

Abstract
Four-coordinate transition metal complexes can adopt a diverse array of coordination geometries, with square planar and tetrahedral coordination being the most prevalent. Previously, we reported the synthesis of a trinuclear Fe(II) complex, Fe3TPM2, supported by a three-fold symmetric 2-pyridylpyrrolide ligand (i.e., tris(5-(pyridin-2-yl)-1H-pyrrol-2-yl)methane), that featured a rare cis-divacant octahedral (CDO) geometry at each Fe(II) center. Here, a series of truncated 2-pyridylpyrrolide ligands is described that support mono- and binuclear Fe(II) complexes that also exhibit CDO geometries. Metallation of tetradentate ligand bis(5-(pyridin-2-yl)-1H-pyrrol-2-yl)methane (H2BPM) in THF results in a binuclear complex Fe2(BPM)2(THF)2 in which both Fe(II) ions are octahedrally coordinated. The coordinated THF solvent ligands are labile: THF dissociation leads to Fe2(BPM)2, which features five-coordinate Fe(II) ions. The Fe–Fe distance in these binuclear complexes can be elongated by ligand methylation. Metalation of bis(5-(6-methylpyridin-2-yl)-1H-pyrrol-2-yl)methane (H2BPMMe) in THF leads to the formation of four-coordinate, CDO Fe(II) centers in Fe(BPMMe)2. Further ligand truncation affords bidentate ligands 2-(1H-pyrrol-2-yl)pyridine (PyrPyrrH) and 2-methyl-6-(1H-pyrrol-2-yl)pyridine (PyrMePyrrH). Metalation of these ligands in THF affords six-coordinate complexes Fe(PyrPyrr)2(THF)2 and Fe(PyrMePyrr)2(THF)2. Dissociation of labile solvent ligands provides access to four-coordinate Fe(II) complexes. Ligand disproportionation at Fe(PyrPyrr)2 results in the formation of Fe(PyrPyrr)3 and Fe(0). Ligand methylation suppresses this disproportionation and enables isolation of Fe(PyrMePyrr)2, which is rigorously CDO. Complete ligand truncation, by separating the 2-pyridylpyrrolide ligands into the constituent monodentate pyridyl and pyrrolide donors, affords Fe(Pyr)2(Pyrr)2 in which the Fe(II) is tetrahedrally coordinated. Computational analysis indicates that the potential energy surface that dictates the coordination geometry in this family of four-coordinate complexes is fairly flat in the vicinity of CDO coordination. These synthetic studies provide the structural basis to explore the implications of CDO geometry on Fe-catalyzed reactions.
Graphical Abstract

INTRODUCTION
Exerting synthetic control over the coordination geometry of transition metal complexes is critical to eliciting specific physical and chemical properties and thus synthetic efforts to access unique coordination geometries provide opportunities to develop new reactivity patterns.1–3 Four-coordinate complexes are of particular interest due to the rich diversity of geometries that are accessible (Figure 1a). Square planar and tetrahedral coordination modes are the most commonly encountered geometries for four coordinate species, and ligand design features that differentiate these geometries have been the topic of extensive investigation.4–6 In general, tetrahedral coordination is sterically preferred and is thus favored for large ligands and small transition metal ions. Square planar coordination is sterically disfavored but can afford significant ligand field stabilization energy. Beyond tetrahedral and square planar geometries, less common seesaw geometries are also available to four-coordinate ions. Cis-divacant octahedral (CDO) and monovacant trigonal bipyramidal (MTB) are see-saw geometries that differ in the bond angle (θ) between the equatorial ligands (CDO = 90° and MTB = 120°). While these geometries are potentially attractive platforms for application in catalysis because they feature sterically accessible coordination sites poised to engage substrates, they are encountered much less often than either tetrahedral or square planar geometries.
Figure 1.

(a) Four-coordinate metal ions most commonly adopt tetrahedral or square planar coordination geometries. Distortions of these geometries gives rise to see-saw structures with limiting cis-divacant octahedral (CDO) or monovacant trigonal bipyramidyl (MTB) geometries. The τ4 parameter utilizes experimentally obtained bond angles to describe the geometry of four-coordination ions. (b) Selected four-coordinate Fe(II) complexes that feature see-saw coordination with the crystallographically derived τ4 values.
Perfect adherence to the idealized structures illustrated in Figure 1a is uncommon and in the context of heteroleptic coordination complexes is not possible. The extent of structural distortion from the idealized geometries can be hard to accurately describe, and thus a number of algorithms have been developed that aim to quantify the nature and extent of geometrical distortion.7–12 Here we utilize two approaches to quantifying the geometries of four-coordinate complexes that are commonly encountered: τ4 values13 and continuous shape analysis.14–16 The τ4 metric is derived from observed metal–ligand (M–L) bond angles to describe the coordination geometry of four-coordinate complexes;13 τ4 values that characterize limiting four-coordinate geometries are listed in Figure 1a. Similarly, continuous shape analysis has been advanced as an approach to evaluate the structural deviations of observed coordination geometries from idealized polyhedra.14–16
In the context of Fe(II) chemistry, a small family of four-coordinate complexes that display highly distorted seesaw geometries have been reported (Figure 1b). Pincer-supported Fe(II) complex 1 displays approximately CDO geometry and participates in two-electron oxidative chemistry upon treatment with adamantyl azide to afford the corresponding Fe(IV) imide complex.17 The observation of CDO coordination in complex 1 was ascribed to minimization of steric interactions that would be encountered in a potential square planar complex. Similarly, trans-chelating bis-alkoxide complex 2 features an Fe(II) center that displays approximately CDO coordination.18 Alkoxide-supported complex 3 also displays see-saw geometry at the Fe(II) center, which is best described as MTB, although this geometry likely arises due to the combination of bridging chloride ligands and the presence of templating Li+ ions.19
In 2020 we reported the synthesis and characterization of a trinuclear Fe(II) complex, Fe3TPM2, assembled from Fe(II) and the three-fold symmetric ligand tris(5-(pyridin-2-yl)-1H-pyrrol-2-yl)methane (H3TPM, Figure 2).20 Each of the Fe(II) ions in this complex displayed close-to-idealized CDO geometry (τ4 = 0.60). Attempts to leverage trinuclear complex Fe3TPM2, which features a central molecular void in which each of the ions presents two vacant coordination sites, for small molecule binding or activation were unsuccessful. To both evaluate the structural origins of the CDO geometry and access more sterically accessible CDO transition metal sites, we have pursued synthetic studies of a series of dimensionally reduced ligands, namely bis(5-(pyridin-2-yl)-1H-pyrrol-2-yl)methane (H2BPM), 2-(1H-pyrrol-2-yl)pyridine (PyrPyrrH) and methylated analogues thereof. Here, we report a family of mono- and binuclear complexes of Fe(II) featuring CDO coordination geometry. We demonstrate the critical role of ligand structure and solvation in accessing the four-coordinate CDO complexes. Further, we demonstrate that the potential energy surface that gives rise to the CDO coordination geometry is fairly flat, which suggests that small structural perturbations can manifest as significant structural changes about this geometry.
