

Abstract
Herein, we expand the current molecular-level understanding of one of the most important and effective additives in iron-catalyzed cross-coupling reactions, N,N,N’,N’-tetramethylethylenediamine (TMEDA). Focusing on relevant phenyl and ethyl Grignard reagents and slow nucleophile addition protocols commonly used in effective catalytic systems, TMEDA-iron(II)-aryl intermediates are identified via in situ spectroscopy, X-ray crystallography, and detailed reaction studies to be a part of an iron(II)/(III)/(I) reaction cycle where radical recombination FePhBr(TMEDA) (2Ph) results in selective product formation in high yield. These results differ from prior studies with mesityl Grignard reagent, where poor product selectivity and low catalytic performance can be attributed to homoleptic iron-ate species. Overall, this study represents a critical advance in how amine additives such as TMEDA can modulate selectivity and reactivity of organoiron species in cross-coupling.
Keywords: iron, homogeneous catalysis, C-C coupling, ligand effects, organometallics
Graphical Abstract

Historically, the role of TMEDA in iron-catalyzed C(sp2)-C(sp3) cross-coupling has been disputed due to a lack of insight into the iron species present during catalysis. In this report, low-temperature synthesis, freeze-trapped 57Fe Mössbauer, X-ray diffraction (XRD), and chemical quenched GC analysis were utilized to identify highly reactive and selective TMEDA-ligated iron species responsible for C(sp2)-C(sp3) cross-coupling catalysis.
Introduction
Iron-catalyzed cross-coupling methods utilizing a variety of ligand and reaction additives have been of broad interest over the past two decades due to their potential as low cost, sustainable systems for C-C bond formation. One additive of particular importance in iron cross-coupling is N,N,N’,N’-tetramethylethylenediamine (TMEDA), originally reported in 2004 by Nakamura and co-workers for the Kumada cross-coupling of C(sp2)-aryl Grignard reagents and C(sp3)-alkyl halides (Scheme 1).[1,2] The effectiveness of TMEDA as an additive in this pioneering study subsequently inspired its use in numerous iron cross-coupling methods including Cahiez’s and Cossy’s cross-couplings of alkenyl Grignard reagents and alkyl halides,[3,4] Füstner’s cross-couplings of aryl Grignard reagents with aryl chloride or allyl halides,[5,6] scalable C-C cross-coupling protocols,[7] and domino catalysis utilizing TMEDA and in situ method for Grignard reagent generation in the presence of iron salts.[8,9] Furthermore, the practical utility of TMEDA-assisted iron-catalyzed cross-coupling has also been demonstrated in several complex organic molecule syntheses.[10–18]
Scheme 1.

Representative examples of iron-catalyzed Kumada cross-coupling reactions using TMEDA.
While TMEDA represents one of the most practical and widely utilized additives in iron cross-coupling methodologies, the molecular-level role of TMEDA that facilitates effective catalysis remains unclear and debated in the literature. The most detailed studies combining organoiron synthesis, in situ iron speciation during catalysis, and organoiron reactivity toward electrophiles have focused on C(sp2)-C(sp3) Kumada cross-couplings with TMEDA and mesityl (Mes) Grignard reagent.[19,20] In 2009, the stoichiometric reactivity of the iron-mesityl complexes, Fe(Mes)(Br)(TMEDA) and Fe(Mes)2(TMEDA), were evaluated, and it was determined that Fe(Mes)2(TMEDA) selectively generates C-C product from alkyl halides leading to the proposal that this TMEDA-bound complex was central to catalysis.[21] More recently, Bedford and co-workers performed a thorough investigation of the reaction with mesityl Grignard reagent by monitoring the iron speciation under catalytically relevant conditions combined with detailed stoichiometric reactivity studies, finding [Fe(Mes)3]- as the catalytically active species.[19,20,22] Combined with additional studies using benzyl Grignard reagent, it was proposed that homoleptic “ate” species, rather than TMEDA-adducts, account for the catalytic activity and that TMEDA is unlikely to play a role in the primary catalytic cycle but, instead, likely traps off-cycle intermediates. It should be noted that subsequent studies have also argued for the role of TMEDA-iron-ate complexes in cross-coupling, though such species were only identified by mass spectrometry and their formation in situ remains unclear.[23]
While these various mechanistic proposals for TMEDA-iron cross-coupling remain widely debated, the homoleptic iron-ate mechanism currently dominates the field due to the broad use of synthesis, spectroscopy, and reactivity studies. However, these mechanistic studies utilizing MesMgBr and TMEDA are unlikely to apply more broadly to the molecular role of TMEDA in cross-couplings with aryl Grignard reagents and the slow nucleophile addition reaction protocols most commonly employed. C(sp2)-C(sp3) Kumada cross-coupling with MesMgBr with TMEDA performs poorly in terms of product yields compared to those with other aryls such as PhMgBr, suggesting that the formation of homoleptic “ate” species identified by Bedford could potentially correlate with less effective catalysis. In fact, such homoleptic complexes may not be prevalent with less sterically bulky and less electron-donating aryl such as phenyl. In fact, previous mechanistic studies have demonstrated a distinct difference in iron speciation in the presence of excess PhMgBr vs. MesMgBr with bisphosphine ligands.[24,25] While it was observed that [Fe(Mes)3]- formed in the presence of excess MesMgBr and bisphosphine ligand, [25] excess PhMgBr did not lead to similar homoleptic iron speciation, and instead, the bis-transmetallated bisphosphine iron species, FePh2(SciOPP), persisted.[24] Additionally, in the original report by Nakamura and co-workers, C(sp2)-C(sp3) Kumada cross-coupling with TMEDA requires strict reaction protocols, including the slow addition of Grignard reagents at low temperature with high TMEDA loading to achieve high catalytic yields.[2] While a feature in nearly all iron cross-coupling methods with TMEDA, this slow Grignard reagent addition effect was not investigated in the previous mesityl studies, yet likely has a significant effect on iron speciation in catalysis since it is critically important for successful cross-coupling.[21] Thus, in order to further develop effective additives such as diamines for iron cross-coupling, the iron speciation and reactivity with more typical and effective aryl Grignard reagents, solvent, and protocols (i.e. slow Grignard addition) are essential to more broadly define the role of TMEDA across the breadth of nucleophiles utilized in cross-coupling.[26]
To address this challenge, the present study focuses on the use of inorganic synthesis, X-ray diffraction (XRD), advanced spectroscopic methods (57Fe Mössbauer, electron paramagnetic resonance(EPR)), and detailed reaction/kinetic studies to define the role of TMEDA in C(sp2)-C(sp3) Kumada cross-couplings with PhMgBr. These studies define the role of TMEDA ligation to iron in the primary catalytic cycle with PhMgBr, along with analogous findings for cross-coupling reactions with EtMgBr. Overall, these studies demonstrate the important role that the nucleophile plays in determining in situ iron speciation (i.e. TMEDA-ligated vs. homoleptic iron “ate” species) to promote effective catalysis and provide a molecular-level framework for the continued development of additives and ligands for iron-catalyzed cross-couplings.
