

SUMMARY
Aberrant expression of programmed death ligand-1 (PD-L1) in tumor cells promotes cancer progression by suppressing cancer immunity. The retinoblastoma protein RB is a tumor suppressor known to regulate the cell cycle, DNA damage response, and differentiation. Here, we demonstrate that RB interacts with nuclear factor κB (NF-κB) protein p65 and that their interaction is primarily dependent on CDK4/6-mediated serine-249/threonine-252 (S249/T252) phosphorylation of RB. RNA-seq analysis shows a subset of NF-κB pathway genes including PD-L1 are selectively upregulated by RB knockdown or CDK4/6 inhibitor. S249/T252-phosphorylated RB inversely correlates with PD-L1 expression in patient samples. Expression of a RB-derived S249/T252 phosphorylation-mimetic peptide suppresses radiotherapy-induced upregulation of PD-L1 and augments therapeutic efficacy of radiation in vivo. Our findings reveal a previously unrecognized tumor suppressor function of hyperphosphorylated RB in suppressing NF-κB activity and PD-L1 expression and suggest that the RB-NF-κB axis can be exploited to overcome cancer immune evasion triggered by conventional or targeted therapies.
Graphical Abstract
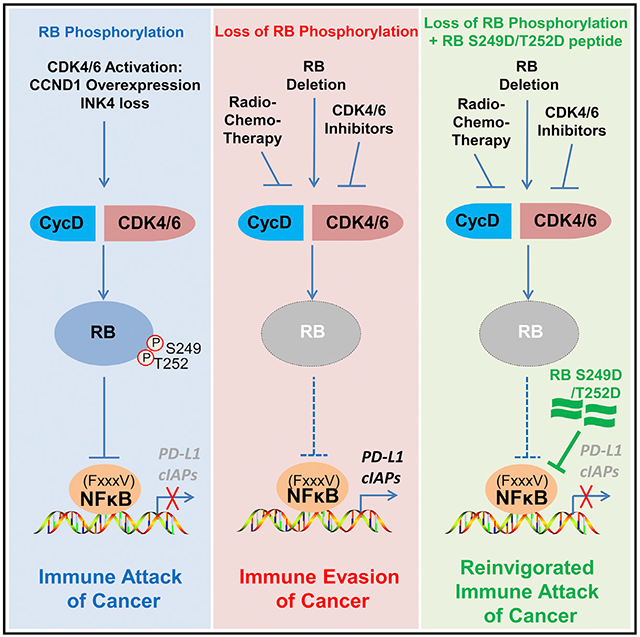
In Brief
RB is known to act as a tumor suppressor by inhibiting E2F transcription factors, and this function is abolished due to its phosphorylation by CDKs. Jin et al. identify a previously uncharacterized tumor suppressor role of CDK4/6-phosphorylated RB in promoting cancer immunity via inhibition of NF-κB transcriptional activity and PD-L1 expression.
INTRODUCTION
Evasion of immune surveillance is a hallmark of human cancers (Hanahan and Weinberg, 2011). While neoantigens generated by cancerous cells are potentially recognizable by the immune system, tumors can often escape from immune attack via distinct mechanisms, including aberrant activation of immune checkpoints that terminates immune responses (Sharma and Allison, 2015).
Programmed death-1 (PD-1) and its ligand PD-L1 (also known as B7-H1) are two major immune checkpoint molecules (Dong et al., 1999, 2002; Freeman et al., 2000; Ishida et al., 1992). PD-L1 expressed in cancer cells binds to its receptor PD-1 in activated T cells, thereby triggering T cell apoptosis, cytotoxic T cell function loss, and cancer immune evasion (Dong et al., 1999, 2002; Freeman et al., 2000). Anti-PD-1 and anti-PD-L1 antibody-based immune checkpoint blockade therapies have been approved for treatment of different cancer types (Iwai et al., 2002; Li et al., 2018; Topalian et al., 2012a; Zhu and Chen, 2003). While the PD-1/PD-L1 blockade can improve patient progress-free survival, the response rate among all patients remains relatively low, and this is often associated with the low expression rate of PD-L1 in patient samples (Herbst et al., 2014; Topalian et al., 2012b). Previous studies have shown that PD-L1 protein abundance is regulated by ubiquitination and proteasome pathway proteins such as the deubiquitinase CSN5 and the ubiquitin E3 ligase adaptor SPOP, and aberrant upregulation of PD-L1 confers immune checkpoint therapy resistance (Lim et al., 2016; Zhang et al., 2018). These studies highlight that further studies are needed to better understand the molecular mechanisms underlying PD-L1 expression to improve patient response to immunotherapy in clinic.
The retinoblastoma protein RB is a well-studied tumor suppressor. It is a multi-functional protein that regulates a number of crucial cellular activities, including cell cycle progression, DNA damage response and checkpoint activation, and differentiation (DeCaprio et al., 1989; Goodrich et al., 1991; Ishak et al., 2016; Manning and Dyson, 2012; Sherr and McCormick, 2002). RB protein primarily exists in three states: unphosphorylated, hypo-phosphorylated (also called under or partially phosphorylated), and hyper-phosphorylated (Burkhart and Sage, 2008; Narasimha et al., 2014). Un- or hypo-phosphorylated RB interacts with E2F transcription factors and inhibits E2F target gene expression and G1/S cell cycle transition. Upon mitogen stimulation or in the late G1 phase, however, RB is hyperphosphorylated at multiple sites by cyclin/CDK complexes such as CYCLIN D/CDK4/6, resulting in release of E2F factors from RB sequestration and progression of the cell cycle (Sherr and Roberts, 2004). Of note, a mouse genetic study suggests that RB also possesses E2F-independent tumor suppressor functions, although the signaling pathways responsible for such a function remain elusive (Sun et al., 2011).
NF-κB is a transcription factor that is highly implicated in cancer. Upon the stimuli of proinflammatory cytokines such as TNF-α, NF-κB can be activated by MAP3K7-IKK signaling axis (Taniguchi and Karin, 2018). In addition to regulating canonical pro-survival genes, NF-κB is known to regulate mRNA expression of PD-L1 (also known as CD274) gene in various cancer types (Bouillez et al., 2017; Peng et al., 2015). A recent CRISPR/Cas9-based screening identifies the NF-κB pathway as one of the key mechanisms that promote cancer cell escape from immune attack of T cells(Manguso et al., 2017; Pan et al., 2018). In the present study, we identify hyperphosphorylated RB as a major suppressor of NF-κB activity and PD-L1 transcription.
RESULTS
Identification of RB as a Negative Regulator of NF-κB Signaling
A recent study identifies the chromatin remodeling factor CHD1 as a positive regulator of NF-κB and shows CHD1 and PTEN tumor suppressor gene are deleted mutually exclusively in human prostate cancers, and their co-deletion is synthetic lethal (Zhao et al., 2017). We demonstrated that the MAP3K7 gene (encoding a kinase also known as TAK1, an upstream activator of NF-κB) and PTEN were almost mutually exclusively deleted in multiple cancer types examined (Figures S1A and S1B), and their co-depletion was also synthetic lethal (Figures S1C-S1H). Therefore, PTEN/MAP3K7- and PTEN/CHD1-dual-deficient cell lines represent two ideal cell systems for us to identify bona fide pathways that can rescue synthetic lethality. MAP3K7 or CHD1 was knocked down (KD) in PTEN null PC-3 cells followed by treatment with an array of small molecule inhibitors for a number of cancer-relevant pathways. As expected, drug sensitivity (IC50) analysis showed that the NF-κB inhibitor JSH-23 further increased viability loss in both MAP3K7- and CHD1-KD PC-3 cells (Figure 1A). Different from other compounds that either exerted opposite responses in MAP3K7- and CHD1-KD cells or had limited protection of cells from being killed, the CDK4/6 inhibitor palbociclib induced a common, large-degree (log2 scale) increase in the viability in both MAP3K7- and CHD1-KD cells (Figure 1A). The protective effect was further evident in PARP and caspase-3 protein cleavage and Annexin-V apoptosis assays (Figures S1I and S1J). Accordingly, expression of NF-κB target genes including TNFA, GADD45B, and JUN was downregulated by MAP3K7 or CHD1 KD alone but was restored to the steady-state level by palbociclib in both cell types (Figure S1K).
Figure 1. RB Functions as a Negative Regulator of NF-κB Signaling.

(A) PC-3 cells infected with lentivirus for indicated shRNAs were treated with different chemicals, and cell viability was measured by MTS assay. Heatmap showing the normalized IC50 ratio (log2[IC50ratio]).
(B-D) PC-3 cells infected with lentivirus expressing indicated shRNAs were harvested for western blots (B), FACS (C), and RT-qPCR (D). All data are shown as mean values ± SD (n = 3). ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
(E and F) PC-3 cells infected with indicated lentivirus were injected s.c. into NSG mice, and tumors were harvested and photographed at day 21 (E). Data in (F) are shown as means ± SD (n = 5). ns, not significant, *p < 0.05.
(G) A model depicting RB functions as a negative regulator of NF-κB (p65/p50), providing a plausible explanation of the finding that depletion of RB offsets the death of PTEN null cells induced by either MAP3K7 or CHD1 deletion.
