

Abstract
Double-stranded RNA (dsRNA) is associated with most viral infections — it either constitutes the viral genome (in the case of dsRNA viruses) or is generated in host cells during viral replication. Hence, nearly all organisms have the capability of recognizing dsRNA and mounting a response, the primary aim of which is to mitigate the potential infection. In vertebrates, a set of innate immune receptors for dsRNA induce a multitude of cell-intrinsic and cell-extrinsic immune responses upon dsRNA recognition. Notably, recent studies showed that vertebrate cells can accumulate self-derived dsRNAs or dsRNA-like species upon dysregulation of several cellular processes, activating the very same immune pathways as in infected cells.
On the one hand, such aberrant immune activation in the absence of infection can lead to pathogenesis of immune disorders, such as Aicardi–Goutières syndrome. On the other hand, the same innate immune reaction can be induced in a controlled setting for a therapeutic benefit, as occurs in immunotherapies. In this Review, we describe mechanisms by which immunostimulatory dsRNAs are generated in mammalian cells, either by viruses or by the host cells, and how cells respond to them, with the focus on recent developments regarding the role of cellular dsRNAs in immune modulation.
Since the original proposal of the RNA world more than 50 years ago, which postulates RNA as the sole type of biopolymer for sustaining primitive forms of life, our understanding of functions of RNA has greatly expanded. These functions range from a simple message bearing a linear array of genetic codes to an active player in transcription regulation, protein synthesis, nutrient sensing and many other biological processes1. These non-coding functions arise from the abilities of RNA molecules to form secondary, tertiary or quaternary structures, and to engage with DNA, proteins, metabolites or other RNA molecules in temporally and spatially controlled manners. The rapid expansion of the RNA world accompanied the increased appreciation of double-stranded RNAs (dsRNAs) as prevalent molecules with diverse functions.
It has long been thought that the presence of dsRNAs is associated solely with viral infection (including both RNA and DNA viruses). One of the most conserved mechanisms by which cells sense viral infection is through detection of dsRNAs by a set of receptors in the innate immune system that trigger antiviral and inflammatory immune responses2 (BOX 1). However, accumulating evidence suggests that dsRNAs are not limited to virally infected cells, but can be produced from endogenous sources, such as retroelements and mitochondrial DNA, in various pathophysiological states. These endogenous dsRNAs activate the same receptors that have evolved to detect viral dsRNAs, often serving as a ‘danger’ signal that alerts misregulated cellular processes. Accordingly, innate immune and inflammatory responses to dsRNAs underlie diverse pathophysiologies from immune disorders to neurodegeneration.
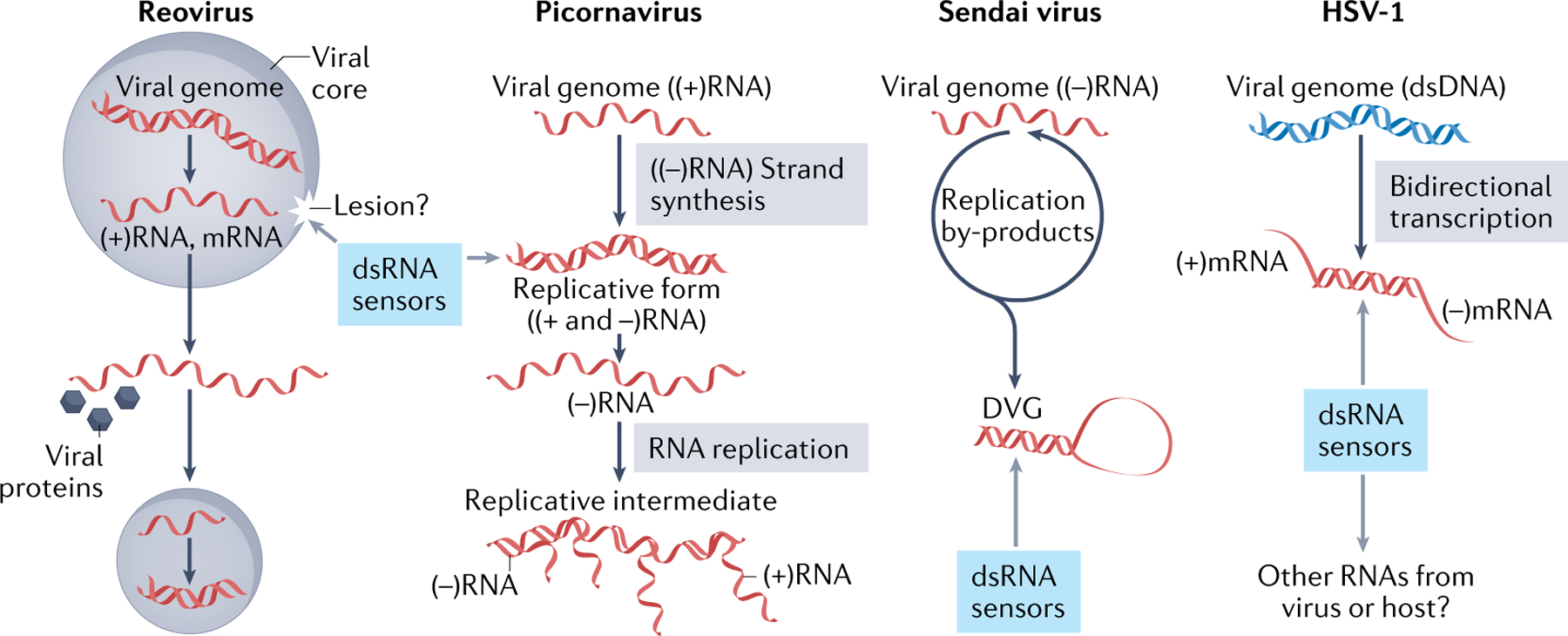
Box 1 |. Activation of dsRNA sensors by viruses.
Most viral infections are associated with the generation of double-stranded RNA (dsRNA), but the source of the dsRNA and its recognition by dsRNA sensors in the cell differ depending on the virus (see the figure).
dsRNA viruses
dsRNA viruses often sequester their duplex genome during the entire replication cycle and use additional mechanisms to prevent innate immune activation. For example, both replication and transcription by reovirus occur entirely within the viral core without exposing the dsRNA genome to the cytosolic compartment and to the host innate immune system184,185. However, many dsRNA viruses, including reovirus, have been shown to activate cellular dsRNA sensors186,187, likely by erroneous or host-mediated leakage of the viral dsRNA genome into the cytoplasm188,189.
Positive-strand RNA viruses
Positive-strand RNA viruses are also known to produce dsRNAs during replication and activate the innate immune system190,191. Upon entry, the positive-strand RNA genome is released into the cytoplasm and is translated by the host ribosomes. Newly synthesized RNA-dependent RNA polymerase then copies the genome (positive strand) to produce the antigenome (negative strand), during which long dsRNA of the genome–antigenome hybrid (that is, the replicative form) is generated. For picornaviruses, the replicative form is ~7–8-kb dsRNA, and has been shown to be the major RNA species activating MDA5, protein kinase R (PKR) and oligoadenylate synthases16,192. The picornavirus replicative form does not activate RIG-I because both the positive strand and the negative strand of viral RNAs lack 5′-triphosphate.
