

Abstract
Classification of myeloid neoplasms with isolated i(17q) [17p deletion with inherent monoallelic TP53 loss plus 17q duplication] is controversial. Most cases fall within the WHO unclassifiable myelodysplastic/myeloproliferative neoplasms (MDS/MPN-U) category. The uniformly dismal outcomes warrant better understanding of this entity. We undertook a multi-institutional retrospective study of 92 adult MDS/MPN-U cases from eight institutions. Twenty-nine (32%) patients had isolated i(17q) [MDS/MPN-i(17q)]. Compared to MDS/MPN without i(17q), MDS/MPN-i(17q) patients were significantly younger, had lower platelet and absolute neutrophil counts, and higher frequency of splenomegaly and circulating blasts. MDS/MPN-i(17q) cases showed frequent bilobed neutrophils (75% vs. 23%; P = 0.03), hypolobated megakaryocytes (62% vs. 20%; P = 0.06), and a higher frequency of SETBP1 (69% vs. 5%; P = 0.002) and SRSF2 (63% vs. 5%; P = 0.006) mutations which were frequently co-existent (44% vs. 0%; P = 0.01). TP53 mutations were rare. The mutation profile of MDS/MPN-U-i(17q) was similar to other myeloid neoplasms with i(17q) including atypical chronic myeloid leukemia, chronic myelomonocytic leukemia, myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis, myelodysplastic syndrome and acute myeloid leukemia, with frequent concomitant SETBP1/SRSF2 mutations observed across all the diagnostic entities. Over a median follow-up of 52 months, patients with MDS/MPN-i(17q) showed a shorter median overall survival (11 vs. 28 months; P < 0.001). The presence of i(17q) retained independent poor prognostic value in multivariable Cox-regression analysis [HR 3.686 (1.17 – 11.6); P = 0.026] along with splenomegaly. We suggest that MDS/MPN-i(17q) warrants recognition as a distinct subtype within the MDS/MPN-U category based on its unique clinico-biologic features and uniformly poor prognosis.
Keywords: MDS/MPN-Unclassifiable, i(17q), aCML, CMML, WHO
Introduction
Isochromosome 17q [i(17q)] is an abnormal chromosome with an unbalanced structural abnormality as a result of deletion of the short arm and duplication of the long arm of chromosome 17. This non-random cytogenetic abnormality is frequently detected within a setting of complex karyotype in medulloblastomas, blast crisis of chronic myeloid leukemia and other myeloid neoplasms. As an isolated abnormality, i(17q) is rare, but it is recurrently identified in Philadelphia chromosome-negative myeloid neoplasms. Studies have shown that the break point region, consistently located on chromosome locus 17p11.2, harbors multiple low copy repeats (LCR)/ segmental duplications, disruption of which can lead to genetic instability and large-scale gene dysregulation [1]. Affected patients have a poor outcome with a high rate of transformation to acute myeloid leukemia (AML) [2–4]. Interestingly, while these cases have an obligatory monoallelic TP53 deletion, we and others have confirmed the absence of multi-hit TP53 alterations involving the remaining allele suggesting other mechanisms for poor prognosis [3,4].
Myeloid neoplasms with isolated i(17q) frequently show hybrid clinical and morphologic findings: myeloproliferative [leukocytosis, splenomegaly and bone marrow (BM) fibrosis] and myelodysplastic (pseudo-Pelger Hüet nuclei in neutrophils, numerous small hypolobated megakaryocytes, cytopenia) features [5,6]. However, using the current World Health Organization (WHO) criteria, which are largely based on laboratory and morphologic features, many of these cases are often difficult to fit into one of the specific subtypes of myelodysplastic/ myeloproliferative neoplasms (MDS/MPNs). A few cases have been reported (mostly in older publications) to meet the criteria for either atypical chronic myeloid leukemia (aCML) or chronic myelomonocytic leukemia (CMML); while the majority are lumped under the heterogeneous category of MDS/MPN-Unclassifiable (MDS/MPN-U) or felt difficult to assign an appropriate categorization.
Since MDS/MPN-U is a diagnosis of exclusion, it includes cases with a variety of different clinicopathologic features that do not fulfill the criteria for other specific MDS/MPN subtypes such as CMML, aCML, juvenile myelomonocytic leukemia (JMML) and myelodysplastic/ myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T) [7]. The presence of a specific molecular profile in combination with shared clinicopathologic features allow refinement in this category. One example is MDS/MPN-RS-T, first as a separate provisional subtype within the MDS/MPN-U group and later a separate disease entity within the general MDS/MPN category due to frequent SF3B1 mutations together with JAK2, MPL, or CALR mutations and the presence of RS and thrombocytosis.
Similarly, in cases of MDS/MPN-U with isolated i(17q), the presence of shared clinicopathologic, mutational and cytogenetic features seem also to suggest a common pathobiology. We question if these combined features could potentially help in recognizing this condition as a novel distinct subtype within the category of MDS/MPN-U. However, published data is still limited due to the extreme rarity of these cases. We therefore undertook a multi-institutional retrospective study in order to better characterize the clinical, morphologic and molecular genetic features of MDS/MPN-U with isolated i(17q). As part of this study, we compared cases of MDS/MPN-U with i(17q) to cases of MDS/MPN-U without i(17q), as well as other well-characterized cohorts of MDS/MPN subtypes (aCML, CMML and MDS/MPN-RS-T classified using the 2016 WHO criteria) in an attempt to identify distinguishing features. Following the similar approach as the definition of MDS with isolated del(5q), we included cases with sole i(17q) or i(17q) associated with one additional cytogenetic abnormality other than del(7q)/−7 to avoid confounding factors that may contribute to adverse outcomes.
Materials and methods
Study cohort
We searched the institutional databases from 8 institutions in the United States: The University of Texas M.D. Anderson Cancer Center, Massachusetts General Hospital, Brigham and Women’s Hospital, Mayo Clinic, Cleveland Clinic, Weill Cornell Medical College, the Hospital of the University of Pennsylvania Hospital, and the University of Utah for all cases that fulfilled the 2016 WHO criteria for MDS/MPN-U. These cases showed overlapping clinical, laboratory and morphologic features of MDS and MPN at the time of diagnosis and did not meet the criteria for any of the specific subtypes of MDS/MPN (aCML, CMML and MDS/MPN-RS-T), MDS or MPN.[7] All cases had <20% blasts in the peripheral blood (PB) and BM. The inclusion criteria for the study group: (1) 2016 WHO diagnosis of MDS/MPN-U; (2) adequate karyotype at baseline showing i(17q) as an isolated abnormality or with 1 additional abnormality (non-complex karyotype) other than del(7q)/−7 [2,3]; if the baseline karyotype was unavailable, karyotype within the first 6 months of diagnosis in the absence of disease-modifying therapy was considered (3) negative for BCR/ABL1 rearrangement. We also identified MDS/MPN-U cases with normal karyotype at baseline that developed isolated i(17q) abnormality over the disease course. Two of these patients received hypomethylating agent therapy prior to development of i(17q), whereas the remaining patients received only supportive care. Upon subset analysis, these cases showed similar clinico-pathologic features as cases of MDS/MPN-U with i(17q) at baseline. Hence, we included these cases within the study group due to the rarity of isolated i(17q). None of the cases had mutations in MPN-associated genes (JAK2/CALR/MPL) at >20% variant allele burden. Control group included MDS/MPN-U cases without i(17q) abnormality in the baseline karyotype, diagnosed over the same time-period, not meeting the WHO criteria for CMML, aCML, MDS/MPN-RS-T, JMML, or any other myeloid neoplasm. We excluded cases with complex karyotype (n=3) and cases with mutation in any of the MPN-associated genes (JAK2/CALR/MPL) [n=9, all had MPN mutation at >25% variant allele frequency (VAF)]. To evaluate the genetic characteristics of isolated i(17q) across all myeloid neoplasms, agnostic of morphologic diagnosis, we collected mutation data on cases of AML, MDS and MDS/MPN subtypes [CMML, aCML and MDS/MPN-RS-T] showing isolated i(17q) or with 1 additional abnormality except del(7q)/−7. This study was approved by the Institutional Review Boards of all the listed institutions in accordance with the Declaration of Helsinki.
