i.p. for 5 days on, 2 days off for 2 weeks. Murine CBC was analyzed from blood collected into EDTA tubes (Greiner Bio-One) and analyzed with a Hemavet (Drew Scientific) analysis system.
Flow Cytometry.
For analysis of MV-4–11 cells expressing the Perceval HR reporter, cells were treated with compounds for 16 hrs in 96 well plates in 100 μL of media. Prior to analysis, propidium iodide was added at a final concentration of 300 ng/mL as a viability stain. Perceval-HR fluorescence was recorded on the YFP and AmCyan channels and intact, single, PI-negative cells were selected for analysis. ATP-ADP ratio was calculated by subtracting the asinh transformed YFP and AmCyan intensities. Two independent clones were analyzed as biological replicates. Cells were analyzed using a 5-laser LSR instrument (Becton Dickinson).
For analysis of pS6 suppression and ATPIF1 expression, cells were analyzed using intracellular flow cytometry multiplexed using fluorescent cell barcoding. For pS6 suppression, cells were treated for 16 hrs with the indicated compound in 96 well plates, ATPIF1 expression was measured in untreated cells. At the time of analysis, Alexa Fluor 700-NHS ester was added to each well as a viability stain (final 20 ng/mL) and allowed to incubate for 15 min, cells were fixed with 1.6% PFA for 10 min, and permeabilized with ice cold methanol for 30 min at −20 °C. Barcoding and staining was performed as described previously58 and demultiplexed using DebarcodeR.
For analysis of mouse xenografts, red blood cells were lysed with EL Buffer on ice (Qiagen), with remaining cells washed and resuspended in 1x PBS with 1% BSA and stained for 15 minutes with the following antibodies: human CD45-APC (Clone 2D1, 0.5 μg/mL) (Biolegend), human CD33-PE-Cy7 (Clone P67.6, 0.125 μg/mL) (Biolegend), murine CD45-PE (Clone 30-F11, 0.5 μg/mL) (Biolegend) and DAPI (Biolegend, 0.25 μg/mL). Cells were washed and submitted for flow cytometric analysis using a 3-laser LSRII (Becton Dickinson).
Histology and Immunohistochemistry.
Tissues were fixed in 4% paraformaldehyde for 48 hours and stored in 70% ethanol before being embedding in paraffin and sectioned at 5 μm. The bone tissue was decalcified prior to being embedded in paraffin. Sections were de-waxed in xylene and rehydrated in successive ethanol baths. Standard Mayer’s Hematoxylin and Eosin (H&E) staining was performed. Antigen retrieval using a standard pH 6 sodium citrate buffer (BioGenex) was performed and sections were stained with Monoclonal Mouse Anti-Human CD45 (Dako, M0701, dilution 1:200) using the M.O.M. Kit (Vector).
Pharmacokinetic Studies.
All animal experiments were conducted in accordance with guidelines approved by the IACUC at Vanderbilt University Medical Center. Pharmacokinetics of Ammocidin in NSGS male mice in biological triplicate were assessed in whole blood after dosing with ammocidin alone i.p. 0.5 mg/kg. Whole blood samples were collected up to in EDTA tubes for analysis of plasma. Blood plasma was mixed 1:1 with an internal standard solution consisting of 1 μM Apop A in PBS. Metabolites were extracted with 200 μL of ethyl acetate, evaporated to dryness and resuspended in 50 μL of MeOH. Ammo A concentration was determined using LC-MS (Thermo TSQ Quantum Access Max) with technical duplicates on a 50 × 1.8mm C18 column, isocratic 60/40 H2O/Acetonitrile + 10 mM ammonium acetate at 250 μL/min in ESI+ mode monitoring Ammo A (1139.7 → 208.8, CE 19 V, RT = 1.00 min) and Apop A (1146.68 → 805.46, CE 19 V, RT = 1.66 min).
Isolation of Apoptolidin A, Apoptolidin H, Ammocidin A. |
Similar methods were used to obtain all three compounds by using different producing strains. Apop A was obtained by cultivation of wild-type Nocardiopsis sp. FU-40. Apop H was obtained by cultivation of Nocardiopsis sp. FU-40 in which ApoGT2 was replaced with an apramycin resistance cassette as previously described21. Ammo A was obtained by cultivation of Saccharothrix sp. AJ9571 provided by Ajinomoto Co., Inc (Kawasaki, Japan). Each organism was plated on Bennett’s agar (0.1% yeast extract, 0.1% beef extract, 0.2% N-Z Amine Type A, 1.0% dextrose, 2.0% agar, pH 7.0) and incubated at 30 °C for 3 – 7 days until sporulation. FU-40 ΔApoGT2 was grown on plates containing Bennet’s media with the addition of apramycin, 80 μg/mL. The seed culture was initiated using spores scraped from the solid culture into 250 mL Erlenmeyer flasks containing 50 mL of seed medium (1.0% soluble starch, 1.0% molasses (Plantation Blackstrap, Unsulfured), 1.0% peptone, 1.0% beef extract, pH 7.0), and incubated for 7 days at 30 °C while shaking at 220 RPM. Production cultures were carried out in multiple 250 mL Erlenmeyer flasks containing 50 mL of production media (2.0% glycerol, 1.0% molasses, 0.5% casamino acids, 0.1% peptone, 0.4% calcium carbonate, pH 7.2) and incubated at 30 °C for 7 days while shaking at 220 RPM.
Apop A, Apop H, and Ammo A were purified using similar methods and differed only in the specific fractions collected according to their retention times. After 7 days of fermentation, the mycelia were separated from the culture broth by centrifugation at 3000 g x 30 min. The culture broth was extracted 3x with 1 volume of ethyl acetate and the combined organic layers were washed with brine, dried with Na2SO4 and concentrated in vacuo. The crude extracts were then subjected to chromatography with LH-20 resin using methanol as the mobile phase and the glycomacrolide containing fractions were identified by thin-layer-chromatography and pooled. The LH-20 fractions were then subjected to reverse phase HPLC using a Waters XBridge Prep C18 19 × 150 mm column with a 20-minute gradient from 70% A / 30% B to 20% A / 80% B, (Buffer A: 95% water, 5% acetonitrile, 10 mM ammonium acetate; Buffer B: 5% water, 95% acetonitrile, 10 mM ammonium acetate). Apop A – RT: 9.5 min, Apop H – RT: 8.0 min, Ammo A – RT 9.0 min. Fractions containing pure compounds were lyophilized using a Genevac HT-6 to yield white solids. Purity of each compound was confirmed by NMR (1H and HSQC) and HPLC/MS.