Negative-strand RNA viruses
Negative-strand RNA viruses also replicate through a dsRNA intermediate, but they do not accumulate a significant level of this intermediate193,194. Instead, immunostimulatory dsRNA from negative-strand viruses, such as Sendai virus, appears to stem from erroneous replication by-products, namely defective viral genomes (DVGs)20,195–198. While there are many types of DVGs, the most immunostimulatory RNAs are copy-back DVGs, which contain a small portion of the genome with two perfectly complementary ends196. Copy-back DVGs contain 5′-triphosphate and activate RIG-I, not MDA5. The longer the complementary ends are, the more potent they are in activating RIG-I, suggesting that it is the DVGs, not their replication intermediates, that activate RIG-I20,197. However, it is worth noting that copy-back DVGs in the virion particles should also be encapsidated for replication, which would protect them from RIG-I recognition. This suggests that a copy-back DVG is either more prone to escape encapsidation, possibly due to the intrinsic stability of the dsRNA hairpin or other, unknown mechanisms199.
For most negative-strand RNA viruses, however, the identity of the RIG-I-stimulatory RNA remains less clear. For influenza virus, for example, RIG-I was proposed to be activated by multiple forms of viral RNAs, including the nucleocapsid200, the full-length genome201, the 3′ untranslated region202 and DVGs203–205. These conflicting reports highlight the challenge of identifying RIG-I ligands.
DNA viruses
DNA viruses, such as vaccinia virus, herpes simplex virus type 1 (HSV-1) and adenovirus, are also known to accumulate significant levels of dsRNA that activate the innate immune system193,194. It has been thought that many DNA viruses generate long sense–antisense hybrids formed by converging bidirectional transcription206,207. However, other types of immunostimulatory dsRNAs were also proposed to accumulate during DNA viral infection. For example, EBER1 and EBER2, non-coding RNAs encoded by Epstein–Barr virus and transcribed by the host RNA polymerase III, were proposed to activate RIG-I208,209. More recently, aberrant endogenous RNAs were also proposed to activate RIG-I during infection by HSV-1, Kaposi sarcoma-associated herpesvirus or Epstein–Barr virus126,128. As with most viruses, however, the precise identity of the dsRNAs activating each of these sensors and the mechanism of activation need further investigation.
In this Review, we summarize recent reports showing endogenous sources of dsRNA (self dsRNA) and cellular responses that result from activation of dsRNA sensors by these cell-derived dsRNA species. We first discuss the dsRNA sensors in the vertebrate innate immune system, with the focus on their signalling pathways, mechanisms and RNA specificity. We then outline the origins and identities of the immunostimulatory endogenous dsRNAs and describe the consequences of self dsRNA-mediated immune activation.
dsRNA sensors
The immune response to viral infection often begins when viral dsRNA is detected by dsRNA-binding proteins in the host (FIG. 1). These sensors include RIG-I-like receptors (RLRs), protein kinase R (PKR), oligoadenylate synthases (OASes), Toll-like receptors (TLRs) and NOD-, LRR- and pyrin domain-containing 1 (NLRP1). These sensors are broadly expressed in a wide array of tissues, including airway, gut and reproductive tract epithelial cells, which are primary entry sites for many viruses. Upon binding to dsRNA, these sensors activate a multitude of immune responses, including activation of antiviral and inflammatory signalling pathways (FIG. 1a), cell growth inhibition (FIG. 1b) and in some cases cell death (FIG. 1c), which altogether inhibit viral replication.
Fig. 1 |. dsRNA sensors and their signalling.
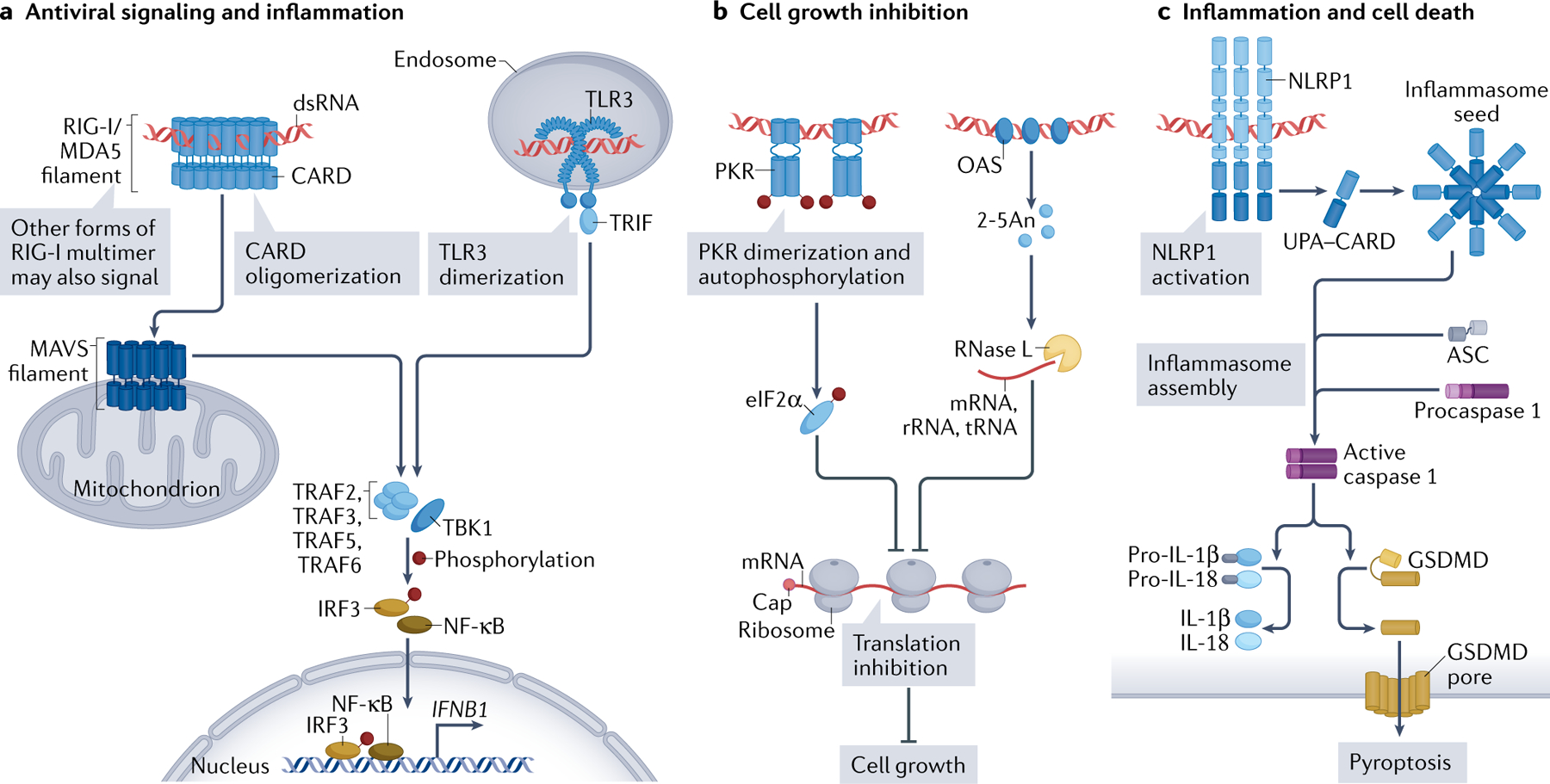
Double stranded RNAs (dsRNAs) from either viral or cellular origins induce three types of cellular responses. They are not mutually exclusive and may occur within the same cell. a | The first type of response is classical antiviral innate immune responses mediated by RIG-I or MDA5 (RIG-I-like receptors (RLRs)) and Toll-like receptor 3 (TLR3). RLRs in the cytoplasm and TLRs in the endosome detect dsRNAs and form signalling-competent oligomers to activate their respective downstream adaptors, mitochondrial antiviral-signalling protein (MAVS) and TRIF. Upon dsRNA binding, RLRs form filaments along the length of dsRNA, which promotes oligomerization of their caspase activation and recruitment domains (CARDs) and triggers MAVS filament formation for downstream signal activation. RIG-I may also be stimulated by RNAs besides long dsRNA by forming non-filamentous multimers, but the structural features of such RNAs and RIG-I multimers are unclear. These pathways then converge by activating the common downstream signalling molecules, such as TNF receptor-associated factors (TRAFs) and TANK-binding kinase 1 (TBK1), culminating in the activation of the transcription factors interferon-regulatory factor 3 (IRF3) and NF-κB for producing type I interferons and other proinflammatory cytokines. b | The second type of response induces global inhibition of protein synthesis and thus cell growth. This response is mediated by protein kinase R (PKR) and oligoadenylate synthases (OASes), which become active upon binding to dsRNA. PKR activation via dimerization and autophosphorylation results in phosphorylation of a key translation initiation factor (eIF2α) and subsequent inhibition of most protein synthesis. Activated OASes synthesize 2′−5′-linked oligoadenylate (2–5An), which serves as a soluble second messenger to activate ribonuclease L (RNase L). RNase L in turn degrades the bulk of cytosolic RNAs, including mRNA, ribosomal RNA (rRNA) and tRNA, resulting in translation inhibition. c | The third type of response to dsRNA is mediated by the NOD-, LRR- and pyrin domain-containing 1 (NLRP1) inflammasome, a macromolecular complex containing the receptor NLRP1, the adaptor ASC and the effector caspase 1. Upon dsRNA binding, NLRP1 triggers release of its UPA and CARD domains, which then assembles the inflammasome seed, inducing inflammasome formation and activating caspase 1. Activated caspase 1 then cleaves precursors of inflammatory cytokines (such as IL-1β and IL-18) and a pore-forming protein gasdermin D (GSDMD). The GSDMD pore forms in the plasma membrane and induces pyroptosis, the inflammatory form of cell death.