Morphologic evaluation
Diagnostic PB and BM specimens (Wright-Giemsa stained aspirate smears and hematoxylin-eosin stained core biopsy/clot sections) were reviewed by at least two hematopathologists including a central review. BM cellularity was estimated on the core biopsy and/or clot section. Blasts percentages were estimated by a 500-cell differential count, or a count of all available cells (if paucicellular). Megakaryocytes were quantified on biopsy and/or clot section. For dysplasia, we used the WHO criteria of a minimum of 10% dysplastic cells in each of the lineages.[7] To evaluate different types of dysplasia in each of the lineages, we adapted the morphologic criteria proposed by Weinberg et al. with further inclusion of ring sideroblasts and pseudo-Pelger-Huet neutrophils.[8,9] Only cases with a minimum of 30 megakaryocytes, 50 erythroid and 50 myeloid precursors were considered evaluable. We quantified the percentage of cells showing dysplasia using the following scoring system: <10% cells showing dysplastic feature=0, 10–25%=1, 26–50%=2, >51%=3. BM fibrosis was quantified using reticulin and trichrome stains per European Myelofibrosis Network (EUMNET) criteria [10,11].
Conventional cytogenetic studies
Karyotyping was performed on metaphase spreads obtained from unstimulated aspirate cultures by G-banding using standard techniques and reported using the International System for Human Cytogenetic Nomenclature. On selected cases, fluorescence in situ hybridization studies were performed for TP53 gene deletion and rearrangements of BCR/ABL1, PDGFRA, PDGFRB, or FGFR1 as indicated.
Somatic gene mutation analysis by next-generation sequencing
Using high-molecular weight genomic DNA isolated from BM aspirates, amplicon-based NGS panel was performed using different institution-specific gene panels that encompassed most myeloid malignancy related genes. All CMML, aCML and MDS/MPN-RS-T cases underwent amplicon-based targeted NGS (Illumina Miseq) using the same 81-gene panel in a CLIA-certified laboratory as described previously.[12] For variant calling, a minimum VAF of 2% with at least 250x coverage was used. Only somatic mutations were called (based on the literature and online databases). Single nucleotide polymorphisms (SNP) in dbSNP, 1000 genome, EXAC and gNOMAD databases were excluded.
Statistical analysis
Data were reported as median and range (continuous variables) and frequencies (categorical variables). Fisher’s exact, chi-square and Kruskal–Wallis tests were used to compare variables between groups. Overall survival (OS) was calculated from the time of diagnosis to death/last follow-up. The distribution of time-to-event was assessed using the Kaplan-Meier method (GraphPad Prism). No adjustments for multiplicity were made. Features in multivariable analysis were selected using one-sided p-values <0.05 from univariate analyses. Cox-proportional hazards regression was fitted to evaluate the association of clinico-pathologic features on outcome. The cohort was randomly divided into 50% training and 50% validation subgroups, the Cox-proportional model generated in the training subgroup was validated in validation subgroup. AUC curves generated for both groups were equivalent at different time-points. The statistical analysis was performed in R version 4.0.3.
Results
Study group characteristics
In aggregate, we identified 92 adult patients that fulfilled the 2016 WHO criteria for MDS/MPN-U: 29 patients with isolated i(17q) (study group) and 63 MDS/MPN-U patients without i(17q) abnormality (control group). MDS/MPN-U with i(17q) included 13 men and 16 women with a median age of 67 years (range, 41–88). At the time of initial presentation of the MDS/MPN-U patients with isolated i(17q), 79% of the patients had leukocytosis (WBC >13 x 109/L) and 25% had thrombocytosis (Platelet count ≥450 x 109/L). All patients had anemia (Hgb <12 g/dL). By definition, none had an absolute monocytosis with ≥10% PB monocytes. Twenty (69%) patients had splenomegaly.
Compared to the control group, MDS/MPN-U with i(17q) patients were younger (median age, 67 vs. 71 yrs; P = 0.005), had lower median hemoglobin (9.4 vs. 10.1 x 109/L, P = 0.037) and absolute neutrophil counts (4.6 vs. 15.8 x 109/L, P = 0.008), a higher frequency of splenomegaly [(20, 69%) vs. (8, 16%); P = 0.001], as well as a trend for higher PB blasts (2% vs. 0%, P = 0.057). The baseline clinical characteristics of both the groups are summarized in Table 1.
Table 1.
Clinical, peripheral blood and bone marrow features of myelodysplastic/ myeloproliferative neoplasms-unclassifiable (MDS/MPN-U) with i(17q) [study group) with MDS/MPN-U cases without isolated i(17q) [ control group].
| MDS/MPN-U with iso i(17q) n = 29 |
MDS/MPN-U without i(17q) n = 63 |
P | |
|---|---|---|---|
| Age yr (median, range) | 67 (41–88) | 71 (60–88) | .005 |
| Male gender | 13 (45%) | 33 (52%) | NS |
| Splenomegaly (frequency) | 20 (69%) | 8 (16%) | .001 |
| Peripheral blood (PB) counts | |||
| Hemoglobin | 9.4 (5.8–15.1) | 10.1 (6.8–13.6) | .037 |
| White Blood Cell counts | 13.6 (1.2–119) | 19.4 (1.5–98.7) | NS |
| Platelet count x 109/L (median, range) | 103 (16–1000) | 142 (11–1040) | NS |
| ANC x 109/L (median, range) | 4.6 (0.3–50) | 15.8 (0.74–79.9) | .008 |
| PB blasts (median, range) | 2 (0–13) | 0 (0–13) | .057 |
| Bone marrow (BM) morphologic features | |||
| BM blasts% (median, range) | 4 (0–18) | 2 (0–17) | NS |
| Dyserythropoiesis | 9/22 (41%) | 29/57 (51%) | NS |
| Dysgranulopoiesis | 13/22 (59) | 38/58 (66%) | NS |
| Dysmegakaryopoiesis | 22/22 (100%) | 41/56 (73%) | .008 |
| Myelofibrosis (MF) grade 2/3 | 9/22 (41%) | 15/57 (26%) | NS |
| Treatment data | n=28 | n=58 | |
| Supportive | 7 (25%) | 12 (21%) | NS |
| Chemotherapy only | 3 (11%) | 5 (9%) | NS |
| Hypomethylating agents (HMA) | 13 (47%) | 25 (43%) | NS |
| HMA plus chemotherapy/ others | 3 (11%) | 9 (14%) | NS |
| Others* | 2 (7%) | 3 (5%) | NS |
| Allogeneic stem cell transplant | 6 of 28 (21%) | 5 of 58 (9%) | NS |
ANC, Absolute Neutrophil Count; PB, peripheral blood; BM, bone marrow
A total of nineteen (66%) patients had baseline i(17q) (at the time of diagnosis or within 6 months of disease onset in the absence of interim therapy if the baseline karyotype was unavailable). Ten cases of MDS/MPN-U had normal karyotype at the time of diagnosis and developed isolated i(17q) over the disease course at a median of 13 months (range, 7–68). Eight patients had received supportive care only while two patients had received hypomethylating agent therapy. Even when MDS/MPN-U cases with only baseline i(17q) (n=19) were considered, and those with subsequent later development of i(17q) (n=10) were excluded, MDS/MPN-U with isolated i(17q) showed a significantly younger age (p=0.003), lower absolute neutrophil count (p=0.042) and a higher frequency of splenomegaly [12, 63% vs. 8, 16%; p=0.0003]. There was still a trend towards lower hemoglobin (p=0.059), with no significant differences in the PB or BM blasts (Supplemental Table S1).
Twenty-six (90%) patients had sole i(17q) while the remaining 3 patients had an additional abnormality, all with trisomy 13 (10%). The median clonal burden of i(17q) was 60% [10–100%]. Array-based comparative genomic hybridization/ single nucleotide polymorphism (aCGH/SNP) was performed on a subset of cases, none showed additional copy number changes beyond the conventional karyotype.
BM morphologic features
The diagnosis of MDS/MPN-U was noted to be challenging in 6 cases with the differential diagnosis considered being primary myelofibrosis in 3 patients, MDS-U in 1 patient and MDS/MPN-RS-T in 2 patients. These cases were reviewed among experts, and a diagnosis of MDS/MPN-U was favored considering the morphology, laboratory data and mutational characteristics. Two cases fulfilled the criteria for oligomonocytic CMML but are still classified as MDS/MPN-U in the current classification. BM cellularity was increased for age in 25 (86%) patients, normal in 2 patients (7%) and decreased for age in 2 (7%) patients. Fourteen (48%) patients had BM blasts <5%; 9 (31%) showed BM blasts 5–9% and 6 (21%) patients had 10–19% blasts. Although the isolated i(17q) group showed a higher median BM blast percentage compared to the control group, the difference was not significant (4 vs. 2%; p=0.106). Using the WHO criterion (at least 10% dysplastic cells), MDS/MPN-U with i(17q) showed a higher frequency of megakaryocytic dysplasia compared to MDS/MPN-U without isolated i(17q) [100% vs. 73%; p=0.008] with no differences in granulocytic and erythroid dysplasia. There was no difference in the degree of BM fibrosis (grade MF-2 or higher) between the groups despite frequent splenomegaly and higher circulating blasts in MDS/MPN-U-i(17q) (Table 1).