Figure 2.
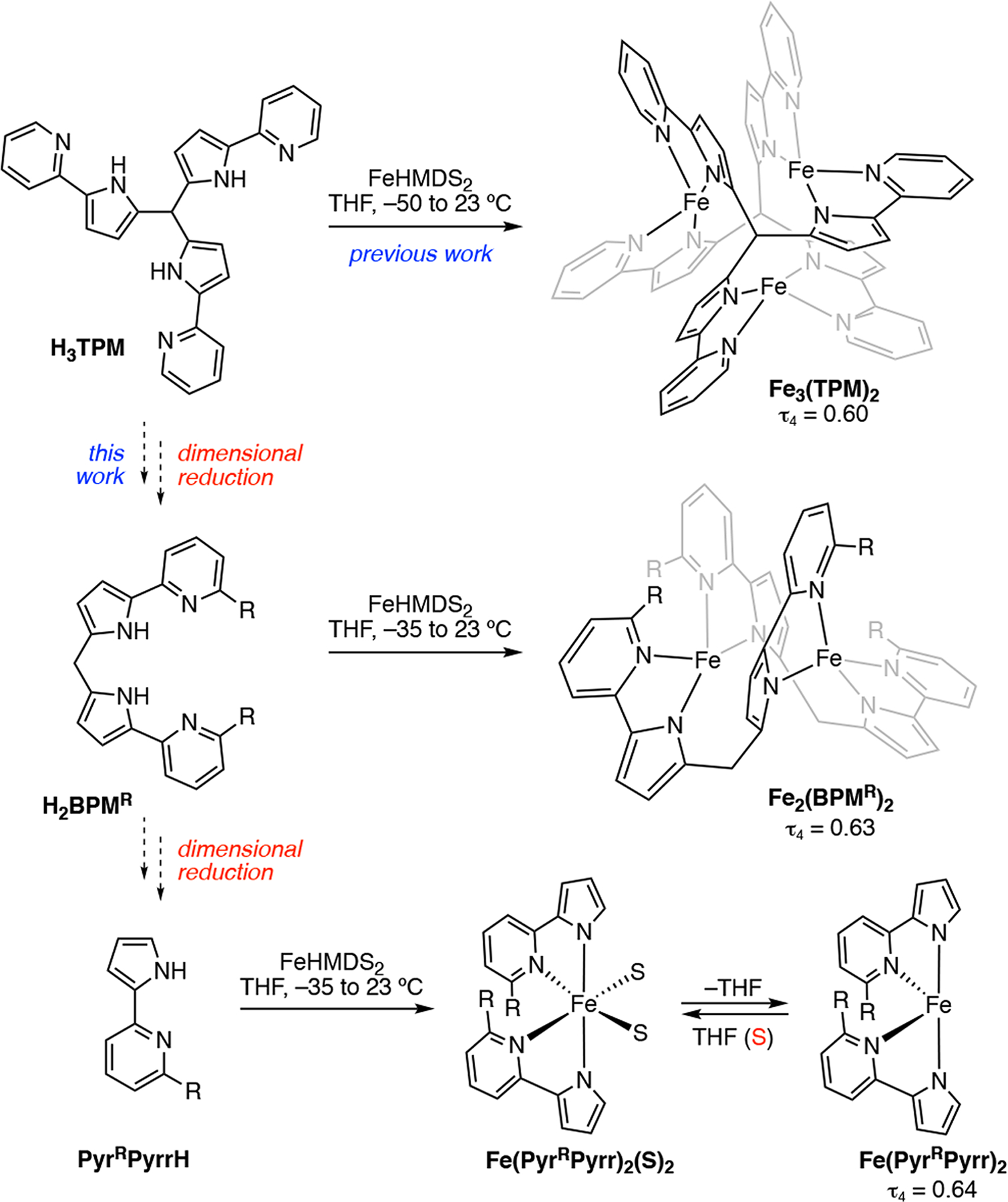
Previously, trinuclear complex Fe3TPM2 was prepared from three-fold symmetric ligand H3TPM and features three CDO Fe(II) centers. In this work, dimensional reduction from H3TPM to two-fold symmetric H2BPM and to 2-(pyridin-2-yl)pyrrole PyrPyrrH was carried out and CDO coordination was maintained across this simpler ligand framework.
RESULTS
Ligand Synthesis.
In order to access a dimensionally reduced family of Fe(II) complexes, we prepared a series of truncated ligands in which pyridylpyrrole arms were removed from H3TPM. Bidentate ligands 2-(1H-pyrrol-2-yl)pyridine (PyrPyrrH) and 2-methyl-6-(1H-pyrrol-2-yl)pyridine (PyrMePyrrH) were prepared by Pd-catalyzed cross coupling of zinc pyrrolide with 2-bromopyridine and 6-methyl-2-bromopyridine, respectively (Figure 3).21 Condensation of each of these compounds with 0.5 equivalents of paraformaldehyde in the presence of InCl3 yielded tetradentate ligands 2-(1H-pyrrol-2-yl)pyridine (H2BPM) and bis(5-(6-methylpyridin-2-yl)-1H-pyrrol-2-yl)methane (H2BPMMe). Each of the ligands was characterized by 1H and 13C NMR spectroscopies as well as high-resolution mass spectrometry (Figure S1–S5).
Figure 3.

Synthesis of 2-(pyridin-2-yl)pyrrole ligands. Bidentate ligands PyrRPyrrH were prepared via Pd-catalyzed coupling of Zn pyrrolide with 2-bromopyridines. Tetradentate ligands H2BPMR were prepared by condensation of PyrRPyrrH with paraformaldehyde in the presence of InCl3.
Synthesis of Binuclear Fe(II) Complexes.
Metallation of H2BPM was accomplished by treating a THF solution of the ligand with FeHMDS2 at −35 °C and then allowing the reaction mixture to slowly warm to 23 °C (Figure 4). Fe2(BPM)2(THF)2 was isolated as an orange crystalline solid following vapor diffusion of pentane into the reaction mixture at −35 °C. The 1H NMR spectrum of Fe2(BPM)2(THF)2 displays eight paramagnetically shifted 1H NMR peaks from 8–135 ppm (Figures S6–S7). Consistent with the dark orange color of the sample, the UV-vis spectrum measured in THF displays a broad absorption at 480 nm (ε = 1.0 × 103 M−1cm−1; Figure S8). High-resolution mass spectrometry was consistent with an Fe2L2 aggregate (HRMS ESI+: calcd for [Fe2(BPM)2]+ 708.1135; expt. 708.1115). The magnetic susceptibility was evaluated by Evans method, which provided μB = 6.9, which is consistent with an S = 3 complex. The 57Fe Mössbauer spectrum obtained for a sample of this compound (Figure S9) could not be adequately simulated with less than three quadrupole doublets. Due to the similarity of the isomer shift and quadrupole coupling of the three doublets, we speculate that the observed spectral features arise from closely related Fe(II) complexes that may be generated by sequential solvent dissociation equilibria (i.e., to generate Fe2(BPM)2THF and Fe2(BPM)2) and associated ligand rearrangements (vide infra).22 Given the integrations attributable to the measured doublets, the two Fe centers in Fe2(BPM)2(THF)2 appear to be indifferentiable by Mössbauer spectroscopy.