RB is a major downstream effector of CDK4/6 signaling. We sought to determine whether RB affects the synthetic lethality caused by PTEN/MAP3K7 or PTEN/CHD1 co-deficiency. RB KD blocked MAP3K7 or CHD1 KD-induced PARP and caspase-3 cleavage, apoptotic cell death, and growth inhibition in PTEN null PC-3 cells (Figures 1B, 1C, S2A, and S2B), and the same was true for NF-κB target gene expression (Figure 1D). MAP3K7 or CHD1 KD also largely inhibited PTEN null PC-3 xenograft tumor growth in mice, but such effects were completely abolished by RB KD (Figures 1E and 1F).
Rescue experiments showed that, similar to RB-WT, restored expression of R661W (R654W in mouse Rb, an E2F1-binding deficient mutant [Sun et al., 2011]) largely blocked MAP3K7 or CHD1 KD-induced inhibition of cell growth and NF-κB target gene expression, although as expected, the growth inhibitory effect of R661W was not as robust as RB-WT in cell culture and in mice (Figures S2A-S2E), suggesting RB-loss-induced protective growth of MAP3K7- or CHD1-deficient tumors is less likely mediated through E2F. These findings are highly disease relevant, since MAP3K7 or CHD1 deletion largely overlapped with RB1 gene deletion in TCGA cohort (Figures S2F and S2G). Genetic depletion of IKKα and IKKβ, two NF-κB activators downstream of MAP3K7/TAK1, or treatment with IKK inhibitors IKK-16 and ACHP decreased NF-κB target gene expression, which was reversed back to the steady-state level by RB KD (Figures S2H and S2I). Thus, our data suggest RB is a major negative regulator of NF-κB (Figure 1G).
RB Interacts with p65, and the Interaction Is Enhanced by RB Phosphorylation
To elucidate the molecular mechanisms by which RB regulates NF-κB function, we examined whether RB interacts with NF-κB. CoIP demonstrated that endogenous RB interacted with endogenous p65, but not other NF-κB/Rel family proteins—RelB, c-Rel, p52, and p50 (Figures 2A and S3A). Reciprocal coIP showed that p65 only interacted with RB, but not other pocket proteins p107 and p130 (Figure 2B), revealing a specific p65-RB interaction. p65-RB binding primarily occurred in the nucleus (Figure S3B). In line with the report that TNF-α increases nuclear accumulation of p65 (Ariga et al., 2002), it increased p65-RB interaction in the nucleus while decreasing p65-IκBα interaction (Figures S3C and S3D). Immuno-depletion assays showed that p65 bound to approximately 11% and 14% of RB and 63% and 72% of IkBα in PC-3 and primary Rb-positive Pten single knockout (SKO Rb+/+) cells (Ku et al., 2017), respectively (Figures S3E and S3F). RB-p65 interaction was also observed in many other cell lines of different cancer types (Figure S3G).
Figure 2. RB Interacts with p65, and Their Interaction Is Enhanced by RB Phosphorylation.

(A and B) Western blot analysis of reciprocally co-immunoprecipitated endogenous RB and p65 proteins in PC-3 cells.
(C) Schematic diagram depicting a set of GST-p65 recombinant protein constructs. RHD, Rel homology domain; TAD, transactivation domain.
(D and E) Western blot analysis of RB proteins in untreated (D) or λ-phosphatase-treated (E) PC-3 whole-cell lysate (WCL) pulled down by GST or GST-p65 recombinant proteins. Asterisks indicate proteins at the expected molecular weight.
(F) Western blot analysis of coIP samples of PC-3 WCL treated with or without λ phosphatase.
(G) Western blot analysis of WCL and coIP samples from DU145 cells 24 hr after transfection with indicated plasmids.
(H) Western blot analysis of WCL and coIP samples from PC-3 cells treated with or without palbociclib (5 mM) for 24 hr.
(I) Western blot analysis of WCL and coIP samples from PC-3 cells 48 hr after infection with indicated shRNAs.
(J) Schematic diagram depicting a set of RB recombinant protein constructs.
(K) Western blot analysis of p65 proteins in PC-3 WCL pulled down by GST or GST-RB recombinant proteins.
GST pull-down assay showed that RHD, the DNA binding domain in the N terminal portion, but not the C terminal region, of p65 specifically bound to RB protein (Figures 2C and 2D). RB is a highly phosphorylated protein. Phosphatase treatment decreased RB interaction with GST-p65-N or endogenous p65 (Figures 2E and 2F). Nonphosphorylatable RB mutant RBΔCDK, in which fifteen major CDK phosphorylation sites were mutated to alanine (Narasimha et al., 2014), only had a minimal interaction with p65 compared to RB-WT, while its interaction with E2F1 was increased as expected (Figure 2G). Thus, RB phosphorylation largely enhances RB-p65 interaction.
While palbociclib treatment substantially reduced the level of hyperphosphorylated RB, it largely decreased RB-p65 interaction but substantially increased RB-E2F1 interaction (Figure 2H). Similarly, CDK4 and CDK6 co-knockdown decreased RB-p65 interaction but increased RB-E2F1 interaction as expected (Figure 2I). However, RB-p65 interaction was not affected by ectopic expression of E2F binding-deficient mutant RB-R661W (Figure S3H), suggesting RB-p65 interaction is independent of RB-E2F complex. This result is not surprising because p65 binds to hyperphosphorylated RB (Figures 2F-2I), whereas E2F1 is primarily bound by un- or-hypo-phosphorylated RB.
Although RB-p65 interaction is largely enhanced by RB phosphorylation, certain basal level interaction between nonphosphorylatable RB and p65 was detectable (Figure 2G). In support of this result, GST pull-down assay showed that GST-RB-N, but not GST-RB-M and GST-RB-C, retains the ability to bind to p65, although the amount of p65 interacted with RB-N was less than the amount of p65 in 1% of input (Figures 2J and 2K). Nevertheless, these data indicate that RB interacts with p65 via its N terminal portion. It is worth noting that the amino acid sequences in the N terminus of RB, p107, and p130 are not conserved, and this region is outside of the E2F-bound pocket domain (Figures S3I and S3J). These results are consistent with the observations that p65 does not interact with p107 and p130, and that p65-RB interaction is independent of RB-E2F1 interaction (Figures 2B and S3H).
RB S249/T252 Phosphorylation and 161FQVTV165 Motif in p65 Are Important for Their Interaction
RB-p65 interaction is dependent on RB phosphorylation, and the interaction was largely diminished by CDK4/6 inhibitors or knockdown (Figures 2G-2I). These observations prompted us to determine whether CDK4/6 phosphorylation of RB is important for RB-p65 interaction. There are four reported CDK4/6 phosphorylation sites in RB-N, two (serine-249 and threonine-252 [S249/T252]) in the arginine-rich linker (R linker) region and the rest (threonine-356 [T356] and threonine-373 [T373]) in the C terminus (Zarkowska and Mittnacht, 1997) (Figure 3A). These phosphorylation sites were mutated individually into aspartic acid (D), an acidic amino acid closely mimicking phosphorylation. GST pull-down assay showed that S249D/T252D, but not the other two mutations, increased RB-N binding with p65 (Figure 3A). In agreement with this observation, in vitro protein binding assay using in vitro transcribed and translated p65 protein showed that the N terminal portion of RB-N (RB-NΔ1 containing S249/T252), but not the C terminal part (RB-NΔ2 containing T356 and T373), specifically bound to p65 (Figures S3K and S3L). Moreover, we performed in vitro kinase assay by inoculating unmutated GST-RB-N or S249/T252 phosphorylation-resistant mutant (S249A/T252A) with the reconstituted CYCLIN D3/CDK4 complex followed by in vitro protein binding assay. S249/T252 phosphorylation on RB-N by CYCLIN D3/CDK4 substantially increased p65-RB-N interaction, but no such effect was observed for S249A/T252A mutant (Figure 3B). Similar results were obtained with the deletion mutant RB-NΔ249SPRT252, in which S249/T252 residues were deleted (Figure S4A). We further generated a RB-N mammalian expression vector containing S, Flag, and Biotin-binding-protein (streptavidin)-binding-peptide (SFB) tags. RB-N strongly bound to p65 in various cell lines, but the binding was substantially diminished by S249A/T252A mutation (Figures S4B-S4E). These data indicate that CDK4/6 phosphorylation of S249/T252 in the R linker region largely enhances RB-N interaction with p65. It is worth noting that the R linker and S249/T252 phosphorylation sites are not conserved in RB homologs p107 and p130 (Figures S3I and S3K), consistent with the finding that p65 does not bind to p107 and p130 (Figure 2B).
Figure 3. S249/T252 Phosphorylation on RB and 161FQVTV165 Motif in p65 Are Important for Their Interaction.

(A) (Top) Schematic diagram depicting CDK4/CYCLIN D phosphorylation sites (P letter) of RB-N. (Bottom) Western blot analysis of p65 proteins in PC-3 whole-cell lysate (WCL) pulled down by GST-RB. Asterisks indicate proteins at the expected molecular weight.
(B) Western blot (top) analysis of in vitro transcribed and translated p65 proteins pulled down by GST-RB (bottom). GST recombinant proteins were inoculated with purified CYCLIN D3 and V5-CDK4 for in vitro kinase assay prior to GST pull-down assay.
(C) Schematic diagram depicting a RB-N interacting FxxxV motif(161FQVTV165) and basic amino acid (positive charge)-containing region in p65-N and an FxxxV motif and acidic amino acid (negative charge)-containing region in EID1.