Based on the reported association with specific dysplastic characteristics such as pseudo-Pelger-Huet neutrophils and increased small monolobated and hypolobated megakaryocytes in previous studies [5,6], we quantified and compared the presence of different types of dysplasia between the study and control groups. Using dysplasia score of 1, MDS/MPN-U with i(17q) cases had a significantly higher frequency of pseudo-Pelger Huet neutrophils, and a trend for increased mono/hypolobated megakaryocytes compared to the control group. Using dysplasia score of 3, MDS/MPN-U with i(17q) showed a trend for increased pseudo-Pelger Huet neutrophils (Table 2). Representative morphologic images are shown in Figure 1.
Table 2.
Comparison of specific types of dysplastic morphologic features of myelodysplastic/ myeloproliferative neoplasms unclassifiable with and without isolated i(17q)
| MDS/MPN-U with iso(17q) | MDS/MPN-U without i(17q) | P | MDS/MPN-U with iso(17q) | MDS/MPN-U without i(17q) | P | |
|---|---|---|---|---|---|---|
| LINEAGE | >10%/ ≥1 | >50%/ ≥3 | ||||
| ERYTHROID | ||||||
| Megaloblastoid changes | 6/8 | 9/13 | 1 | 0/8 | 2/13 | .5048 |
| Multinucleation | 0/8 | 0/13 | 1 | 0/8 | 0/13 | 1 |
| Nuclear contour irregularities | 3/8 | 5/13 | 1 | 0/8 | 0/13 | 1 |
| Pyknosis | 0/8 | 2/13 | .505 | 0/8 | 0/13 | 1 |
| Ring Sideroblasts | 1/8 | 3/13 | 1 | 0/8 | 2/13 | .5048 |
| MYELOID | ||||||
| Abnormal nuclear shape | 6/8 | 5/13 | .183 | 3/8 | 1/13 | .3002 |
| Pseudo-Pelger Huet nuclei | 6/8 | 3/13 | .032 | 3/8 | 0/13 | .0815 |
| Cytoplasmic hypogranulation | 6/8 | 12/13 | .531 | 2/8 | 2/13 | .5308 |
| MEGAKARYOCYTIC | ||||||
| Micromegakaryocytes | 2/8 | 1/12 | .537 | 0/8 | 0/12 | 1 |
| Hypolobated/ monolobated nuclei | 5/8 | 2/10 | .062 | 0/8 | 0/12 | 1 |
| Separated nuclear lobes | 1/8 | 3/12 | .642 | 0/8 | 1/12 | 1 |
MDS/MPN-U, myelodysplastic/ myeloproliferative neoplasm-unclassified; iso(17q), isochromosome (17q)
Figure 1.
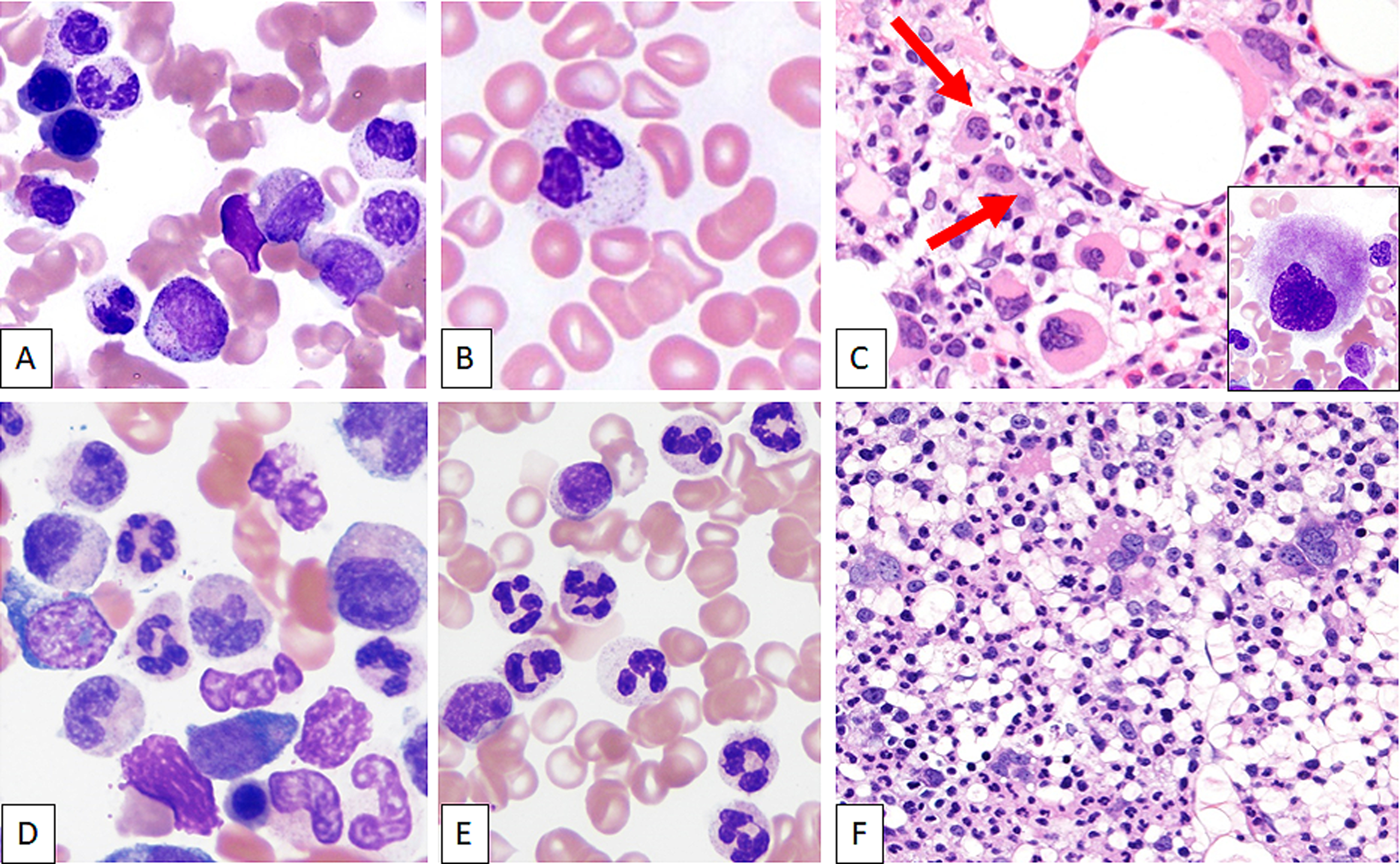
Peripheral Blood and bone marrow morphologic features of MDS/MPN-U with and without isolated i(17q). Top panel: Representative images of typical BM morphological features of MDS/MPN-U with i(17q) [top panel] showing frequent pseudo-Pelger Huet neutrophils on bone marrow aspirate smear (A) and peripheral blood (B) smears with frequent dysplastic small hypolobated megakaryocytes (C, biopsy; inset: aspirate smear). Bottom panel: In contrast, cases of MDS/MPN-U without isolated i(17q) showed dysgranulopoiesis of all types such as hypogranulation of cytoplasm and neutrophilic nuclear hyperlobulation on bone marrow aspirate smear (D) and peripheral blood (E) smears. Megakaryocytic dysplasia was less frequent (F).
