

Abstract
Histatins are a group of small cationic peptides in human saliva which are well known for their antibacterial and antifungal activities. In a previous study we demonstrated that histatin 5 kills both blastoconidia and germ tubes of Candida albicans in a time- and concentration-dependent manner at 37°C, whereas no killing was detected at 4°C. This indicated that killing activity depends on cellular energy. To test histatin 5 killing activity at lower cellular ATP levels at 37°C, respiratory mutants, or so-called petite mutants, of C. albicans were prepared. These mutants are deficient in respiration due to mutations in mitochondrial DNA. Mutants were initially identified by their small colony size and were further characterized with respect to colony morphology, growth characteristics, respiratory activity, and cytochrome spectra. The killing activity of histatin 5 at the highest concentration was only 28 to 30% against respiratory mutants, whereas 98% of the wild-type cells were killed. Furthermore, histatin 5 killing activity was also tested on wild-type cells in the presence of the respiratory inhibitor sodium azide or, alternatively, the uncoupler carbonyl cyanide m-chlorophenylhydrazone. In both cases histatin 5 killing activity was significantly reduced. Additionally, supernatants and pellets of cells incubated with histatin 5 in the presence or absence of inhibitors of mitochondrial ATP synthesis were analyzed by sodium dodecyl sulfate gel electrophoresis. It was observed that wild-type cells accumulated large amounts of histatin 5, while wild-type cells treated with inhibitors or petite mutants did not accumulate significant amounts of the peptide. These data showed first that cellular accumulation of histatin 5 is necessary for killing activity and second that accumulation of histatin 5 depends on the availability of cellular energy. Therefore, mitochondrial ATP synthesis is required for effective killing activity of histatin 5.
Candida albicans is a dimorphic yeast which can switch between the blastospore form and one of several filamentous forms referred to as germ tubes, pseudohyphae, and hyphae (24, 25). This fungus is frequently found in the oral flora of healthy individuals. A comprehensive analysis of publications on oral yeast carriage revealed that the frequency of C. albicans in the oral cavity ranges between 2 and 69% (median, 24.5%) in healthy adults and between 41 and 54% (median, 44.0%) in infants of ages between 1 week and 18 months (24). Various systemic and local factors such as malnutrition, immunodeficiencies, endocrine disorders, malignant diseases, radiation therapy, xerostomia, and denture wearing can predispose humans to Candida infections (24, 26, 33).
The fact that a majority of the individuals carrying Candida in the oral cavity do not develop candidiasis demonstrates that host defense systems prevent development of this disease under normal conditions. Saliva appears to play a crucial role in this function, because patients with decreased saliva secretion, due to Sjögren's syndrome or as a consequence of radiation treatment for head and neck cancer, often develop oral candidiasis (1, 4, 22, 30, 31). Among the many proteins secreted into human parotid and submandibular-sublingual saliva, the family of histatins may play an important role in maintaining oral health. The major members of the histatin family are histatins 1, 3, and 5, containing 38, 32, and 24 amino acids, respectively (27, 37). They are cationic due to a high content of the basic amino acids lysine and arginine. In a recent clinical study, histatin concentrations in salivary secretions of individuals not harboring oral C. albicans were found to be significantly higher than those of healthy carriers (15). This suggests that histatins may prevent candidiasis in vivo, and indeed, in vitro studies have shown that these peptides kill C. albicans very effectively (27, 29, 43). Therefore, elucidation of the antifungal mechanism of histatins may contribute to understanding the role of saliva in oral homeostasis.
In a previous study, we found that histatin 5, which is the most active of all histatins, killed both C. albicans blastoconidia and germ tubes in a time- and concentration-dependent manner at 37°C but not at 4°C (C. Gyurko, U. Lendenmann, M. S. Lamkin, C. Champagne, R. F. Troxler, and F. G. Oppenheim, J. Dent. Res., abstr. 1443, 77:286, 1998). These results suggested that killing activity may depend on metabolic activity of the cells. Therefore, carrying out C. albicans killing assays under conditions where mitochondrial ATP synthesis cannot take place could be a powerful tool to determine the role of cellular energy in the antifungal activity of histatin 5. In yeast, oxidative phosphorylation can be abolished by suppressing mitochondrial ATP synthesis with specific inhibitors or by inducing respiratory mutations. Sodium azide inhibits cytochrome oxidase, and therefore, the cells are restricted to nonoxidative pathways for ATP synthesis (16, 42). Alternatively, carbonyl cyanide m-chlorophenylhydrazone (CCCP), an uncoupler of membrane proton gradients, stops ATP synthesis without blocking oxygen uptake by the cells (13, 41). Indeed, it was recently described that sodium azide and CCCP protected C. albicans from the antifungal activity of histatin 5. However, the results could not conclusively answer whether the protection was due to mitochondrial dysfunction, because cellular ATP levels were not reduced by treatment of cells with these inhibitors (18). An additional indication of a mitochondrial role in killing activity was provided by colocalization experiments with fluorescently labeled histatin 5 and a mitochondrion-specific probe that identified the mitochondria as an intracellular target of histatin 5 (12). Limitations on the use of metabolic inhibitors or fluorescent probes are their toxic side effects on microbial cells. Therefore, the role of mitochondria in the susceptibility of C. albicans to histatin 5 can be identified only if their function is abolished without treating cells with metabolic inhibitors.