Results and Discussion
Synthesis and Characterization of Well-Defined TMEDA-Fe-Phenyl Complexes
The initial focus of this study was to synthesize and isolate TMEDA-ligated iron-phenyl complexes and evaluate their role and relevance to C-C cross-coupling catalysis. Since to our knowledge, the synthetically convenient phenyl analog Fe2(Mes)4 is not available for the reaction with TMEDA to generate FePh2(TMEDA),[21] a well-defined TMEDA-iron bis-halide complex that could subsequently act as a convenient starting material for subsequent transmetallation with PhMgBr was targeted. The reaction of 2.0 equiv of TMEDA and FeBr2 in toluene at room temperature (RT) for 1 h, followed by filtration over Celite and storage at −30 °C for 16 h, yielded colorless needles. These were analyzed and identified by XRD as FeBr2(TMEDA) (1), the desired distorted tetrahedral bis-halide complex (Figure 1). Solid-state 80 K Mössbauer spectroscopy of crystalline 1 displayed an isomer shift (δ) = 0.89 mm/s and electric quadrupole splitting (|ΔEQ|) = 2.89 mm/s (Figure S1) and a µeff = 5.9(1) B.M. by Evans method NMR. Notably, 1 in THF displays an additional iron species by Mössbauer (δ = 1.08 mm/s and |ΔEQ| = 2.86 mm/s) (Figure S2). Redissolving 57Fe-labeled 1 in 2-Me-THF led to the disappearance of this species confirming it as a THF-adduct (1THF; Figure S3). With complex 1 in hand, we pursued reactions with 1.0 and 2.0 equiv of PhMgBr to generate mono- and bis-phenylated TEMDA-iron complexes, respectively.
Figure 1.

Synthesis of FeBr2(TMEDA) (1). Crystal structure of 1 with selected bond lengths and angles. Note: Hydrogen atoms omitted for clarity and ellipsoids drawn at 50% probability level.
The reaction of 1.0 equiv of PhMgBr with FeBr2(TMEDA) in THF at 0 °C was initially interrogated using 57Fe Mössbauer spectroscopy. Following the addition of PhMgBr, the FeBr2(TMEDA) solution changed from pale green to colorless within 30 s. 80 K Mössbauer spectroscopy revealed the generation of one major iron species (Figure 2A, left) with parameters of δ = 0.68 mm/s and |ΔEQ| = 2.04 mm/s that are consistent with a high-spin FeII species. Crystallization efforts were pursued at low temperature (−80 °C) using THF/pentane solvent mixtures. Colorless plates crystallized over several days at −80 °C. XRD analysis confirmed the identity of the crystallized species as the distorted tetrahedral complex, FePhBr(TMEDA) (2Ph) with a µeff = 5.8(1) B.M. (Figure 2A, right). Crystals of 2Ph were found to be highly air-sensitive and rapidly decomposed upon transfer to the diffractometer even with careful handling at cryogenic temperatures (see SI for handling of sensitive crystals). However, crystalline 2Ph was determined to be indefinitely stable at −30 °C under N2 atmosphere. Solid-state Mössbauer spectroscopy of crystals of 2Ph confirmed the in situ generated iron species parameters at 0 °C (δ = 0.68 mm/s and |ΔEQ| = 2.01 mm/s; Figure S4).
Figure 2.

80 K Mössbauer spectra of in situ generated A) FePhBr(TMEDA) (2Ph) with δ = 0.68 mm/s and |ΔEQ| = 2.04 mm/s and B) FePh2(TMEDA) (3Ph) with δ = 0.52 mm/s and |ΔEQ| = 1.06 mm/s. Crystal structures and bond metrics of 2Ph (top) and 3Ph (bottom). Note: Hydrogen atoms omitted for clarity and ellipsoids drawn at 50 % probability level.