Figure 4.

Top: Synthesis of binuclear Fe(II) complexes. Metallation of H2BPM with Fe(HMDS)2 affords Fe2(BPM)2(THF)2 in which both Fe(II) centers are six-coordinate. Recrystallization from toluene affords Fe2(BPM)2 in which the THF ligands have dissociated and both Fe(II) centers are five-coordinate. Metallation of H2BPMMe with Fe(HMDS)2 affords Fe2(BPMMe)2(THF)2 in which both Fe(II) centers are four-coordinate. Ligands are color-coded gray and blue to highlight the coordination mode of each ligand. Bottom: Displacement ellipsoid plots (50% probability) of the prepared binuclear Fe(II) complexes. Selected bond distances (Å): Fe2(BPM)2 Fe1–Fe1’ = 2.963(2), Fe1–N3’ = 2.031(4), and Fe1–N3 = 2.381(6); Fe2(BPM)2(THF)2 Fe1–Fe2 = 3.1551(6), Fe1–N7 = 2.105(4), and Fe2–N7 = 2.496(3); Fe2(BPMMe)2 Fe1–Fe2 = 3.6343(4), Fe1–N2 = 1.992(2), Fe1–N3 = 3.060(2).
Crystallization of Fe2(BPM)2(THF)2 by vapor diffusion of pentane into a THF solution at −35 °C afforded single crystals of Fe2(BPM)2(THF)2 (Figure 4). Fe2(BPM)2(THF)2 crystallizes in the triclinic space group P–1 with two whole molecules and three THF molecules residing in the crystallographically independent unit. The Fe(II) centers in Fe2(BPM)2(THF)2 are both six-coordinate. Fe1 is ligated by pyridylpyrrolide moieties of two different ligands, such that the pyridyl donors are mutually trans, and by two THF donors. The pyrrolide donors to Fe1 are σ-bound and the Fe1–Npyrr distance is 2.118(2) Å (Table 1; i.e., average Fe–N of Fe1–N2 and Fe1–N7), which is consistent with the Fe–N distance expected of a high-spin Fe(II) ion.23, 24 Fe2 is also supported by pyridylpyrrolide donors of two different ligands, although unlike Fe1, the pyridyl donors to Fe2 are mutually cis. The remaining two coordination sites appear to be occupied by long, π-interactions with the pyrrolide ligands that are σ-bound to Fe1; Fe2–Npyrr = 2.448(1) Å (i.e., average of Fe2–N2 and Fe2–N7).25 The Fe–Fe distance in Fe2(BPM)2(THF)2 is 3.1551(6) Å. Based on this distance, the formal shortness ratio (fsr),26, 27 which is the ratio of the interatomic distance divided by the sum of the covalent radii,28 is 1.35. Because the value is significantly greater than 1 we do not formulate any Fe–Fe bonding in Fe2(BPM)2(THF)2.
Table 1.
Summary of selected bond lengths (Å) for binuclear Fe(II) complexes.
| Fe2(BPM)2(THF)2 | Fe2(BPM)2 | Fe2(BPMMe)2 | |
|---|---|---|---|
| Fe•••Fe | 3.1551(6) | 2.963(2) | 3.6343(4) |
| Fe1–Npyrra | 2.118(2) | 2.031(4) | 1.9972(7) |
| Fe2–Npyrrb | 2.029(1) | ||
| Fe–Npyrr,π | 2.448(1)c | 2.381(6)d | --- |
| Fe1–Npyre | 2.202(1) | 2.176(4) | 2.1304(8) |
| Fe2–Npyrf | 2.149(2) |
Average bond distance of Fe1–N2 and Fe1–N7 (Fe1–N3’).
Average bond distance of Fe2–N3 (Fe1’–N3) and Fe2–N6 (Fe1’–N2’).
Average bond distance of Fe2–N2 and Fe2–N7.
Average bond distance of Fe1–N3’ and Fe1’–N3.
Average bond distance of Fe1–N1 and Fe1–N8 (Fe1–N4’).
Average bond distance of Fe2–N4 (Fe1’–N4) and Fe2–N5 (Fe1’–N1’).
The THF ligands of Fe2(BPM)2(THF)2 are labile. Recrystallization of Fe2(BPM)2(THF)2 from toluene afforded a single crystal of Fe2(BPM)2, in which the binuclear core in conserved but the previously coordinated THF ligands are no longer present (Figure 4). Loss of coordinated solvent is accompanied by ligand rearrangement: The coordination sphere is comprised of pyridylpyrrolide moieties from two different ligand molecules with one of the pyrrolide donors adopting a bridging position. The Fe centers in Fe2(BPM)2 are symmetry equivalent and five-coordinate. Both the τ5 parameter of 0.15629,30 and continuous shape measurement (CShM) calculations,14–16 are consistent with distorted square pyramidal coordination of both Fe centers (τ5 for an idealized square pyramid is 0).13, 29 The Fe–Fe separation in Fe2(BPM)2 (2.963(2) Å) is slightly shorter than that observed in Fe2(BPM)2(THF)2, but the formal shortness ratio (fsr = 1.27) is again inconsistent with meaningful Fe–Fe bonding. Consistent with symmetry equivalent Fe centers, the 57Fe Mössbauer spectrum of Fe2(BPM)2 displays a single quadrupole doublet with isomer shift of δ = 0.962 and a quadrupole splitting of |ΔEQ| = 3.054 (Figure 5a). In addition, UV-vis spectroscopy of Fe2(BPM)2 measured in toluene shows absorption at 493 and 525 nm (ε = 1.0 × 103 and 8.1 × 102 M−1cm−1, respectively), which reflects a red shift of d-d transition band upon removal of coordinated solvent. The removal of THF is reversible: Recrystallization of Fe2(BPM)2 from THF affords Fe2(BPM)2(THF)2.
Figure 5.

57Fe Mössbauer spectra of binuclear Fe(II) complexes measured at 5 K. (a) Fe2(BPM)2 displays a single quadrupole doublet with δ = 0.962, |ΔEQ| = 3.054, γ = 0.314. (b) Fe2(BPMMe)2 displays a single quadrupole doublet with δ = 0.868, |ΔEQ| = 2.339, γ = 0.373. δ, isomer shift; |ΔEQ|, quadrupole splitting; γ, linewidth.