(D) Western blot (top) analysis of proteins pulled down by GST-p65 recombinant proteins (bottom) from WCL of PC-3 cells transfected with HA-RB protein.
(E) Schematic diagram depicting a model wherein introduction of negative charge by S249/T252 phosphorylation (P letter) allows otherwise fully positively charged RB-N to bind to the positively charged 161FQVTV165 motif-containing region in p65.
(F) Schematic diagram depicting a model wherein introduction of negative charge by S249/T252 phosphorylation allows otherwise fully positively charged RB-N to disassociate from the negatively charged 166FQVTV171 motif in EID1.
(G) Schematic diagram depicting a working model wherein introduction of negative charge by S249/T252 phosphorylation (P letter) allows otherwise fully positively charged RB-N to bind to the positively charged 161FQVTV165 motif-containing region in p65; this process can be reversed by CDK4/6 inhibition.
(H) (Top) GST-tagged mammalian expression vectors for the N terminus of wild-type (WT) p65 and alanine (A) and arginine (R) mutants. (Bottom) PC-3 cells were transfected with indicated plasmids followed by western blot analysis of PC-3 WCL pulled down by GST or GST-p65 recombinant proteins.
The amino acids surrounding S249/T252 in the R linker region are evolutionally conserved from human to mouse (Figure S4F), further highlighting the functional importance of the p65-interaction region in RB-N. To test this notion, we constructed an SFB-tagged expression vector for a S249/T252-containing peptide constituted of twenty-one amino acids in the R linker region (termed as R-linker (RL) peptide) (Figure S4F). p65 was effectively immunoprecipitated by SFB-tagged RL-S249D/T252D, but not SFB alone, and such effect was largely diminished by nonphosphorylatable mutant RL-S249A/T252A (Figure S4G). In vitro protein binding assays demonstrated that the binding between in vitro transcribed and translated RL-S249D/T252D peptide and GST-p65-N was much stronger compared to phosphorylation-resistant mutant RL-S249/T252A (Figure S4H). Similar to a previous report that osmotic shock induces p38-dependent S249/T252 phosphorylation of RB and enhances RB-N interaction with E2F1 (Gubern et al., 2016), sodium chloride treatment also increased p65-RB interaction in a S249/T252-phosphorylation-dependent manner (Figures S4I-S4K). Together, we identified a small RB-N-derived phospho-mimetic peptide that can strongly bind to p65.
A previous study identifies an FxxxV (166FSLMV170) motif-centered region in a protein termed E1A-like inhibitor of differentiation-1 (EID1) that is important for its interaction with RB-N (Hassler et al., 2007). We found p65 also harbors an evolutionally conserved FxxxV (161FQVTV165) motif within the RB-N-binding region (Figure 3C). Like EID1, deletion of 161FQVTV165 in p65-N (p65-NΔFV) largely diminished RB-p65 interaction (Figure 3D). However, different from the previous report that the EID1-RB-N interaction is abrogated by CDK-mediated S249/T252 phosphorylation (Hassler et al., 2007), p65-RB-N interaction was largely enhanced by S249/T252 phosphorylation (Figure 3B). After carefully examining the amino acid sequences in the interaction motifs in these three proteins, we noticed that the R linker in RB-N is an arginine (positive charge)-rich region, and there are several positively charged arginine residues surrounding the 161FQVTV165 motif in p65, whereas more negatively charged (e.g., glutamic acid) than positively charged amino acids are present adjacent to the 166FSLMV170 motif in EID1 (Figures 3E and 3F). Based upon these observations, we envision a model wherein introduction of negative charge by S249/T252 phosphorylation may allow otherwise fully positively charged RB-N to bind to the positively charged 161FQVTV165-containing region in p65. In contrast, the same phosphorylation in the R linker impedes RB-N interaction with the negatively charged 166FSLMV170-centered region in EID1 (Figures 3E-3G). To experimentally test this hypothesis, we converted positively charged arginine residues surrounding the 161FQVTV165 motif in p65-N to alanine (A mutant) or negatively charged aspartic acid (D mutant), but mutated negatively charged glutamic acid residues next to the 166FSLMV170 motif in EID1-N to alanine (A mutant) or positively charged arginine (R mutant) (Figures 3H and S4L, top panels). GST recombinant proteins of these mutants and the WT control were purified for pull-down assay. The interaction between p65-N D mutant and RB-N was much stronger than that of WT or A mutant (Figure 3H, lower panel). In striking contrast, the interaction of EID1-N D mutant with RB-N was much weaker than that of WT or A mutant (Figure S4L, lower panel). These data indicate that charge composition of amino acids in the RB-interacting region in client proteins such as p65 and EID1 is the determinant for RB binding.
RB Regulates Expression of a Subset of NF-κB Target Genes
Specific interaction of RB with p65, but not other NF-κB proteins, suggests that RB may partially regulate NF-κB transcription program. RNA-seq analysis was performed in RB-knockdown and palbociclib-treated PC-3 cells. We identified a subset of genes whose expression was commonly down- or upregulated by RB knockdown and palbociclib in three replicates (one inconsistent replicate in shControl cells was excluded from analysis) (Figures 4A and S5A). Ingenuity pathway analysis (IPA) of 897 commonly upregulated genes showed that among the signaling pathways significantly affected by both RB knockdown and palbociclib were T cell regulatory pathway genes including CD274 (gene symbol for PD-L1 or B7-H1), expression of which is known to be regulated by NF-κB (Bouillez et al., 2017; Peng et al., 2015) (Figures 4A-4C). Gene set enrichment analysis (GSEA) revealed that the upregulated genes also significantly overlapped with TNF-α-NF-κB pathway, epithelial-to-mesenchymal transition (EMT), hypoxia, and ultraviolet (UV) response genes (Figures 4C and 4D). RNA-seq results for the genes commonly upregulated by RB knockdown and palbociclib are exemplified by the NF-κB targets CD274 (PD-L1), GADD45B, NR4A2, and CD83 (Figure 4E). Upregulation of these genes was further verified by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) (Figure 4F). These data indicate that expression of a subset of NF-κB target genes including PD-L1 is selectively regulated by RB.
Figure 4. RB Partially Regulates NF-κB Transcription Target Genes in Cells.

(A) RNA-seq analysis indicates the genes commonly upregulated by RB1 knockdown and palbociclib treatment in PC-3 cells.
(B) Ingenuity pathway analysis (IPA) of common targets shown in (A) (ranked by p-values).
(C) Heatmap showing a subset of RB knockdown and palbociclib-coregulated genes involved in T cell regulation and TNF-α-NF-κB pathways.
(D) Gene set enrichment analysis (GSEA) of the common targets shown in (A).
(E) UCSC Genome Browser screen shot of RNA-seq tracks for T cell/NF-κB pathway genes (PD-L1, GADD45B, NR4A2, and CD83) commonly upregulated by RB knockdown and palbociclib treatment.
(F and G) PC-3 cells treated with or without palbociclib (5 μM) for 24 hr (left) or infected with lentivirus expressing control or RB-specific shRNAs (right) were harvested for RT-qPCR (F) and ChIP-qPCR (G) analysis of p65 binding at the promotor of PD-L1, GADD45B, NR4A2, and CD83 genes. All data are shown as mean values ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001.
Chromatin immunoprecipitation-coupled quantitative PCR (ChIP-qPCR) analysis revealed that p65 protein readily bound to the genomic loci of the RB-affected NF-κB target genes including PD-L1, and p65 occupancy at these loci was substantially increased by RB knockdown or palbociclib in PC-3 cells (Figure 4G). Transcription factor DNA-binding sequence analysis revealed that there is a putative NF-κB binding sequence (NBS) in the promoter of PD-L1 and other NF-κB target genes examined (Figures S5B and S5C). We performed electrophoretic mobility shift assay (EMSA) using biotin-labeled NBS from the PD-L1 promoter (PD-L1-Prom) as probe (Figures S5B, S5D, and S5E). A DNA-protein complex (DPC) was detected in nuclear extract of PC-3 cells treated with vehicle or control shRNA (Figures S5D and S5E). The DPC level was largely augmented by RB knockdown or palbociclib treatment alone and further enhanced by TNF-α (Figures S5D and S5E). A super-shifted DPC was detected in reactions inoculated with anti-p65 or anti-p50 anti-bodies, but not non-specific IgG (Figure S5E). Thus, we identify a functional NF-κB binding site in the PD-L1 promoter at which p65 binding is regulated by the CDK4/6-RB signaling axis.
Importance of RB S249/T252 Phosphorylation in Regulation of PD-L1 Expression
T cell responses can be reactivated by blockade of PD-1/PD-L1 interaction with agents such as PD-1 and PD-L1 antibodies and utilized for cancer treatment (Iwai et al., 2002; Topalian et al., 2012a; Zhu and Chen, 2003). We sought to determine whether RB regulation of PD-L1 expression is affected by RB phosphorylation and whether such regulatory mechanism can be harnessed for cancer therapy. Consistent with the finding that RB depletion upregulated PD-L1 mRNA expression (Figures 4E and 4F), RB knockdown by two independent shRNAs in PC-3 cells invariably increased PD-L1 protein expression as demonstrated by western blot, FACS, and immunofluorescent cytochemistry (Figures 5A-5C), and similar results were obtained in another prostate cancer cell line LNCaP (Figures S6A-S6C). We further examined Pd-l1 expression in murine prostate cancer cell lines established from prostate-specific Rb1, Pten double knockout (DKO), and Pten single knockout (SKO) mice (Ku et al., 2017). Pd-l1 expression at both mRNA and protein level was much higher in Rb-deficient DKO cells than that in Rb-proficient SKO cells (Figures S6D and S6E). The findings from cell lines are fully supported by the data in a cohort of metastatic castration-resistant prostate cancer (mCRPC) patients that PD-L1 mRNA levels were significantly higher in RB1 homozygously deleted prostate tumors compared with tumors without RB1 deletion (Figure S6F). Notably, RB knockdown-induced upregulation of PD-L1 was abrogated by p65 knockdown in PC-3 cells (Figures 5D and 5E). These data suggest that RB represses PD-L1 expression in an NF-κB-dependent manner.