Yeasts offer the unique opportunity that mutants deficient in mitochondrial respiration can be isolated. For Saccharomyces cerevisiae such petite mutants have been known for a long time (8), but petite mutants of C. albicans were thought not to be viable (5). However, recent work indicates that petite mutants of C. albicans can be used to study a host of biological questions related to mitochondrial function (2, 3, 14, 32). Such mutants form only tiny, so-called petite, colonies because their cell division rates are lower than that of the normal cells and their biomass yield on glucose is decreased (8, 34, 36). The growth of petite mutants is limited to fermentable carbon sources such as glucose or sucrose, but they fail to grow on glycerol and ethanol (2, 34). Cytochrome determinations show that petite mutants are lacking either cytochrome b or cytochrome aa3 or both but always retain cytochrome c (6, 10, 35). The molecular basis of the petite phenotype is mutation or deletion of mitochondrial genes (9).
In this study we successfully induced petite mutation by culturing C. albicans in the presence of acriflavine at an elevated temperature of 42°C. The mutants obtained were thoroughly characterized with respect to growth pattern, cytochrome spectrum, and respiratory activity. Subsequently, the killing activity of histatin 5 was tested with the petite mutants, and the results were compared to those obtained with wild-type cells in the presence of sodium azide or CCCP. Additionally, the cellular uptake of histatin 5 was determined with wild-type, sodium azide-treated wild-type, and petite cells.
MATERIALS AND METHODS
Yeast culture.
The strains used in this study were C. albicans ATCC 44505 and petite mutants 1 and 2, which were isolated following mutagenesis of C. albicans ATCC 44505. Cells were maintained on Sabouraud dextrose agar (SDA) (Difco, Detroit, Mich.).
Induction and isolation of respiratory mutants.
Petite mutants were obtained according to a procedure published by Aoki and Ito-Kuwa in 1987 (3). C. albicans wild-type cells were grown on an SDA plate overnight at 37°C. A colony was transferred into an Erlenmeyer flask containing 100 ml of liquid yeast-peptone-glucose (YPG) medium consisting of (per liter) 10 g of yeast extract, 20 g of peptone (both from Difco), and 20 g of glucose. Acriflavine was added to the cell suspension at a final concentration of 100 μg · ml−1, and subsequently, the flask was incubated at 42°C. After cells reached stationary phase, the culture was centrifuged, washed, plated onto glucose-limited agar plates, and incubated at 37°C for 5 days. Small colonies were selected and transferred onto glucose- and glycerol-limited agar plates. Glucose-limited agar plates contained (per liter) 20 g of peptone, 10 g of yeast extract, 10 g of glucose, 10 g of glycerol, and 15 g of agar, while in glycerol-limited plates glucose was omitted and the glycerol concentration was increased to 40 g · liter−1. After incubation at 37°C for 5 days, colonies growing only on glucose- but not on glycerol-limited agar plates were selected and used for further studies.
Growth and colony characteristics.
The growth patterns of the petite mutants and the wild-type parent strain were compared in YPG medium at 30°C with constant shaking at 200 rpm. At various time intervals, growth was measured spectrophotometrically (Spectronic 1201; Milton-Roy Londonderry, N.H.) at 600 nm. Detection of respiration-deficient cells was carried out with glucose-limited and glycerol-limited agar plates which were incubated at 37°C for 72 h. For color-based identification of respiration-deficient mutants, cells were grown on indicator plates containing (per liter) 1.5 g of KH2PO4, 1.5 g of (NH4)2SO4, 1 g of MgSO4 · H2O, 1.5 g of peptone, 1.5 g of yeast extract, 20 g of glucose, 0.01 g of eosin Y, 0.01 g of trypan blue, and 15 g of agar (23).
Cytochrome spectra.
Yeast cells were grown in 500 ml of YPG medium at 37°C, collected by centrifugation, washed with distilled water, and resuspended in 6 to 10 ml of water to give a dense cell slurry. Aliquots of cell suspensions were used for spectral analysis with a dual-beam U-2000 spectrophotometer (Hitachi Instruments Inc., Stoughton, Mass.) as described previously (6).
Respiration measurements.
C. albicans wild-type cells and mutant cells deficient in respiration were grown on SDA plates at 37°C for 24 and 72 h, respectively. Colonies were collected, washed with sterile water, and centrifuged, and the pellet was resuspended in 5 ml of 10 mM potassium phosphate buffer (pH 7.4) (PPB) to give a cell density of 5 × 106 cells · ml−1. Oxygen consumption by the cells was measured at 37°C with a biological oxygen monitor (model-5300; Yellow Springs Instrument, Yellow Springs, Ohio). Initially, endogenous oxygen consumption was measured, and subsequently, effects of addition of glucose, sodium azide (Fisher Scientific, Fair Lawn, N.J.), and CCCP (Sigma, St. Louis, Mo.) were measured at final concentrations of 30 mM, 25 mM, and 100 μM, respectively. Respiration rates were calculated from triplicate experiments and are expressed as nanomoles of O2 · milliliter−1 · minute−1.
Killing assay.