We hypothesized that ligand methylation (i.e., PyrMePyrrH) would introduce interligand steric interactions that would result in Fe–Fe elongation that could eliminate the Fe–Npyrr π-interactions present in Fe2(BPM)2 and give rise to four-coordinate Fe(II) ions. We validated this hypothesis by preparing Fe2(BPMMe)2 from H2BPMMe and Fe(HMDS)2 (Figure 4). Crystallization via vapor diffusion of pentane into the THF solution yielded a dark orange crystalline solid which displayed 7 paramagenetically shifted 1H NMR peaks (Figure S10–S11); Evans method provides μB = 6.8 which is consistent with an S = 3 complex. The UV-vis spectrum measured in THF displayed absorptions at 492 and 523 nm (ε = 1.1 × 103 and 9.1 × 102 M−1cm−1, respectively; Figure S12), which are similar to those observed for Fe2(BPM)2 in toluene. High-resolution mass spectrometry was consistent with binuclear iron compound Fe2(BPMMe)2 (HRMS-ESI+ [M]+ calcd. 764.1756; expt. 764.1736; [M+H]+ calcd 765.1835; expt. 765.1807). The 57Fe Mössbauer spectrum of Fe2(BPMMe)2 displays a single quadrupole doublet with δ = 0.868 and |ΔEQ| = 2.339 (Figure 5b). These parameters are similar to those obtained for trinuclear complex Fe3(TPM)2 (δ = 0.835 and |ΔEQ| = 2.171) and suggested the two compounds may feature similar CDO coordination at the Fe(II) sites.
A single crystal of Fe2(BPMMe)2 was obtained by a vapor diffusion of pentane into the THF solution.31 Refinement of X-ray diffraction data collected at 110 K reveals a binuclear complex in which each of the Fe centers is four-coordinate. The Fe–Fe distance is significantly elongated (3.6342(4) Å, fsr = 1.56) relative to the unmethylated analogue Fe2(BPM)2 (2.963(2) Å). The Fe–Fe elongation results in the removal of the π-interactions that are observed in Fe2(BPM)2; in Fe2(BPMMe)2 the Fe–N distances are 3.064(1) Å (average of Fe1–N3 and Fe2–N7) and 4.383(1) Å (average of Fe1–N6 and Fe2–N2) Å (Table 1). CShM analysis indicate each Fe(II) center adopts a slightly distorted CDO geometry (Table 2), and τ4 values of 0.62 and 0.63 for each metal center are also consistent with CDO geometry (τ4 = 0.640 for an idealized CDO geometry).13
Table 2.
CShM for four-coordinate iron(II) complexes using SHAPE program, which calculates deviations of a set of atomic positions relative to the vertices of idealized polygons or polyhedra on the definition of minimal distortion paths and generalized interconversion coordinates.14, 33, 34
| Idealized Geometry | Calculated Deviation | |||
|---|---|---|---|---|
| Fe2(BPMMe)2 | Fe(PyrMePyrr)2 | Fe(Pyr)2(Pyrr)2 | ||
| Fe1 | Fe2 | |||
| Sq. Planar | 23.132 | 25.339 | 23.982 | 31.655 |
| Tetrahedral | 6.398 | 6.204 | 5.958 | 0.356 |
| CDO | 5.054 | 5.204 | 5.215 | 6.992 |
| MTB | 6.802 | 6.587 | 6.733 | 2.804 |
| τ4 | 0.621 | 0.630 | 0.640 | 0.902 |
Synthesis of Mononuclear Fe(II) Complexes.
Mononuclear complex Fe(PyrPyrr)2(THF)2 was obtained as a yellow crystalline solid by metallation of PyrPyrrH with Fe(HMDS)2 in THF following by vapor diffusion of pentane into the crude reaction mixture (Figure 6). The 1H NMR spectrum of Fe(PyrPyrr)2(THF)2 displays six paramagnetically shifted 1H NMR signals from 28 to 140 ppm in d8-THF (Figures S13–S14). The UV-vis spectrum of Fe(PyrPyrr)2(THF)2 measured in THF displays a broad tailing UV-vis absorption that ends at ~530 nm and no distinct lower energy absorptions (Figure S15). Evans method affords a magnetic moment of 4.5 μB, which is consistent with an S = 2 spin state. The 57Fe Mössbauer spectrum displays a single quadrupole doublet with δ = 1.141 and |ΔEQ| = 2.450 (Figure 7a). A single crystal of Fe(PyrPyrr)2(THF)2 was obtained from a concentrated THF/pentane solution. Refinement of the single-crystal X-ray diffraction data provides the structure illustrated in Figure 6 (selected metrical parameters are summarized in Table 3). The octahedra coordination sphere of the Fe(II) center is composed of two pyridylpyrrolide ligands and two THF molecules. The Fe–Npyrr distances (avg. 2.089(2) Å) are consistent with an S = 2 complex.32
Figure 6.

Top: Synthesis of mononuclear Fe(II) complexes. Metallation of PyrPyrrH with Fe(HMDS)2 affords Fe(PyrPyrr)2(THF)2. Attempts to access the corresponding 4-coordinate complex Fe(PyrPyrr)2 resulted in disproportionation to Fe(PyrPyrr)3 and Fe(0). Metallation of PyrMePyrrH with Fe(HMDS)2 affords Fe(PyrMePyrr)2(THF)2. Recrystallization from toluene affords 4-coordinate complex Fe(PyrMePyrr)2, which features cis-divacant octahedral coordination at the Fe(II) center. Bottom: Displacement ellipsoid plots (50% probability) of the prepared binuclear Fe(II) complexes. Selected bond distances (Å): Fe(PyrPyrr)3 Fe1–N(1,3) = 1.905(2); Fe(PyrPyrr)2(THF)2 Fe1–N(1,3) = 2.089(2); Fe(PyrMePyrr)2(THF)2, Fe1–N(1,3) = 2.034(1); Fe(PyrMePyrr)2, Fe1–N(1,3) = 1.985(2).
Figure 7.

57Fe Mössbauer spectra of mononuclear Fe(II) complexes measured at 5 K. All compounds display a single quadrupole doublet with 100% area of following parameters. (a) Fe(PyrPyrr)2(THF)2: δ = 1.141, |ΔEQ| = 2.450, γ = 0.334. (b) Fe(PyrMePyrr)2(THF)2: δ = 1.020, |ΔEQ| = 3.20, γ = 0.443. (c) Fe(PyrMePyrr)2: δ = 0.807, | ΔEQ | = 3.427, γ = 0.455. (d) Fe(Pyr)2(Pyrr)2: δ = 0.839, |ΔEQ| = 2.606, γ = 0.300. δ, isomer shift; |ΔEQ|, quadrupole splitting; γ, linewidth.