Figure 5. The Importance of RB S249/T252 Phosphorylation in Regulation of PD-L1 Expression.

(A–C) PC-3 cells were infected with lentivirus expressing indicated shRNAs, and PD-L1 expression was determined by western blots (A), FACS (B), and IFC (C). (D and E) PC-3 cells infected with lentivirus expressing indicated shRNAs were harvested for western blotting (D) and RT-qPCR (E). All data are shown as mean values ± SD (n = 3). ns, not significant; ***p < 0.001.
(F and G) DU145 cells transfected with indicated plasmids were harvested for western blotting (F) and RT-qPCR (G). All data are shown as mean values ± SD (n = 3). *p < 0.05, **p < 0.01.
(H and I) DU145 cells transfected with indicated plasmids were harvested for western blotting (H) and RT-qPCR (I). All data are shown as mean values ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001.
(J and K) PC-3 cells were infected with indicated lentivirus and subject to western blot (J) and RT-qPCR (K). All data are shown as mean values ± SD (n = 3).ns, not significant, ***p < 0.001.
(L) Representative images of IHC analysis with anti-PD-L1 and anti-pRB-S249/T252 antibodies on TMA (n = 145 TMA elements) tissue sections.
(M) Correlation analysis of IHC staining of PD-L1 and pRB-S249/T252 proteins in prostate cancer patient specimens.
We further examined whether PD-L1 expression is regulated by RB phosphorylation. Ectopic expression of RB WT substantially decreased PD-L1 expression, and this effect was largely attenuated in cells expressing the phosphorylation-resistant mutant RBDCDK (Figures 5F and 5G). Notably, PD-L1 expression level was lower in RBDCDK-expressing cells than in control cells (Figures 5F and 5G), which is consistent with the presence of the certain basal level interaction between RBΔCDK and p65 (Figure 2G). Expression of RB-N, the p65-interacting region, was also sufficient to inhibit PD-L1 expression at both mRNA and protein levels, but this effect was largely diminished by S249A/T252A mutation (Figures 5H and 5I), highlighting the importance of CDK4/6 phosphorylation of RB in repression of PD-L1 expression. PD-L1 protein expression was much lower in cells at G1/S phase compared to that in cells at G2/M phase (Figures S6G and S6H). Similar to the previous finding in B cells (Inoue et al., 2016), expression of NF-κB target genes including PD-L1 was cell cycle regulated and occurred at mRNA level (Figures S6I-S6K), but the alteration was not mediated by RNAst ability, at least in G1/S and G2/M phases examined (Figure S6L). In support of the protein binding results (Figures S4G and S4H), expression of the small phospho-mimetic peptide derived from the p65-binding motif in RB-N (RL-S249/T252D peptide) largely inhibited PD-L1 expression in p65-proficient cells, but little or no effect was observed in p65-depleted cells (Figures 5J and 5K). Expression of this phospho-mimetic peptide also blocked TNF-α-induced expression of NF-κB target genes (Figures S6M and S6N), and such effect was not due to its sequestration of p65 proteins in the cytoplasm, but rather mediated by its inhibition of p65 binding to the cognate DNA in the PD-L1 promoter (Figures S6O and S6P). This peptide also inhibited PD-L1 expression in several cell lines of different cancer types (Figures S7A and S7B), indicating that this peptide is bioactive in a broad spectrum of cancer types. Together, we identify a small phospho-mimetic peptide of RB-N that can inhibit PD-L1 expression in a p65-dependent manner.
RB phosphorylation at S249/T252, but not total RB protein, was generally corrected with PD-L1 protein expression in an array of prostatic cell lines (Figures S7C). We further examined S249/T252-phosphorylated RB (pRB-S249/T252) and PD-L1 expression in patient samples by performing immunohistochemistry (IHC) on a tissue microarray (TMA) containing a cohort of metastatic castration-resistant prostate cancer (mCRPC) specimens (n = 145 TMA elements). IHC staining was evaluated by measuring both staining intensity and percentage of positive cells (Table S1). Representative IHC images displaying high and low/lost staining of PD-L1 and pRB-S249/T252 are shown in Figure 5L. In this cohort, 65 out of 145 (44.8%) TMA specimens were PD-L1 positive, but only 17 out of 145 (11.7%) TMA specimens expressed moderate to high levels of PD-L1 protein on cell surface. Detection of low rate expression of PD-L1 in this cohort is consistent with previous reports (Gevensleben et al., 2016; Massari et al., 2016). Further analysis showed that PD-L1 protein expression inversely correlated with pRB-S249/T252 level among these patients (Pearson’s product-moment correlation r = −0.389, p = 1.33 × 106) (Figure 5M). These data suggest regulation of PD-L1 expression by S249/T252-phosphorylated RB may also occur in mCRPC patients.
RL-S249/T252D Blocks Radiation-Induced PD-L1 Expression and Cancer Immunity
Given that radiation inhibits RB phosphorylation by inducing cell cycle arrest, we examined if radiation increases PD-L1 expression and if this effect can be reversed by RL-S249/T252D treatment. Gamma radiation inhibited RB phosphorylation at S249/T252 and markedly increased PD-L1 expression in a time-dependent manner (Figures S7D and S7E), and such effect was abrogated by p65 knockdown (Figures 6A and 6B). Gamma radiation not only decreased S249/T252 phosphorylation and p65 binding of RB, but also increased p65 binding at the PD-L1 gene promoter (Figures 6C and 6D). Ectopic expression of RL-S249/T252D decreased PD-L1 basal level expression and largely diminished gamma radiation-induced upregulation of PD-L1 mRNA and protein (Figures 6E and 6F). These data suggest gamma radiation-induced upregulation of PD-L1 is primarily mediated through the RB-NF-κB signaling pathway.
Figure 6. The Small RB-Derived S249/T252 Phosphorylation-Mimicking Peptide Blocks Radiation-Induced PD-L1 Expression and Promotes Cancer Immunity.

(A and B) PC-3 cells infected with lentivirus expressing indicated shRNAs were treated with or without gamma radiation (IR, 12 Gy) followed by western blotting (A) and RT-qPCR (B). All data are shown as mean values ± SD (n = 3). ns, not significant; **p < 0.01.
(C) Western blot analysis of WCL and coIP samples of PC-3 cells treated with or without gamma radiation (12 Gy).
(D) ChIP-qPCR analysis of p65 binding at the PD-L1 promotor in PC-3 cells treated with gamma radiation (12 Gy). All data are shown as mean values ± SD (n = 3). **p < 0.01.
(E and F) PC-3 cells treated with or without gamma radiation (12 Gy) were transfected with indicated constructs followed by western blotting (E) and RT-qPCR (F). All data are shown as mean values ± SD (n = 3). ns, not significant, ***p < 0.001.
(G and H) PTEN-CaP8 murine prostate cancer cells (5 × 106) transduced with indicated lentivirus were injected subcutaneously into C57BL/6 mice. Mice (n = 8/group) were treated with gamma radiation (12 Gy), anti-PD-L1 (200 μg), or non-specific IgG. Tumor growth with different treatments is shown in (G). At the end of treatment, the numbers of infiltrated T and myeloid cells in tumors were analyzed by FACS (H). All data are shown as mean values ± SD (n = 8). ns, not significant; *p < 0.05, ***p < 0.001.
Next, we examined whether administration of the bioactive RL-S249/T252D peptide of RB could enhance the anti-tumor efficacy of radiotherapy. PTEN-CaP8 murine prostate cancer cells infected with lentivirus of Tsin empty vector (EV) or Tsin-RL-S249D/T252D peptide were injected subcutaneously into immune-proficient mice. PTEN-CaP8 tumor-bearing mice were treated with gamma radiation (12 Gy) in combination with anti-PD-L1 antibody or non-specific control IgG (Figures S7F and S7G). While anti-PD-L1 antibody alone did slightly, but not dramatically, inhibit tumor growth, gamma radiation alone substantially decreased tumor growth; tumor growth was completely blocked by radiation and anti-PD-L1 co-administration in the first 15 days, although tumor re-growth was observed afterward (Figure 6G). Consistent with the result that RL-S249/T252D peptide completely blocked Pd-l1 expression in PTEN-CaP8 cells (Figure S7F), RL-S249/T252D treatment markedly inhibited tumor growth regardless of anti-PD-L1 treatment (Figure 6G). Most importantly, tumors regressed when the small peptide was co-administrated with gamma radiation (Figure 6G). Notably, while inhibiting Pd-l1 expression, RL-S249D/T252D peptide treatment alone also blocked expression of other NF-κB targets, including a few pro-survival genes (Figures S7H and S7I), providing a plausible explanation as to how this peptide outperforms the anti-PD-L1 antibody in tumor suppression. Moreover, radiation alone, but not anti-PD-L1 antibody alone, significantly increased tumor infiltration of immune effectors including CD45+CD8+ T cells and CD45+CD4+ T cells, but decreased the infiltration of CD11b+Gr1+ myeloid cells in tumors (Figure 6H). The combination of radiation and anti-PD-L1 antibody additively increased CD45+CD8+ and CD45+CD4+ T cell infiltration in tumors (Figure 6H). In agreement with the effect on tumor regression, co-treatment of radiation and the RL-S249/T252D peptide induced a dramatic increase in CD45+CD8+ and CD45+CD4+ T cell infiltration, but a sharp decrease of CD11b+Gr1+ myeloid cells in tumors (Figure 6H).