Somatic gene mutation analysis
NGS-based mutation data were collected from multiple gene panels, each specific to the institution, but encompassing most of the genes implicated in myeloid malignancies. Comprehensive mutation data that included recurrently mutated genes in MDS/MPN-U-i(17q) (ASXL1, SRSF2 and SETBP1) was available in 16 patients with MDS/MPN-U with i(17q)] and 21 MDS/MPN-U patients without i(17q). The highest frequency of mutations in MDS/MPN with i(17q) included SETBP1 (11/16, 69%), ASXL1 (12/18, 67%), SRSF2 (10/16, 63%), followed by mutations in one of the RAS-MAPK pathway genes, in accord with the MDS/MPN phenotype. Among these, mutations in SETBP1 (69% vs. 14%; P = 0.002) and SRSF2 (63% vs. 24%; P = 0.023) were more frequent in MDS/MPN-U with i(17q) compared to MDS/MPN-U without i(17q). Double mutations involving SRSF2/SETBP1 were significantly more frequent in MDS/MPN-U with i(17q) [44% vs. 5%; P = 0.012] and a similar trend was noted with triple mutations involving SRSF2/SETBP1/ASXL1 [31% vs. 5%; P = 0.066]. No significant differences were seen with respect to mutations in ASXL1 (67% vs. 43%; p = NS) or TET2 (19% vs. 33%; p = NS). Notable negative findings were follows: TP53 mutations were rare [seen in only 1 patient at low (4.8%) VAF] even though the other TP53 allele was deleted in all cases due to formation of i(17q), signifying monoallelic alterations. None of the cases had mutations in MPN-associated genes at a high allele burden: JAK2 mutation noted in 1 patient at 2.1%, and CALR mutation was noted in 1 patient at ~20% VAF. MPL mutation was absent. The mutational heatmap of both groups MDS/MPN-U is shown in Figure 2.
Figure 2.
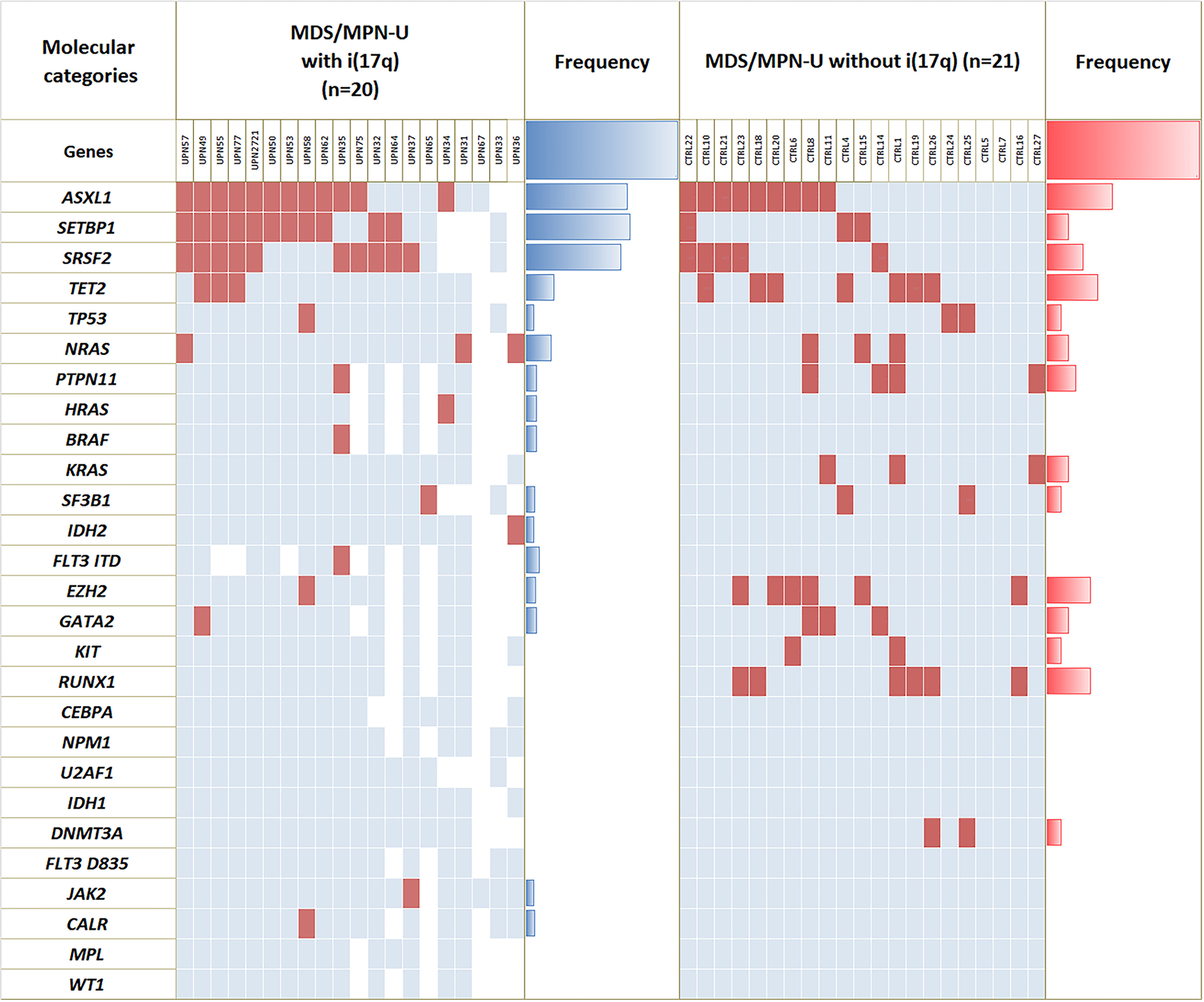
Mutational landscape. Cases of MDS/MPN-U with isolated i(17q) shows significantly higher frequency of mutations in SETBP1 and SRSF2 compared to cases of MDS/MPN-U without i(17q). There were no significant differences in mutational frequencies in other genes such as ASXL1 and TET2. TP53 mutations were rare. Cases of MDS/MPN-U with clonal mutations in one of the MPN-associated genes that included JAK2, CALR and MPL were excluded.
Comparison of mutation profile of MDS/MPN-U with isolated i(17q) with other MDS/MPN subtypes
The baseline mutational profile of other MDS/MPN subtypes that included consecutively diagnosed cases of CMML (n=185), aCML (n=19) and MDS/MPN-RS-T (n=23), presenting to a single center that underwent NGS-based mutation analysis is shown in (Supplemental Figure S1). All cases were diagnosed and sub-classified using 2016 WHO-defined criteria. The mutation signatures of CMML, aCML and MDS/MPN-RS-T were consistent with reports in the literature. With the exception of MDS/MPN-RS-T, all the other subtypes of MDS/MPN showed a significant degree of overlap in their mutational profiles. We compared the frequencies of the characteristic mutated gene(s) and their combinations that we observed in MDS/MPN-U with i(17q) [ASXL1, SETBP1, SRSF2, SRSF2/SETBP1 (double mutant), SRSF2/SETBP1/ASXL1 (triple mutant) and TET2] with those of CMML, aCML and MDS/MPN-RS-T. The overall mutation landscape of MDS/MPN-U with i(17q) overlapped with that of aCML and partly with CMML, but was distinctively different from MDS/MPN-RS-T. MDS/MPN-U with i(17q) showed significantly higher frequency of ASXL1 and SRSF2 than MDS/MPN-RS-T (P = 0.0022 and P = 0.0009 respectively) and higher SETBP1 than CMML (P < 0.0001), with the SRSF2/SETBP1 double mutant and SRSF2/SETBP1/ASXL1 (triple mutant) combinations significantly higher than both CMML and MDS/MPN-RS-T (P < 0.0001) (Figure 3). Overlapping gene mutation(s) among different MDS/MPN subtypes with slight differences further reiterate the importance of evaluating genomic profile within the context of clinical, laboratory and morphologic features and not as a standalone approach for the purpose of diagnostic categorization.
Figure 3.