Candida killing assays were performed with wild-type blastoconidia and petite mutants 1 and 2 as described previously (43). Briefly, C. albicans wild-type and petite cells were grown on SDA plates at 37°C for 24 and 72 h, respectively. Colonies were suspended in 10 ml of 10 mM PPB (pH 7.4) and diluted to give a cell density of approximately 105 cells · ml−1. Aliquots of 50 μl of cell suspension were added to the wells of a microtiter plate, and cells were allowed to attach for 15 min. Subsequently, 50-μl portions of histatin test solutions (in PPB) were added to the wells, and the microtiter plate was incubated for 60 min at 37°C. To test the effect of inhibitors of mitochondrial ATP synthesis, cells were suspended in PPB containing sodium azide (25 mM) or CCCP (100 μM) and killing assays were carried out with histatin test solutions containing the same concentrations of these inhibitors. In separate control experiments sodium azide (25 mM) and CCCP (100 μM) were evaluated for their toxicity towards C. albicans in the absence of histatin 5. After 60 min of incubation, wells were washed three times with PPB and 100 μl of molten Sabouraud dextrose broth (45°C) containing 2% agarose were added to each well. Subsequently, wild-type cells were incubated for 5 to 6 h and petite mutant cells were incubated for 16 to 18 h at 30°C. Under these conditions, surviving cells divide and form colonies, while dead cells remain as single cells. A colony was defined as a cluster of more than five contiguous cells. To estimate killing activity, a total of 100 dead cells or live colonies were counted with an inverted microscope and the results were expressed as percentage of killed cells per total number of cells. Killing assays were carried out in triplicates.
Interaction of wild-type and petite cells with histatin 5.
Wild-type and petite cells were grown in YPG liquid medium at 30°C. Exponentially growing cells were centrifuged, washed three times with distilled water, and resuspended in 10 mM PPB to a concentration of approximately 107 cells · ml−1 as measured by optical density at 530 nm. Wild-type cells were also resuspended in 10 mM PPB containing 25 mM sodium azide to test effects of respiration inhibitors on the interactions of histatin 5 with cells. Cell suspensions of 1 ml were incubated with histatin 5 at 65 nmol · ml−1 (200 μg · ml−1) at 37°C for 60 min. In control experiments, cells were incubated with buffer alone. Cells were pelleted at 10,000 × g for 2 min, and the supernatant was collected, recentrifuged, and filtered (type HV filter, 0.45-μm pore size; Millipore, Bedford, Mass.). The pellets were washed twice with PPB and suspended in 200 μl of PPB containing a protease inhibitor cocktail consisting of AEBSF [4-(2-aminoethyl)-benzenesulfonylfluoride], aprotinin, E-64, EDTA, and leupeptin (Calbiochem-Novabiochem Corp., La Jolla, Calif.). Glass beads (425 to 600 μm; Sigma) were added to the cell suspension, and cells were disrupted by three 1-min bursts in a Bead Beater-8 Chamber (BioSpec Products, Bartlesville, Okla.). Subsequently, proteins in supernatants and lysates of cell pellets were examined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 16% gels and stained with 0.4% Coomassie brilliant blue R250.
RESULTS
Characterization of petite mutants.
Petite mutation was induced by growing C. albicans cells at 42°C to stationary phase in the presence of acriflavine. A sample of the culture was plated on glucose-limited agar plates, and several hundred colonies that were significantly smaller than average were selected and tested for their ability to grow on glucose- and glycerol-limited agar plates. Approximately 0.5% of the colonies that grew with glucose failed to grow on glycerol-limited agar plates, indicating a petite phenotype. Two of these colonies were designated petites 1 and 2 and were used for further studies.
The growth patterns of wild-type and petite 1 and 2 cells were examined on glucose- and glycerol-limited agar plates. Within 24-h, wild-type cells grew well on both glucose-limited (Fig. 1A) and glycerol-limited (Fig. 1B) plates. In contrast, even after 72 h, petites 1 and 2 formed only tiny colonies on glucose-limited agar (Fig. 1C) and completely failed to grow on plates containing only glycerol as a carbon source (Fig. 1D). Further confirmation of respiratory deficiency in petites 1 and 2 was obtained on indicator plates containing eosin Y and trypan blue. On this medium, wild-type cells formed large, pale-bluish colonies, whereas the petite mutants formed only small, deep-violet colonies (Fig. 2). The specific growth rates for the wild type and petites 1 and 2 in liquid YPG medium were 0.54, 0.07, and 0.06 h−1, respectively. The corresponding optical densities at 600 nm of the cultures at stationary phase were approximately 5 times higher for the wild type than for mutant cells. These results showed that petite mutants grow approximately 10 times slower and form 5 times less biomass than wild-type cells. This growth pattern is similar to that of petite mutants of S. cerevisiae (36).
FIG. 1.

Growth characteristics of C. albicans blastoconidia and petites 1 and 2 on agar plates. Cells were incubated at 37°C on glucose-limited agar plates for 24 h (A) and 72 h (C) or on glycerol-limited agar plates for 24 h (B) and 72 h (D). wt, wild type.
FIG. 2.