DISCUSSION
A major finding in the current study is the identification of a previously unappreciated tumor suppressor function of hyperphosphorylated RB. We demonstrate that RB specifically binds to p65, but not other NF-κB family proteins, and RB S249/T252 phosphorylation is required for such interaction. These findings help explain our observation that S249/T252-phosphorylated RB suppresses NF-κB transcriptional activity and PD-L1 mRNA expression. Importantly, we provide evidence suggesting this appears to be a general regulatory mechanism occurring in various cell lines of different cancer types examined.
Phosphorylation in the R linker, pocket linker, and interdomain linker is essential for RB to bind to its client proteins such as E2F and EID1 (Burke et al., 2010; Hassler et al., 2007). Like EID1 (Hassler et al., 2007), we identified a similar FxxxV motif (161FQVTV165) in p65 that is important for RB-N binding. CDK phosphorylation (adding negative charge) of RB-N at S249/T252 in the R linker enhanced RB-p65 interaction while decreasing RB-EID1 interaction as reported (Hassler et al., 2007). We further uncovered that there are a number of acidic (negative charge) amino acids adjacent to the FxxxV (166FSLMV170) motif in EID1, but quite few basic (positive charge) amino acids surrounding 161FQVTV165 in p65. Importantly, we found the charge differences in the RB-interacting regions in p65 and EID1 are the determinants for the different abilities of the phosphorylated RB-N to bind with different client proteins. Thus, our findings provide a framework for discovery of more FxxxV motif-containing proteins that are functionally influenced by RB-N phosphorylation.
Response to immune checkpoint therapy in patients seems highly correlated with PD-L1 expression in tumor cells (Herbst et al., 2014; Topalian et al., 2012b). Our RNA-seq analysis showed that expression of a subset of NF-κB target genes including PD-L1 was selectively upregulated by RB knockdown or treatment of CDK4/6 inhibitor palbociclib. Importantly, we identified a small bioactive S249/T252 phosphorylation-mimetic peptide of RB that not only blocked PD-L1 expression but also enhanced the anti-cancer efficacy of radiotherapy. While our study was ongoing, a most recent report shows that palbociclib promotes tumor immune evasion and drug resistance by inducing PD-L1 protein stabilization in a manner independent of PD-L1 mRNA regulation and RB function (Zhang et al., 2018). Therefore, our study identifies a previously uncharacterized role of S249/T252-phosphorylated RB in suppressing NF-κB activity and PD-L1 transcription (Figure 7A). Intriguingly, similar to a previous report (Gubern et al., 2016), we also found that osmotic shock (e.g., sodium chloride treatment) can induce RB S249/T252 phosphorylation in the cell systems we studied. Importantly, sodium chloride treatment can also enhance RB-p65 interaction in a S249/T252 phosphorylation-dependent manner. Thus, future investigation is warranted to determine whether PD-L1 expression and cancer immunity are influenced by osmotic shock in a manner dependent on RB S249/T252 phosphorylation.
Figure 7. Models Depicting RB S249/T252 Phosphorylation-Mediated Regulation of NF-κB Activity and PD-L1 Expression.

(A) CYCLIN D/CDK4/6-mediated RB S249/T252 phosphorylation promotes specific RB-p65 interaction, thereby triggering inhibition of expression of NF-κB target genes such as PD-L1 and cancer immunity.
(B) Deletion of RB or radiotherapy or CDK4/6 inhibitor-induced loss of RB S249/T252 phosphorylation promotes undesired immune evasion of cancer cells by inducing PD-L1 expression.
(C) Cancer immunity can be reinvigorated by co-administration of radiotherapy in combination with a RB S249/T252 phosphorylation-mimicking peptide (RL S249D/T252D).
It is well known that CDK phosphorylation of RB at many residues including S249/T252 can be abolished by radio- or chemotherapy-induced cell cycle arrest or CDK4/6 inhibitors. In agreement with the essential role of RB S249/T252 phosphorylation in repressing PD-L1 expression, we demonstrate that inhibition of S249/T252 phosphorylation by radiation or CDK4/6 inhibitor or RB depletion can largely enhance PD-L1 expression. Thus, our findings support a model in which radiation therapy, CDK4/6 inhibitor administration, or RB deletion may trigger undesirable upregulation of PD-L1, thereby causing immune evasion of cancer cells (Figure 7B). These findings are highly relevant, particularly in prostate cancer, since RB deletion associates with high-level expression of PD-L1 in metastatic prostate cancer (Figure S6F) as well as acquisition of castration resistance (Akamatsu et al., 2015). Most importantly, we identify a small RB-derived S249/T252 phosphorylation-mimetic peptide that can overcome radiation-induced immune tolerance by suppressing PD-L1 expression (Figure 7C). Mechanistically, we show that such effect of the peptide is mediated by its blockade of p65 binding to the cognate DNA sequence in the PD-L1 promoter rather than sequestration of p65 in the cytoplasm.
In summary, our present study identifies a previously uncharacterized tumor suppressor function of hyperphosphorylated RB that promotes tumor immunity by suppressing NF-κB transcription activity and PD-L1 expression. Our findings also suggest that this activity of RB can be exploited to overcome cancer immune tolerance associated with current therapeutics, including radio- and chemotherapies and targeted therapies such as CDK4/6 small molecule inhibitors.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Haojie Huang (huang.haojie@mayo.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and transfection
LNCaP, C4-2, PC-3, 22Rv1, H1299, SK-Hep1, PANC-1and DU145 cells were cultured in RPMI 1640 supplemented with 10% FBS. LNCaP-RF cells were cultured in RPM11640 supplemented with 10% charcoal-stripped FBS (CSS). VCaP cells were maintained in DMEM supplemented with 20% FBS. PTEN-CaP8 and 293T cells were maintained in DMEM supplemented with 10% FBS. RWPE-1 cells were cultured in keratinocyte serum-free medium supplied with recombinant human EGF (Invitrogen). Cells were cultured at 37°C supplied with 5% CO2. LAPC-4 cells were maintained in Iscove’s Modified Dulbecco’s Media with 10% FBS. BPH-1 cell line was cultured RPMI 1640 medium supplemented with 10% FBS. Rb-proficient SKO (Cre+:Ptenf/f:Rb1+/+) and castration resistant Rb-deficient DKO (Cre+:Ptenf/f:Rb1f/f) were maintained in DMEM supplemented with 2.5% charcoal-stripped FBS, 5 μg/mL of insulin/ transferring/selenium (Thermo Fisher Scientific), 10 μg/mL of bovine pituitary extract (Sigma), 10 μg/mL of epidermal growth factor (Collaborative Research), 1 μg/mL of cholera toxin (Sigma). All cell lines were kept in a 37°C incubator at 5% CO2. Transfections were performed by using Lipofectamine 2000 (Thermo Fisher Scientific).
Prostate cancer patient samples and tissue microarray
The advanced prostate cancer dataset was generated from patients undergoing standard of care clinical biopsies at Mayo Clinic. A tissue microarray was constructed from the formalin-fixed, paraffin-embedded (FFPE) samples of metastatic prostate cancer, identified after a search of pathologic and clinical databases of archival tissues. The Mayo Clinic institutional review board (IRB) approved the experimental protocols for retrieving pathology blocks/slides and for accessing electronic medical records. The human tissue microarray (TMA) contained 157 cores (16 0.6 mm and 141 1.0 mm cores) resulting from 53 samples (20 bone metastases and 33 non-bone metastases) from 51 patients. FFPE tissue was used for IHC analysis. 145 cores were used for IHC data analysis after cores with lost tissue greater than 50% were excluded.
RNA interference
Lentivirus-based control and gene-specific small hairpin RNAs (shRNAs) were purchased from Sigma-Aldrich. Viral packaging plasmids (pEXQV and pVSV-G) and shRNA plasmid were transfected to 293T cells by using Lipofectamine 2000. After 24 h, virus culture medium was replaced with DMEM containing 10% FBS with 1:100 of sodium Pyruvate. 48 h post transfection, medium was collected and added to prostate cancer cells added with 12 μg/ml of polybrene. Prostate cancer cells were harvested 48 h after puromycin selection. shRNA sequence information is provided in Table S2.
Generation of prostate cancer xenografts in mice
6-week-old NOD-SCID IL-2-receptor gamma null (NSG) mice were generated in house and used for animal experiments. The animal study was approved by the IACUC at Mayo Clinic. All mice were housed in standard conditions with a 12 h light/dark cycle and access to food and water ad libitum. PC-3 cells (5 × 106) infected with lentivirus (in 100 μl 1 × PBS plus 100 μl Matrigel (BD Biosciences)) were injected s.c. into the right flank of mice. The volume of xenografts was measured every other day for 21 days and calculated using the formula LxW2×0.5. Upon the completion of measurement, tumor grafts were harvested.