Similarly, the reaction of 2.0 equiv of PhMgBr with FeBr2(TMEDA) in THF at 0 °C revealed the presence of one major iron species by Mössbauer spectroscopy (Figure 2B, left) with parameters of δ = 0.52 mm/s and |ΔEQ| = 1.06 mm/s. The decrease in the δ and ΔEQ values is consistent with a second transmetallation event with PhMgBr. Low-temperature crystallographic investigations, similar to that of 2Ph, revealed the formation of pale-yellow blocks identified as FePh2(TMEDA) (3Ph) (Figure 2B, right) by XRD analysis (µeff = 6.2(2) B.M.).[27] Complex 3Ph was determined to have markedly decreased thermal stability, decomposing at −30 °C after 24 h in both solution and in the solid-state. The 80 K Mössbauer spectroscopy of crystalline 3Ph displayed Mössbauer parameters consistent with those in solution (δ = 0.52 mm/s and |ΔEQ| = 1.24 mm/s; Figure S5). A slightly perturbed ΔEQ-value is consistent with the small structural distortion of complex 3Ph in THF. It should be noted that previously reported Mössbauer studies by Koszinowski and co-workers of the reaction of 57FeCl2 with 4.0 equiv of PhMgBr and 4.0 equiv of TMEDA in THF demonstrated the formation of a major iron species (78% of total iron) with parameters of δ = 0.49 mm/s and |ΔEQ| = 1.03 mm/s, assigned as [FePh4]- in combination with ESI-MS with gas-phase fragmentation studies.[23] However, based on our current study, this major iron species observed by Mössbauer appears to correlate to the frozen solution parameters for 3Ph. Lastly, it should be noted, that additional studies have previously suggested the formation of homoleptic iron-aryl species which may be in equilibrium with TMEDA-bound species,[19,23,28–30] though we have not observed such homoleptic complexes in the current spectroscopic studies.
The TMEDA-ligated iron-phenylated complexes, 2Ph and 3Ph, join a small collection of 4-coordinate iron-TMEDA hydrocarbyl complexes, which include the mesityl and, the more recently reported, ethyl analogs.[21,31] The bond lengths of the mono- and bis-phenylated complexes, 2Ph and 3Ph, closely resemble those of the ethyl analogs, FeEtBr(TMEDA) (2Et) and FeEt2(TMEDA) (3Et), respectively (Table 1). However, complexes 2Ph and 3Ph exhibit significantly shorter Fe-Cipso bond lengths (> 0.02 Å) than the corresponding mesityl species. The difference in overall molecular geometries of the TMEDA-ligated complexes is best illustrated by their calculated 4-coordinate geometric indices, τ4.[32] Both the bis-phenyl and bis-ethyl complexes maintain slightly distorted tetrahedral geometries (τ4 = 0.89 and 0.84, respectively), while the bis-mesityl complex exhibits significant structural distortion toward square planar geometry (τ4 = 0.62). A less pronounced but similar τ4-trend was observed in the mono-transmetallated complexes as well. The significant deviation from tetrahedral geometry present in the mesityl complexes is attributable to the steric encumbrance of the mesityl substituents. If this distortion effect was due to the electron-donor strength of the substituent, the ethyl analogs would likely display a similar magnitude of distortion away from tetrahedral.
Table 1.
Summary of bond metrics of FeRBr(TMEDA) and FeR2(TMEDA) (where R = Ph, Et, or Mes).
| Complex | Fe-N(1) (Å) | Fe-C(1) (Å) | Fe-C(7) (Å) | τ4 |
|---|---|---|---|---|
| FePhBr(TMEDA) (2Ph) | 2.194(2) | 2.050(3) | --- | 0.86 |
| FeEtBr(TMEDA) (2Et)35 | 2.195(3) | 2.046(4) | --- | 0.84 |
| FeMesBr(TMEDA)19 | 2.242(3) | 2.072(4) | --- | 0.79 |
|
| ||||
| FePh2(TMEDA) (3Ph) | 2.2235(8) | 2.7062(9) | 2.0806(9) | 0.89 |
| FeEt2(TMEDA) (3Et)35 | 2.2437(15) | 2.0821(15) | 2.0821(15) | 0.84 |
| FeMes2(TMEDA)21 | 2.304(3) | 2.097(3) | 2.101(2) | 0.69 |
Further characterization of complexes 1–3Ph was performed including NMR and MCD spectroscopy. Unfortunately, the NMR spectra do not provide a good descriptor of the individual iron species but may prove useful as an in situ identifier for future studies (see SI). Additional insight into the electronic structure of the TMEDA-ligated iron species 1–3Ph was obtained using 5 K, 7 T near-infrared (NIR) magnetic circular dichroism (MCD) spectroscopy (Figure S6). Most notably, solution-state characterization of 3Ph in THF/2-MeTHF (1:1) revealed the presence of two major transitions centered at circa 5680 cm−1 and circa 6300 cm−1 (Figure S6C) consistent with the previously reported NIR MCD spectrum of 3Et (ca. 5940 cm−1 and ca. 6900 cm−1).[31] The similarity in spectral features between the ethyl and phenyl complexes is consistent with minimal ligand field differences as a function of nucleophile for these complexes.