RIG-I-like receptors.
RLRs encompass three proteins, RIG-I, MDA5 and LGP2, with all three sharing similar RNA-binding domains, including the conserved DExD/H helicase domain3,4. Upon viral dsRNA recognition, RIG-I and MDA5 activate their shared downstream adaptor molecule, mitochondrial antiviral-signalling protein (MAVS) (FIG. 1a). Activated MAVS then recruits multiple signalling molecules, including TNF receptor-associated factors (TRAFs), TANK-binding kinase 1 (TBK1) and interferon-regulatory factor 3/7 (IRF3/7), which in turn induce transcription of type I interferons and other proinflamamtory cytokines3,4. Unlike RIG-I and MDA5, LGP2 lacks the ability to directly control the antiviral signalling pathway, but is thought to positively and negatively regulate MDA5 and RIG-I, respectively5–7.
Type I interferons.
A large subgroup of secreted proteins that bind to interferon receptors and stimulate the expression of antiviral genes.
MDA5 and RIG-I both use RNA duplex length as a key criterion to distinguish viral RNA from cellular RNA that may have local secondary structure, and they do so by forming filaments along the length of dsRNA4 (FIG. 1a; TABLE 1). For both receptors, filament formation is important for robust antiviral signalling as it increases signalling capacity, which relies on the interaction between the filaments and their activating E3 ligases (RIPLET for RIG-I and TRIM65 for MDA5)8,9. These E3 ligases conjugate K63-linked ubiquitin chains, which act as ‘molecular glues’ to help oligomerize their receptor signalling domains — caspase activation and recruitment domains (CARDs)10,11 — which occurs more readily when the receptors are preoligomerized on RNA (FIG. 1a).
Table 1 |.
dsRNA length sensitivity of dsRNA sensors
| Mechanism of dsRNA length discrimination | ||||
|---|---|---|---|---|
| At the receptor level | By downstream pathway | |||
| RLRs | RIG-I | ~22–500 bp with peak activity for ~50–200-bp dsRNA | Filament formation (through ATP-driven translocation of the receptor) | RIPLET binding |
| MDA5 | ~500–1,000 bp; activity progressively increases with dsRNA length | Filament formation (through ATP-independent cooperative binding) | TRIM65 binding | |
| TLR3 | 40–50 bp; activity progressively increases with dsRNA length | Dimerization or multimerization Activity progressively increases with dsRNA length |
ND | |
| PKR | >~33 bp; activity progressively increases with dsRNA length | Proximity of at least two PKR dimers (interdimeric phosphorylation) | ND | |
| OASes | OAS1 | <20 bp | ND | ND |
| OAS2 | Unclear | ND | ND | |
| OAS3 | >50 bp | Binding in an extended conformation (?) | ND | |
| NLRP1 | >500 bp | ND | ND | |
dsRNA, double-stranded RNA; ND, not determined; NLRP1, NOD-, LRR- and pyrin domain-containing 1; OAS, oligoadenylate synthase; PKR, protein kinase R; RIPLET, E3 ubiquitin-protein ligase RNF135; RLR, RIG-I-like receptor; TLR3, Toll-like receptor 3; TRIM65, tripartite motif-containing protein 65.
Caspase activation and recruitment domains.
(CARDs). interaction motifs found in a wide array of proteins, typically those involved in processes relating to inflammation and apoptosis.
Despite these similarities, RIG-I and MDA5 use different mechanisms to assemble filaments and regulate filament stability, resulting in their divergence in RNA and viral specificities. For MDA5, filaments form directly within the RNA duplex stem in a highly cooperative manner with little dependence on dsRNA sequence12–15. Concurrent to filament formation is ATP hydrolysis within the helicase domain, which in turn stimulates filament disassembly from filament termini12,13,15. Therefore, MDA5 filament continuously undergoes cycles of end disassembly and elongation, where the dynamic equilibrium between the two processes determines the stability of the MDA5 filament13,14. Since filament disassembly occurs from the filament end, the MDA5 filament stability increases with its increasing length, which is in turn dictated by dsRNA length. As a result, MDA5 signalling increases with dsRNA length and requires an ~0.5–1-kb duplex region13,16. This RNA-length dependency also enables detection of duplex structural irregularities introduced by mismatches and bulges, which are common among cellular dsRNAs and interfere with MDA5 filament assembly, allowing it to discriminate between viral and cellular RNAs17. Additionally, RNA modifications (FIG. 2), also present in many cellular RNAs, can regulate MDA5 filament formation in a manner dependent on the physicochemical property of the specific modifications. For example, adenosine-to-inosine (A-to-I) modification, which affects Watson–Crick base pairing (discussed further in the section entitled Endogenous sources of RNA), strongly interferes with MDA5 filament assembly17, while pseudouridylation and N6-methylation of adenosine have little impact18 (FIG. 2).
Fig. 2 |. RNA modifications affect the RNA’s secondary structures and interaction with immune sensors.