Table 3.
Summary of selected metrical parameters for mononuclear Fe(II) complexes.
| Fe–Npyrr / Å | Fe–Npyr / Å | |
|---|---|---|
| Fe(PyrPyrr)2(THF)2 | 2.089(2) | 2.214(1) |
| Fe(PyrMePyrr)2(THF)2 | 2.034(1) | 2.222(1) |
| Fe(PyrMePyrr)2 | 1.985(1) | 2.113(1) |
| Fe(Pyr)2(Pyrr)2 | 1.990(2) | 2.101(2) |
| Fe(PyrPyrr)3 | 1.905(2) | 1.978(2) |
Dissolution of Fe(PyrPyrr)2(THF)2 in benzene or toluene provide 1H NMR and UV-vis spectra that are consistent with THF dissociation to afford Fe(PyrPyrr)2 (Figures S14 and S15). Attempts to crystallize Fe(PyrPyrr)2 have been unsuccessful. Over the course of ~24 hours, disproportionation afforded dark red crystals of Fe(III) complex Fe(PyrPyrr)3 and black amorphous powder (presumed to be Fe(0)); the spectral data of Fe(PyrPyrr)3 prepared by disproportionation of Fe(PyrPyrr)2(THF)2 are consistent with those obtained for a sample of Fe(PyrPyrr)3 that was independently synthesized from PyrPyrrH, FeCl3, and NaHMDS. Single crystals obtained following disproportionation of Fe(PyrPyrr)2(THF)2 and from independently prepared Fe(PyrPyrr)3 displayed identical unit cells. The Fe–Npyrr distances determined from the X-ray structure of Fe(PyrPyrr)3 (1.905(2) Å) are comparable to reported Fe(III) complexes.35 For solution-phase characterization of Fe(PyrPyrr)3, see Supporting Information (Figure S16–S18).
Metallation of PyrMePyrrH with Fe(HMDS)2 followed by vapor diffusion of pentane into the crude metalation reaction mixture at −35 °C afforded Fe(PyrMePyrr)2(THF)2 as a yellow solid. Solution-phase characterization of Fe(PyrMePyrr)2(THF)2 is similar to that obtained for Fe(PyrPyrr)2(THF)2: The 1H NMR spectrum displays 7 paramagnetic peaks from −25 to 64 ppm and the UV-vis spectrum displays weak absorptions at 470 and 496 nm (ε = 3.7 × 102 and 2.2 × 102 M−1cm−1, respectively; Figure S19–S21). Evans method (4.9 μB) is consistent with an S = 2 complex and the 57Fe Mössbauer spectrum displays a single quadrupole doublet with δ = 1.020 (|ΔEQ| = 3.20) (Figure 7b). The structure of Fe(PyrMePyrr)2(THF)2 was determined by refinement of X-ray diffraction data collected at 296 K and reveals an octahedral coordination sphere composed of two pyridylpyrrolide ligands and two coordinated THF ligands.36 Similar to Fe(PyrPyrr)2(THF)2, the Fe–Npyrr distances in Fe(PyrMePyrr)2(THF)2 (avg. 2.034(1) Å) are consistent with an S = 2 complex.
Dissolution of Fe(PyrMePyrr)2(THF)2 in toluene results in a red shift of the UV-vis spectral features from 470 and 496 nm to 485 and 511 nm. This observation mirrors a similar red shift observed in the conversion of Fe2(BPM)2(THF)2 (480 nm, br) to Fe2(BPM)2 (493 and 525 nm), suggesting that a similar ligand dissociation is observed with the methylated complex. A single crystal of Fe(PyrMePyrr)2 was obtained from a toluene/pentane solution and refinement of the single-crystal X-ray data indicates that Fe(PyrMePyrr)2 displays a nearly ideal CDO geometry (τ4 = 0.640). The 57Fe Mössbauer spectrum displays a single quadrupole doublet with δ = 0.807 (|ΔEQ| = 3.42) (Figure 7c).
Synthesis of Model Systems.
For comparison, we prepared an Fe(II) complex in which the pyrrolide and pyridyl ligands were separated (i.e., Fe(Pyr)2(Pyrr)2, Figure 8). Fe(Pyr)2(Pyrr)2 was prepared by combination of Fe(Pyr)2Cl2 and sodium pyrrolide in THF at 23 °C. The 1H NMR spectrum of Fe(Pyr)2(Pyrr)2 in d8-THF displays three broad peaks from 11 to 27 ppm and two sharper peaks at 6.64 and 6.00 ppm (Figures S22–S23). Similar to the other monomeric Fe(II) complexes described, Evans method provide a magnetic moment of 4.6 μB, consistent with an S = 2 complex. The 57Fe Mössbauer spectrum displays a single quadrupole doublet with δ = 0.839 (|ΔEQ| = 2.606) (Figure 7d). A single crystal was obtained by vapor diffusion of pentane into the THF solution at −35 °C. Refinement of X-ray diffraction data provides the solid-state structure illustrated in Figure 8, which reveals a tetrahedral coordination geometry for Fe(Pyr)2(Pyrr)2. CShM analysis (Table 2) and τ4 value (τ4 = 0.902) confirm tetrahedral coordination sphere of Fe(II) center.
Figure 8.

Synthesis of Fe(Pyr)2(Pyrr)2. Displacement ellipsoid plot drawn at 50% probability illustrates the tetrahedral coordination geometry at the Fe(II) center. Selected bond distances (Å): Fe1–N1 = 1.990(2).
Electrochemistry.
To examine the redox properties of the prepared Fe(II) complexes, cyclic voltammetry data was collected in 1,2-difluorobenzene (THF for Fe(Pyr)2(Pyrr)2 due to limited solubility in 1,2-difluorobenzene) with NBu4PF6 as the supporting electrolyte under an N2 atmosphere.
In general, the family of binuclear Fe(II) complexes featured poorly defined features by cyclic voltammetry in both oxidative and reductive directions (Figures S24–S28). These observations mirror those made for Fe3(TPM)220 and other Fe(II) pyrrolide complexes.37 Analysis of the mononuclear complexes reveals that Fe(PyrPyrr)2 displays a partially reversible [Fe(PyrPyrr)2]/[Fe(PyrPyrr)2]− couple at −1.22 V (vs. Fc+/Fc); the observed feature is only observed when the scan is initiated in the cathodic direction (Figure 9a; see Supporting Information for additional CV data). The CDO four-coordinated Fe(PyrMePyrr)2 displays a partially reversible oxidation event at −0.47 V vs. Fc+/Fc when the sweep begins anodically (Figure 9b).
Figure 9.