Reaction of FePhBr(TMEDA) (2Ph) and FePh2(TMEDA) (3Ph) with Bromocycloheptane (Chp-Br)
We probed the stoichiometric reactivity and selectivity of 2Ph and 3Ph with bromocycloheptane (Chp-Br) using a combination of time-resolved, freeze-trapped 57Fe Mössbauer and chemical-quench GC analysis. Complex 2Ph or 3Ph was generated in situ from FeBr2(TMEDA) (0.1 Μ Fe) and PhMgBr (1.0 or 2.0 equiv, respectively) in THF at 0 °C. The reaction of in situ generated 2Ph with 1.0 equiv of Chp-Br resulted in full recovery of Chp-Br after 5 min. This result is consistent with the reactivity of the mono-mesityl TMEDA-iron complex studied by Nagashima,[21] though, contrasts significantly with previous studies into the reactivity of iron-SciOPP chemistry in which the mono-phenylated complex Fe(Ph)Br(SciOPP) was determined to be both reactive and selective toward Chp-Br to generate cross-coupled product.[24]
Unlike complex 2Ph, stoichiometric reactions of the bis-phenylated complex 3Ph with 1.0 equiv of Chp-Br readily produced cross-coupled product. Reaction of in situ generated 3Ph and 1.0 equiv of Chp-Br led to the rapid consumption of 3Ph and generation of cross-coupled product, Chp-Ph, in 58% yield after 150 s. Cycloheptene and cycloheptane side products were formed in 31% and 11% yield, respectively. The remaining iron species was determined to be 2Ph (69% of total Fe) and 3Ph (21% of total Fe) by Mössbauer (Figure 3), consistent with some organic radical escape to react with solvent or Chp-Br. The low selectivity of 3Ph for cross-coupled product formation was surprising as the bis-mesityl analog was previously determined to be highly selective for C-C product formation. Interestingly, addition of 2.0 equiv of Chp-Br to 3Ph led to rapid consumption of 3Ph within 150 s, but with a marked increase in selectivity for Chp-Ph formation (Figure 4). Notably, the remaining iron species were complex 2Ph (32%) and two other iron components consistent with FeX2(TMEDA)2 (X = halide) species formed in the presence of the excess TMEDA in the reaction mixture (Figure 4B and C), gold and light blue traces, 32% and 34%, respectively; see Figures S7 and S8 for further details).[33] GC analysis revealed the generation of Chp-Ph in 145% yield with respect to (w.r.t.) Fe. The organic side products cycloheptene and cycloheptane were generated in 37% and 6% yield, respectively. In both cases where Chp-Br is reacted with 2Ph or 3Ph, trace amounts of phenyl byproducts (PhH and biphenyl) were observed, however, it should be noted that phenyl counting can be misleading due to not knowing how much can be attributed to byproducts versus from the quenching phenylated-iron(II) species as been previously reported.[24]
Figure 3.

80 K Mössbauer spectra of frozen solution of reaction of in situ generated FePh2(TMEDA) (3Ph) with 1.0 equiv of Chp-Br after 150 s; raw data (black dots), total fit (black trace), individual fit components are shown.
Figure 4.

80 K Mössbauer spectra of frozen solution time points of A) in situ generated FePh2(TMEDA) (3Ph) and reaction of 2.0 equiv of Chp-Br after B) 60 s and C) 150 s; raw data (black dots), total fit (black trace), individual fit components are shown.
The observed reactivities in these stoichiometric reactions are reminiscent of recent published work with bisphosphines and are suggestive of a radical reaction in which 3Ph is responsible for initial radical generation upon reaction with electrophile followed by radical recombination with either 3Ph or 2Ph to form product. The observation that higher selectivities are obtained for the 2.0 equiv electrophile reaction would be consistent with more selective product formation in the second turnover when the average transmetallation of iron is reduced (i.e. upon recombination with 2Ph). This would also provide an explanation for one role of the slow Grignard reagent addition required for high selectivity – to form just enough 3Ph to initial radical formation but maintain predominantly undertransmetalled 2Ph as the main species in situ during catalysis in order to favor recombination with 2Ph (note that 2Ph is observed as the major species in situ during catalysis, vide infra). It should be acknowledged that while radical generation with homoleptic iron complexes where the radical ultimately recombines with TMEDA-bound iron species could also be led to selective product formation, no experimental evidence for the in situ formation of such homoleptics was observed in our studies.
To further evaluate whether 3Ph reactivity with Chp-Br is relevant to catalysis, pseudo first-order kinetic experiments reflective of catalytic conditions were performed. In the presence of 20.0 equiv of Chp-Br and 3Ph, Chp-Ph was generated within 10 s (kobs > 6 min−1). No induction period was observed prior to reaction of 3Ph with Chp-Br. Compared with the average turnover frequency (TOF) of catalysis (~ 0.28 min−1),[2] the rapid consumption of Chp-Br to produce Chp-Ph is consistent with 3Ph being kinetically competent for catalysis. Notably, the first turnover of 3Ph with Chp-Br is unselective with Chp-Ph being generated in 43% yield with concomitant generation of side products cycloheptene and cycloheptane (34% and 9% yield, respectively). However, GC analysis after 30 s revealed a significant increase in Chp-Ph to 145% yield (w.r.t. Fe) with only a small increase in cycloheptene (37%). The 80 K Mössbauer spectrum revealed the full consumption of 3Ph to 2Ph and FeBr2(TMEDA)2 (Figure 4, S8, and S9B, vida supra). The rapid consumption of Chp-Br by 3Ph compared to the rate of Grignard addition during catalysis (0.02 equiv/min)[2] is indicative that 3Ph is a catalytically relevant reactive iron species.
Iron Speciation in the Iron-Catalyzed Cross-Coupling of PhMgBr and Bromocycloheptane (Chp-Br)
The rapid reaction of Chp-Br and 3Ph at catalytically relevant rates motivated further investigations into the iron speciation present during catalytic turnover to determine whether TMEDA-ligated iron complexes 2Ph and 3Ph are relevant to catalysis. We first determined whether complex 1 could serve as a suitable pre-catalyst for the cross-coupling of PhMgBr and Chp-Br at 0 °C in THF. Following an adapted Nakamura catalytic protocol (see SI),[2] slow addition of a mixture of PhMgBr (1.3 equiv) and TMEDA to Chp-Br in the presence of FeBr2(TMEDA) (5 mol %) was performed and resulted in the selective production of Chp-Ph in 92% yield. The success of 1 as a pre-catalyst encouraged us to utilize freeze-trapped Mössbauer spectroscopy to monitor catalysis with 57Fe-labelled 1. Aliquots were freeze-trapped at the beginning and approximately halfway through the catalytic reaction using 57FeBr2(TMEDA) (5 mol %) (t = 5 and 20 min, respectively) with corresponding chemically quenched GC analysis to trace cross-coupled product formation. After 5 min, the iron speciation closely resembled that of the stoichiometric reaction of FePh2(TMEDA) and 2.0 equiv of Chp-Br (Figure 5A). The major iron species present in solution is the mono-phenylated TMEDA-iron species, 2Ph (72% total Fe), at which time Chp-Ph is produced (9% yield). A similar iron speciation and continued production of cross-coupled product (45% yield) at an extended time (t = 20 min, Figure 5B) is consistent with 2Ph acting as a pre-catalytic species in the Fe-catalyzed cross-coupling of PhMgBr and Chp-Br. Furthermore, no other iron species were detected by 10 K EPR spectroscopy (Figure S10).