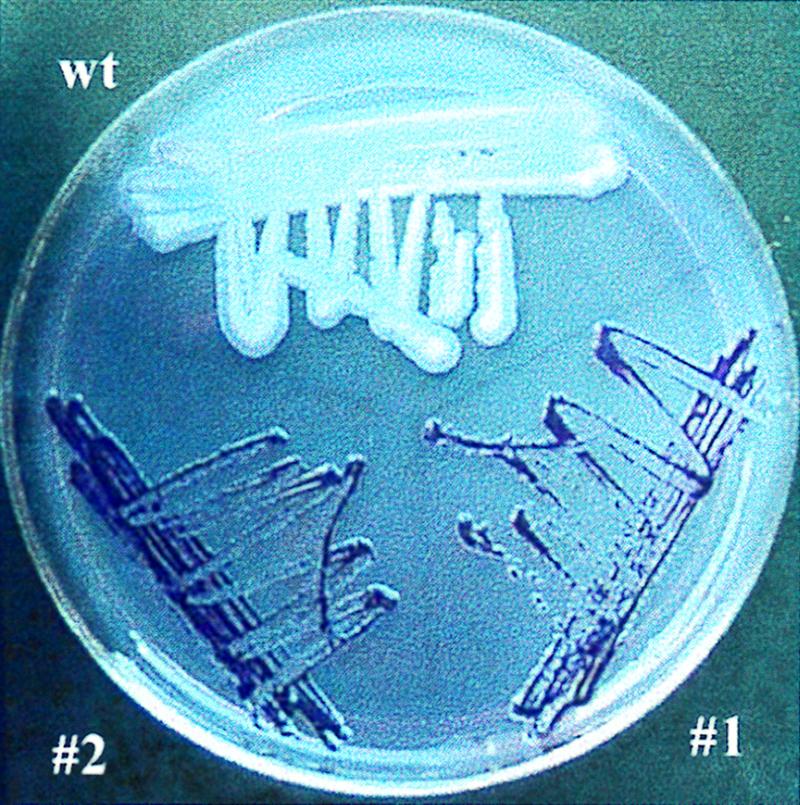
Color differentiation of C. albicans blastoconidia and petites 1 and 2 on an indicator plate containing eosin Y and trypan blue. On this medium metabolically active wild-type (wt) cells from large, pale-bluish colonies, while petite mutants grow as small, deep-violet colonies.
Cytochrome spectra of C. albicans wild-type cells and petites 1 and 2 were analyzed in a dual-beam spectrophotometer at room temperature (Fig. 3). The spectra of wild-type cells showed peaks characteristic for cytochrome c (550 nm), cytochrome b (560 nm), and cytochrome aa3 (600 nm). The spectra of petites 1 and 2 lacked absorption bands for cytochromes b and aa3 and indicated reduced amounts of cytochrome c. This result was confirmed by spectroscopic cytochrome analysis at −190°C (35).
FIG. 3.

Cytochrome spectra of wild-type (wt) and petite mutant cells. Spectra were recorded with a thick cell slurry at room temperature in a cuvette with a 1-cm light path. AU, absorption units.
Oxygen consumption measurements.
Respiration rates of petites 1 and 2 were compared with that of C. albicans wild-type cells in an oxygraph at 37°C by using cell suspensions containing 5 × 106 cells · ml−1. Figure 4 shows the results of a typical experiment. Initially, the endogenous oxygen consumption rate was measured. During the first 5 min, wild-type cells exhibited a relatively high endogenous oxygen consumption rate of 7.0 ± 0.45 nmol · ml−1 · min−1, which is characteristic for C. albicans (39). With time, endogenous oxygen consumption decreased slightly to 3.8 ± 1.0 nmol · ml−1 · min−1 and could be stimulated again to 5.6 ± 1.5 nmol · ml−1 · min−1 by the addition of glucose after 10 min. Compared to that of wild-type cells, the oxygen consumption by petites 1 and 2 was negligible and the addition of glucose could not stimulate oxygen consumption. These results provide further evidence that petites 1 and 2 are deficient in respiration and therefore are incapable of mitochondrial ATP synthesis.
FIG. 4.

Oxygen consumption by wild-type (wt) and petite mutant cells. (A) Comparison of rates of oxygen consumption by wild-type and petite mutant cells; (B) effect of inhibitors on wild-type cells. █, glucose was added to a final concentration of 30 mM after 10 min; ⧫, either sodium azide or CCCP was added to a final concentration of 25 mM or 100 μM, respectively. The decrease in oxygen concentration (conc.) prior to additions represents endogenous consumption.
To test the effect of sodium azide and CCCP on mitochondrial activity of C. albicans, oxygen consumption measurements were carried out in the presence of these inhibitors (Fig. 4B). After the endogenous oxygen consumption rate was measured for the first 5 min (6.9 ± 0.44 nmol · ml−1 · min−1), sodium azide was added to a final concentration of 25 mM, which reduced the respiration rate to 1.5 ± 0.44 nmol · ml−1 · min−1. In contrast, at a concentration of 100 μM, CCCP increased the respiration rate approximately twofold to 14.7 ± 3.2 nmol · ml−1 · min−1. This is significantly higher than the endogenous oxygen consumption. The addition of glucose after 10 min stimulated oxygen consumption in untreated wild-type cells, while it had no detectable effect on sodium azide- or CCCP-treated cells. This indicated that the concentrations of sodium azide and CCCP were high enough to achieve maximum inhibition or stimulation of respiration, respectively.
Killing activity of histatin 5.
Histatin 5 killing efficacy was tested with C. albicans wild-type and petite mutant 1 and 2 cells. Killing assays were carried out at 37°C with histatin 5 concentrations ranging from 0 to 131 nmol · ml−1 (0 to 400 μg · ml−1). Exposure of wild-type cells to histatin 5 resulted in a typical dose-response curve which reached a maximum of 98% killing at the highest histatin 5 concentration (Fig. 5A). Under the same experimental conditions, histatin 5 killing activity was significantly reduced to 28% for petite 1 and 32% for petite 2 at the highest concentration tested (Fig. 5A).
FIG. 5.