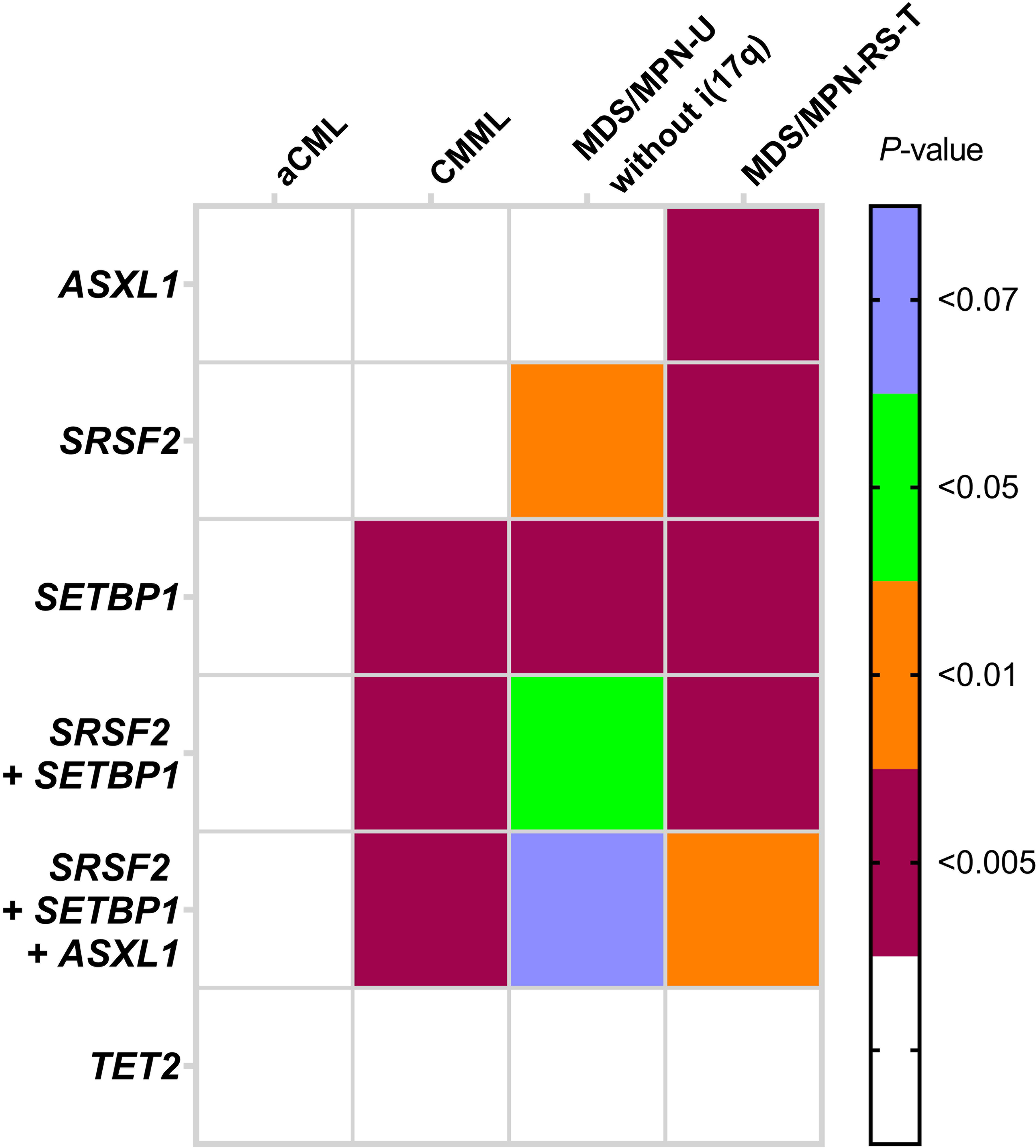
Differences in mutational frequencies (single and combination of gene mutations) between MDS/MPN-U with i(17q) and other specific WHO-defined subgroups of MDS/MPN show similarities and differences. The significant differences are highlighted in color (P value represented using different colors).
We then sought to assess the gene mutation spectrum associated with i(17q) across all types of myeloid neoplasms. There were 36 patients with isolated i(17q) with available mutation data: AML-MRC (n=16), aCML (n=2), CMML (n=5) and MDS (n=13). Similar to MDS/MPN-U with i(17q), these cases showed frequent mutations in SETBP1 and SRSF2 genes. SETBP1 was mutated in 10/16 (63%) AML-MRC, 2/2 (100%) aCML, 4/5 (80%) CMML and 8/13 (62%) MDS. SRSF2 was mutated in 7/16 (54%) AML-MRC, 2/2 (100%) aCML; 5/5 (100%) CMML and 8/13 (62%) MDS]. Concomitant SETBP1/SRSF2 mutations (double mutant) were noted in >50% of cases across all the diagnostic entities: 7/16 (54%), 2/2 (100%), 4/5 (80%) and 8/13 (62%) cases in AML-MRC, aCML, CMML and MDS respectively. These similarities to the mutation profile of MDS/MPN-U with i(17q) across a variety of phenotypic manifestations, agnostic of morphologic diagnoses, suggest that i(17q) may be the driver of a similar underlying disease biology in all of these entities. Supplemental Figure S2).
Outcomes
Over a median duration of 52 months, follow-up data was available in 28 MDS/MPN-U patients with i(17q) and all patients without i(17q). Twenty-three (82%) MDS/MPN-U patients with i(17q) died. Four (14%) patients transformed to AML. The median time to AML transformation was 6 months from the time of i(17q) detected. Treatment information was available in 28 MDS/MPN-U patients with i(17q) and 58 MDS/MPN-U patients without i(17q). There were no significant differences in treatment characteristics between the two groups (Table 1). Over the follow-up period, 10 patients with i(17q) showed karyotypic evolution, while the remaining patients did not show any new additional karyotypic alterations.
Patients with MDS/MPN-U with baseline i(17q) had a significantly shorter median overall survival (OS) than MDS/MPN-U without i(17q) [10.6 vs. 27.6 months; P < 0.001] (Figure 4A). Univariate analysis of the entire MDS/MPN-U cohort showed that i(17q), higher BM and PB blast percentages, and splenomegaly associated with shorter OS (Table 3). By multivariable analysis, i(17q) [HR: 1.69; P = 0.026] and splenomegaly [HR: 6.98; P = 0.001] retained independent predictive value while PB blast percentage became only borderline significant [HR: 1.2; P = 0.073] (Table 4; Figure 4B). Univariate analysis on the subset of patients with available mutation data (n=36) showed that SETBP1 mutation had a trend for worse outcome (P = 0.099). SRSF2 and ASXL1 mutations did not impact outcome (Supplemental Table S2).
Figure 4.
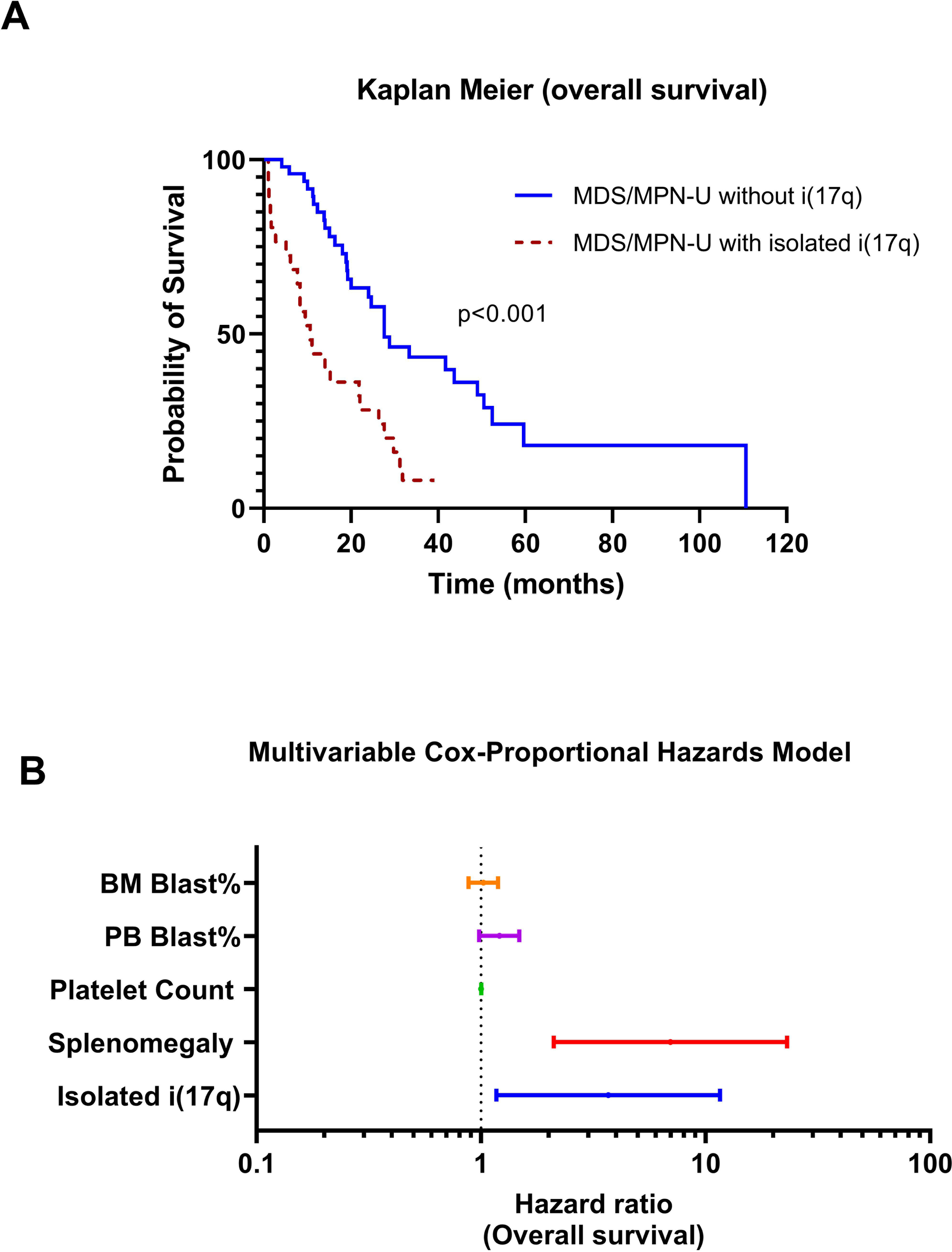
Overall survival analysis. A. Kaplan-Meier curve showing significantly different overall survival of myelodysplastic/ myeloproliferative neoplasms with and without isolated i(17q). B. Multivariable analysis demonstrating that both i(17q) [HR: 1.3; P = 0.026] and splenomegaly [HR: 1.94; P = 0.001] retained independent predictive value for overall survival.