Quantitative RT-PCR
Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific). The NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) was used to assess RNA yield and quality. RNA was reversely transcribed using Superscript reverse transcriptase (Thermo Fisher Scientific) following manufacturer’s instructions. Quantitative real-time PCR was performed by mixing cDNA, gene-specific primers and IQ SYRB Green Supermix and detected by iCycler QTX detection system (Bio-Rad). The 2-ρCt method was used to quantitate fold changes by normalizing to GAPDH. Sequence information for primers used for RT-qPCR is provided in Table S3.
Co-immunoprecipitation (coIP)
Cells were harvested and lysed by IP buffer (50 mM Tris-HCl, pH 7.4,150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate and 1% protease inhibitor cocktails) on ice for more than 30 min. Cell lysate was centrifuged for 15 min at 13,000 rpm at 4°C, and the supernatant was incubated with primary antibodies and protein A/G agarose beads (Thermo Fisher Scientific) with rotating at 4°C overnight. The next day, the beads were washed at least six times with IP buffer on ice, and then subjected to western blotting analysis.
Western blotting
Cells were harvested and lysed by IP buffer, the supernatant was quantified by BCA protein quantification assay. Equal amounts of protein sample were added into 4 × sample buffer and boiled for 5min. The sample was subjected to SDS-PAGE analysis and transferred to nitrocellulose membrane. The membrane was blocked by 5% milk for 1 hr at room temperature and incubated with primary antibody at 4°C overnight. The next day, the membrane was washed three times with 1 × TBST and incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The protein bands were visualized by SuperSignal West Pico Stable Peroxide Solution (Thermo Fisher Scientific).
Detection of apoptosis using Annexin V assay and flow cytometry
Cells were washed two times with PBS and resuspended in 1 × Binding Buffer. 1 × 105 cells were stained with PE Annexin V and 7-amino-actinomycin following the manufacturer’s instruction of PE Annexin V Apoptosis Detection Kit I (BD Biosciences). Cells were incubated for 15 min at room temperature and analyzed on a flow cytometer. Data was analyzed with FlowJo analysis software.
MTS assay
Cell viability was measured using the MTS assay according to manufacturer’s instructions (Promega). PC-3 cells were seeded in 96-well plates with 100 μl of culture medium. Each well was added with 20 μl of CellTiter 96R AQueous One Solution Reagent (Promega) and absorbance was measured in a microplate reader at 490 nm.
Immunofluorescent cytochemistry (IFC)
IFC was performed as previously described (Zhang et al., 2017). Briefly, cells were fixed in 4% paraformaldehyde for 15 min. After washed in PBS three times, fixed cells were permeabilized with 0.2% Triton X-100 for 20 min, washed in PBS and then blocked in PBS supplemented with 10% goat serum. Cells were incubated with indicated primary antibody at 4°C overnight. After washed three times with PBS, cells were incubated with secondary antibody that was conjugated with Alexa Fluor 488 dye or Alexa Fluor 594 dye (Thermo Fisher Scientific) for 1 hr at room temperature. After washed three times with PBS, cells were counterstained with Vectashield (Vector Laboratories) containing DAPI (4’, 6-diamidino-2-phenylindole). Images were captured using Zeiss laser confocal microscope (LSM780).
Glutathione S-transferase (GST) pull down assay
Cells were lysed with IP buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate and 1% protease inhibitor cocktails) on ice for more than 30 min. GST fusion proteins were immobilized on glutathione-Sepharose beads (GE Health-care Lifesciences). After washing with lysis buffer, the beads were incubated with cell lysates at 4°C overnight. The beads were then washed six times with binding buffer and re-suspended in sample buffer. The bound proteins were subjected to western blotting analysis.
In vitro kinase assay
Plasmid DNA (pcDNA3.1-V5-CDK4 or pCMV4-p65) was add to the TNT T7 Quick Master Mix and add 1 μl methionine (1 mM), by following the manufacturer’s instruction of TNT Quick Coupled Transcription/Translation Systems (Promega). GST or GST-RB-N recombinant proteins (GST-RB-N, GST-RB-N S249A/T252A or GST-RB-NΔ249SPRT252) were immobilized on glutathione-Sepharose beads. After washing with PBS, the beads were incubated with in vitro transcribed and translated CDK4, human recombinant CYCLIN D3 (Promega) and reaction buffer (40 mM Tris,7.5, 20 mM MgCl2, 0.1 mg/ml BSA, 50 mM DTT) at room temperature for 60 min. After washing with PBS, the beads were incubated with in vitro transcribed and translated p65 for 4 h. The beads were then washed six times with PBS and re-suspended in sample buffer. The bound proteins were subjected to western blotting analysis.
RNA-seq and data analysis
PC-3 cells were treated with or without palbociclib (5 μM) for 24 h or infected with lentivirus expressing control or RB1-specific shRNAs followed by puromycin selection for 48 h. Total RNA was isolated from cells using the RNeasy Plus Mini Kit (QIAGEN). High-quality (Agilent Bioanalyzer RIN > 7.0) total RNA was employed for the preparation of sequencing libraries using the Illumina TruSeq Stranded Total RNA/Ribo-Zero Sample Prep Kit. A total of 500–1,000 ng of riboRNA-depleted total RNA was fragmented by RNase III treatment at 37°C for 10–18 min, and RNase III was inactivated at 65°C for 10 min. Size selection (50- to 150-bp fragments) was performed using the FlashPAGE denaturing PAGE-fractionator (Thermo Fisher Scientific) before ethanol precipitation overnight. The resulting RNA was directionally ligated, reverse-transcribed and treated with RNase H.
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed as described previously (Zhang et al., 2017). Cell lysate was sonicated and subjected to immunoprecipitation using anti-p65 antibody or nonspecific IgG. After extensive wash, immunoprecipitated DNA was amplified by real-time PCR. Sequence information for ChIP primers is provided in Table S4.
Nuclear extracts preparation and electrophoretic mobility shift assay (EMSA)
Cells were collected and cell pellet was resuspend in 1 mL of Buffer A (10 mM HEPES-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.1% NP-40) to lyse the cells on ice for 10 min. Samples were spined down at 6,500 rpm 4°C for 3 min to pellet the nuclei. Nuclei pellet was washed with Buffer A, and spined down at 3,500 rpm for 5 min at 4°C. Cell pellet was lysed by IP buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate and 1% protease inhibitor cocktails) on ice for more than 30 min. Protein concentration was determined by BCA protein quantification assay.
DNA oligonucleotide was labeled with biotin by following the manufacturer’s instruction of Pierce Biotin 3′ End DNA Labeling Kit (Thermo Fisher Scientific). Complimentary 3′ end biotin-labeled oligos were annealed prior to use. Five microgram nuclear extracts was added to the binding reaction system (10X Binding Buffer, 1 μg/μL Poly (dI·dC), 1% NP-40, 100 mM MgCl2) and incubated at room temperature for 5min. Then, biotin-labeled double strand PD-L1-prom oligonucleotide DNA (5′-GGTCAGGAAAGTCCAACGCC-3′) was added to binding reactions and incubated at room temperature for 20 min. Competitive assays were also performed by addition of 200-fold excess of unlabeled unmutated probe (5′-GGTCAGGAAAGTCCAACGCC-3′) or unlabeled mutant probe (5′-GGTCATTCCCTGAACACGCC-3′) to nuclear extracts at room temperature for 5 min before the addition of the labeled probe. The shift was performed by following the manufacturer’s instruction of LightShift Chemiluminescent EMSA Kit (Thermo Fisher Scientific). Briefly, bound complexes were separated on 6% nondenaturating polyacrylamide gels and transferred to nylon membrane. The transferred DNA was crosslinked to membrane using UV-light, detected with HPR-conjugated streptavidin and chemiluminescence.
Generation and treatment of PTEN-CaP8 xenografts in mice
The 6-week-old C57BL/6 mice (Jackson Lab) were used for animal experiments. The animal study was approved by the IACUC at Mayo Clinic. All mice were housed in standard conditions with a 12 h light/dark cycle and access to food and water ad libitum. PTEN-CaP8 cells (5 × 106 in 50 ml 1 × PBS plus 50 ×l Matrigel (BD Biosciences)) infected with lentivirus of Tsin empty vector (Tsin-EV) or Tsin-RL S249D/T252D peptide were injected s.c. into the right flank of mice. The volume of xenografts was measured every other day and calculated using the formula LxW2×0.5. After xenografts reached a size of approximately 40 mm3, mice carrying similar type of tumors were randomized into different groups and treated with IR (12 Gy initiated at day 1) and anti-PD-L1 (200 μg, i.p., given at days 0, 3, 6, and 9) alone or combination of IR. Mice were euthanized and tumors collected from all animals once tumors reached a volume of 200 mm3.
Flow cytometry analysis
PC-3 cells infected with control or RB1-specific shRNAs were harvested and washed with PBS. Cells were fixed in 4% paraformaldehyde for 15 min. After washed with PBS, cells were incubated with ice-cold 100% methanol 30 min on ice. Cells were washed with PBS and incubated with PD-L1 antibody (Cell Signaling, 13684S, working dilution 1:400) or isotype IgG for 1h at room temperature. Then, cells were washed with PBS and incubated with secondary antibody that was conjugated with Alexa Fluor (Thermo Fisher Scientific) for 1 h at room temperature. After washed three times with PBS, cells were resuspended with PBS and analyzed on flow cytometer.