Cyclic voltammograms (CVs) measured in 1,2-difluorobenzne under N2 atmosphere (Potential vs. Fc/Fc+). (a) The CV of Fe(PyrPyrr)2 displays a partially reversible reduction event at −1.22 V, and (b) the CV of Fe(PyrMePyrr)2 displays a partially reversible oxidation event at −0.47 V.
The computed HOMO and LUMO of Fe(PyrMePyrr)2 are consistent with significant ligand contribution to the oxidation and reduction chemistry of this complex (Figure 10, other relevant orbitals are collected in Figure S30). The HOMO and LUMO of Fe(PyrMePyrr)2 are predominantly pyrrollide and pyridyl centered, respectively. Similar orbital distributions have previously been described for pyridylpyrrolide complexes of first-row metals.37
Figure 10.

HOMO and LUMO of Fe(PyrMePyrr)2 are pyrrole- and pyridyl-centered, respectively.
Computational Evaluation of Isomerization of Four-Coordinate Fe(II) Centers.
To evaluate the ligand-dependent geometrical preferences of the Fe(II) complexes described above, we have evaluated the potential energy surfaces (PES) that govern isomerization of the primary coordination sphere in Fe(PyrMePyrr)2 and Fe(Pyr)2(Pyrr)2. Structures were calculated using the UB3LYP functional combined with the 6-311+G basis set on all atoms and the MDF10 effective core potential on Fe. Constrained coordinates were set for the N1–Fe–N3 angle (the angle between pyrrolide donors) and N2–Fe–N4 angle (the angle between the pyridyl donors) to give a range of τ4 values while allowing for relaxation of the bond lengths and angles of the remainder of the molecule. The results of these calculations are plotted in Figure 11. The energies for the optimized structures of Fe(PyrMePyrr)2 (τ4 = 0.70) and Fe(Pyr)2(Pyrr)2 (τ4 = 0.82) are set to 0 kcal/mol. These calculations indicate a strong preference for the cis-divacant geometry in Fe2(PyrMePyrr)2 with τ4 values in the 0.6–0.7 range being the lowest in energy. As the complex approaches a square planar geometry (τ4 = 0), the energy rapidly increases. This is most likely due to a combination of a strong electronic preference for Fe to stay in a geometry conducive to high spin and large steric repulsions between ligands as they are forced into the same plane. Values for τ4 significantly larger than 0.7 were inaccessible due to the 82.7° bite angle of the PyrMePyrr ligand preventing tetrahedral-like geometries. In contrast, Fe(Pyr)2(Pyrr)2 shows a preference for a geometry closer to tetrahedral.
Figure 11.

Computed PESs for the isomerization of Fe(PyrMePyrr)2 and Fe(Pyr)2(Pyrr)2. Structures were generated by fixing N–Fe–N bond angles to give rise to the indicated τ4 values and optimizing the remainder of the structure.
DISCUSSION AND CONCLUDING REMARKS
The intimate relationship between transition metal ion coordination geometry and reactivity provides constant impetus to develop platforms that exhibit new or uncommon geometries. In the context of four-coordinate complexes, cis-divacant octahedral coordination is rare, although the presence of cis-divacant coordination sites available for potential substrate binding and activation renders CDO complexes attractive for potential catalyst development (Figure 1). We had previously observed CDO coordination at Fe(II) sites in the trinuclear complex Fe3(TPM)2 assembled by metalation of the three-fold symmetric H3TPM ligand. The origin of the peculiar coordination geometry in this trinuclear complex — ligand enforced, aggregation induced, or electronically preferred — was not clear. Further, the vacant coordination sites of the CDO Fe(II) ions in Fe3(TPM)2 were inaccessible to substrate due to confinement within the core of the trinuclear array. To better evaluate the origins of the observed CDO geometry and to develop a suite of complexes that would provide a structural basis to evaluate the reactivity of this unique structure class, we have pursued exhaustive ligand truncation studies that have resulted in the isolation of a family of mono- and binuclear complexes that feature isostructural CDO Fe(II) ions.
Excision of one of the three 2-pyridylpyrrole arms of the H3TPM ligand afforded H2BPM, which supports binuclear Fe(II) complexes (Figures 3 and 4). While metalation of this ligand in THF afforded a binuclear complex Fe2(BPM)2(THF)2 with two six-coordinate Fe(II) ions, the coordinated solvent ligands are labile. Dissolution in a non-coordinating solvent, such as toluene, resulted in a binuclear complex with two five-coordinate Fe(II) ions. In this complex, rearrangement of the ligand results in bridging pyrrolide ligands. Ligand methylation results in expansion of the Fe–Fe distance and enforcement of CDO geometry at both Fe(II) centers; the local geometry at each of the Fe centers in Fe2(BPMMe)2 are highlighted in Figure 12. Analysis of the local geometry using the τ4 metric was consistent with CDO coordination at each of the Fe(II) sites, which is corroborated by continuous shape analysis (Table 2).
Figure 12.

Displacement ellipsoid plot of Fe2(BPMMe)2. Highlight of the primary coordination sphere featuring CDO coordination at each Fe(II) center.
Further ligand truncation by removal of a 2-pyridylpyrrole arm from the H2BPM ligand series provides access to bidentate ligands PyrPyrrH and PyrMePyrrH. Metallation of each of these in coordinating solvents provides access to monomeric, six-coordinate Fe(II) species in which solvent ligands occupy mutually cis coordination sites (Figure 6). Again, dissolution of these complexes in non-coordinating solvents results in labilization of the bound THF ligands to generate unsaturated Fe(II) complexes. In the case of Fe(PyrPyrr)2, disproportionation to Fe(PyrPyrr)3 and Fe(0) prevents isolation of the unsaturated monomer. In contrast, THF dissociation from Fe(PyrMePyrr)2(THF)2 affords four-coordinate CDO complex Fe(PyrMePyrr)2. We hypothesize that the difference in stability of Fe(PyrPyrr)2 and Fe(PyrMePyrr)2 toward disproportionation results from steric protection in the case of the methylated ligand, which prevents the bimolecular reaction needed for disproportionation to proceed.
Comparison of the primary coordination sphere before and after THF dissociation reveals remarkably little structural rearrangement accompanied by ligand dissociation (Figure 13). The Fe(II) center in Fe(PyrMePyrr)2 is rigorously CDO by analysis of the τ4 metric. In addition, the Fe(II) ion in Fe(PyrMePyrr)2 shows similar coordination structure of Fe to the four-coordinate binuclear complex, Fe2(BPMMe)2: Both complexes feature CDO coordination and the Fe–Npyrr distances in Fe(PyrMePyrr)2 (1.985(2) Å) are comparable to that of Fe2(BPMMe)2 (1.997(1) Å). The formation of two vacant sites by solvent dissociation is notable because previous studies in related pyrrolide complexes has resulted in five-coordinate, mono-solvento complexes.38
Figure 13.