Isolation of Aurovertins B, D |
Aurovertin was obtained by cultivation of Calcarisporium arbuscula, NRRL-3705, obtained from the ARS Culture Collection, Peoria, IL, using the approach described by Baldwin 59. Spores of NRRL-3705 were plated on modified Czapek agar (3.0% sucrose, 0.5% corn steep solids, 0.1% KH2PO4, 0.05% MgSO4, 0.05% KCl, 0.01% FeSO4) and incubated at 24 °C for 10 days. The production cultures were inoculated from a single colony of fungal mycelia into Roux bottles containing 70 mL of Baldwin production medium (3.0% Dextrose, 1.0% peptone, 0.5% NaCl, 0.5% dried corn steep, 0.5% yeast extract, 0.3% beef extract, pH 6.0) and incubated at room temperature for 14 days in the dark.
The fungal mycelia were separated from the broth by centrifugation and extracted with 250 mL of acetone for 2 hrs. The acetone was concentrated in vacuo, until only water remained. The aqueous layer was extracted 3 x with CHCl3, the organic layer was washed with brine, and dried with Na2SO4 and concentrated to ~5 mL. The desired product was precipitated by the addition of pentane, filtered over Celite, and redissolved in methanol. The resulting semi-purified extract was then subjected to reverse phase HPLC using a Waters XBridge Prep C18 19 × 150 mm column with the following gradient at 10 mL /min : 0 min – 75 % A / 25 % B; 20 min – 60 % A/40 % B; 25 min – 60 % A/40 % B, 27 min – 5 %A/95 % B, 39 min – 5 % A/95 % B (Buffer A: 95 % water, 5 % acetonitrile, 10 mM ammonium acetate; Buffer B: 5% water, 95% acetonitrile, 10 mM ammonium acetate). Aurovertin D – RT: 15 min; Aurovertin B – RT: 30 min. Fractions containing pure compounds were lyophilized using a Genevac HT-6 to yield yellow solids. Purity of each compound was confirmed by NMR (H1 and HSQC) and HPLC/MS.
Isolation of Efrapeptins |
Efrapeptins were obtained by cultivation of Tolypocladium cyclindrosporium, ARSEF 962 (ATCC 42438) obtained from the American Type Tissue Culture Collection (ATCC, Manassas, VA). ARSEF 962 was plated on Sabouraud agar + yeast extract (4.0 % peptone, 1.0 % peptone, 1.0 %, 2.0 % agar, 1.0 % yeast extract, pH 5.6) and incubated at room temperature for 14 days. Production cultures were inoculated into 250 mL flasks containing 100 mL of Czapek Dox + peptone media (3.0% sucrose, 0.3 % NaNO3, 0.1 % KH2PO4, 0.05 % MgSO4, 0.05 % KCl, 0.01 % FeSO4, 0.5 % peptone, pH 5.6) 60. The fungal mycelia were separated from the broth by centrifugation and discarded. The broth was extracted 3x with dichloromethane, washed with brine, and dried with Na2SO4. The crude extract was concentrated in vacuuo, resuspended in 1 mL of methanol, and subjected to size exclusion chromatography using methanol as the mobile phase. The efrapeptin containing fractions were combined and subjected to reverse phase HPLC using a Waters XBridge Prep C18 19 × 150 mm column with the following gradient at 10 mL /min: 0 min – 70 % A / 30 % B; 20 min – 20 % A/80 %B (Buffer A: 95% water, 5 % acetonitrile, 10 mM ammonium acetate; Buffer B: 5% water, 95% acetonitrile, 10 mM ammonium acetate). Efrapeptin E – RT 16.8 min; efrapeptin F – RT 17.7 min. Fractions containing pure compounds were lyophilized using a Genevac HT-6 to yield white solids. Purity of each compound was confirmed by NMR (H1 and HSQC) and HPLC/MS.
Statistics and Reproducibility |
All samples sizes and statistical tests are described in the figure legends and accompanying methods. All enzymatic assays represent n = 3 technical replicates and representative data from one of two biological replicates. All viability assays represent n = 3 technical replicates and representative data from two biological replicates except when independent clones were tested in parallel. All gel based profiling and immunoblots were carried out as single biological replicates and the following key gels and immunoblots were verified by two or more independent biological replicates: Main Fig. 2b [lanes 1, 2, 4, 6, 7, 9]; Fig. 2f [VDAC and ATB5B], Extended data Fig. 1d [lanes 1, 4, 6]; Fig. 1e [lanes 1, 4, 7]; Fig. 1g; Fig. 2a; Fig. 3c [lanes 1, 2, 3, 4, 7]; Fig. 3f [lanes 1,2,3,4]; Fig. 3h, Fig. 5b.
Extended Data
Extended Data Fig. 1: Extended validation of glycomacrolide photoaffinity probes for target identification.