For flow cytometry analysis of mouse tissue samples, tumors were cut into small pieces and digested with 2 mg/ml collagenase (Sigma) in DMEM for 1 h at 37°C. Cells were filtered through 70 μm nylon strainer and resuspended in red blood cell lysis buffer (Biolegend) for 3 min at room temperature. Cells were then suspended in PBS with 2% BSA and co-stained with the following antibodies: CD45, CD4, CD8, CD11b (Biolegend, 101212, APC conjugated), Gr1. After incubated with antibody for 30 min, cells were washed with PBS and analyzed on flow cytometer.
STATISTICAL ANALYSIS
Statistical analyses were performed with one-sided or two-sided paired Student t test for single comparison and One-way ANOVA and a post hoc test for multiple comparisons. P values < 0.05 are considered statistically significant. All values were expressed as means ± SD. Pearson’s product-moment correlation was used to calculate the correlation between PD-L1 and pRB-S249/T252 staining index in prostate cancer TMAs.
DATA AND SOFTWARE AVAILABILITY
Original images of western blot data are available at Mendeley Data under the following link: https://doi.org/10.17632/sz4bt84x5v.1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-RB | BD Biosciences | Cat# 554136; RRID: AB_395259 |
| Mouse monoclonal anti-RB | Cell Signaling Technology | Cat# 9309; RRID: AB_331745 |
| Rabbit monoclonal anti-p65 | Cell Signaling Technology | Cat# 8242; RRID: AB_10859369 |
| Mouse monoclonal anti-p65 | Cell Signaling Technology | Cat# 6956; RRID: AB_10828935 |
| Rabbit monoclonal anti-PD-L1 | Cell Signaling Technology | Cat# 13684; RRID: AB_2687655 |
| Mouse polyclonal anti-PD-L1 | Proteintech Group | Cat# 17952-1-AP; RRID: AB_10597552 |
| Rabbit monoclonal anti-p105/50 | Cell Signaling Technology | Cat# 13586; RRID: AB_2665516 |
| Rabbit polyclonal anti-Phospho-Rb (Ser795) | Cell Signaling Technology | Cat# 9301S; RRID: AB_330013 |
| Rabbit monoclonal anti- Phospho-Rb (Ser249,T252) | Thermo Fisher Scientific | Cat# 701059; RRID: AB_2532363 |
| Rabbit polyclonal anti-p107 | Santa Cruz | Cat# sc-318; RRID: AB_2175428 |
| Rabbit polyclonal anti-p130 | Santa Cruz | Cat# sc-317; RRID: AB_632093 |
| Rabbit polyclonal anti-E2F-1 | Santa Cruz | Cat# sc-193; RRID: AB_631394 |
| Rabbit polyclonal anti-CDK4 | Santa Cruz | Cat# sc-601; RRID: AB_631221 |
| Rabbit polyclonal anti-CDK6 | Santa Cruz | Cat# sc-177; RRID: AB_631225 |
| Mouse monoclonal anti-p52 | Santa Cruz | Cat# sc-7386; RRID: AB_2267131 |
| Mouse monoclonal anti-c-Rel | Santa Cruz | Cat# sc-6955; RRID: AB_670194 |
| Mouse monoclonal anti- RelB | Santa Cruz | Cat# sc-48366; RRID: AB_628212 |
| Rabbit monoclonal anti-MAP3K7 | Cell Signaling Technology | Cat# 5206S; RRID: AB_10694079 |
| Rabbit monoclonal anti-CHD1 | Cell Signaling Technology | Cat# 4351S; RRID: AB_11179073 |
| Rabbit monoclonal anti-PARP | Cell Signaling Technology | Cat# 9532S; RRID: AB_10695538 |
| Rabbit monoclonal anti- Cleaved Caspase-3 | Cell Signaling Technology | Cat# 9579S; RRID: AB_10897512 |
| Rabbit polyclonal anti-Phospho-NF-kappaB p65 (Ser536) | Cell Signaling Technology | Cat# 3031; RRID: AB_330559 |
| Mouse monoclonal anti-IκBα | Cell Signaling Technology | Cat# 4814; RRID: AB_390781 |
| Mouse monoclonal anti-IKK alpha | Santa Cruz | Cat# sc-71333; RRID:AB_1124857 |
| Mouse monoclonal anti-IKK beta | Santa Cruz | Cat# sc-8014; RRID:AB_627785 |
| Rabbit polyclonal anti-Histone H3 | Abcam | Cat# ab1791; RRID: AB_302613 |
| Rabbit monoclonal anti-beta-Tubulin | Cell Signaling Technology | Cat# 2128; RRID: AB_823664 |
| Mouse monoclonal anti-ERK2 | Santa Cruz | Cat# sc-135900; RRID: AB_2141283 |
| Mouse monoclonal anti-c-Myc | Santa Cruz | Cat# sc-40; RRID: AB_627268 |
| Mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat# F-3165; RRID: AB_259529 |
| Mouse monoclonal anti-HA.11 | Covance | Cat# MMS-101R; RRID: AB_291262 |
| Peroxidase IgG Fraction Monoclonal Mouse Anti-Rabbit IgG | Jackson ImmunoResearch | Cat# 211-032-171; RRID: AB_2339149 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG | Jackson ImmunoResearch | Cat# 115-035-174; RRID: AB_2338512 |
| APC monoclonal anti-mouse CD45 | BioLegend | Cat# 103112; RRID: AB_312977 |
| PE monoclonal anti-mouse CD8 | BioLegend | Cat# 100708; RRID: AB_312747 |
| APC monoclonal anti-mouse/human CD11b | BioLegend | Cat# 101212; RRID: AB_312795 |
| FITC monoclonal anti-mouse Ly-6G/Ly-6C (Gr-1) | BioLegend | Cat# 108406; RRID: AB_313371 |
| FITC anti-mouse CD4 | BioLegend | Cat# 100510; RRID: AB_312713 |
| Bacterial and Virus Strains | ||
| E. coli DH5α | Thermo Fisher | Cat# 18258012 |
| E. coli BL21 | Thermo Fisher | Cat# C600003 |
| lentivirus-expressing Rb1-shRNAs (mouse) | Sigma-Aldrich | SHCLNG-NM_009029 |
| lentivirus-expressing RB1-shRNAs (human) | Sigma-Aldrich | SHCLNG-NM_000321 |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| lentivirus-expressing CHD1-shRNAs (human) | Sigma-Aldrich | SHCLNG-NM_001270 |
| lentivirus-expressing MAP3K7-shRNAs (human) | Sigma-Aldrich | SHCLNG-NM_145332 |
| lentivirus-expressing RELA-shRNAs (human) | Sigma-Aldrich | SHCLNG-NM_021975 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Temsirolimus | AbMole BioScience | Cat# M3722 |
| Palbociclib (PD0332991) | Selleckchem | Cat# S1116 |
| VE-822 | Selleckchem | Cat# S7102 |
| I-CBP112 | Cayman Chemical | Cat# 14468 |
| Paclitaxel | Sigma-Aldrich | Cat# T7402 |
| SB203580 | ApexBio | Cat# A8254 |
| JSH-23 | Millipore | Cat# 481408 |
| GSK1120212 | Selleckchem | Cat# S2673 |
| GDC-0068 | MedChem Express | Cat# HY-15186A |
| Cisplatin | Selleckchem | Cat# S1166 |
| IKK-16 | Selleckchem | Cat# S2882 |
| JQ1 | Sigma-Aldrich | Cat# SML1524 |
| Lipofectamine 2000 reagent | Thermo Fisher | Cat# 11668500 |
| ACHP | TOCRIS | Cat# 4547 |
| Insulin-Transferrin-Selenium | Thermo Fisher Scientific | Cat# 41400045 |
| Anti-mouse PD-L1 mAb (clone 10B5) | Dr. Haidong Dong | Dong et al., 2002 |
| InVivoMAb mouse IgG1 isotype control (MOPC-21) | Bio X Cell | Cat# BE0083 |
| Critical Commercial Assays | ||
| KOD Plus Mutagenesis Kit | Toyobo | Cat# F0936K |
| PE Annexin V Apoptosis Detection Kit I | BD Biosciences | Cat# 557963 |
| CDK6/CyclinD3 Kinase Assay | Promega | Cat# V4510 |
| Lambda Protein Phosphatase | NEB | Cat# P0753S |
| Deposited Data | ||
| Raw and analyzed sequencing data | This paper | GEO: GSE109724 |
| Raw data and images | This paper and Mendeley Data | https://doi.org/10.17632/sz4bt84x5v.1 |
| Experimental Models: Cell Lines | ||
| Human: C4-2 | Uro Corporation | N/A |
| Human: PC-3 | ATCC | CRL-1435 |
| Human: DU145 | ATCC | HTB-81 |
| Human: HEK293T | ATCC | CRL-11268 |
| Human: LNCaP | ATCC | CRL-1740 |
| Human:22Rv1 | ATCC | CRL-2505 |
| Human: RWPE-1 | ATCC | CRL-11609 |
| Huamn: VCaP | ATCC | CRL-2876 |
| Human: SK-Hep1 | ATCC | HTB-52 |
| Human: H1299 | ATCC | CRL-5803 |
| Human: PANC-1 | ATCC | CRL-1469 |
| Human: LAPC-4 | Dr. C. L. Sawyers | Craft et al., 1999 |
| Human: BPH-1 | Dr. S. W. Hayward | Hayward et al., 1995 |
| Mouse: PTEN-CaP8 | Dr. Hong Wu | Jiao et al., 2007 |
| Mouse: RB−/− PrE | Dr. M. L. Da | Day et al., 2002 |
| Mouse: Rb-proficient SKO (Cre+:Ptenf/f:Rb1+/+) | Dr. D. W. Goodrich | Ku et al., 2017 |
| Mouse: castration resistant Rb-deficient DKO (Cre+:Ptenf/f:Rb1f/f) | Dr. D. W. Goodrich | Ku et al., 2017 |
| Oligonucleotides | ||
| See Tables S3 and S4 for primer sequences | ||
| Recombinant DNA | ||
| pCMV4-p65 | Addgene | Cat# 21966 |
| pCMV-HA-hRB-WT | Addgene | Liu et al., 2017; Cat# 58905 |
| pCMV-HA-hRBDCDK | Addgene | LeRoy et al., 2008; Cat# 58906 |
| Software and Algorithms | ||
| ImageJ | NIH | N/A |
| FlowJo | FlowJo LLC | N/A |
| GraphPad Prism 5.0 | Graphpad, Inc | N/A |
Highlights.