Killing activity of histatin 5. (A) Comparison of histatin 5 activity towards wild-type (⧫) and petite mutant 1 (▴) and 2 (■) cells. (B) Killing of wild-type cells incubated in the presence of 10 mM PPB only (⧫), 25 mM sodium chloride (○), 25 mM sodium azide (▴), or 100 μM CCCP (■). The data shown are means and standard deviations from at least two experiments performed in triplicates.
Killing of blastoconidia by histatin 5 was also tested with wild-type cells in the presence and absence of sodium azide and CCCP. Initial control experiments carried out to test whether sodium azide (25 mM) and CCCP (100 μM) were themselves toxic showed that these inhibitors do not kill C. albicans at the concentrations employed. Therefore, these compounds could be used to test whether inhibition of mitochondrial ATP synthesis would alter the susceptibility of C. albicans to histatin 5. When sodium azide or CCCP was included in the histatin 5 killing assay, the effect of histatin 5 on C. albicans was significantly reduced (Fig. 5B). At the highest histatin 5 concentration, only 49% of the sodium azide-treated and 56% of the CCCP-treated cells were killed, while 98% killing occurred in the absence of these inhibitors. To exclude the possibility that a salt effect could account for the reduced killing activity observed with sodium azide, a control experiment was carried out in the presence of sodium chloride at the same concentration (25 mM). Sodium chloride reduced killing at low histatin concentrations but caused no reduction at 65 nmol · ml−1 (Fig. 5B). These data show that the reduced killing observed with sodium azide was due to its action as a respiratory inhibitor. In summary, our results obtained with wild-type and petite cells revealed that reduction of metabolic activity decreased the susceptibility of C. albicans to histatin 5.
Interaction of the cells with histatin 5.
Blastoconidia of C. albicans wild-type cells, wild-type cells treated with 25 mM sodium azide or 100 μM CCCP, and petites 1 and 2 were incubated with 65 nmol · ml−1 of histatin 5 at 37°C for 10, 30, and 60 min. After incubation, cell suspensions were centrifuged and pellets and supernatants were separately analyzed by SDS-PAGE on 16% gels. Comparing the electrophoretograms of the supernatants and pellets, we found that a band with an electrophoretic mobility identical to that of histatin 5 disappeared from the suspending medium and accumulated in the cell pellets of untreated wild-type cells in a time-dependent manner (Fig. 6A and B). In contrast, when sodium azide was included in the assay, the vast majority of histatin 5 remained in the supernatant even after 60 min of incubation, and only an extremely small amount of histatin 5 could be found in cell pellets (Fig. 6C and D). When wild-type cells were treated with CCCP, histatin 5 partially disappeared from the supernatant and a small amount accumulated intracellularly in a time-dependent fashion (Fig. 6E and F). Comparing the results obtained with CCCP (Fig. 6E and F) and untreated wild-type cells (Fig. 6A and B), it becomes clear that CCCP significantly reduced, but did not completely abolish, cellular uptake of histatin 5. This is in agreement with the killing data obtained under the same conditions (Fig. 5B). The vast majority of histatin 5 remained in the supernatant when petite 1 was incubated with this peptide. However, a small but clearly detectable amount of histatin 5 could be found in the pellet after 10 min and showed only a minor increase in staining intensity after 60 min (Fig. 6G and H). Identical results were obtained with petite 2 (Fig. 6I and J). These results show that only metabolically active wild-type cells were able to completely take up histatin 5 from the suspending medium.
FIG. 6.

SDS-PAGE analysis of C. albicans incubated with histatin 5. Cells were incubated for 10 min (lanes 1), 30 min (lanes 2), or 60 min (lanes 3) with histatin 5 (65 nmol · ml−1) and centrifuged. The resulting supernatants (A, C, E, G, and I) and pellets (B, D, F, H, and J) were analyzed by SDS-PAGE on 16% gels. Lanes 4, 60-min control without histatin 5; lanes 5, histatin 5 standard (10 μg). (A and B) Untreated wild-type cells; (C and D) wild-type cells treated with 25 mM sodium azide; (E and F) wild-type cells treated with 100 μM CCCP; (G and H) petite 1; (I and J) petite 2. Gels were stained with Coomassie brilliant blue R250.
DISCUSSION
Petite mutants of S. cerevisiae have been known for many years. They occur spontaneously at a rate of 0.5% and can be easily identified and isolated by their small colony size and inability to grow on nonfermentable substrates (34). In contrast, for a long time it was thought that petite mutants of C. albicans form only microcolonies which die before becoming visible (5). We have successfully isolated petite mutants from C. albicans which exhibited the same phenotypic characteristics as the well-known petite mutants of S. cerevisiae with respect to growth pattern, oxygen consumption rates, and cytochrome spectra. Petite mutants are unable to carry out mitochondrial ATP synthesis because they lack cytochromes a and b and therefore are limited to substrate-level phosphorylation to generate ATP. This reduces the amount of cellular energy that can be derived from glucose to a small fraction of that generated by wild-type cells.
It is interesting that in the presence of sodium azide, oxygen consumption by C. albicans wild-type cells was reduced to almost the same extent as in petite mutants (Fig. 4). Sodium azide is a specific inhibitor of cytochrome oxidase (42). This enzyme catalyzes the reduction of O2 to water, and therefore, oxygen consumption by C. albicans cells was almost completely abolished upon addition of this inhibitor (Fig. 4B). In petite mutants, cytochrome oxidase is also not functional due to the absence of cytochrome a. In contrast, CCCP is an uncoupler of the membrane proton gradients and thus inhibits mitochondrial ATP synthesis without interrupting the respiratory chain and oxygen consumption. This results in an increased rate of oxygen consumption by C. albicans cells upon addition of CCCP (Fig. 4B).