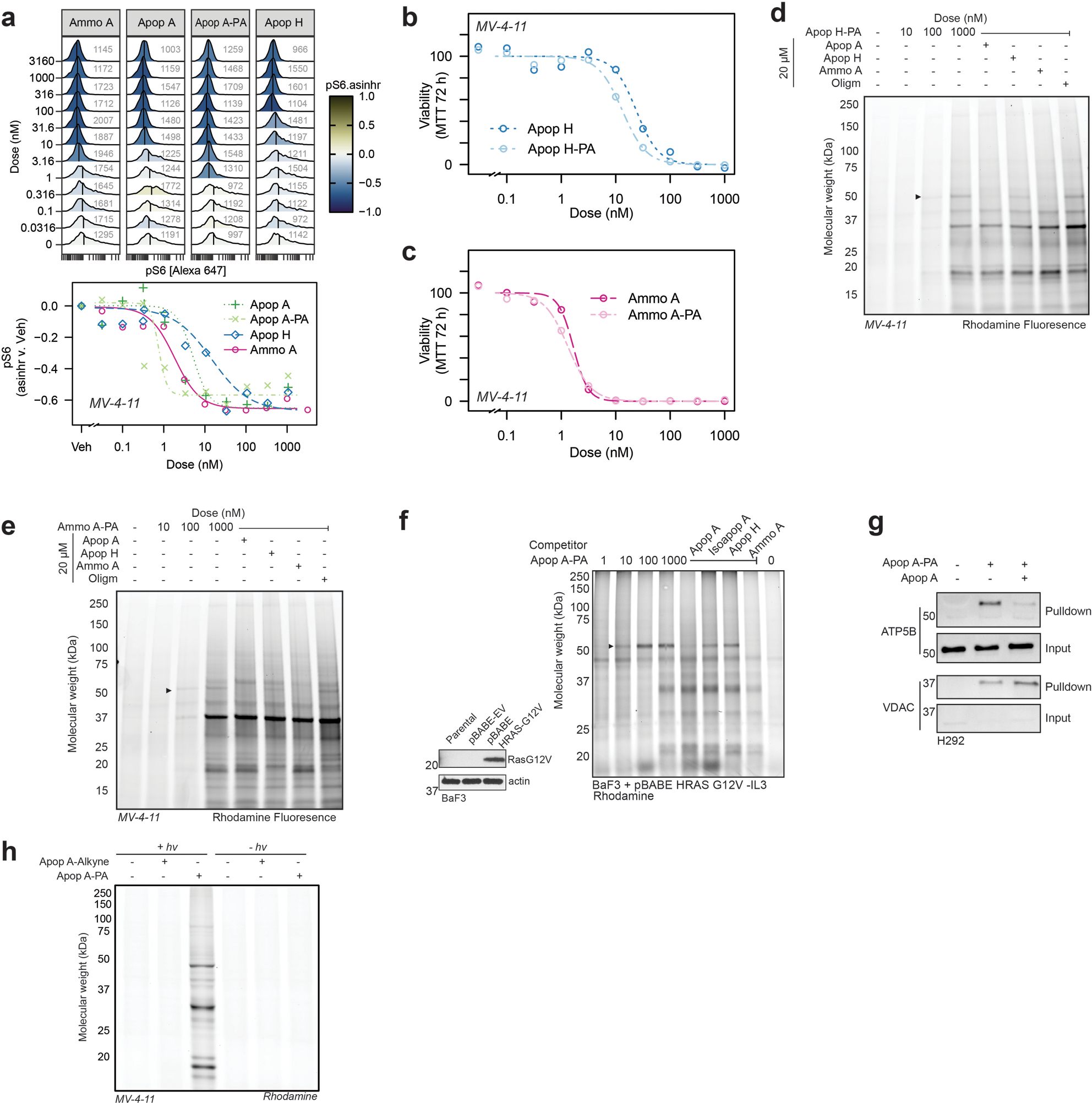
a, Analysis of pS6 phosphorylation using FCB barcoded flow cytometry of MV-4–11 cells treated for 16 h with glycomacrolides or probes; b, Concentrations response curves in MV-4–11 cells treated for 72 h with Apop H/Apop H-PA and c, Ammo A/Ammo A-PA; d, Gel-based profiling of Apop H PA or e, Ammo A-PA adducts in MV-4–11 cells; f, Gel-based profiling of Apop A-PA targets in BaF3 cells transformed with HRas-G12V or; g Immunoblot showing specific enrichment of ATP5B in H292 cells treated with Apop A-PA; h; Gel-based analysis of diazirine and UV light dependent labeling of Apop A-PA at 1 μM.
Extended Data Fig. 2: Identification of ATP5B as the target of apoptolidin A using comparative proteomics.

a, Gel-based profiling of Apop A-PA targets in MV-4–11 with ± 1 μM Apop A-PA, ± 20 μM Apop A after affinity enrichment using streptavidin resin prior to digestion and TMT labeling imaged by rhodamine fluorescence and after silver staining; b, schematic of the TMT multiplexing strategy for to combine vehicle, probe, and competition, conditions from two separate biological replicates; c, Volcano plot of proteins significantly enriched in ‘Probe’ condition compared to ‘Vehicle’ analyzed by TMT-multiplexed MuDPIT proteomics; d, Volcano plot of proteins significantly enriched in ‘Competition’ condition compared to ‘Vehicle’ analyzed by TMT-multiplexed MuDPIT proteomics; e, Coomassie stained gel-based profiling of Apop A-PA targets in MV-4–11 cells – 50 kDa band from probe and competition conditions were cut and subjected to in-gel digestion (see Supplementary Table 1); f, Immunoblot showing pulldown of FLAG tagged ATP5B expressed in H293 landing pad cells.
Extended Data Fig. 3: Analysis of apoptolidin A binding mode via photoaffinity labeling, and enzymatic assays.

a, Concentration response-curves showing inhibition of ATPase activity in purified yeast FOF1 ATP synthase, measured using PK/LDH coupled assay; b, Gel-based analysis of Apop A competition against Apop A-PA at a fixed concentration; c, Gel-based analysis of Apop A-PA in the presence of known ATP synthase inhibitors Aurovertin B (Auro B), Efrapeptin F (Efra F), Citreoviridin (Citr), and Oligomycin A (Oligm); d, Gel-based profiling of Apop A-PA in MV-4–11 cells treated with or without uncoupling agents (1 μM). e, Analysis of ADP concentration dependence on inhibition of ATP synthesis by Apop A in isolated mouse liver mitochondria using HK/G6PDH coupled assay; f, Analysis of ATP concentration dependence on inhibition of ATP hydrolysis by Apop A and Ammo A in isolated yeast F1 ATPase using a PK/LDH coupled assay; g, confirmation of ATPIF1 KO in two independent K562 clones after knockout using PX458 with two independent ATP5IF1 targeting gRNAs; h, Gel-based profiling of membrane potential dependent adduction of Apop A-PA in WT or IF1 KO K562 cells; i, comparison of ammocidin bound (left) to IF1 bound F1 ATPase.
Extended Data Fig. 4: Workflow for cryoEM image analysis and validation for ammocidin-bound and apoptolidin-bound yeast ATP synthase.