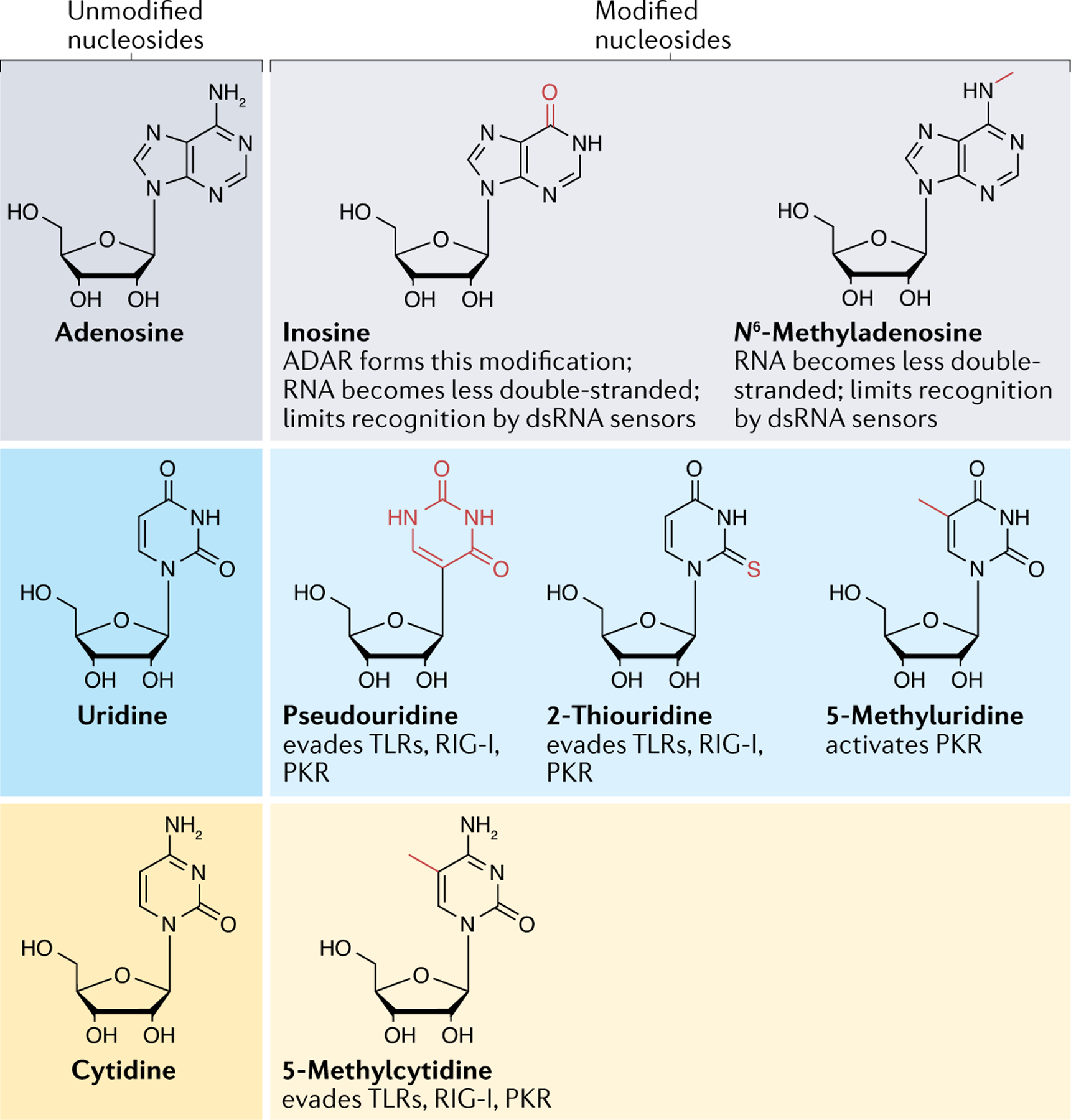
The canonical nucleosides adenosine, uridine and cytidine can be modified by enzymes that install new chemical groups (shown in red). The RNA modifications can change base-pairing interactions, protein binding and secondary structures, which can prevent the modified RNAs from forming immunogenic structures, such as double-stranded RNAs (dsRNAs), and evading detection by immune sensors, including Toll-like receptors (TLRs), RIG-I and protein kinase R (PKR). ADAR, double-stranded RNA-specific adenosine deaminase.
Unlike MDA5, RIG-I filament assembles from a dsRNA end with a 5′-triphosphate group (5′ppp) or a 5′-diphosphate group19–21. 5′ppp is present in all nascent transcripts, but is removed from host RNAs during normal 5′ processing1. Viral RNAs, especially those generated in the cytoplasm of the host cell, often do not undergo such 5′ processing and thus retain 5′ppp. Binding of an individual RIG-I molecule to a 5′ppp-containing dsRNA end stimulates the ATPase activity in the RIG-I helicase domain. This then drives RIG-I to translocate from the dsRNA end to the interior22 (the activity not shared with MDA5), re-exposing the 5′ppp-containing dsRNA end and allowing recruitment of another RIG-I molecule. Iterations of end recruitment and translocation lead to the formation of a filamentous oligomer of RIG-I, albeit not as long as MDA5 filaments19,20. Even though filamentous oligomerization is also important for RIG-I, the importance of the dsRNA end limits the ability of RIG-I to form filaments on very long dsRNA (longer than ~500-bp dsRNA)8,19 (TABLE 1). Thus, the preference of RIG-I for dsRNA displays a bell-shaped curve with respect to dsRNA length8. This exemplifies how RIG-I, unlike MDA5, utilizes two distinct pathogen-associated molecular patterns (PAMPs) − the presence of 5′ppp and RNA duplex length − to recognize viral dsRNAs that differ from those stimulating MDA5. Some reports suggest that monomeric RIG-I stimulated by a short (~14-bp) hairpin RNA is sufficient for activation23. However, RIG-I signalling requires the E3 ligase RIPLET, which in turn needs at least dimeric RIG-I for bivalent binding and ubiquitin-mediated tetramerization of the RIG-I CARD. It is possible that the short hairpin multimerizes to form longer duplex RNA (as shown before)24, which may then support RIG-I multimerization and signalling (FIG. 1a). It should also be noted that while RIG-I filament is the only RNA-bound multimer characterized to date, other forms of RIG-I multimer may exist on unknown RNA and activate signalling. In general, there is a limited understanding of the diversity of RNA structure (besides the simple duplex stem structure) for RIG-I stimulation, which is an intriguing area for future studies.
Pathogen-associated molecular patterns.
(PAMPs). Conserved motifs that are associated with pathogen infection that serve as ligands for host pattern recognition receptors.
TLR3.
TLR3 is another dsRNA receptor that activates the antiviral signalling pathway that transcriptionally induces interferon-β (IFNβ) and proinflammatory cytokines. Unlike RLRs, which are cytosolic, TLR3 is anchored to the endosomal membrane, surveying the presence of dsRNA in the lumen of the endosome (FIG. 1a). In some cell types, TLR3 is also present on the cell surface25, but dsRNA binding requires an acidic environment26, suggesting that recognition occurs within the endosome. For this reason, it is generally believed that TLR3 detects dsRNAs that are released from infected and dying cells through endocytosis (cell-extrinsic sensing). This differs from cytosolic dsRNA sensors (such as RLRs), which sense dsRNAs originating within the cell (cell-intrinsic sensing). TLR3 deficiency results in increased susceptibility to a broad range of viruses, including poliovirus27 and herpes simplex virus type 1 (HSV-1)28. In humans, TLR3 deficiency causes HSV-1 encephalitis in children due to failure to restrict the virus in the central nervous system (CNS)29. Interestingly, such patients display normal immunity against HSV-1 in non-CNS tissues and against most other microorganisms in general. This suggests that TLR3 has a unique role in host defence against HSV-1 in the CNS, while its function is redundant with those of other receptors (such as RLRs) in most other tissues.