Treatment of cells with inhibitors or petite mutation dramatically reduced the killing activity of histatin 5. This showed that the effectiveness of histatin 5 depends on cellular energy. Previous studies have shown that the activity of the cationic antimicrobial peptides defensins against tumor cells (21), bacteria (38), and C. albicans (20) is energy dependent. This energy dependence is explained by our current understanding of the mechanism of cationic antimicrobial peptides, in which a positively charged peptide first binds to the external surface of the negatively charged phospholipid bilayer. Subsequently, two to four peptide molecules form a channel-like structure under the influence of the electric field induced by the membrane potential, leading to leakage of cytoplasmic molecules and cell death (11, 40).
If the interaction of histatins with the cytoplasmic membrane of C. albicans occurs in a similar fashion, then treatment of wild-type cells with inhibitors of ATP synthesis or petite mutations would result in reduced cellular accumulation of this peptide. This was indeed found when cells incubated with histatin 5 were analyzed by SDS-PAGE (Fig. 6). At a concentration of 65 nmol · ml−1 histatin 5, wild-type cells not treated with inhibitors accumulated the largest amount of the peptide, and 98% of the cells were killed (Fig. 5B and 6B). On the other hand, wild-type cells treated with CCCP and sodium azide were killed at 40 and 10%, respectively, and accumulated proportionally less histatin 5. Petites 1 and 2 displayed intermediate uptake and killing (Fig. 5A and 6G to J).
Even though the above findings imply similarities between defensins and histatins, it cannot be concluded that the two proteins kill microorganisms with the same mechanism. Histatins release potassium (29) magnesium (44), and ATP (18) from C. albicans, but liberation of large molecules, e.g., proteins and nucleic acids, has not yet been reported. Therefore, it is unlikely that histatin 5 can form stable membrane pores. This notion is further supported by the finding that histatin 5 treatment did not depolarize the membrane of C. albicans (18). Additionally, specific histatin binding proteins on the cell membrane of C. albicans have been described (7, 44) and it has been suggested that such receptors may be involved in histatin transport (7). The role of receptors for histatin killing activity needs further exploration, since a histatin 5 fragment synthesized from d-amino acids kills C. albicans as effectively as the l-isomer (28).
In summary, our results show that inhibition of mitochondrial ATP synthesis protected C. albicans from the fungicidal activity of histatin 5 and that this protection appeared to be due to reduced cellular accumulation of the peptide. This is in agreement with recent findings that fluorescently labeled histatin 3 accumulated in cells of C. albicans at 30°C but not at 0°C (44). Therefore, it can be considered as established that energy-dependent uptake of histatins is necessary for killing of C. albicans. Whether histatin autonomously penetrates the cytoplasmic membrane or uptake is mediated by a specific receptor remains unclear. The correlation between killing and uptake of both histatin 5 (this study) and histatin 3 (44) in the absence of obvious membrane damage suggests that the final events in killing occur intracellularly. For instance, studies have shown that the energized mitochondrion may be the intracellular target of histatin 5 (12). Furthermore, petite mutants contain only degenerated mitochondria without typical cristae (3, 17, 19). Therefore, petite mutants lack this type of intracellular target, which may reduce equilibrium-driven uptake of histatin 5. Further research is necessary to determine whether mitochondrial energy is only a preliminary requirement for histatin 5 uptake or whether the mitochondrion is indeed the primary intracellular target of histatin 5.
ACKNOWLEDGMENTS
We thank Fred Sherman, University of Rochester, for examining cytochrome expression in our petite mutants at −190°C.
This work was supported in part by NIH/NICDR grants DE05672 and DE07652.