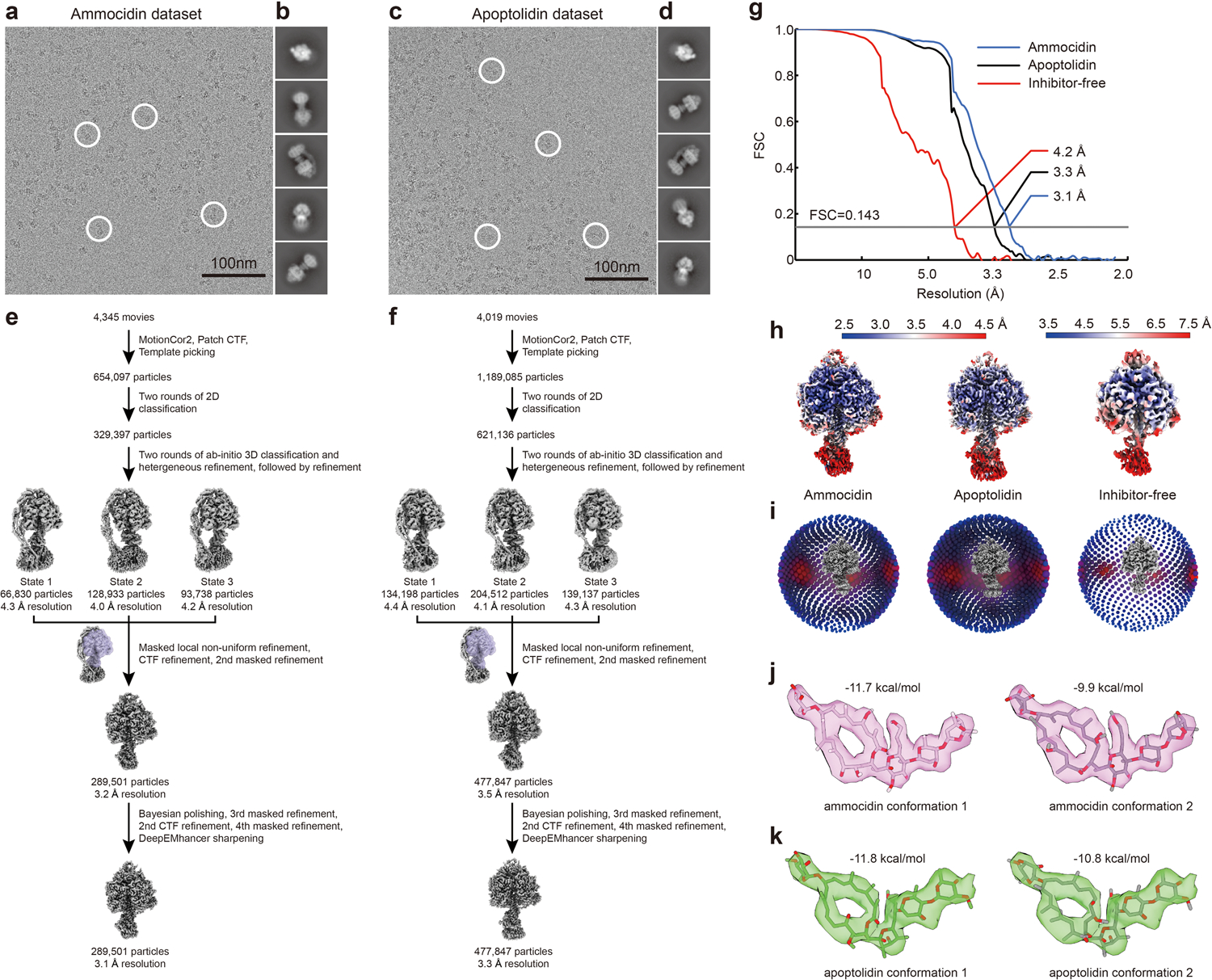
Example micrographs (a, c), 2D class average images (b, d), and workflow for obtaining maps of the F1 regions (e, f) for ammocidin-bound, and apoptolidin-bound ATP synthases, respectively. Corrected Fourier shell correlation curves after gold-standard refinement g, local resolution maps h, and orientation distribution plots i, are shown for the maps of the F1 regions of the ammocidin-bound, apoptolidin-bound, and inhibitor-free datasets. Alternative conformations of ammocidin (j) and apoptolidin (k) atomic models.
Extended Data Fig. 5: ATP5B-L394F mutation has opposing effects on apoptolidin and ammocidin sensitivity.
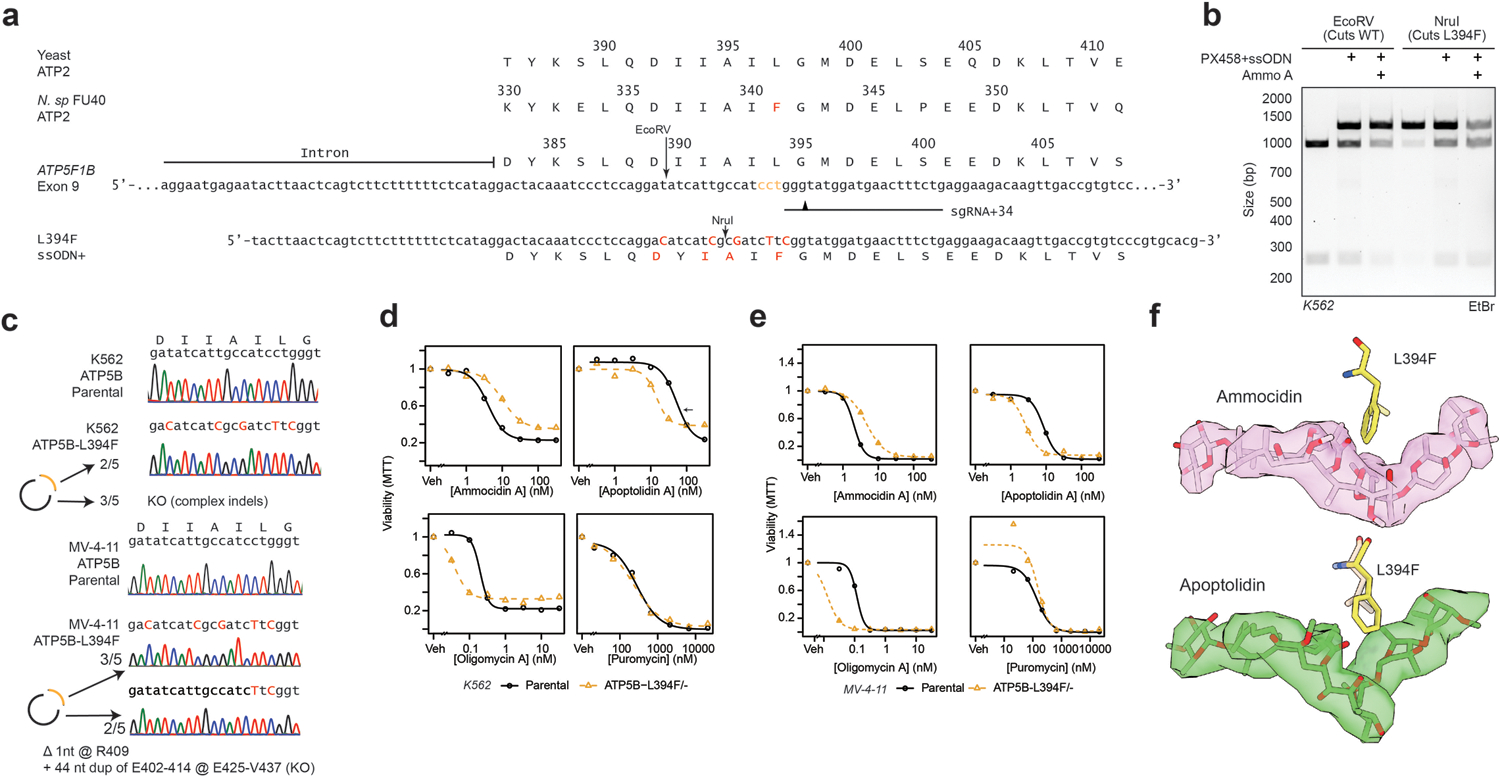
a, Multiple sequence alignment of ATP synthase β subunit C-terminal domain and CRISPR/Cas9 HDR editing strategy for human exon 9 and using plasmid (PX458) encoding sgRNA and Cas9 and ssODN repair template, PAM sites labeled in yellow, cut locations noted with arrowhead, edits noted in red, restriction sites noted; b, Restriction digest of ATP5F1B Exon 9/10 amplicons from parental K562s or PX458 + ssODN treated K562s treated with Ammo A (150 nM x 5 d) showing loss of wildtype allele and retention of edited alleles with ammocidin treatment; c, Confirmation of introduction of ATP5B-L394F mutation and KO of wildtype ATP5B in K562s and MV-4–11 cells by Sanger sequencing; d, ATP5B-L394F sensitizes cells to Apop A and provides some resistance against ammocidin in K562 and e, MV-4–11 cells as measured by MTT assay at 72 h; f, Modeling of the L394 (yeast L397) mutation on the ammocidin (top) and apoptolidin (bottom) structures showing proximity to the hemiketal.