Like for RLRs, dsRNA recognition by TLR3 also depends on the RNA duplex length, with a minimal requirement of 40–50 bp (REFS26,30) (TABLE 1). Structural and biochemical studies showed that this length sensitivity stems from the requirement of TLR3 to form a dimer26 or a multimer29. Ectodomains of two TLR3 molecules cooperatively bind one dsRNA molecule in a way that the cytoplasmic carboxy-terminal signalling domains are juxtaposed to allow recruitment and activation of the downstream adaptor molecule TRIF30. TLR3 interacts with the ribose phosphate backbone of dsRNA and thus has no RNA sequence specificity. Nucleoside modification can suppress recognition by TLR3 (REF.31). Some studies, however, reported that incomplete duplex structures within single-stranded RNA (ssRNA) molecules can also activate TLR3 (REF.32), although the mechanistic basis for this observation is unclear.
PKR.
PKR is a dsRNA-dependent protein kinase. Upon dsRNA binding, PKR dimerizes, autophosphorylates and becomes an active kinase that can phosphorylate the α-subunit of the translation initiation factor eIF2 (REF.2) (FIG. 1b). Phosphorylation of eIF2α prevents functions of eIF2, leading to the global shutdown of protein synthesis and cell growth inhibition. Since all viruses depend on the host machineries for viral protein synthesis, PKR-mediated eIF2 restriction has a broad antiviral function, although some viruses evade this restriction mechanism through eIF2-independent translation33.
PKR was also found to modulate RLR signalling, although the detailed mechanism remains controversial. First, PKR activation and the consequent inhibition of translation initiation induce stress granules. Stress granule formation has been proposed to amplify RLR signalling by serving as a signalling hub34,35, although the opposite effect was also reported in a recent preprint36. PKR was also reported to promote production of IFNα and IFNβ by stabilizing their mRNAs, rather than by upregulating their transcription37. Finally, PKR was proposed to modulate RLR signalling simply by suppressing global protein synthesis, as some inhibitors of the RLR pathway, such as IκB, have a short half-life, requiring continuous protein synthesis to maintain its level and inhibitory function38,39.
Stress granules.
Molecular condensates that form in response to various cellular stresses. They often result from accumulation of stalled translation initiation complexes that expose mRNAs.
PKR contains two tandem repeats of dsRNA-binding domains and a kinase domain40,41. Data to date suggest that PKR activation requires stable dimerization, which is induced by dsRNA binding and stabilized by autophosphorylation41,42 (FIG. 1b). The PKR dimeric structure has a back-to-back dimeric configuration where the two active sites face away from each other41,42. Therefore, autophosphorylation likely occurs through interdimeric phosphorylation rather than intradimeric or intramolecular phosphorylation43, although intradimeric phosphorylation has also been proposed44. It has been reported that the minimal length of dsRNA required for PKR activation is ~33 bp (REF.45) (TABLE 1), likely reflecting the requirement for clustering of at least two PKR dimers on a single RNA molecule to facilitate interdimeric phosphorylation. While other RNA features, such as 5′ppp and single-stranded regions, were also proposed to activate PKR46, the molecular details remain unclear. It is also worth noting that certain RNAs, such as adenovirus-associated RNA 1 (REF.47), were shown to inhibit PKR through competition with other activating dsRNAs and by preventing PKR dimerization or higher-order clustering.
Oligoadenylate synthases.
As with PKR, OASes are enzymes whose catalytic activities are regulated by dsRNA. Upon dsRNA binding, OASes undergo a conformational change in the active site, allowing joining of two ATP molecules through the 2′−5′ linkage. Iteration of ATP joining leads to production of 2′−5′-linked oligomers of adenosines48,49, which then serve as second messengers to activate the downstream effector ribonuclease L (RNase L)50 (FIG. 1b). Activated RNase L degrades both cellular and viral ssRNA with limited sequence specificity, which culminates in global suppression of protein synthesis, cellular proliferation and viral replication50,51.
2′−5′-linked oligomers of adenosines.
The polymerized form of ATP with the general formula pppa(2′p5′A)n that is catalysed by 2′−5′-oligoadenylate synthase. The oligomers activate ribonuclease L (RNase L), which mediates RNA degradation.
Ribonuclease L.
(RNase L). an interferon-induced endoribonuclease that degrades RNA when activated.
The use of non-canonical oligonucleotide as a signalling molecule during antiviral response is analogous to that of cyclic GMP–AMP synthase (cGAS), an innate immune receptor that synthesizes cyclic GMP–AMP (cGAMP) upon binding foreign dsDNA. OASes and cGAS share close structural homology and belong to a family of template-independent nucleotidyltransferases (NTase). They also use a similar activation mechanism where the binding of the substrate nucleic acid induces an active site conformational change to stimulate the catalytic activity52.
Cyclic GMP–AMP synthase.
(cGAS). a cytosolic DNA sensor that activates a type I interferon response, part of the cGAS–STING DNA sensing pathway.
In humans, there are four isoforms of OASes; OAS1, OAS2 and OAS3 are enzymatically active, while OASL is not. Accordingly, OAS1, OAS2 and OAS3 have the ability to activate RNase L, while OASL does not (FIG. 1b). Instead, OASL is thought to regulate other antiviral immune pathways through diverse mechanisms, including direct binding to and activation of RIG-I53,54. OAS1, OAS2 and OAS3 respectively harbour one, two and three tandem repeats of the NTase domain. For OAS2 and OAS3, only one NTase domain is catalytically active and the other NTase domains (pseudo-NTase domains) lack the catalytic triad in the active site55,56. The pseudo-NTase domains, however, appear to play an important function as a dsRNA-binding domain, explaining the higher dsRNA affinity of OAS3 than the other two isoforms57,58. Consistent with this observation, OAS3 is the primary receptor for activating RNase L during viral infection59. OAS3 was also found to preferentially recognize long dsRNAs (more than 50 bp)55 (TABLE 1), but the mechanism of dsRNA length recognition is unclear. It also remains to be investigated what functions OAS1 and OAS2 have and whether they recognize different types of dsRNA for non-redundant antiviral functions.
NLRP1.
NLRP1 is an inflammasome-forming sensor that detects microorganisms and activates the cytokines IL-1β and IL-18 or promotes gasdermin D-mediated pore formation in the plasma membrane60 (FIG. 1c). In humans, NLRP1 relies on both RNA secondary structure and RNA length for sensing61. The carboxy-terminal leucine-rich repeat (LRR) domain and the amino-terminal death-fold domain that engages caspase 1 (NACHT) region on NLRP1 directly sense dsRNAs longer than 500 bp to activate the inflammasome pathway61 (TABLE 1). The strength of NLRP1 activation positively correlates with dsRNA length. Duplex RNA structure is required for the activity, as even long (2,500-bp) ssRNAs do not induce NLRP1 inflammasome activity61. Collectively, dsRNA length dependence appears to be a shared property among many dsRNA sensors, suggesting its importance in self versus non-self discrimination (TABLE 1).
Inflammasome.
A multiprotein complex that forms in response to a variety of inflammatory triggers (both pathogen derived and host derived). Assembly of the inflammasome often leads to activation of caspase 1, which then processes inflammatory cytokines into their mature forms and induces gasdermin pore formation.
Gasdermin D.