Table 3.
Univariate analysis of all patients with myelodysplastic/ myeloproliferative neoplasms-unclassifiable
| Overall Survival | ||
|---|---|---|
| Parameters | Hazard Ratio (95% CI) | P |
| Isolated isochromosome i(17q) | 3.18 (1.773 – 5.689) | < .001 |
| PB Blast% | 1.16 (1.075 – 1.254) | < .001 |
| BM Blast% | 1.13 (1.064 – 1.196) | < .001 |
| Splenomegaly | 2.1 (1.177 – 3.734) | .012 |
| Platelet count | 1 (0.998 – 1) | .055 |
| Age | 1.02 (0.986 – 1.048) | .297 |
| White Blood Cell count | 1.01 (0.994 – 1.017) | .366 |
| Hemoglobin | 0.94 (0.818 – 1.086) | .415 |
| Gender | 0.81 (0.466 – 1.392) | .438 |
| Absolute Neutrophil Count | 1.01 (0.985 – 1.029) | .549 |
| Mean Corpuscular Volume | 1 (0.9772 – 1.0334) | .731 |
PB, peripheral blood; BM, bone marrow
Table 4.
Multivariable analysis (cox-proportional hazards model) of patients with myelodysplastic/ myeloproliferative neoplasms-unclassifiable
| Overall Survival | ||
|---|---|---|
| Parameters | Hazard Ratio (95% CI) | P |
| Isolated isochromosome i(17q) | 3.686 (1.17 – 11.6) | .026 |
| Splenomegaly | 6.98 (2.11 – 23.06) | .001 |
| Platelet Count | 1.001 (0.99 – 1.00) | .179 |
| PB Blast% | 1.207 (0.98 – 1.48) | .073 |
| BM Blast% | 1.023 (0.88 – 1.19) | .7673 |
PB, peripheral blood; BM, bone marrow
Due to the identification of double mutant (SRSF2/SETBP1) genotype in MDS/MPN-U patients with i(17q), we performed a sub-analysis within the 16 MDS/MPN-U with i(17q) group comparing double mutant SRSF2/SETBP1 cases with the rest. Although double mutant patients had higher frequency of splenomegaly (100% vs. 67%; P = 0.2125) and shorter OS (8 vs. 21 months, P = 0.18), these differences were not statistically significant.
Discussion
Improved understanding of disease biology by genomic profiling has facilitated identification of distinct subsets within the category of MDS/MPN-U. The prototypic example is the recognition of MDS/MPN-RS-T, initially as a provisional entity and subsequently as a separate category of MDS/MPN by the 2016 WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue as a separate entity based on unique mutational signature of concomitant JAK2 and SF3B1 mutations in the majority of cases [13–18]. More recently, genomic studies have uncovered additional diagnostic entities that were “lumped” within the category of MDS/MPN-U on the basis of laboratory and morphologic features alone that did not qualify for another specific myeloid neoplasm. These include NPM1 mutated non-acute myeloid neoplasms, the majority of which are classified as MDS or as one of the MDS/MPN entities, although these cases are more akin to NPM1 mutated AML based on the disease biology, evolution and therapy response [12,19]. Oligomonocytic CMML, despite not meeting the currently proposed WHO criteria for CMML, shows a similar underlying genomic and immunophenotypic profile and outcome to CMML [20–23]. One study has also recognized MDS/MPN-U with ≥15% ring sideroblasts but not fulfilling the hematologic criteria for MDS/MPN-RS-T to be within the spectrum of the same disease [24]. Hence, there is an increasing need to refine this so-called “waste-basket” category based on genomic studies [25].
We demonstrate that MDS/MPN-U with isolated i(17q) represents a homogeneous disease with characteristic clinical and morphologic features and a molecular profile that fits well with what is seen in other MDS/MPN adult disease entities (except for MDS/MPN-RS-T), i.e. resembling that of aCML and to a certain extent CMML. To the best of our knowledge, this is the largest study of MDS/MPN-U with isolated i(17q) describing the comprehensive clinicopathologic and genomic features. These cases are rare but recurrent and pose a significant diagnostic challenge to pathologists. Until now, most of the large-scale studies that have investigated clinical and molecular landscape of MDS/MPN have failed to recognize cases with isolated i(17q) for reasons that include (1) rarity and (2) the presence of the same cytogenetic abnormality across a range of myeloid neoplasms currently classified as CMML, aCML, MDS, “triple-negative” MPN cases (lacking the canonical JAK2, MPL or CALR mutations) and AML, as also shown in this study [24,26–28]. When all cases of myeloid neoplasms with i(17q) were reviewed and the WHO classification criteria carefully applied, this study showed that the majority (~67%) of them were classified under the nondescript MDS/MPN-U.
The current WHO classification recognizes specific genetic alterations among the diagnostic criteria for two disease entities: JMML (PTPN11, KRAS, NRAS, NF1, CBL) and MDS/MPN-RS-T (SF3B1). Characteristic molecular patterns have also been described in other specific subtypes of MDS/MPN, although there is significant overlap [29]. Most cases of CMML are characterized by mutations in TET2, with a second hit in SRSF2, ASXL1 or CBL [30]. Co-mutations in TET2 and SRSF2 are highly specific to CMML [22,27,31,32]. The mutation profile of aCML includes concomitant mutations in SETBP1 with either SRSF2 or ASXL1, and ASXL1 with SRSF2 or EZH2 [26–28,33–35]. In contrast to these better defined MDS/MPN subtypes, the molecular landscape of MDS/MPN-U is diverse, as expected, since the diagnosis of MDS/MPN-U is based on the “absence” of specific defining criteria [25,26,29,36]. The enrichment of MPN-associated mutations noted by a few authors in MDS/MPN-U is due to the under recognition of an underlying MPN that is inherent to the definition of MDS/MPN-U, which we addressed in this study by excluding MDS/MPN-U cases with JAK2 and CALR mutations [28,36]. There is conflicting data regarding the frequencies of mutations in the RAS-MAPK pathway in MDS/MPN-U [24,36,37]. Recent whole genome sequencing study of a large number of MDS/MPN-U cases by Palomo and colleagues has shown subgroups with different molecular patterns that the authors have termed CMML-like (17%), aCML-like (33%), MDS/MPN-RS-T-like (11%), TP53 positive (13%) and “others” [26]. The authors showed that these molecular subgroups may correlate with corresponding hematologic phenotypes and outcomes. Using this approach, the findings from this study demonstrate enrichment of SRSF2/SETBP1 double mutant pattern in MDS/MPN with i(17q). Although a similar profile can be seen in aCML and less frequently in CMML, the notable absence of other clinicopathologic features needed for the diagnosis of aCML or CMML allows for diagnostic distinction. Hence, it is important to characterize these entities not just by mutation(s) alone, but also incorporating clinical, laboratory and morphologic features. Overall, the mutations seen alone or in combination among MDS/MPN-U with i(17q) cases fit well with adult MDS/MPNs as a group. The other characteristics such as morphology, complex patterns of prognostically adverse mutations, lack of TP53 mutation and generally poor prognosis define this as a distinct MDS/MPN entity. Of interest, cases with i(17q) classified as CMML, aCML, MDS and AML showed the same mutational profile as those classified as MDS/MPN-U. On the other hand, although the focus of this study is on the MDS/MPN-U patients, we found challenges in classifying some of these cases due to overlapping features with PMF and MDS-F. It is reasonable to suggest that among chronic myeloid neoplasm with i(17q), the molecular genetic features and commonly observed adverse prognosis are largely driven by these shared genomic aberrations.