Overlay of the primary coordination spheres of Fe(PyrMePyrr)2(THF)2 (faded) and Fe(PyrMePyrr)2, which highlights the minimal structural reorganization that accompanies THF dissociation and formation of the CDO Fe(II) ion.
To evaluate the origin of the observed CDO geometry, we prepared Fe(Pyr)2(Pyrr)2 in which the aforementioned 2-pyridylpyrrolide ligands were separated into their constituent monodentate donors (Figure 8). In contrast to the suite of complexes prepared from chelating ligands, Fe(Pyr)2(Pyrr)2 displays tetrahedral coordination (τ4 = 0.902) about the Fe(II) ion. This observation indicates that the electronically preferred geometry of Fe(II) with two pyridyl and two pyrrolide donors is tetrahedral and suggests that ligand chelation is responsible for the observed geometrical preferences in the obtained CDO complexes. The relative coordination preferences of monodentate and chelating bidentate donors was evaluated by examining the potential energy surfaces that dictate the ligand-dependent coordination geometries of Fe(II) (Figure 11). Consistent with experiment, the potential energy surface for monodentate donors prefers tetrahedral coordination whereas the bidentate potential energy surface prefers CDO. The consistency of these computations with experiment indicates that the observed geometries do not arise due to crystal packing. Further, in the case of complex Fe(PyrMePyrr)2, the potential energy surface is fairly flat in the vicinity of CDO coordination, which indicates that small structural perturbations have a significant impact of the observed coordination geometry. We speculate that the disparity in geometry between monodentate donors (i.e., Fe(Pyr)2(Pyrr)2) and bidentate donors (i.e., Fe(PyrMePyrr)2 arises because the chelating ligand has a narrow bite angle (~83°) that cannot be accommodated in more tetrahedral arrangements.
Access to a family of Fe complexes that display rigorous CDO coordination about the metal center provides an opportunity to evaluate the activity of these sites towards substrate binding and activation. Of particular note is the observation that while Fe pyrrolides often display ligand-dominated, irreversible electrochemistry, Fe(PyrMePyrr)2 displays a reversible Fe(III)/Fe(II) couple. Combined with the available open coordination sites, this observation suggests Fe(PyrMePyrr)2 may be a good candidate for substrate engagement. Ongoing work is aimed at pursuing both the broader synthetic chemistry of CDO geometries with other transition metals and to the development of group transfer catalysis with CDO Fe(II) catalysts.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge Nattamai Bhuvanesh and Gayle Bornovski for assistance with crystallographic and electrochemical experiments, respectively. In addition, the authors acknowledge financial support from the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Catalysis Program (DE-SC0018977), the Welch Foundation (A-1907), and an Alfred P. Sloan Fellowship to DCP and the National Institutes of Health (GM127021) to PAL.
Footnotes
Supporting Information
Experimental procedures and spectroscopic data (PDF). The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
REFERENCES
- 1.Kochem A; Carrillo A; Philouze C; van Gastel M; du Moulinet d’Hardemare A; Thomas F, Copper(II)-Coordinated α-Azophenols: Effect of the Metal-Ion Geometry on Phenoxyl/Phenolate Oxidation Potential and Reactivity. Eur. J. Inorg. Chem 2014, 2014, 4263–4267. [Google Scholar]
- 2.Muthuramalingam S; Anandababu K; Velusamy M; Mayilmurugan R, Benzene Hydroxylation by Bioinspired Copper(II) Complexes: Coordination Geometry versus Reactivity. Inorg. Chem 2020, 59, 5918–5928. [DOI] [PubMed] [Google Scholar]
- 3.Combariza MY; Vachet RW, Effect of Coordination Geometry on the Gas-Phase Reactivity of Four-Coordinate Divalent Metal Ion Complexes. J. Phys. Chem. A 2004, 108, 1757–1763. [DOI] [PubMed] [Google Scholar]
- 4.Favas MC; Kepert DL, Aspects of the Stereochemistry of Four-Coordination and Five-Coordination. In Progress in Inorganic Chemistry, 1980; pp 325–463. [Google Scholar]
- 5.Cirera J; Ruiz E; Alvarez S, Stereochemistry and Spin State in Four-Coordinate Transition Metal Compounds. Inorg. Chem 2008, 47, 2871–2889. [DOI] [PubMed] [Google Scholar]
- 6.Hawrelak EJ; Bernskoetter WH; Lobkovsky E; Yee GT; Bill E; Chirik PJ, Square Planar vs Tetrahedral Geometry in Four Coordinate Iron(II) Complexes. Inorg. Chem 2005, 44, 3103–3111. [DOI] [PubMed] [Google Scholar]
- 7.Vela J; Cirera J; Smith JM; Lachicotte RJ; Flaschenriem CJ; Alvarez S; Holland PL, Quantitative Geometric Descriptions of the Belt Iron Atoms of the Iron–Molybdenum Cofactor of Nitrogenase and Synthetic Iron(II) Model Complexes. Inorg. Chem 2007, 46, 60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muetterties EL; Guggenberger LJ, Idealized Polytopal Forms. Description of Real Molecules Referenced to Idealized Polygons or Polyhedra in Geometric Reaction Path Form. J. Am. Chem. Soc 1974, 96, 1748–1756. [Google Scholar]
- 9.Murray-Rust P; Buergi HB; Dunitz JD, Chemical Reaction Paths. V. SN1 Reaction of Tetrahedral Molecules. J. Am. Chem. Soc 1975, 97, 921–922. [Google Scholar]
- 10.Vela J; Stoian S; Flaschenriem CJ; Münck E; Holland PL, A Sulfido-Bridged Diiron(II) Compound and Its Reactions with Nitrogenase-Relevant Substrates. J. Am. Chem. Soc 2004, 126, 4522–4523. [DOI] [PubMed] [Google Scholar]
- 11.Shinji T; Michinori O, Structure of Intramolecular Boron–Amine Complexes and Proposal of Tetrahedral Character for Correlation between Molecular Structure and Barrier to Dissociation of the N–B Bonds. Bull. Chem. Soc. Jpn 1992, 65, 1832–1840. [Google Scholar]