A member of the gasdermin family, which form pores in response to inflammasome activation. Gasdermin D pore formation leads to release of inflammatory cytokines and causes a highly inflammatory form of programmed cell death known as pyroptosis
Upon dsRNA binding, NLRP1 hydrolyses ATP using the NACHT domain, which was shown to be crucial for inflammasome activation62. Strikingly, a 15-bp dsRNA was sufficient for ATP hydrolysis by NLRP1 (REF.61). The difference in dsRNA length dependence for NLRP1 activation (500 bp) and for ATPase activity (15 bp) is of interest. How the extra length beyond 15 bp contributes to NLRP1 inflammasome activity and antiviral response, as well as the tolerance for imperfect base pairing within the dsRNAs, is worthy of further investigation. There is still an incomplete understanding of the impact of Watson–Crick base mismatches (see the section entitled Endogenous sources of RNA) or bulges or other duplex RNA structural irregularities on NLRP1 activation. In addition, it is not clear whether multiple NLRP1 molecules oligomerize on a single, long dsRNA to ensure activation. Finally, investigation of the potential contributions of other cellular factors would be useful to illuminate the complete mechanisms of NLRP1 activation.
Endogenous sources of dsRNA
dsRNAs were originally considered as PAMPs and their generation was associated solely with infection by pathogens (BOX 1). However, studies in the past two decades point to diverse cellular sources for dsRNAs and their frequent occurrence either during normal physiological process or upon various types of physiological perturbations. Just like viral dsRNA, self-derived dsRNAs activate cellular dsRNA sensors and activate antiviral innate immune pathways and cell stress responses (FIG. 3). The presence of endogenous dsRNA has been understood largely on the basis of the observation that dsRNA sensors can be activated in the absence of viral infection. Since the precise identities of the endogenous dsRNAs responsible for activation of dsRNA sensors are only beginning to be understood, we discuss here circumstances where endogenous dsRNA-mediated activation of these sensors were reported, the current understanding of the identity of these immunostimulatory endogenous dsRNAs and how they are recognized to mount the immunostimulatory response.
Fig. 3 |. Endogenous sources of dsRNA and cellular regulatory processes.
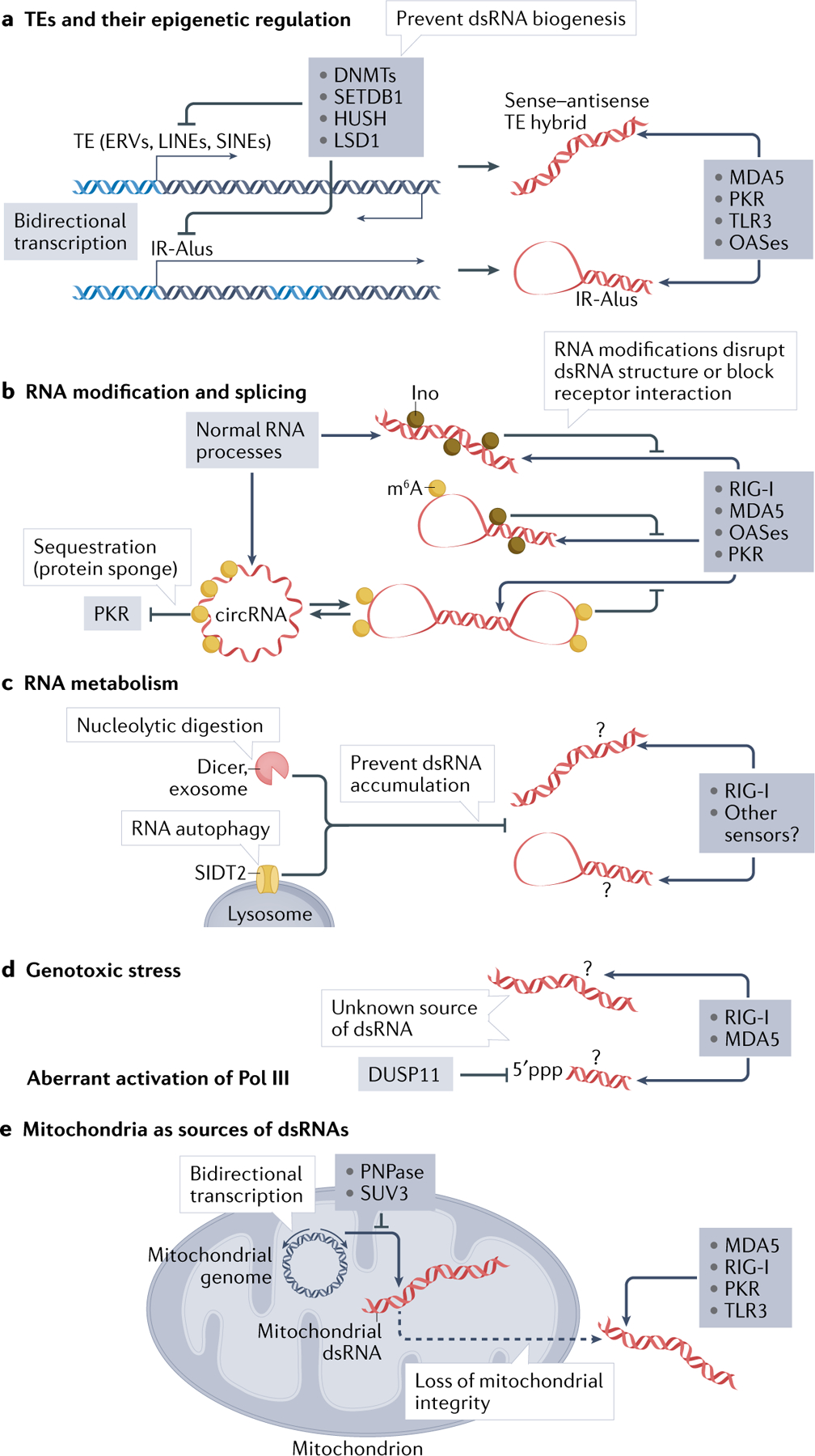
Cells have diverse endogenous sources of double-stranded RNA (dsRNA) and utilize multiple mechanisms to suppress its biogenesis and accumulation. a | Epigenetic derepression of transposable elements (TEs), such as endogenous retroviruses (ERVs), long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs), can lead to dsRNA generation. These elements can be transcribed in a bidirectional manner or as an inverted repeat, forming sense–antisense hybrid or fold-back hairpin dsRNA. Biogenesis of TE-based dsRNAs is normally suppressed by epigenetic silencing mechanisms involving DNA methyltransferases (DNMTs), the histone H3 K9 methyltransferase SETDB1, its partner the human silencing hub (HUSH) and the histone demethylase LSD1. The only exception is inverted repeat Alus (IR-Alus), some of which are constitutively produced within the 3′ untranslated region of many mRNAs. b | Once transcribed, cellular RNAs are regulated by post-transcriptional modifications, such as adenosine deamination to inosine (Ino) and N6-adenosine methylation (to produce N6-methyladenosine (m6A)), both of which disrupt the structure of dsRNA and lower its immunogenicity (shown by inhibitory arrows). Deregulation of these (and potentially other) RNA modifications can result in the recognition of normal cellular transcripts as foreign, owing to the formation of local duplex structures. Splicing inhibition can lead to an increase in the levels of dsRNAs, as a result of the increase in the levels of transcripts with retained introns, which may form double-stranded structures. Splicing is also associated with the generation of circular RNAs (circRNAs), which can form dsRNA structures more easily than their linear counterparts. On the one hand, these dsRNA structures can be recognized by RIG-I, but this is negatively regulated by m6A modification, normally present in circRNAs. On the other hand, circRNAs can also act as protein sponges and sequester protein kinase R (PKR) and prevent its activation in sterile conditions. c | RNA degradation mechanisms, such as those involving Dicer, RNA exosome complex and the lysosomal RNA transporter SIDT2, may prevent excessive accumulation of dsRNA through poorly understood mechanisms (question marks). d | Genotoxic stress (for example, resulting from exposure to ionizing radiation) and aberrant activation of RNA polymerase III (Pol III; for example when MYC is activated in cancer) can promote biogenesis of RLR-stimulatory dsRNAs, but the precise nature of these dsRNAs remains unclear (question marks). These may be aberrantly processed RNA components of the spliceosome (U1 and U2 small nuclear RNAs) or products of Pol III which contain 5′-triphosphate (5′ppp). The recognition of the latter could be regulated by the phosphatase DUSP11, which can remove 5′ppp. e | Mitochondria are a rich source of dsRNA as mitochondrial RNAs are produced by bidirectional transcription of the circular DNA. Normally, the level of mitochondrial dsRNA is regulated by the mitochondrial RNA degradosome, which includes the nuclease polynucleotide phosphorylase (PNPase) and the helicase SUV3. During mitochondrial dysregulation, mitochondrial dsRNA can gain access to cytosolic dsRNA sensors and activate them through a poorly understood mechanism. OAS, oligoadenylate synthase; TLR3, Toll-like receptor 3.