Extended Data Fig. 6: Deep mutational scanning of the ammocidin binding site on ATP Synthase.
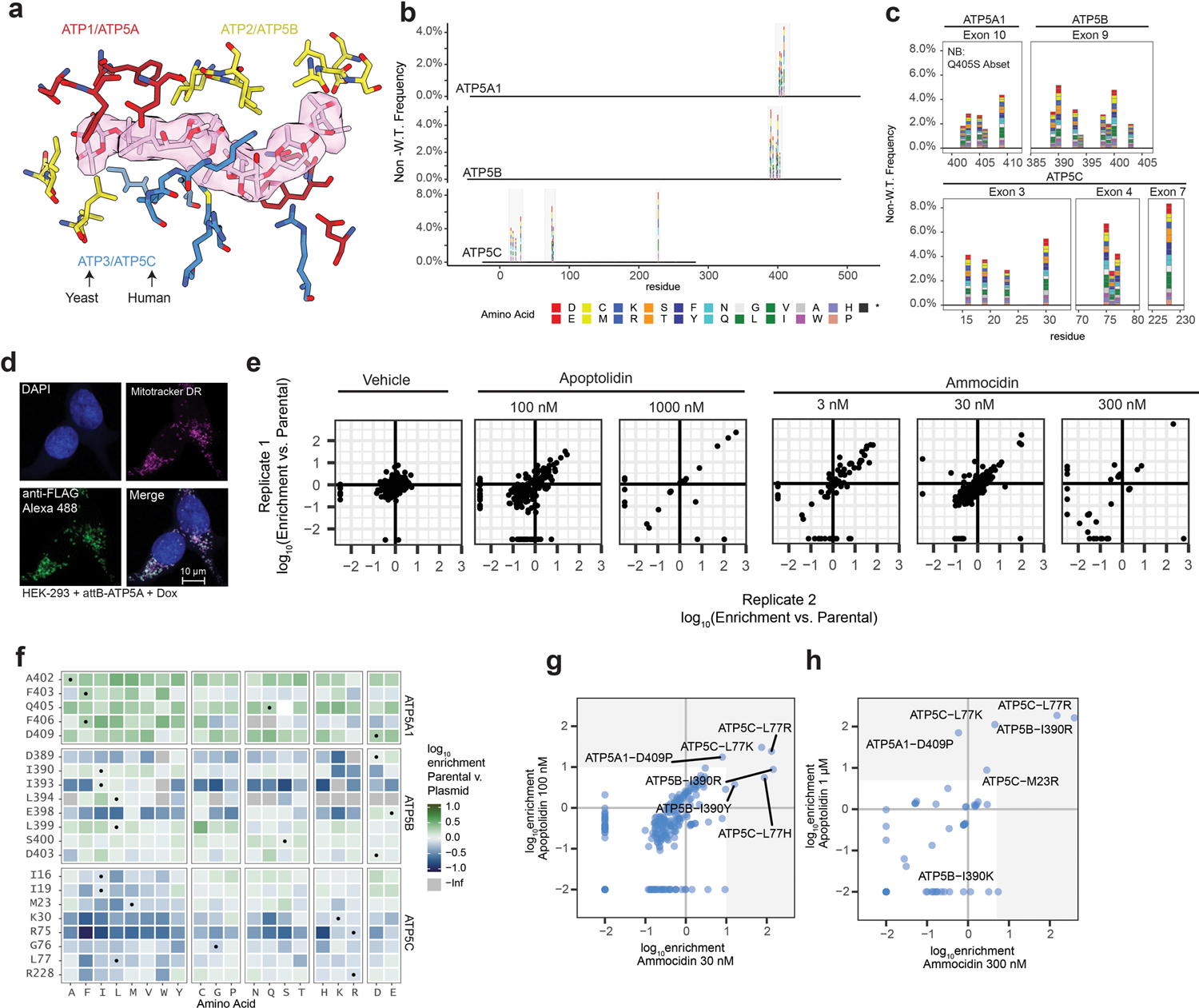
a, Selection of residues for mutagenesis on α (Yeast ATP1, Human ATP5A – red), β (Yeast ATP2, Human ATP5B – yellow), and γ (Yeast ATP3, Human ATP5C – blue) subunits within 4.5 Å of ammocidin or apoptolidin; b, c, Validation of ATP synthase variant library after nicking mutagenesis showing the frequency of non-wildtype variants at each position, with residue 1 corresponding to the first residue of the mature peptide; d, Assessment of reproducibility between biological replicates showing consistent enrichment of resistance mutations, variants which were not observed were set to −2.5; e, Confirmation of proper mitochondrial localization of the landing-pad expressed ATP5A by immunofluorescence at 63x in HEK-293T landing pad cells; f, Comparison of variant frequencies observed after selection for successful integration compared to the original library, variants which were observed in the original library but not detected after integration were set to -Inf. Variants which exhibit negative enrichment or complete loss are thought to exhibit decreased fitness relative to the WT alleles; g, Comparison of variants enriched by apoptolidin and ammocidin at medium dose (100 nM / 30 nM) or h, high (1 μM / 300 nM) demonstrating that apoptolidin and ammocidin select for similar resistance mutations.