REFERENCES
- 1.Abraham C M, Al-Hashimi I, Haghighat N. Evaluation of the levels of oral Candida in patients with Sjögren's syndrome. Oral Surg Oral Med Oral Radiol Endod. 1998;86:65–68. doi: 10.1016/s1079-2104(98)90151-2. [DOI] [PubMed] [Google Scholar]
- 2.Abu Hatab M A, Whittaker P A. Isolation and characterization of respiration-deficient mutants from the pathogenic yeast Candida albicans. Antonie Leeuwenhoek. 1992;61:207–219. doi: 10.1007/BF00584227. [DOI] [PubMed] [Google Scholar]
- 3.Aoki S, Ito-Kuwa S. Induction of petite mutation with acriflavine and elevated temperature in Candida albicans. J Med Vet Mycol. 1987;25:269–277. doi: 10.1080/02681218780000611. [DOI] [PubMed] [Google Scholar]
- 4.Atkinson J C, Fox P C. Salivary gland dysfunction. Clin Geriat Med. 1992;8:499–511. [PubMed] [Google Scholar]
- 5.Bulder C J E A. Lethality of the petite mutation in petite negative yeasts. Antonie Leeuwenhoek. 1964;30:442–454. doi: 10.1007/BF02046758. [DOI] [PubMed] [Google Scholar]
- 6.Claisse M L, Pere-Aubert G A, Clavilier L P, Slonimski P P. Methode d'estimation de la concentration des cytochromes dans les cellules entieres de levure. Eur J Biochem. 1970;16:430–438. doi: 10.1111/j.1432-1033.1970.tb01098.x. [DOI] [PubMed] [Google Scholar]
- 7.Edgerton M, Koshlukova S E, Lo T E, Chrzan B G, Straubinger R M, Raj P A. Candidacidal activity of salivary histatins. Identification of a histatin 5-binding protein on Candida albicans. J Biol Chem. 1998;273:20438–20447. doi: 10.1074/jbc.273.32.20438. [DOI] [PubMed] [Google Scholar]
- 8.Ephrussi B, Hottinguer H, Chimenes A-M. Action de l'acriflavine sur les levures. I. La mutation “petite colonie.”. Ann Inst Pasteur (Paris) 1949;76:351–367. [Google Scholar]
- 9.Faye G, Fukuhara H, Grandchamp C, Lazowska J, Michel F, Casey J, Getz G S, Locker J, Rabinowitz M, Bolotin-Fukuhara M, Coen D, Deutsch J, Dujon B, Netter P, Slonimski P P. Mitochondrial nucleic acids in the petite colonie mutants: deletions and repetitions of genes. Biochimie. 1973;55:779–792. doi: 10.1016/s0300-9084(73)80030-6. [DOI] [PubMed] [Google Scholar]
- 10.Flury U, Mahler H R, Feldman F. A novel respiration-deficient mutant of Saccharomyces cerevisiae. J Biol Chem. 1974;249:6130–6137. [PubMed] [Google Scholar]
- 11.Hancock R E W. Peptide antibiotics. Lancet. 1997;349:418–422. doi: 10.1016/S0140-6736(97)80051-7. [DOI] [PubMed] [Google Scholar]
- 12.Helmerhorst E J, Breeuwer P, van't Hof W, Walgreen-Weterings E, Oomen L C J M, Veerman E C I, van Niew Amerongen A, Abee T. The cellular target of histatin 5 on Candida albicans is the energized mitochondrion. J Biol Chem. 1999;274:7286–7291. doi: 10.1074/jbc.274.11.7286. [DOI] [PubMed] [Google Scholar]
- 13.Heytler P G, Prichard W W. A new class of uncoupling agents—carbonyl cyanide phenylhydrazones. Biochem Biophys Res Commun. 1962;7:272–275. doi: 10.1016/0006-291x(62)90189-4. [DOI] [PubMed] [Google Scholar]
- 14.Ito-Kuwa S, Aoki S, Watanabe T, Ehara T, Osafune T. Fluorescence microscopic studies on mitochondria and mitochondrial nucleotids in a wild-type strain and respiratory mutants of Candida albicans. J Med Vet Mycol. 1988;26:207–217. doi: 10.1080/02681218880000301. [DOI] [PubMed] [Google Scholar]
- 15.Jainkittivong A, Johnson D A, Yeh C-K. The relationship between salivary histatin levels and oral yeast carriage. Oral Microbiol Immunol. 1998;13:181–187. doi: 10.1111/j.1399-302x.1998.tb00730.x. [DOI] [PubMed] [Google Scholar]
- 16.Keilin D, Hartree E F. Cytochrome and cytochrome oxidase. Proc R Soc B. 1939;127:167–191. [Google Scholar]
- 17.Kimura A, Tatsutomi Y, Fukuda H, Morioka H. Effect of acriflavine on the hexokinase isoenzyme pattern of a yeast, Hansenula jadinii. Biochim Biophys Acta. 1980;629:217–224. doi: 10.1016/0304-4165(80)90095-1. [DOI] [PubMed] [Google Scholar]
- 18.Koshlukova S E, Lloyd T L, Araujo M W B, Edgerton M. Salivary histatin 5 induced non-lytic release of ATP from Candida albicans leading to cell death. J Biol Chem. 1999;274:18872–18879. doi: 10.1074/jbc.274.27.18872. [DOI] [PubMed] [Google Scholar]
- 19.Kot E J, Rolewic L J, Olson V L, McClary D O. Growth, respiration and cytology of acetate-negative mutants of Candida albicans. Antonie Leeuwenhoek. 1975;41:229–238. doi: 10.1007/BF02565058. [DOI] [PubMed] [Google Scholar]
- 20.Lehrer R I, Ganz T, Szklarek D, Selsted M E. Modulation of the in vitro candidacidal activity of human neutrophil defensins by target cell metabolism and divalent cations. J Clin Invest. 1988;81:1829–1835. doi: 10.1172/JCI113527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lichtenstein A K, Ganz T, Nguyen T-M, Selsted M E, Lehrer R I. Mechanism of target cytolysis by peptide defensins. Target cell metabolic activities, possibly involving endocytosis, are crucial for expression of cytotoxicity. J Immunol. 1988;140:2686–2694. [PubMed] [Google Scholar]
- 22.Liu R P, Fleming T J, Toth B B, Keene H H. Salivary flow rates in patients with head and neck cancer 0.5 and 25 years after radiotherapy. Oral Surg Oral Med Oral Radiol Endod. 1990;70:724–729. doi: 10.1016/0030-4220(90)90008-g. [DOI] [PubMed] [Google Scholar]
- 23.Nagai S. Diagnostic color differentiation plates for hereditary respiration deficiency in yeast. J Bacteriol. 1963;86:299–302. doi: 10.1128/jb.86.2.299-302.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Odds F C. Candida and Candidiosis. 2nd ed. London, United Kingdom: Bailliere Tindall; 1988. [Google Scholar]
- 25.Odds F C. Morphogenesis in Candida albicans. Crit Rev Microbiol. 1991;12:45–93. doi: 10.3109/10408418509104425. [DOI] [PubMed] [Google Scholar]
- 26.Oksala E. Factors predisposing to oral yeast infections. Acta Odontol Scand. 1990;48:71–74. doi: 10.3109/00016359009012736. [DOI] [PubMed] [Google Scholar]
- 27.Oppenheim F G, Xu T, McMillian F M, Levitz S M, Diamond R D, Offner G D, Troxler R F. Histatins, a novel family of histidine-rich proteins in human parotid secretion. Isolation, characterization, primary structure and fungistatic effects on Candida albicans. J Biol Chem. 1988;263:7472–7477. [PubMed] [Google Scholar]
- 28.Oppenheim, F. G., T. Xu, and F. D. Roberts. May 1997. U.S. patent 5,631,228.