One point of interest is a paucity of mutations in TP53. The formation of i(17q) results in an inherent loss of TP53 allele located on 17p13.1. However, as it is clear from this study and others, the remaining TP53 allele is wild-type [2,4,34,38]. FISH and aCGH/SNP studies performed on a subset of cases confirmed the heterozygous nature of the TP53 deletion and absence of copy-neutral loss-of-heterozygosity, confirming monoallelic TP53 alteration [39]. Recent study has suggested that myeloid neoplasms with monoallelic TP53 alteration are distinct from multi-hit TP53 alterations and show favorable outcomes similar to those with wild-type TP53 [39]. However, the particularly adverse outcome noted in these cases are contradictory to these observations. The current study findings allude to a different pathogenic mechanism (outside of TP53) for the adverse phenotype involving SRSF2 and SETBP1. The association between i(17q) and SETBP1 and SRSF2 mutations are also supported by prior studies [2–4,34]. The reasons for the noted associations, the molecular mechanisms underlying i(17q) and the consequences thereof, and the difference between 17p deletion vs. i(17q) [17p deletion plus 17q duplication] are still unclear. Of note, SRSF2 gene is located on 17q which is duplicated, suggesting the added contribution of dosage effect to the mRNA mis-splicing resulting from this spliceosome gene mutation.
The reported survival of MDS/MPN-U patients in the literature is highly variable, ranging between 12–32 months and is dependent on various clinical variables [24–26,37,40]. In the current study, i(17q) was an independent predictor of worse outcome in MDS/MPN-U, further underscoring the need to annotate these cases separately. Complementing the presence of isolated i(17q) with mutation data (double mutations involving SRSF2/SETBP1) can further identify those patients with worse outcomes. In addition to poor prognosis, the unique clinico-morphologic and mutational characteristics associated with MDS/MPN with i(17q) support its recognition as a distinct entity. The proposed criteria for the diagnosis are described in Table 5. The inclusion criteria of i(17q) as an isolated or with 1 additional abnormality other than del(7q)/−7 are along the similar lines as the WHO entity: MDS with isolated del(5q). Although we set the initial criteria to exclude del(7q)/−7 to avoid confounding factors contributing to poor outcome, in our search, we did not find any cases with both i(17q) and del(7q)/−7 outside the context of a complex karyotype.
Table 5.
Proposed criteria for MDS/MPN with isolated isochromosome (17q)
| Myeloid neoplasm with mixed myeloproliferative and myelodysplastic features at onset, not meeting the WHO criteria for any other myelodysplastic/myeloproliferative neoplasm, myelodysplastic syndrome or myeloproliferative neoplasm |
| - < 20% blastsa in the peripheral blood and bone marrow |
| - Clinical and morphological features of myelodysplastic syndrome in addition to clinical and morphologic myeloproliferative features manifesting as a platelet count of ≥ 450 x 109/L associated with bone marrow megakaryocytic proliferation and/or a white blood cell count of ≥13 x 109 /L |
| - Presence of isolated isochromosome i(17q) or with 1 additional abnormality [other than del(7q)/−7] |
| - No BCR/ABL1 fusion; no PDGFRA, PDGFRB, or FGFR1 rearrangement; no PCM1-JAK2 |
| - Absence of MPN-associated mutations (JAK2, CALR and MPL)b |
| - No history of recent cytotoxic or growth factor therapy that could explain the myelodysplastic/myeloproliferative features |
Blasts and blast equivalents include myeloblasts, monoblasts and promonocytes
Presence of MPN features in the bone marrow, and/or MPN-associated mutations (in JAK2, CALR or MPL) suggests progression of an underlying MPN that was not diagnosed, and should be excluded; conversely, in the appropriate clinical context, mutations in SRSF2 and SETBP1 genes further supports the diagnosis. However, these must be interpreted with caution since some of these mutations can be age-related or present in other neoplasms.
The study has limitations: although this is the largest number of MDS/MPN-U cases with i(17q) in the literature, it is limited by the retrospective and multi-institutional nature of the study and a small number of cases due to the rarity. Hence, further confirmation of the findings in a validation cohort and prospective studies in these patients using uniform therapy within a setting of clinical trial is needed. Standard criteria proposed here in Table 5 will help with uniform characterization of this entity, patient accrual within MDS/MPN clinical trials and prospective validation. While dissecting out these MDS/MPN-U cases defined by a common cytogenetic abnormality, i(17q) from the highly heterogeneous MDS/MPN-U cases demonstrated distinctive genotypic and phenotypic attributes, therapeutic options are limited at this time. Despite this, the characteristic molecular signature (SETBP1/SRSF2 mutations) with relatively intact TP53 pathway provides opportunities to explore potential strategies targeting SETBP1-induced transcriptional activation or spliceosome modulators [41,42].
In summary, MDS/MPN-U with i(17q) represents a disease entity based on its distinct clinical and morphologic features that do not fulfill WHO criteria for other MDS/MPN entities, poor outcome, and mutational profile that befits the MDS/MPN disease group. Hence, recognition of MDS/MPN-U with i(17q) as a distinct entity, is warranted. Doing so seems congruent with the overarching strategy of the WHO classification system to increasingly incorporate molecular genotypes. Accurate diagnosis has significant implications for enrollment in clinical trials and for the development of appropriately target therapeutic interventions.
Supplementary Material
Funding
This work was supported in part by institutional start-up funds, institutional research grant and leukemia SPORE career development grant awarded to R.K-S. KAD was partially supported by Cancer Center Support Grant NCI Grant P30 CA016672, NIH grant UL1TR00316, and the Moon Shots funding at MD Anderson Cancer Center.
Footnotes
Ethical Approval
This study was approved by the Institutional Review Boards of all the participating institutions in accordance with the Declaration of Helsinki.
Conflicts of Interest
R.K-S declares research support from Novartis and scientific advisory role for with Novartis, Amgen, Aptitude Health and Physicians Education Resource for work performed outside of the current study. E.D.H declares research funding from Eli Lilly and AbbVie and serves on the advisory boards of Curio Sciences, Cytomx, and Eli Lilly for work performed outside of the current study. G.G-M declares research support and an advisory role with Amphivena, Astex, and Celgene, and research support from AbbVie, H3 Biomedicine, Helsin, Onconova, Merck, and Novartis for work performed outside of the current study. C.B-R declares research support and advisory role with Amgen and Incyte. The remaining authors declare no competing financial interests.
Data Availability Statement
The datasets generated for the current study are not publicly available due to patient privacy concerns but are available from the corresponding author on reasonable request.