Extended Data Fig. 7: Apoptolidin and ammocidin select for similar resistance mutations.

Tile maps of amino acid enrichment relative to the parental library at each position at each dose of ammocidin or apoptolidin; a, Vehicle treated cells were allowed to grow for one additional passage after selection, while remaining wells were passaged with Apoptolidin A at concentrations of b, 100 nM or c, 1 μM or Ammocidin A at concentrations of d, 3 nM, e, 30 nM, f, 300 nM. White squares indicate variants which were not detected (<100 counts) by deep sequencing, black circles represent WT alleles.
Extended Data Fig. 8: Validation of ATP synthase resistance mutations using CRISPR/Cas9 genome editing.

a, Validation of resistance mutations using transgenic mutant ATP synthase in HEK293LP cells at 72 h by SRB assay for mutations in the alpha, b, beta, and c, gamma subunits; d, CRISPR/Cas9 HDR editing strategy for ATP5B exon 9 and e, ATP5C exon 4 using plasmid (PX458) encoding sgRNA and Cas9 and ssODN repair template, PAM sites labeled in yellow, cut locations noted with arrowhead, edits noted in red; f, Confirmation of introduction of ATP5B-I390R in K562 cells; g, ATP5C-L77R in MV-4–11 cells, or h, ATP5C-L77R in K562 cells by sanger sequencing of single-cell clones via PCR amplicons and/or PCR cloning to resolve heterozygous edits; i, Confirmation of unchanged expression of ATP synthase genes in CRISPR/Cas9 edited clones by immunoblot; j, Secondary validation of ATP5C-L77R mutant in CRISPR edited MV-4–11 cell line using MTT-assay at 48 h; k, Gel-based profiling of Apop A-PA targets in K562s Parental or CRISPR/Cas9 edited cell lines showing loss of binding to ATP5B visualized using TAMRA-azide-desthiobiotin.
Extended Data Fig. 9: Evaluation of ammocidin dosing, pharmacokinetics and toxicity in NSGS mice and efficacy in NSGS MV-4–11 xenograft.

a, Preliminary (non-GLP) evaluation of PK profile of Ammo A in NSGS mice with a single dose of Ammo A (technical duplicate); b, Gross histology of H&E sections from organs of NSGS mice 2 weeks after treatment with Ammo A (0.1 mg/kg IP QD for 5 on, 2 off) revealing/reveal no tissue damage related toxicity; c, Engrafted mice (n = 4 per group) were treated from day 7 to day 18 (pink brackets) and therapy was then held. Mouse survival was measured by Kaplan-Meier analysis (P value calculated using two-sided log-rank test.)
Extended Data Fig. 10: Gating schemes for flow cytometry data.

Gating schemes for a. Perceval-HR reporter MV-4–11s; b, multiplexed activity profiling in MV-4–11 cells; c, ATPIF1 KO K562 cells; and d, murine xenograft chimerism experiments.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements |
We are grateful to Kenneth Matreyek, PhD of Case Western Reserve University for providing the Bxb1 landing pad cells and plasmids for the mutagenesis experiments as well as Ayesha Muhammad and Andrew Glazer, PhD for assistance applying the landing pad system. We are grateful to David Mueller, PhD of Rosalind Franklin University for providing the USY006 yeast strain and protocols for handling isolated mitochondria, as well as Pankaj Sharma, PhD for his assistance isolating F1 ATPase for enzymatic assays. We thank Kerry Brown, PhD and Hye-Young Kim, PhD for their helpful advice regarding synthesis and affinity enrichment, as well as Agnes Gorska for her assistance with immunohistochemistry experiments. The ammocidin producing strain AJ9571 was provided by Ajinomoto Co., Inc., Tokyo, Japan under a material transfer agreement.
Funding |
This work was supported by research grants from the National Institutes of Health for research (R01 GM092218 [B.O.B.], R01 CA226833 [B.J.R., B.O.B.], R35 GM133552 [L.P.], K23 HL138291 [P.B.F.]), and training support (T32 GM065086 [B.J.R., M.T.W.], T32 GM007347 [B.J.R., B.I.R.], F30 CA236131 [B.J.R.], F30 CA247202 [B.I.R.]). This work was also supported by a Canadian Institutes of Health grant PJT162186 [J.L.R.]. J.L.R. was supported by the Canada Research Chairs program and H.G. by an International Student Ontario Graduate Scholarship. M.T.W was supported by the National Science Foundation Graduate Research Fellowship Program. We are thankful for the resources provided by the following core facilities: Vanderbilt Flow Cytometry Shared Resource [supported by the Vanderbilt Ingram Cancer Center (P30 CA68485), Vanderbilt VANTAGE [VANTAGE is supported in part by CTSA Grant (5UL1 RR024975-03)] genomics core, the Vanderbilt Cell Imaging Shared Resource (CISR is supported by NIH grant DK020593), Vanderbilt Small Molecule NMR Facility core (supported in part by NIH grant S10 RR019022), and Center for Innovation Technology at Vanderbilt University. CryoEM data was collected at the Toronto High-Resolution High-Throughput cryoEM facility, supported by the Canada Foundation for Innovation and Ontario Research Fund.