Figure 5.

Freeze-trapped 80 K Mössbauer spectrum of the iron speciation in the iron-catalyzed cross-coupling of PhMgBr and Chp-Br at A) 5 min and B) 20 min; raw data (black dots), total fit (black trace), individual fit components are shown.
The iron speciation observed in both the stoichiometric reactivity of 3Ph with electrophile and during catalysis (Figure 4 and 5, respectively) led us to explore whether a phenyl ligand redistribution between 1 and 3Ph was possible. Ligand redistribution reactions of sterically unencumbered, unsaturated nucleophiles have been previously observed in iron-SciOPP chemistry in which a collection of mono-, bis-, and tris-alkynylated iron-SciOPP complexes formed from 1.0 equiv of alkynyl nucleophile and iron-SciOPP bis-halide.[34] 57Fe Mössbauer experiments of in situ formation and addition of 1.0 equiv of 3Ph and 1 revealed a phenyl ligand redistribution to generate 2Ph in 73% yield within 30 s (Figure S11). While facile, this ligand redistribution reaction is only 73% complete in 30 s compared to 93% formation of cross-coupled product from the reaction of 3Ph with Chp-Br in high yields and rapid formation (10 s) with excess electrophile. Note again that 2Ph has no observed reactivity towards electrophile even over a 5 min reaction timeframe.
The combination of the observed catalytic iron speciation and the previously described stoichiometric reactivity contrasts significantly with current mechanistic proposals for the TMEDA-assisted aryl-alkyl cross-coupling in which homoleptic iron(II)-aryl monomers are the proposed active catalytic species.[19] These results suggest that the role of TMEDA-ligated iron-aryl species in the primary catalytic cycle is nucleophile and reaction protocol dependent.[2] Thus, the role of a reaction additive such as TMEDA may vary from one nucleophile to the next or from one protocol to the next (ex. solvent, nucleophile addition rate). Future studies of additional systems that utilize TMEDA, including aryl-alkyl cross-coupling with TMEDA with PhMgBr under refluxing conditions in Et2O reported by Bedford and co-workers, can further refine the potential use of homoleptic versus TMEDA-bound iron reaction manifolds for cross-couplings with TMEDA and aryl nucleophiles.
Iron Speciation in the Iron-catalyzed Cross-Coupling of EtMgBr and 4-chloromethyl benzoate (ArCl)
While the above studies have demonstrated the importance of TMEDA-ligated iron-aryl complexes, including 2Ph and 3Ph, as catalytically relevant species for the cross-coupling of aryl Grignard reagents and alkyl halides, the use of TMEDA in iron cross-coupling is not limited to systems using aryl Grignard reagents. For example, Fox and co-workers in 2013 reported cross-couplings with alkyl Grignard reagents, iron, and TMEDA that employed a similar catalytic protocol to Nakamura’s 2004 report with aryl Grignard reagents.[2,12] Notable minor differences between the two catalytic protocols include the catalytic loading of TMEDA (10 mol% in Fox protocol vs. 2 mol% in Nakamura protocol) and the multiportion addition of Grignard reagent of Fox employed to simulate the syringe pump slow addition by Nakamura. Thus, we were motivated to broaden our studies to evaluate whether analogous TMEDA-bound iron intermediates are more broadly relevant across different classes of Grignard reagents. As a representative example, we focused our studies on cross-coupling of EtMgBr and aryl chlorides in the Fox protocol and the potential roles of the previously reported complexes, FeEtBr(TMEDA) (2Et) and FeEt2(TMEDA) (3Et),[35] in catalysis.
Initial studies focused on determining the minimum amount of TMEDA necessary to achieve the same catalytic performance originally reported by Fox and co-workers.[12] It was determined that 2.0 equiv of TMEDA (2 mol%) yielded 93% cross-coupled product compared to > 99% yield with the original 10 mol% TMEDA. Similar to investigations into the reactivity of complexes 2Ph and 3Ph, the reaction of in situ generated ethyl analogs, 2Et and 3Et, and varying equivalents of 4-chloromethyl benzoate (ArCl) to generate 4-ethylmethyl benzoate (ArEt) were undertaken. Using the minimum amount of TMEDA possible, complex 3Et was generated by reaction of FeCl2 with 2.0 equiv of TMEDA at RT in THF, followed by cooling to −25 °C and dropwise addition of 2.0 equiv of EtMgBr. After stirring at RT for 60 s, 1.0 equiv of ArCl was added in one portion resulting in the formation of cross-coupled product, ArEt, in 86% yield within 60 s. Analogous to the phenyl study, we wanted to examine whether 3Et with electrophile is kinetically relevant to catalysis by performing pseudo first-order kinetic reactions. To mimic conditions relevant to catalysis, 3Et was reacted with 20.0 equiv ArCl, giving a rate constant comparable to the phenyl studies (kobs > 5 min−1). Knowing that 3Et is reactive towards electrophile, we then wanted to determine if it was a catalytically relevant species. Following the protocol from Fox and co-workers (see SI), EtMgBr was added in portions every 30 s to a solution of ArEt, FeCl2 (1 mol %) and TMEDA (10 mol%) which resulted in the formation of 85% cross-coupled product. An aliquot for Mössbauer spectroscopy was taken approximately halfway through catalysis using 57FeCl2 (1 mol %) (t = 20 min) with corresponding chemical quenched GC analysis to get % of cross-coupled product formation (Figure 6). The speciation 20 min into the catalytic reaction shows iron speciation of the mono- & bis-ethylated TMEDA-iron species (71% 2Et and 29% 3Et), at which 45% cross-coupled product is formed, showing that TMEDA-bound iron species are present during the catalytic reaction despite excess nucleophile being present. Cross-coupling with EtMgBr likely follows a similar reaction pathway to that with PhMgBr, via radical generation upon reaction of 3Ph with electrophile followed by recombination with either 2Ph or 3Ph, though the recombination with 3Et is more selective than it was for 3Ph. Lastly, the observed reactivity of 3Ph with electrophile contrasts the lack of reactivity previously reported for the benzyl analog of this complex, further demonstrating the role of nucleophile in modulating the role of TMEDA-bound iron species in the primary catalytic cycle.