References
- 1.Barbouti A, Stankiewicz P, Nusbaum C, Cuomo C, Cook A, Höglund M et al. The breakpoint region of the most common isochromosome, i (17q), in human neoplasia is characterized by a complex genomic architecture with large, palindromic, low-copy repeats. Am J Hum Genet 74, 1–10 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Visconte V, Tabarroki A, Zhang L, Hasrouni E, Gerace C, Frum R et al. Clinicopathologic and molecular characterization of myeloid neoplasms harboring isochromosome 17(q10). Am J Hematol 89, 862 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Meggendorfer M, Haferlach C, Zenger M, Macijewski K, Kern W & Haferlach T The landscape of myeloid neoplasms with isochromosome 17q discloses a specific mutation profile and is characterized by an accumulation of prognostically adverse molecular markers. Leukemia 30, 1624–1627 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Kanagal-Shamanna R, Luthra R, Yin CC, Patel KP, Takahashi K, Lu X et al. Myeloid neoplasms with isolated isochromosome 17q demonstrate a high frequency of mutations in SETBP1, SRSF2, ASXL1 and NRAS. Oncotarget 7, 14251–14258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McClure RF, Dewald GW, Hoyer JD & Hanson CA Isolated isochromosome 17q: a distinct type of mixed myeloproliferative disorder/myelodysplastic syndrome with an aggressive clinical course. Br J Haematol 106, 445–454 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Kanagal-Shamanna R, Bueso-Ramos CE, Barkoh B, Lu G, Wang S, Garcia-Manero G et al. Myeloid neoplasms with isolated isochromosome 17q represent a clinicopathologic entity associated with myelodysplastic/myeloproliferative features, a high risk of leukemic transformation, and wild-type TP53. Cancer 118, 2879–2888 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Weinberg OK, Pozdnyakova O, Campigotto F, DeAngelo DJ, Stone RM, Neuberg D et al. Reproducibility and prognostic significance of morphologic dysplasia in de novo acute myeloid leukemia. Mod Pathol 28, 965–976 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Della Porta MG, Travaglino E, Boveri E, Ponzoni M, Malcovati L, Papaemmanuil E et al. Minimal morphological criteria for defining bone marrow dysplasia: a basis for clinical implementation of WHO classification of myelodysplastic syndromes. Leukemia 29, 66–75 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J & Orazi A European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 90, 1128–1132 (2005). [PubMed] [Google Scholar]
- 11.Barosi G, Bordessoule D, Briere J, Cervantes F, Demory JL, Dupriez B et al. Response criteria for myelofibrosis with myeloid metaplasia: results of an initiative of the European Myelofibrosis Network (EUMNET). Blood 106, 2849–2853 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Montalban-Bravo G, Kanagal-Shamanna R, Sasaki K, Patel K, Ganan-Gomez I, Jabbour E et al. NPM1 mutations define a specific subgroup of MDS and MDS/MPN patients with favorable outcomes with intensive chemotherapy. Blood Adv 3, 922–933 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S, Hasserjian R, Loew J, Sechman E, Jones D, Hao S et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis harbors JAK2 mutation and shows overlapping myeloproliferative and myelodysplastic features. Leukemia 20, 1641–1644 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Cannella L, Breccia M, Latagliata R, Frustaci A & Alimena G Clinical and prognostic features of patients with myelodysplastic/myeloproliferative syndrome categorized as unclassified (MDS/MPD-U) by WHO classification. Leuk Res 32, 514–516 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Malcovati L, Della Porta MG, Pietra D, Boveri E, Pellagatti A, Galli A et al. Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood 114, 3538–3545 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Jeromin S, Haferlach T, Grossmann V, Alpermann T, Kowarsch A, Haferlach C et al. High frequencies of SF3B1 and JAK2 mutations in refractory anemia with ring sideroblasts associated with marked thrombocytosis strengthen the assignment to the category of myelodysplastic/myeloproliferative neoplasms. Haematologica 98, e15–17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 26, 542–545 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Jeromin S, Haferlach T, Weissmann S, Meggendorfer M, Eder C, Nadarajah N et al. Refractory anemia with ring sideroblasts and marked thrombocytosis cases harbor mutations in SF3B1 or other spliceosome genes accompanied by JAK2V617F and ASXL1 mutations. Haematologica 100, e125–127 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel SS, Ho C, Ptashkin RN, Sadigh S, Bagg A, Geyer JT et al. Clinicopathologic and genetic characterization of nonacute NPM1-mutated myeloid neoplasms. Blood Adv 3, 1540–1545 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geyer JT, Tam W, Liu Y-C, Chen Z, Wang SA, Bueso-Ramos C et al. Oligomonocytic chronic myelomonocytic leukemia (chronic myelomonocytic leukemia without absolute monocytosis) displays a similar clinicopathologic and mutational profile to classical chronic myelomonocytic leukemia. Mod Pathol 30, 1213–1222 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Valent P, Orazi A, Savona MR, Patnaik MM, Onida F, van de Loosdrecht AA et al. Proposed diagnostic criteria for classical chronic myelomonocytic leukemia (CMML), CMML variants and pre-CMML conditions. Haematologica 104, 1935–1949 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montalban-Bravo G, Kanagal-Shamanna R, Guerra V, Ramos-Perez J, Hammond D, Shilpa P et al. Clinical outcomes and influence of mutation clonal dominance in oligomonocytic and classical chronic myelomonocytic leukemia. Am J Hematol 96, E50–E53 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Calvo X, Garcia-Gisbert N, Parraga I, Gibert J, Florensa L, Andrade-Campos M et al. Oligomonocytic and overt chronic myelomonocytic leukemia show similar clinical, genomic, and immunophenotypic features. Blood Adv 4, 5285–5296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mangaonkar AA, Swoboda DM, Coltro G, Lasho TL, Novotny PJ, Pophali P et al. Clinicopathologic characteristics, prognostication and treatment outcomes for myelodysplastic/myeloproliferative neoplasm, unclassifiable (MDS/MPN-U): Mayo Clinic-Moffitt Cancer Center study of 135 consecutive patients. Leukemia 34, 656–661 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Shallis RM & Zeidan AM Myelodysplastic/myeloproliferative neoplasm, unclassifiable (MDS/MPN-U): More than just a “catch-all” term? Best Pract Res Clin Haematol, 101132 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Palomo L, Meggendorfer M, Hutter S, Twardziok S, Adema V, Fuhrmann I et al. Molecular landscape and clonal architecture of adult myelodysplastic/myeloproliferative neoplasms. Blood 136, 1851–1862 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meggendorfer M, Haferlach T, Alpermann T, Jeromin S, Haferlach C, Kern W et al. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia. Haematologica 99, e244–246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meggendorfer M, Jeromin S, Haferlach C, Kern W & Haferlach T The mutational landscape of 18 investigated genes clearly separates four subtypes of myelodysplastic/myeloproliferative neoplasms. Haematologica 103, e192–e195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Wilmot B, Bottomly D, Dao KT, Stevens E, Eide CA et al. Genomic landscape of neutrophilic leukemias of ambiguous diagnosis. Blood 134, 867–879 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coltro G, Mangaonkar AA, Lasho TL, Finke CM, Pophali P, Carr R et al. Clinical, molecular, and prognostic correlates of number, type, and functional localization of TET2 mutations in chronic myelomonocytic leukemia (CMML)—a study of 1084 patients. Leukemia 34, 1407–1421 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Patnaik M, Lasho T, Vijayvargiya P, Finke C, Hanson C, Ketterling R et al. Prognostic interaction between ASXL1 and TET2 mutations in chronic myelomonocytic leukemia. Blood Cancer J 6, e385–e385 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patnaik M, Zahid M, Lasho T, Finke C, Ketterling R, Gangat N et al. Number and type of TET2 mutations in chronic myelomonocytic leukemia and their clinical relevance. Blood Cancer J 6, e472–e472 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piazza R, Redaelli S, Valletta S, Pirola A, Spinelli R, Magistroni V et al. SETBP1 and CSF3R Mutations In Atypical Chronic Myeloid Leukemia. In: (American Society of Hematology; Washington, DC, 2013). [Google Scholar]
- 34.Meggendorfer M, Bacher U, Alpermann T, Haferlach C, Kern W, Gambacorti-Passerini C et al. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia 27, 1852–1860 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Faisal M, Stark H, Busche G, Schlue J, Teiken K, Kreipe HH et al. Comprehensive mutation profiling and mRNA expression analysis in atypical chronic myeloid leukemia in comparison with chronic myelomonocytic leukemia. Cancer Med 8, 742–750 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bose P, Nazha A, Komrokji RS, Patel KP, Pierce SA, Al-Ali N et al. Mutational landscape of myelodysplastic/myeloproliferative neoplasm-unclassifiable. Blood 132, 2100–2103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Al-Kali A, Abou Hussein AK, Patnaik M, Zblewski D, Gangat N, Hashmi S et al. Hypomethylating agents (HMAs) effect on myelodysplastic/myeloproliferative neoplasm unclassifiable (MDS/MPN-U): single institution experience. Leuk Lymphoma 59, 2737–2739 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Adema V, Larrayoz MJ, Calasanz MJ, Palomo L, Patino-Garcia A, Agirre X et al. Correlation of myelodysplastic syndromes with i(17)(q10) and TP53 and SETBP1 mutations. Br J Haematol 171, 137–141 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Bernard E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-Martinez JS et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med 26, 1549–1556 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiNardo CD, Daver N, Jain N, Pemmaraju N, Bueso-Ramos C, Yin CC et al. Myelodysplastic/myeloproliferative neoplasms, unclassifiable (MDS/MPN, U): natural history and clinical outcome by treatment strategy. Leukemia 28, 958–961 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen N, Oakley K, Han Y, Kwok M, Crouch G & Du Y Interaction with XPO1 is essential for SETBP1 to induce myeloid transformation. Leukemia 33, 2758–2762 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Steensma DP, Wermke M, Klimek VM, Greenberg PL, Font P, Komrokji RS et al. Phase I First-in-Human Dose Escalation Study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia, 1–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated for the current study are not publicly available due to patient privacy concerns but are available from the corresponding author on reasonable request.