- 29.Pollock J J, Denepitiya L, MacKay B J, Iacono V J. Fungistatic and fungicidal activity of human parotid saliva histidine-rich polypeptides on Candida albicans. Infect Immun. 1984;44:702–707. doi: 10.1128/iai.44.3.702-707.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramirez-Amador V, Silverman S, Mayer P, Tyler M, Quivey J. Candidal colonization and oral candidiasis in patients undergoing oral and pharyngeal radiation therapy. Oral Surg Oral Med Oral Radiol Endod. 1997;84:149–153. doi: 10.1016/s1079-2104(97)90061-5. [DOI] [PubMed] [Google Scholar]
- 31.Rossie K M, Taylor J, Beck F M, Hodgson S E, Blozis G G. Influence of radiation therapy on oral Candida albicans colonization: a quantitative assessment. Oral Surg Oral Med Oral Radiol Endod. 1987;64:698–701. doi: 10.1016/0030-4220(87)90171-x. [DOI] [PubMed] [Google Scholar]
- 32.Roth-Ben Arie Z, Altboum Z, Berdicevsky I, Segal E. Isolation of a petite mutant from a histidine auxotroph of Candida albicans and its characterization. Mycopathologia. 1998;141:127–135. doi: 10.1023/a:1006988119891. [DOI] [PubMed] [Google Scholar]
- 33.Scully C, El-Kabir M, Samaranayake L P. Candida and oral candidosis: a review. Crit Rev Oral Biol Med. 1994;5:125–157. doi: 10.1177/10454411940050020101. [DOI] [PubMed] [Google Scholar]
- 34.Sherman F. The preparation of cytochrome-deficient mutants of yeast. Methods Enzymol. 1967;10:9610–9616. [Google Scholar]
- 35.Sherman F, Slonimski P P. Respiration-deficient mutants of yeast. II Biochemistry Biochim Biophys Acta. 1964;90:1–15. doi: 10.1016/0304-4165(64)90113-8. [DOI] [PubMed] [Google Scholar]
- 36.Tavlitzki J. Action de l'acriflavine sur les levures. III. Etude de la croissance des mutants “petite colonies.”. Ann Inst Pasteur (Paris) 1949;76:497–509. [Google Scholar]
- 37.Troxler R F, Offner G D, Xu T, van der Speck J C, Oppenheim F G. Structural relationship between human salivary histatins. J Dent Res. 1990;69:2–6. doi: 10.1177/00220345900690010101. [DOI] [PubMed] [Google Scholar]
- 38.Walton E, Gladstone G P. Factors affecting the susceptibility of staphylococci to killing by the cationic proteins from rabbit polymorphonuclear leucocytes: the effect of alteration of cellular energetics and of various iron compounds. Br J Exp Pathol. 1976;57:560–570. [PMC free article] [PubMed] [Google Scholar]
- 39.Ward J M, Nickerson W J. Respiratory metabolism of normal and divisionless strains of Candida albicans. J Gen Physiol. 1958;41:703–724. doi: 10.1085/jgp.41.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weinberg A, Krisanaprakornkit S, Dale B A. Epithelial antimicrobial peptides: review and significance for oral applications. Crit Rev Oral Biol Med. 1998;9:399–414. doi: 10.1177/10454411980090040201. [DOI] [PubMed] [Google Scholar]
- 41.Wilson D F. The stoichiometry and site specificity of the uncoupling of mitochondrial oxidative phosphorylation by salicylanilide derivatives. Biochemistry. 1969;8:2475–2481. doi: 10.1021/bi00834a033. [DOI] [PubMed] [Google Scholar]
- 42.Wilson D F, Chance B. Azide inhibition of mitochondrial electron transport. I. The aerobic state of succinate oxidation. Biochim Biophys Acta. 1967;131:421–430. doi: 10.1016/0005-2728(67)90002-3. [DOI] [PubMed] [Google Scholar]
- 43.Xu T, Levitz S M, Diamond R D, Oppenheim F G. Anticandidal activity of major human salivary histatins. Infect Immun. 1991;59:2549–2554. doi: 10.1128/iai.59.8.2549-2554.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu Y, Ambudkar I, Yamagishi H, Swain W, Walsh T J, O'Connell B C. Histatin 3-mediated killing of Candida albicans: effect of extracellular salt concentration on binding and internalization. Antimicrob Agents Chemother. 1999;43:2256–2262. doi: 10.1128/aac.43.9.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]