Dysregulated epigenetic control.
Cancer chemotherapy has long been known to trigger antiviral signalling pathways, but its mechanism of immune activation had been unclear. Multiple studies with 5-aza-2′-deoxycytidine (5-aza-CdR), an inhibitor of DNA methyltransferases (DNMTs) and a common chemotherapeutic agent, showed that its therapeutic efficacy is in part mediated by activation of a broad range of dsRNA sensors (including MDA5, TLR3 and OASes), indicating that derepression of transcription achieved by suppressing DNA methylation can lead to dsRNA formation63–66 (FIG. 3a). Indeed, 5-aza-CdR strongly stimulates transcriptional upregulation of various types of transposable elements (TEs) that are otherwise epigenetically silenced. These include endogenous retroviruses (ERVs), long interspersed nuclear elements (LINEs) and short interspersed nuclear elements. Some studies proposed that dysregulated, bidirectional transcription of these genomic regions leads to production of sense and antisense transcripts that can then hybridize to form dsRNA63,64. However, the concomitant presence of sense and antisense transcripts does not necessarily result in RNA duplex formation. In a more recent study17, MDA5 activation was found to be driven largely by Alu repeats that are juxtaposed in the inverted configuration (inverted repeat Alu elements (IR-Alus)), which fold back and form long (~300-bp) hairpins65. While ~300 bp is below the usual length threshold for MDA5 stimulation, it is likely that the abundance of Alu dsRNA in cells compensates for the suboptimal length for MDA5 activation.
Transposable elements.
(TEs). DNA sequences that can change their position within a genome, sometimes creating or reversing mutations and altering the cell’s genetic identity and genome size.
Endogenous retroviruses.
(ERVs). endogenous viral elements in the genome that closely resemble and can be derived from retroviruses. These elements constitute up to 8% of the human genome.
Long interspersed nuclear elements.
(LINEs). a group of retrotransposons that are not long terminal repeats. LINEs constitute ~21% of the human genome. They are transcribed into mRNA and translated into a protein that acts as a reverse transcriptase, which makes a DNA copy of the LINE RNA, which then can be integrated into the genome at a new site.
Short interspersed nuclear elements.
Non-autonomous, non-coding retrotransposons that are capable of copying and pasting themselves into another region of the genome via an RNA intermediate and the action of reverse transcriptase. They constitute ~15% of the human genome.
Alu repeats.
A clustered arrangement of Alu (a kind of short interspersed nuclear element), which can often be transcribed within a single transcript.
Alu is a primate-specific TE, constituting ~10% of the genome. While most Alus are transcriptionally silenced through various epigenetic means, including DNA methylation, a significant number of Alus are normally transcribed within untranslated regions (UTRs) of many common mRNAs. Earlier investigations of MDA5 with gain-of-function mutations causing inflammatory diseases showed that IR-Alus in 3′ UTRs of endogenous mRNAs are the main source of dsRNA for stimulating MDA5 in the absence of infection17. In the case of 5-aza-CdR treatment, IR-Alus that stimulate MDA5 originate from intergenic and intronic regions downstream of CpG islands, rather than those embedded within mRNA UTRs65. Regardless of the specific origin of IR-Alus, these studies together suggest that duplex formation associated with IR-Alus is mediated largely by in cis hairpin formation between adjacent Alus, rather than in trans hybridization between separate Alus or sense/antisense transcripts containing these elements (FIG. 3a). Similarly, IR-Alus have been linked to PKR activity in the absence of infection67, suggesting that IR-Alus may be the major source of dsRNA molecules that activate a broad range of dsRNA sensors.
Similarly to the inhibition of DNMTs, perturbation of the activity of other epigenetic modifiers has been shown to lead to the activation of dsRNA sensors. For example, trimethylation of histone H3 K9 (H3K9) is an epigenetic mark typically associated with transcriptional suppression of TEs and heterochromatin regions. Genetic depletion of an H3K9 methyltransferase (SETDB1)68 or its partner (the human silencing hub (HUSH))69 causes induction of a broad range of TEs and concomitant activation of RLRs signalling (FIG. 3a). Similarly, inhibition of the histone demethylase LSD1 (also known as KDM1A) activates TLR3 and RLRs70 (FIG. 3a). LSD1 can act as a transcription co-repressor of ERVs through removal of methyl groups on H3K4 (REF.71), but can also function as a transcription activator by demethylating H3K9 (the suppressive mark)72. This raises the question of whether the effect of the LSD1 inhibition on the dsRNA sensors is through increased methylation of H3K4, increased methylation of H3K9 or both. Regardless, these studies collectively suggest that epigenetic regulation is a key mechanism for suppressing biogenesis of endogenous dsRNAs, in particular those formed by TEs (FIG. 3a).
Changes to RNA modification.
RNA modifications have been shown to regulate RNA metabolism, secondary structure and protein association. Defects in the installation or recognition of RNA modifications cause self RNAs to appear foreign, which induces immune signalling (FIG. 3b). Here, we discuss two main types of RNA modifications — adenosine deamination and methylation — both of which affect local dsRNA structure and decrease the recognition of endogenous RNAs by dsRNA sensors.
ADAR1 is a dsRNA-specific adenosine deaminase and one of the three adenosine deaminases in vertebrates (ADAR1, ADAR2 and ADAR3)73–75. Unlike ADAR2 and ADAR3, ADAR1 is ubiquitously expressed and has an important role in suppressing basal innate immune activity. Upon dsRNA binding, ADAR1 deaminates adenosines within the duplex structure with minimal sequence dependence. Deaminated adenosine (inosine) can impact RNA structure and function in at least two major ways (FIG. 3b). First, inosine is recognized as guanosine by reverse transcriptase and by the ribosome, and is therefore mutagenic. By introducing mutations, ADAR1 can thus antagonize viruses in the case of infection76. However, the role of ADAR1 in host RNA biology seems largely independent of this mutagenic effect as most ADAR1 modifications in host RNAs occur in non-coding regions77. One of the major functions of ADAR1-mediated A-to-I editing seems to be through its effect on dsRNA structure78,79. A-to-I editing replaces the A•U pairs by less stable I•U pairs, destabilizing the dsRNA structure and thereby limiting recognition by dsRNA sensors. Therefore, ADAR1 deficiency leads to constitutive activation of MDA5, PKR and OASes67,80–83 (discussed further in the section entitled Consequences of endogenous dsRNA).