Figure 6.
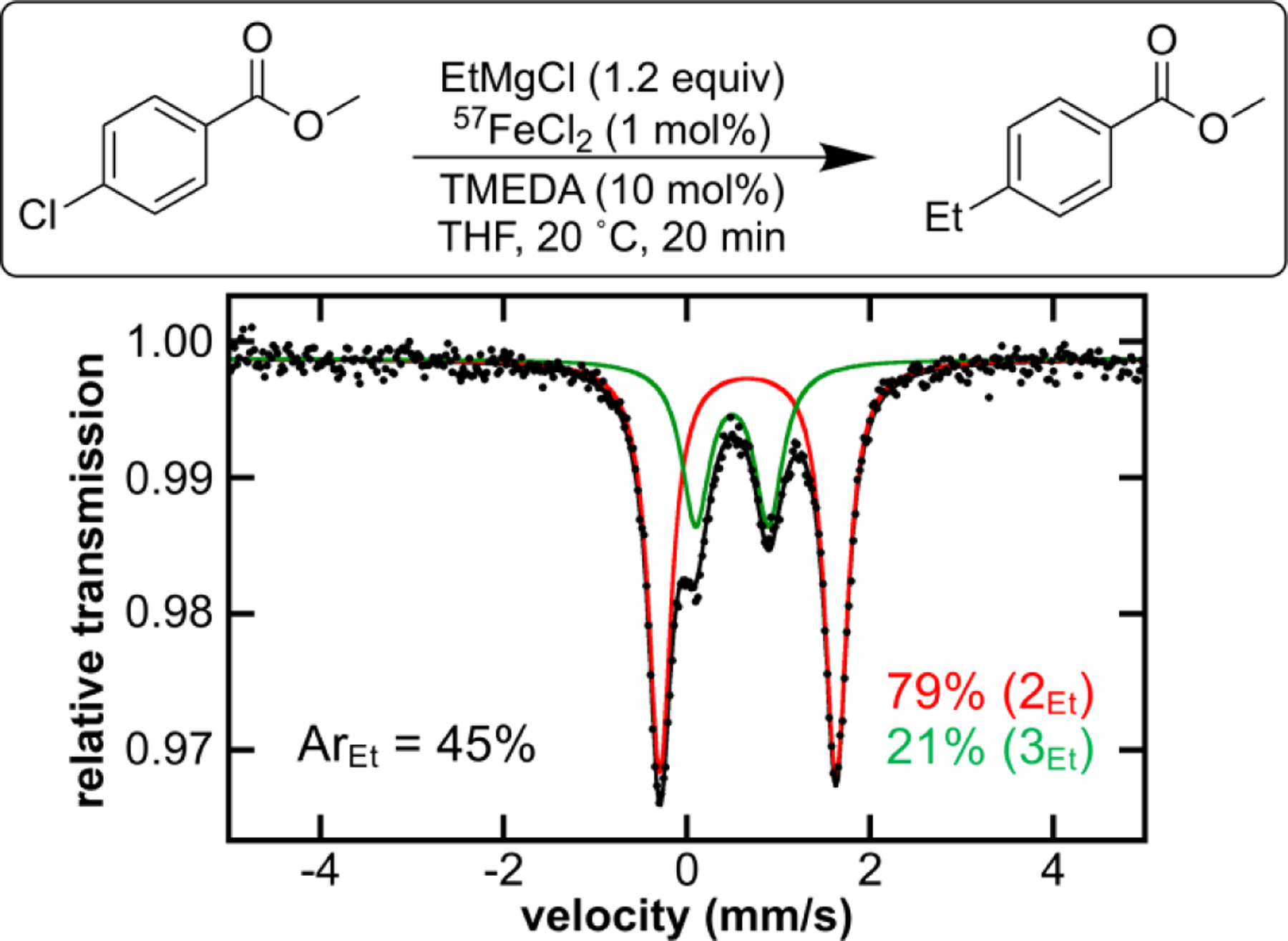
Freeze-trapped 80 K Mössbauer spectrum of Fox catalysis at 20 min (ca. halfway through catalysis); raw data (black dots), total fit (black trace), individual fit components are shown.
Conclusion
While TMEDA is a highly effective additive for iron-catalyzed cross-couplings, the molecular-level role of TMEDA in promoting effective catalysis remains ambiguous, especially as a function of the nucleophiles and reaction protocols most commonly employed in catalysis. While previous studies by Bedford and co-workers demonstrated the importance of homoleptic iron complexes with mesityl and benzyl Grignard reagents (with TMEDA proposed to bind only to off-cycle species), the current study demonstrates that for less sterically bulky aryls such as phenyl that TMEDA-bound iron intermediates play a central role in the primary catalytic cycle for cross-coupling. The present study has addressed a central ambiguity in the literature regarding the role of TMEDA in iron-catalyzed C-C cross-coupling by identifying, isolating, and testing the reactivity of well-defined TMEDA-iron complexes. Essential insight into the in situ iron speciation was obtained through freeze-quenched 57Fe Mössbauer spectroscopy. It was observed that TMEDA stabilizes monomeric iron species as a ligand with less sterically bulky aryls (i.e. phenyl). By correlating 57Fe Mössbauer and chemical-quenched GC analysis of organic products, stoichiometric reaction studies with electrophile identified the bis-phenylated TMEDA-iron complex of 3Ph as a radical initiator in reactions with excess electrophile, while selective generation of C-C product was determined to proceed through the mono-phenylated complex, 2Ph. Together these results also provide insight into why slow addition is so important in these reactions: there needs to be just enough 3Ph iron to initiate catalysis while maintaining most of the iron in a lower transmetallated state (2Ph) to favor selective product formation. Beyond phenyl, analogous results were found upon extension of these studies to ethyl Grignard reagent, where TMEDA-ligated species were also found to be involved in the primary catalytic cycle.