- 12.Hutchison AR; Mitra A; Atwood DA, The Four Coordinate Geometric Parameter: A New Quantification of Geometry for Four Coordinate Aluminum and Gallium. Main Group Chemistry 2005, 187–200. [Google Scholar]
- 13.Yang L; Powell DR; Houser RP, Structural Variation in Copper(I) Complexes with Pyridylmethylamide Ligands: Structural Analysis with a New Four-Coordinate Geometry Index, τ4. Dalton Trans. 2007, 955–964. [DOI] [PubMed] [Google Scholar]
- 14.Pinsky M; Avnir D, Continuous Symmetry Measures. 5. The Classical Polyhedra. Inorg. Chem 1998, 37, 5575–5582. [DOI] [PubMed] [Google Scholar]
- 15.Keinan S; Avnir D, Continuous Symmetry Analysis of Tetrahedral/Planar Distortions. Copper Chlorides and Other AB4 Species. Inorg. Chem 2001, 40, 318–323. [DOI] [PubMed] [Google Scholar]
- 16.Keinan S; Avnir D, Studies in Copper(II) Complexes: Correlations Between Quantitative Symmetry and Physical Properties. J. Chem. Soc., Dalton Trans 2001, 941–947. [Google Scholar]
- 17.Searles K; Fortier S; Khusniyarov MM; Carroll PJ; Sutter J; Meyer K; Mindiola DJ; Caulton KG, A cis-Divacant Octahedral and Mononuclear Iron(IV) Imide. Angew. Chem. Int. Ed 2014, 53, 14139–14143. [DOI] [PubMed] [Google Scholar]
- 18.Kurup SS; Wannipurage D; Lord RL; Groysman S, Tying the Alkoxides Together: an Iron Complex of a New Chelating bulky Bis(alkoxide) Demonstrates Selectivity for Coupling of Non-Bulky Aryl Nitrenes. Chem. Commun 2019, 55, 10780–10783. [DOI] [PubMed] [Google Scholar]
- 19.Bellow JA; Fang D; Kovacevic N; Martin PD; Shearer J; Cisneros GA; Groysman S, Novel Alkoxide Cluster Topologies Featuring Rare Seesaw Geometry at Transition Metal Centers. Chem. Eur. J 2013, 19, 12225–12228. [DOI] [PubMed] [Google Scholar]
- 20.Hyun S-M; Upadhyay A; Das A; Burns CP; Sung S; Beaty JD; Bhuvanesh N; Nippe M; Powers DC, Kinetic versus Thermodynamic Metalation Enables Synthesis of Isostructural Homo- and Heterometallic Trinuclear Clusters. Chem. Commun 2020, 56, 5893–5896. [DOI] [PubMed] [Google Scholar]
- 21.Rieth RD; Mankad NP; Calimano E; Sadighi JP, Palladium-Catalyzed Cross-Coupling of Pyrrole Anions with Aryl Chlorides, Bromides, and Iodides. Org. Lett 2004, 6, 3981–3983. [DOI] [PubMed] [Google Scholar]
- 22. See Figure S9 for details of data fitting, including attempts to fit the observed spectral data with two components in-stead of three.
- 23.Oliver JD; Mullica DF; Hutchinson BB; Milligan WO, Iron-Nitrogen Bond Lengths in Low-Spin and High-Spin Iron(II) Complexes with Poly(pyrazolyl)borate Ligands. Inorg. Chem 1980, 19, 165–169. [Google Scholar]
- 24.Greenaway AM; Sinn E, High-Spin and Low-Spin α-Picolylamine Iron(II) Complexes. Effect of Ligand Reversal on Spin State. J. Am. Chem. Soc 1978, 100, 8080–8084. [Google Scholar]
- 25.These Fe–N interactions are significantly longer than other cases of asymmetrically bridging pyrrolide ligands. See for example,; Crewdson P; Gambarotta S; Yap GPA; Thompson LK, Dinuclear and Octanuclear Mn(II) Complexes with μ2-C, μ2-N(Pyrrolide), and μ-η1:η5-(Pyrrolide) Bridges: A Structural and Magnetic Study. Inorg. Chem 2003, 42, 8579–8584. [DOI] [PubMed] [Google Scholar]
- 26.Eisenhart RJ; Rudd PA; Planas N; Boyce DW; Carlson RK; Tolman WB; Bill E; Gagliardi L; Lu CC, Pushing the Limits of Delta Bonding in Metal–Chromium Complexes with Rdox Changes and Metal Swapping. Inorg. Chem 2015, 54, 7579–7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cotton FA; Murillo CA; Walton RA, Multiple Bonds between Metal Atoms. Springer Science: New York, 2005. [Google Scholar]
- 28.Pauling L, Atomic Radii and Interatomic Distances in Metals. J. Am. Chem. Soc 1947, 69, 542–553. [Google Scholar]
- 29.Addison AW; Rao TN; Reedijk J; van Rijn J; Verschoor GC, Synthesis, Structure, and Spectroscopic Properties of Copper(Ii) Compounds Containing Nitrogen–Sulphur Donor Ligands; The Crystal and Molecular Structure of Aqua [1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc., Dalton Trans 1984, 1349–1356. [Google Scholar]
- 30. Conceptually similar to τ4 values, τ5 values are descriptors of the coordination geometries of 5-coordinate complexes.
- 31. A molecule of PhCH3 was located in the unit cell. This solvent molecule was carried over from previous synthetic manipulations.
- 32.Werncke CG; Bouammali M-A; Baumard J; Suaud N; Martins C; Guihéry N; Vendier L; Zheng J; Sortais J-B; Darcel C; Sabo-Etienne S; Sutter J-P; Bontemps S; Pichon C, Ising-type Magnetic Anisotropy and Slow Relaxation of the Magnetization in Four-Coordinate Amido-Pyridine FeII Complexes. Inorg. Chem 2016, 55, 10968–10977. [DOI] [PubMed] [Google Scholar]
- 33.Casanova D; Cirera J; Llunell M; Alemany P; Avnir D; Alvarez S, Minimal Distortion Pathways in Polyhedral Rearrangements. J. Am. Chem. Soc 2004, 126, 1755–1763. [DOI] [PubMed] [Google Scholar]
- 34.Cirera J; Ruiz E; Alvarez S, Shape and Spin State in Four-Coordinate Transition-Metal Complexes: The Case of the d6 Configuration. Chem. Eur. J 2006, 12, 3162–3167. [DOI] [PubMed] [Google Scholar]
- 35.Nishida Y; Kino K; Kida S, Crystal Structures of Low- and High-Spin Iron(III) Complexes with Quadridentate Schiff Bases. J. Chem. Soc., Dalton Trans 1987, 1157–1161. [Google Scholar]
- 36. Determination of the structure of Fe(PyrMePyrr)2(THF)2 was carried out at 296 K because loss of crystallinity was observed during cooling on single crystals below ~150 K.
- 37.Sazama GT; Betley TA, Ligand-Centered Redox Activity: Redox Properties of 3d Transition Metal Ions Ligated by the Weak-Field Tris(pyrrolyl)ethane Trianion. Inorg. Chem 2010, 49, 2512–2524. [DOI] [PubMed] [Google Scholar]
- 38.Searles K; Das AK; Buell RW; Pink M; Chen C-H; Pal K; Morgan DG; Mindiola DJ; Caulton KG, 2,2′-Pyridylpyrrolide Ligand Redistribution Following Reduction. Inorg. Chem 2013, 52, 5611–5619. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.