Competing interests | M.R.S. receives research funding from Astex, Incyte, Takeda, and TG Therapeutics; has equity with Karyopharm; serves as an advisor or consultant to AbbVie, Astex, BMS, Celgene, Incyte, Karyopharm, Ryvu, Sierra Oncology, Takeda, and TG Therapeutics. P.B.F currently receives research funding from Incyte, and has received research funding from Astex and Forma Therapeutics in the past. Within the past 2 years, W.K.R. has received unrelated clinical research support Bristol-Meyers Squib, Merck, Pfizer, Peloton, Calithera, and Incyte
Footnotes
Code Avalibility | Code used for data analysis generation of figures is avalible at github.com/bjreisman/apoptolidin
Material Avalibility | Materials and compounds are available from the corresponding author upon request.
Data Availability |
The atomic coordinates have been deposited in the Protein Data Bank (PDB) with the accession codes 7MD2 and 7MD3. The EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) with the accession codes 23763, 23764 and 23765. The sequencing data and variant counts for the deep mutational scanning experiments have been deposited in the Gene Expression Omnibus (GEO) database under accession code GSE171362. The data generated in this study are contained in the published article including its supplementary information.
References
- 1.Akimov Y & Aittokallio T Re-defining synthetic lethality by phenotypic profiling for precision oncology. Cell Chem Biol 28, 246–256, doi: 10.1016/j.chembiol.2021.01.026 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Pavlova NN & Thompson CB The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47, doi: 10.1016/j.cmet.2015.12.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674, doi: 10.1016/j.cell.2011.02.013 (2011). [DOI] [PubMed] [Google Scholar]
- 4.DeBerardinis RJ & Chandel NS We need to talk about the Warburg effect. Nat Metab 2, 127–129, doi: 10.1038/s42255-020-0172-2 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Xu Y, Xue D, Bankhead A 3rd & Neamati N Why All the Fuss about Oxidative Phosphorylation (OXPHOS)? J Med Chem 63, 14276–14307, doi: 10.1021/acs.jmedchem.0c01013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Reyes I et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292, doi: 10.1038/s41586-020-2475-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones CL, Inguva A & Jordan CT Targeting Energy Metabolism in Cancer Stem Cells: Progress and Challenges in Leukemia and Solid Tumors. Cell Stem Cell 28, 378–393, doi: 10.1016/j.stem.2021.02.013 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter JL et al. Targeting mitochondrial respiration for the treatment of acute myeloid leukemia. Biochem Pharmacol 182, 114253, doi: 10.1016/j.bcp.2020.114253 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lagadinou ED et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12, 329–341, doi: 10.1016/j.stem.2012.12.013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pei S et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov 10, 536–551, doi: 10.1158/2159-8290.CD-19-0710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sighel D et al. Inhibition of mitochondrial translation suppresses glioblastoma stem cell growth. Cell Rep 35, 109024, doi: 10.1016/j.celrep.2021.109024 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang F et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626, doi: 10.1126/science.1234769 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Faubert B, Solmonson A & Deberardinis RJ Metabolic reprogramming and cancer progression. Science 368, eaaw5473, doi: 10.1126/science.aaw5473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JW, Adachi H, Shin-ya K, Hayakawa Y & Seto H Apoptolidin, a new apoptosis inducer in transformed cells from Nocardiopsis sp. J Antibiot (Tokyo) 50, 628–630, doi: 10.7164/antibiotics.50.628 (1997). [DOI] [PubMed] [Google Scholar]
- 15.Murakami R et al. Ammocidin, a new apoptosis inducer in Ras-dependent cells from Saccharothrix sp. I. Production, isolation and biological activity. J Antibiot (Tokyo) 54, 710–713, doi: 10.7164/antibiotics.54.710 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Salomon AR, Voehringer DW, Herzenberg LA & Khosla C Apoptolidin, a selective cytotoxic agent, is an inhibitor of F0F1-ATPase. Chem Biol 8, 71–80, doi: 10.1016/s1074-5521(00)00057-0 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Wender PA et al. Correlation of F0F1-ATPase inhibition and antiproliferative activity of apoptolidin analogues. Org. Lett 8, 589–592, doi: 10.1021/ol052800q (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serrill JD et al. Apoptolidins A and C activate AMPK in metabolically sensitive cell types and are mechanistically distinct from oligomycin A. Biochem Pharmacol 93, 251–265, doi: 10.1016/j.bcp.2014.11.015 (2015). [DOI] [PubMed] [Google Scholar]
- 19.DeGuire SM et al. Fluorescent probes of the apoptolidins and their utility in cellular localization studies. Angew. Chem., Int. Ed 54, 961–964, doi: 10.1002/anie.201408906 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mackinnon AL & Taunton J Target Identification by Diazirine Photo-Cross-linking and Click Chemistry. Curr Protoc Chem Biol 1, 55–73, doi: 10.1002/9780470559277.ch090167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du Y et al. Biosynthesis of the Apoptolidins in Nocardiopsis sp. FU 40. Tetrahedron 67, 6568–6575, doi: 10.1016/j.tet.2011.05.106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salomon AR, Voehringer DW, Herzenberg LA & Khosla C Understanding and exploiting the mechanistic basis for selectivity of polyketide inhibitors of F(0)F(1)-ATPase. Proc. Natl. Acad. Sci. U. S. A 97, 14766–14771, doi: 10.1073/pnas.97.26.14766 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wender PA, Jankowski OD, Tabet EA & Seto H Toward a structure-activity relationship for apoptolidin: selective functionalization of the hydroxyl group array. Org. Lett 5, 487–490, doi: 10.1021/ol027366w (2003). [DOI] [PubMed] [Google Scholar]
- 24.Abrahams JP, Leslie AG, Lutter R & Walker JE Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628, doi: 10.1038/370621a0 (1994). [DOI] [PubMed] [Google Scholar]
- 25.Guo H & Rubinstein JL Cryo-EM of ATP synthases. Curr Opin Struct Biol 52, 71–79, doi: 10.1016/j.sbi.2018.08.005 (2018). [DOI] [PubMed] [Google Scholar]
- 26.West AV et al. Labeling Preferences of Diazirines with Protein Biomolecules. J. Am. Chem. Soc 143, 6691–6700, doi: 10.1021/jacs.1c02509 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salomon AR, Zhang Y, Seto H & Khosla C Structure-activity relationships within a family of selectively cytotoxic macrolide natural products. Org. Lett 3, 57–59, doi: 10.1021/ol006767d (2001). [DOI] [PubMed] [Google Scholar]