A possible iron(II)/iron(III)/iron(I) reaction cycle for the TMEDA-iron reaction manifold consistent with these experimental results, as well as recent mechanistic studies of iron cross-coupling reactions, is shown in Scheme 2.[36] These experimental results of the present study are consistent with an iron(II)/iron(III)/iron(I) reaction cycle for the TMEDA-iron reaction related to recent proposals for iron cross-coupling reactions (Scheme 2).[36] Catalysis is initiated by radical generation from reaction of electrophile with 3Ph. This organoradical can then recombine with 2Ph to generate a five-coordinate iron(III) intermediate which subsequently undergoes reductive elimination to generate cross-coupled product and an TMEDA-iron(I) species. This iron(I) complex can then react with an additional equivalent of electrophile to generate additional organoradical to react with 2Ph. Note that the iron(III) species formed upon reaction of 3Ph with electrophile can re-enter the catalytic cycle upon reductive elimination to form the iron(I) intermediate and biphenyl (a side product observed in catalysis). A ligand comproportionation reaction of this iron(III) species and iron(I) intermediate also could potentially reform 2Ph, though less favored due to low concentrations of these intermediates during catalysis. This represents a distinct mechanism of catalysis from those previously proposed for TMEDA-iron cross-coupling. Lastly, while not observed in the present study, if very minor amounts of potentially reactive homoleptic iron-phenyl species are formed, they could contribute to the initiation of catalysis in addition to 3Ph. However, from the reactivity studies presented herein, a TMEDA-bound iron species would still be responsible for the generation of C-C product upon subsequent reaction with the generated organoradical.
Scheme 2.

Proposed mechanism of iron-catalyzed C(sp2)-C(sp3) cross-coupling using TMEDA and phenyl Grignard reagent, main cycle is guided by bolded blue arrows.
Based upon the current studies and resulting mechanistic findings, the poor catalytic performance of MesMgBr in cross-couplings using iron and TMEDA can now be directly attributed to the absence on TMEDA-iron(II)-mesityl species in situ during catalysis, where radical recombination with [Fe(Mes)3]- is likely less effective and selective. It should be noted that a similar observation has previously been made in iron-SciOPP cross-coupling with MesMgBr where slow nucleophile addition was found to be essential to avoiding the formation of [Fe(Mes)3]- to maximize cross-coupled product yields.[24] Lastly, while benzyl Grignard reagents was previously shown to be effective for iron-TMEDA cross-coupling with homoleptic species accessible synthetically, the catalytic reaction were run at very low temperature (- 40 ˚C) and no in situ speciation measurements were reported.[19] In fact, 80 K 57Fe Mössbauer studies of the iron speciation during low temperature catalysis in this system support the formation of significant monobenzyl-TMEDA-iron(II) complex as part of a broader mixture of iron species (see Figure S12), consistent with the improved cross-coupling yields with this nucleophile compared to MesMgBr.
Overall, this study identifies a TMEDA-bound adduct reaction manifold which enables the selective formation of C(sp2)-C(sp3) cross-coupled product in iron-catalyzed cross-coupling. The results address a longstanding and debated aspect of iron catalysis and provide a broader mechanistic framework from which new amine additives can be developed for iron catalysis. Furthermore, this study highlights the limitations that can exist from utilizing model nucleophiles in mechanistic studies as well as investigations that do not fully reproduce key aspects of catalytic protocols such as slow nucleophile addition. These results critically expand our understanding of the role of reaction additives and iron speciation in achieving effective and selective cross-coupling reactions with iron, providing a fundamental platform for the development of bespoke ligand, additive, and reaction methods for iron cross-coupling.
Supplementary Material
Acknowledgements
This work was supported by a grant from the National Institutes of Health (R01GM111480 to M.L.N.). The National Science Foundation is gratefully acknowledged for support for the acquisition of an X-ray diffractometer (CHE-1725028).
Footnotes
Supporting information for this article is given via a link at the end of the document.
Institute and/or researcher Twitter usernames: @URochesterChem, @TheNeidigLab
Contributor Information
Nikki J. Bakas, Department of Chemistry, B31 Hutchison Hall, University of Rochester, 120 Trustee Rd, Rochester, NY, 14627 (USA).
Jeffrey D. Sears, Department of Chemistry, B31 Hutchison Hall, University of Rochester, 120 Trustee Rd, Rochester, NY, 14627 (USA).