RB specifically binds to p65, but not other NF-κB/Rel family proteins
RB-p65 interaction relies on CDK4/6 S249/T252 phosphorylation of RB
S249/T252-phosphorylated RB inhibits NF-κB activity and PD-L1 expression
S249/T252 phospho-mimetic peptide promotes cancer immunity via PD-L1 suppression
ACKNOWLEDGMENTS
We thank the patients and their families for their altruism in participating in research studies. This work was supported in part by the Mayo Clinic Foundation (to H.H.) and the National Natural Science Foundation of China (81702374 to X.J.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and four tables and can be found online at https://doi.org/10.1016/j.molcel.2018.10.034.
DECLARATION OF INTERESTS
Mayo Clinic has filed a patent related to this work.
REFERENCES
- Akamatsu S, Wyatt AW, Lin D, Lysakowski S, Zhang F, Kim S, Tse C, Wang K, Mo F, Haegert A, et al. (2015). The placental gene PEG10 promotes progression of neuroendocrine prostate cancer. Cell Rep. 12, 922–936. [DOI] [PubMed] [Google Scholar]
- Ariga A, Namekawa J, Matsumoto N, Inoue J, and Umezawa K (2002). Inhibition of tumor necrosis factor-alpha -induced nuclear translocation and activation of NF-kappa B by dehydroxymethylepoxyquinomicin. J. Biol. Chem 277, 24625–24630. [DOI] [PubMed] [Google Scholar]
- Bouillez A, Rajabi H, Jin C, Samur M, Tagde A, Alam M, Hiraki M, Maeda T, Hu X, Adeegbe D, et al. (2017). MUC1-C integrates PD-L1 induction with repression of immune effectors in non-small-cell lung cancer. Oncogene 36, 4037–046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JR, Deshong AJ, Pelton JG, and Rubin SM (2010). Phosphorylation-induced conformational changes in the retinoblastoma protein inhibit E2F transactivation domain binding. J. Biol. Chem 285, 16286–16293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart DL, and Sage J (2008). Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 8, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft N, Shostak Y, Carey M, and Sawyers CL (1999). A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat. Med 5, 280–285. [DOI] [PubMed] [Google Scholar]
- Day KC, McCabe MT, Zhao X, Wang Y, Davis JN, Phillips J, Von Geldern M, Ried T, KuKuruga MA, Cunha GR, et al. (2002). Rescue of embryonic epithelium reveals that the homozygous deletion of the retinoblastoma gene confers growth factor independence and immortality but does not influence epithelial differentiation or tissue morphogenesis. J. Biol. Chem 277, 44475–44484. [DOI] [PubMed] [Google Scholar]
- DeCaprio JA, Ludlow JW, Lynch D, Furukawa Y, Griffin J, Piwnica-Worms H, Huang CM, and Livingston DM (1989). The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell 58, 1085–1095. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, and Chen L (1999). B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med 5, 1365–1369. [DOI] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med 8, 793–800. [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med 192, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevensleben H, Dietrich D, Golletz C, Steiner S, Jung M, Thiesler T, Majores M, Stein J, Uhl B, Müller S, et al. (2016). The immune checkpoint regulator PD-L1 is highly expressed in aggressive primary prostate cancer. Clin. Cancer Res. 22, 1969–1977. [DOI] [PubMed] [Google Scholar]
- Goodrich DW, Wang NP, Qian YW, Lee EY, and Lee WH (1991). The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell 67, 293–302. [DOI] [PubMed] [Google Scholar]
- Gubern A, Joaquin M, Marquès M, Maseres P, Garcia-Garcia J, Amat R, González-Nuñez D, Oliva B, Real FX, de Nadal E, and Posas F (2016). The N-terminal phosphorylation of RB by p38 bypasses its inactivation by CDKs and prevents proliferation in cancer cells. Mol. Cell 64, 25–36. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hassler M, Singh S, Yue WW, Luczynski M, Lakbir R, Sanchez-Sanchez F, Bader T, Pearl LH, and Mittnacht S (2007). Crystal structure of the retinoblastoma protein N domain provides insight into tumor suppression, ligand interaction, and holoprotein architecture. Mol. Cell 28, 371–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward SW, Dahiya R, Cunha GR, Bartek J, Deshpande N, and Narayan P (1995). Establishment and characterization of an immortalized but non-transformed human prostate epithelial cell line: BPH-1. In Vitro Cell. Dev. Biol. Anim 31, 14–24. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Shinohara H, Behar M, Yumoto N, Tanaka G, Hoffmann A, Aihara K, and Okada-Hatakeyama M (2016). Oscillation dynamics underlie functional switching of NF-κB for B-cell activation. NPJ Syst. Biol. Appl 2, 16024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishak CA, Marshall AE, Passos DT, White CR, Kim SJ, Cecchini MJ, Ferwati S, MacDonald WA, Howlett CJ, Welch ID, et al. (2016). An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol. Cell 64, 1074–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Agata Y, Shibahara K, and Honjo T (1992). Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, and Minato N (2002). Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 99, 12293–12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, Wang S, Qiao R, Vivanco I, Watson PA, Sawyers CL, and Wu H (2007). Murine cell lines derived from Pten null prostate cancer show the critical role of PTEN in hormone refractory prostate cancer development. Cancer Res. 67, 6083–6091. [DOI] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbé DP, Gomez EC, Wang J, et al. (2017). Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy G, Rickards B, and Flint SJ (2008). The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 30, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, Cha JH, Xia W, Chan LC, Kim T, et al. (2018). Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell 33, 187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, Chang SS, Lin WC, Hsu JM, Hsu YH, et al. (2016). Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 30, 925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Yu J, Deng M, Yin Y, Zhang H, Luo K, Qin B, Li Y, Wu C, Ren T, et al. (2017). CDK4/6-dependent activation of DUB3 regulates cancer metastasis through SNAIL1. Nat. Commun 8, 13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AL, and Dyson NJ (2012). RB: mitotic implications of a tumour suppressor. Nat. Rev. Cancer 12, 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari F, Ciccarese C, Calicò A, Munari E, Cima L, Porcaro AB, Novella G, Artibani W, Sava T, Eccher A, et al. (2016). Magnitude of PD-1, PD-L1 and T lymphocyte expression on tissue from castration-resistant prostate adenocarcinoma: an exploratory analysis. Target. Oncol 11, 345–351. [DOI] [PubMed] [Google Scholar]
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, and Dowdy SF (2014). Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 3, pii: e02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Hamanishi J, Matsumura N, Abiko K, Murat K, Baba T, Yamaguchi K, Horikawa N, Hosoe Y, Murphy SK, et al. (2015). Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-kB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. 75, 5034–5045. [DOI] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015). Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, and McCormick F (2002). The RB and p53 pathways in cancer. Cancer Cell 2, 103–112. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, and Roberts JM (2004). Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 18, 2699–2711. [DOI] [PubMed] [Google Scholar]
- Sun H, Wang Y, Chinnam M, Zhang X, Hayward SW, Foster BA, Nikitin AY, Wills M, and Goodrich DW (2011). E2f binding-deficient Rb1 protein suppresses prostate tumor progression in vivo. Proc. Natl. Acad. Sci. USA 108, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, and Karin M (2018). NF-κB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol 18, 309–324. [DOI] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, and Pardoll DM (2012a). Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol 24, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012b). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkowska T, and Mittnacht S (1997). Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J. Biol. Chem 272, 12738–12746. [DOI] [PubMed] [Google Scholar]
- Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z, Wang S, Pan CW, Zhu Y, Yan Y, et al. (2017). Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat. Med 23, 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, Tan Y, Ci Y, Wu F, Dai X, et al. (2018). Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Lu X, Wang G, Lan Z, Liao W, Li J, Liang X, Chen JR, Shah S, Shang X, et al. (2017). Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature 542, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, and Chen L (2003). Cancer therapeutic monoclonal antibodies targeting lymphocyte co-stimulatory pathways. Curr. Opin. Investig. Drugs 4, 691–695. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original images of western blot data are available at Mendeley Data under the following link: https://doi.org/10.17632/sz4bt84x5v.1.