- 28.Gledhill JR & Walker JE Inhibitors of the catalytic domain of mitochondrial ATP synthase. Biochemical Society transactions 34, 989–992, doi: 10.1042/BST0340989 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Symersky J, Osowski D, Walters DE & Mueller DM Oligomycin frames a common drug-binding site in the ATP synthase. Proc. Natl. Acad. Sci. U. S. A 109, 13961–13965, doi: 10.1073/pnas.1207912109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tantama M, Martinez-Francois JR, Mongeon R & Yellen G Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat. Commun 4, 2550, doi: 10.1038/ncomms3550 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gledhill JR, Montgomery MG, Leslie AG & Walker JE How the regulatory protein, IF(1), inhibits F(1)-ATPase from bovine mitochondria. Proc. Natl. Acad. Sci. U. S. A 104, 15671–15676, doi: 10.1073/pnas.0707326104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308, doi: 10.1038/nprot.2013.143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matreyek KA, Stephany JJ, Chiasson MA, Hasle N & Fowler DM An improved platform for functional assessment of large protein libraries in mammalian cells. Nucleic Acids Res 48, e1, doi: 10.1093/nar/gkz910 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wrenbeck EE et al. Plasmid-based one-pot saturation mutagenesis. Nat. Methods 13, 928–930, doi: 10.1038/nmeth.4029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith RM Biological and chemical studies on a new antibiotic, oligomycin, University of Wisconsin--Madison, (1953). [Google Scholar]
- 36.Pennington JD, Williams HJ, Salomon AR & Sulikowski GA Toward a stable apoptolidin derivative: identification of isoapoptolidin and selective deglycosylation of apoptolidin. Org. Lett 4, 3823–3825, doi: 10.1021/ol026829v (2002). [DOI] [PubMed] [Google Scholar]
- 37.Ramsey HE et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov 8, 1566–1581, doi: 10.1158/2159-8290.CD-18-0140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baccelli I et al. Mubritinib Targets the Electron Transport Chain Complex I and Reveals the Landscape of OXPHOS Dependency in Acute Myeloid Leukemia. Cancer Cell 36, 84–99 e88, doi: 10.1016/j.ccell.2019.06.003 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Jones CL et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 34, 724–740 e724, doi: 10.1016/j.ccell.2018.10.005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pollyea DA et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med 24, 1859–1866, doi: 10.1038/s41591-018-0233-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sriskanthadevan S et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 125, 2120–2130, doi: 10.1182/blood-2014-08-594408 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farge T et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov 7, 716–735, doi: 10.1158/2159-8290.cd-16-0441 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ashton TM, McKenna WG, Kunz-Schughart LA & Higgins GS Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin Cancer Res 24, 2482–2490, doi: 10.1158/1078-0432.CCR-17-3070 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Molina JR et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 24, 1036–1046, doi: 10.1038/s41591-018-0052-4 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Sharon D et al. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci Transl Med 11, eaax2863, doi: 10.1126/scitranslmed.aax2863 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Guieze R et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 36, 369–384 e313, doi: 10.1016/j.ccell.2019.08.005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gorelick AN et al. Respiratory complex and tissue lineage drive recurrent mutations in tumour mtDNA. Nat Metab 3, 558–570, doi: 10.1038/s42255-021-00378-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arrowsmith CH et al. The promise and peril of chemical probes. Nat. Chem. Biol 11, 536–541, doi: 10.1038/nchembio.1867 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
References (Methods):
- 49.Moulton MJ & Letsou A Modeling congenital disease and inborn errors of development in Drosophila melanogaster. Dis Model Mech 9, 253–269, doi: 10.1242/dmm.023564 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mueller DM et al. Ni-chelate-affinity purification and crystallization of the yeast mitochondrial F1-ATPase. Protein Expr Purif 37, 479–485, doi: 10.1016/j.pep.2004.06.035 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Rubinstein JL, Dickson VK, Runswick MJ & Walker JE ATP synthase from Saccharomyces cerevisiae: location of subunit h in the peripheral stalk region. J Mol Biol 345, 513–520, doi: 10.1016/j.jmb.2004.10.060 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Marr CR, Benlekbir S & Rubinstein JL Fabrication of carbon films with approximately 500nm holes for cryo-EM with a direct detector device. J Struct Biol 185, 42–47, doi: 10.1016/j.jsb.2013.11.002 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Tivol WF, Briegel A & Jensen GJ An improved cryogen for plunge freezing. Microsc Microanal 14, 375–379, doi: 10.1017/S1431927608080781 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296, doi: 10.1038/nmeth.4169 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332, doi: 10.1038/nmeth.4193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Punjani A, Zhang H & Fleet DJ Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221, doi: 10.1038/s41592-020-00990-8 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Medina-Cucurella AV et al. User-defined single pot mutagenesis using unamplified oligo pools. Protein Eng Des Sel 32, 41–45, doi: 10.1093/protein/gzz013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boyce JH, Reisman BJ, Bachmann BO & Porco JA Jr. Synthesis and Multiplexed Activity Profiling of Synthetic Acylphloroglucinol Scaffolds. Angew. Chem., Int. Ed 60, 1263–1272, doi: 10.1002/anie.202010338 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baldwin C Biological and chemical properties of aurovertin, a metabolic product of Calcarisporium abuscula. Lloydia 27, 88–95 (1964). [Google Scholar]
- 60.Krasnoff SB & Gupta S Identification and directed biosynthesis of efrapeptins in the fungus Tolypocladium geodes gams (Deuteromycotina: Hyphomycetes). J Chem Ecol 17, 1953–1962, doi: 10.1007/BF00992580 (1991). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates have been deposited in the Protein Data Bank (PDB) with the accession codes 7MD2 and 7MD3. The EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) with the accession codes 23763, 23764 and 23765. The sequencing data and variant counts for the deep mutational scanning experiments have been deposited in the Gene Expression Omnibus (GEO) database under accession code GSE171362. The data generated in this study are contained in the published article including its supplementary information.