William W. Brennessel, Department of Chemistry, B31 Hutchison Hall, University of Rochester, 120 Trustee Rd, Rochester, NY, 14627 (USA)
Michael L. Neidig, Department of Chemistry, B31 Hutchison Hall, University of Rochester, 120 Trustee Rd, Rochester, NY, 14627 (USA)
References
- [1].Nakamura M, Matsuo K, Inoue T, Nakamura E, Org. Lett 2003, 5, 1373–1375. [DOI] [PubMed] [Google Scholar]
- [2].Nakamura M, Matsuo K, Ito S, Nakamura E, J. Am. Chem. Soc 2004, 126, 3686–3687. [DOI] [PubMed] [Google Scholar]
- [3].Cahiez G, Duplais C, Moyeux A, Org. Lett 2007, 9, 3253–3254. [DOI] [PubMed] [Google Scholar]
- [4].Guérinot A, Reymond S, Cossy J, Angew. Chemie Int. Ed 2007, 46, 6521–6524. [DOI] [PubMed] [Google Scholar]
- [5].Fürstner A, Martin R, Krause H, Seidel G, Goddard R, Lehmann CW, J. Am. Chem. Soc 2008, 130, 8773–8787. [DOI] [PubMed] [Google Scholar]
- [6].Casitas A, Krause H, Lutz S, Goddard R, Bill E, Fürstner A, Organometallics 2018, 37, 729–739. [Google Scholar]
- [7].Cahiez G, Habiak V, Duplais C, Moyeux A, Angew. Chemie Int. Ed 2007, 46, 4364–4366. [DOI] [PubMed] [Google Scholar]
- [8].Nagano T, Hayashi T, Org. Lett 2004, 6, 1297–1299. [DOI] [PubMed] [Google Scholar]
- [9].Bedford RB, Bruce DW, Frost RM, Hird M, Chem. Commun 2005, 4161–4163. [DOI] [PubMed]
- [10].Berardi F, Abate C, Ferorelli S, de Robertis AF, Leopoldo M, Colabufo NA, Niso M, Perrone R, J. Med. Chem 2008, 51, 7523–7531. [DOI] [PubMed] [Google Scholar]
- [11].Denmark SE, Cresswell AJ, J. Org. Chem 2013, 78, 12593–12628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rushworth PJ, Hulcoop DG, Fox DJ, J. Org. Chem 2013, 78, 9517–9521. [DOI] [PubMed] [Google Scholar]
- [13].Parmar D, Henkel L, Dib J, Rueping M, Chem. Commun 2015, 51, 2111–2113. [DOI] [PubMed] [Google Scholar]
- [14].Guérinot A, Cossy J, in Top. Curr. Chem, 2016, p. 49. [DOI] [PubMed]
- [15].Fürstner A, ACS Cent. Sci 2016, 2, 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jia Z, Liu Q, Peng X-S, Wong HNC, Nat. Commun 2016, 7, 10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Miao W, Zhao Y, Ni C, Gao B, Zhang W, Hu J, J. Am. Chem. Soc 2018, 140, 880–883. [DOI] [PubMed] [Google Scholar]
- [18].An L, Xiao Y-L, Zhang S, Zhang X, Angew. Chemie Int. Ed 2018, 57, 6921–6925. [DOI] [PubMed] [Google Scholar]
- [19].Bedford RB, Brenner PB, Carter E, Cogswell PM, Haddow MF, Harvey JN, Murphy DM, Nunn J, Woodall CH, Angew. Chemie - Int. Ed 2014, 53, 1804–1808. [DOI] [PubMed] [Google Scholar]
- [20].Bedford RB, Acc. Chem. Res 2015, 48, 1485–1493. [DOI] [PubMed] [Google Scholar]
- [21].Noda D, Sunada Y, Hatakeyama T, Nakamura M, Nagashima H, J. Am. Chem. Soc 2009, 131, 6078–6079. [DOI] [PubMed] [Google Scholar]
- [22].Bedford RB, Betham M, Bruce DW, Danopoulos AA, Frost RM, Hird M, J. Org. Chem 2006, 71, 1104–1110. [DOI] [PubMed] [Google Scholar]
- [23].Parchomyk T, Demeshko S, Meyer F, Koszinowski K, J. Am. Chem. Soc 2018, 140, 9709–9720. [DOI] [PubMed] [Google Scholar]
- [24].Daifuku SL, Kneebone JL, Snyder BER, Neidig ML, J. Am. Chem. Soc 2015, 137, 11432–11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Daifuku SL, Al-Afyouni MH, Snyder BER, Kneebone JL, Neidig ML, J. Am. Chem. Soc 2014, 136, 9132–9143. [DOI] [PubMed] [Google Scholar]
- [26].Bakas NJ, Neidig ML, ACS Catal 2021, 11, 8493–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]. Deposition Numbers 2094072, 2094073, and 2094074 contain the supplementary crystallographic data for this paper. These data are provided for free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- [28].Parchomyk T, Koszinowski K, Chem. - A Eur. J 2016, 22, 15609–15613. [DOI] [PubMed] [Google Scholar]
- [29].Trost BM, Tanoury GJ, Lautens M, Chan C, MacPherson DT, J. Am. Chem. Soc 1994, 116, 4255–4267. [Google Scholar]
- [30].Bazhenova TA, Lobkovskaya RM, Shibaeva RP, Shilov AE, Shilova AK, Gruselle M, Leny G, Tchoubar B, J. Organomet. Chem 1983, 244, 265–272. [Google Scholar]
- [31].Sears JD, Neate PGN, Neidig ML, J. Am. Chem. Soc 2018, 140, 11872–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sears JD, Muñoz SB, Daifuku SL, Shaps AA, Carpenter SH, Brennessel WW, Neidig ML, Angew. Chemie - Int. Ed 2019, 58, 2769–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Davies SC, Hughes DL, Jeffery Leigh G, Roger Sanders J, de Souza JS, J. Chem. Soc. Dalt. Trans 1997, 1981–1988.
- [34].Kneebone JL, Brennessel WW, Neidig ML, J. Am. Chem. Soc 2017, 139, 6988–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Neate PGN, Greenhalgh M, Brennessel W, Thomas SP, Neidig M, Angew. Chemie Int. Ed 2020, 59, 17070–17076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu L, Aguilera MC, Lee W, Youshaw CR, Neidig ML, Gutierrez O, Science 2021, 374, 432–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.