Studies showed that IR-Alus in 3′ UTRs are the major substrates for ADAR1 in human cells77,84,85. Some IR-Alus are highly edited, with more than ~50% of the adenosines capable of being edited85. The correlation between the editing efficiency and proximity between adjacent Alus in the inverted orientation is consistent with the notion that IR-Alus form a duplex through intrastrand Alu–Alu interaction as discussed earlier herein84. Similarly to the response of MDA5 with gain-of-function mutations, IR-Alus within the mRNA 3′ UTR were found to be responsible for activation of wild-type MDA5 under ADAR1 deficiency17. IR-Alus were also proposed to be primarily responsible for constitutive activation of PKR and OASes upon ADAR1 deletion67,82. Collectively, these results suggest that IR-Alus are an important source of dsRNA that can breach the immunological threshold, and that ADAR1 is one of the major regulators that suppresses immunostimulatory activities of IR-Alus and guards immune homeostasis.
N6-Methyladenosine (m6A) is the most abundant internal modification on mRNAs and long non-coding RNAs in eukaryotes86,87 (FIG. 2). m6A can regulate RNA splicing, stability, cellular localization, translation, immune stimulation and secondary structure86,88–91 (FIG. 3b). The diverse effects of m6A are caused by the modification being recognized by a plethora of ‘reader’ proteins. Other m6A-specific machinery includes ‘writers’ that install and ‘erasers’ that remove the modification. Dysregulation of m6A modification levels has been correlated with various diseases, including autoimmune disorders, cancer, metabolic disorders and neurological disorders88,92.
The m6A RNA modification is essential to prevent haematopoietic failure and perinatal lethality during murine fetal development93. The lack of m6A induces the formation of dsRNAs and activation of the dsRNA sensors (FIG. 3b). The dsRNAs are predominantly protein coding and have extensive m6A modifications in their native state. How the regulation of m6A modification contributes to the secondary structure for the dsRNAs remains to be investigated. Potentially, m6A could act as a structural switch on these RNAs where the modification inhibits base pairing; in the absence of m6A, the transcript becomes double-stranded91.
m6A could also regulate TE expression, but the effects of m6A on TE levels and function differ depending on the species and cell type. In mouse embryonic stem cells, the m6A modification on ERVs associates with the m6A-reader protein YTHDF2, which leads to the destabilization of ERVs and prevention of dsRNA accumulation in the cell. However, TEs in murine fetal liver had similar expression in both low-m6A-level and wild-type conditions93. Another study in murine embryonic stem cells found that m6A on TEs interacts with YTHDC1 (REF.94) and the modified RNAs associate with chromatin to affect downstream gene expression and the transcription rate. Differences between these studies could be attributed to cell-specific m6A modification patterns or the different approaches to deplete cellular m6A levels.
In addition to modifying host transcripts, m6A also modifies viral RNAs. Positive-strand RNA viruses (for example, HIV, hepatitis C virus, Zika virus and enterovirus 71), negative-strand segmented RNA viruses (for example, influenza virus) and negative-strand non-segmented RNA viruses (for example, human metapneumovirus, Sendai virus and vesicular stomatitis virus) have all been characterized to contain the m6A modification on their RNAs95–98. The m6A modification on viral RNAs enables them to evade host immune surveillance by weakening the association between the immune sensor and the modified RNA, which leads to lowered immune signalling and interferon production. Typically, RIG-I is the main cytosolic receptor that senses viral dsRNA, in a manner dependent on m6A, while MDA5 also seems to be involved, albeit to a lesser extent95. Similarly to viral dsRNAs, m6A also prevents recognition of endogenous circular RNAs (circRNAs) by RIG-I (see the section entitled Splicing inhibition).
Circular RNAs.
(circRNAs). single-stranded RNas where the 5′ and 3′ ends are joined through a phosphodiester bond.
In addition to inosine and m6A, more than 170 other RNA modifications have been identified thus far, but their roles are still mostly unknown99. Some of these RNA modifications have the potential to affect the formation of dsRNAs and/or their recognition by immune receptors. The incorporation of pseudouridine, 2-thiouridine or 5-methylcytidine (FIG. 2) onto exogenously introduced RNAs (such as therapeutic mRNA) allows them to evade detection by TLRs, RIG-I and PKR100. By contrast, 5-methyluridine activates PKR, which reveals that there are differences in how immune receptors respond to the various RNA modifications100.
Splicing inhibition.
Similarly to the use of DNMT inhibitors that induce the expression of ERVs, use of small molecules that block proper spliceosome activity in conjunction with MYC hyperactivation leads to dsRNA accumulation in the cytoplasm and promotes antiviral-like responses101. Cells treated with spliceosome inhibitors increase the levels of transcripts with retained introns, and those transcripts form double-stranded structures. Both RIG-I and MDA5 detect the induced dsRNAs101. Anti-J2 enrichment, which captures dsRNAs greater than 40 bp in length102,103, revealed that both transcripts with introns that lack retrotransposons and those that have Alus form double-stranded secondary structures. A major question is how do the transcripts with retained introns bypass cellular quality control, including nuclear retention and decay as well as cytoplasmic nonsense-mediated decay? One possibility could be that the stress of inhibiting an essential cellular process, splicing, on top of MYC amplification significantly impacts the cell’s ability to monitor and degrade mis-spliced transcripts. An alternative and mutually inclusive possibility could be that the transcripts that are inappropriately spliced may encode proteins that are involved in the quality control of cytoplasmic RNAs. Dysregulation of splicing could affect both potential pathways to enable the transcripts with retained introns to escape and accumulate in the cytoplasm.
MYC.
A family of regulatory genes and proto-oncogenes that code for transcription factors. Many types of cancers have dysregulated MYC levels and activity.
Anti-J2 enrichment.
Use of the anti-double-stranded RNA (dsRNA) J2 antibody to capture dsRNAs that are longer than 40 bp.
Nonsense-mediated decay.
An evolutionarily conserved mechanism of degradation of mRNA species with a premature termination codon.
Deregulation of circRNAs.
circRNAs are a class of endogenous transcripts found throughout the eukaryotic kingdom. The spliceosome produces circRNAs from pre-mRNAs by back-splicing the end of one exon to the beginning of a previous exon to form a covalent loop104. Even though circRNAs are identical in primary sequence to their linear RNA counterparts, except for the back-splice junction, emerging studies show that endogenous circRNAs and linear RNAs have different secondary structures and RNA modification patterns, which lead to differential recognition by nucleic acid sensors and immune receptors105–107.
Endogenous circRNAs are more likely to form imperfect short RNA duplexes (16–26 bp) than their corresponding linear RNAs107. circRNAs have been shown to function as targets of RIG-I in the presence of K63-linked polyubiquitin chains to initiate immune signalling. However, the m6A modification — normally present on endogenous circRNAs — interacts with the reader protein YTHDF2, which prevents the recognition by RIG-I105 (FIG. 3b). Therefore, even though mammalian cells contain up to thousands of endogenous circRNAs108, they are not under a state of constant inflammation and immune stress because RIG-I does not associate with m6A-modified circRNAs (FIG. 3b). Indeed, the addition of the m6A modification to exogenous circRNAs masks their ‘non-selfness’ to significantly reduce their binding to RIG-I and subsequent immune signalling105.
The increase in dsRNA structure as compared with the linear transcripts enables the circRNAs to associate with PKR. Remarkably, this interaction does not lead to PKR activation, presumably due to the limited size of the duplexed secondary structure. Instead, the circRNA–PKR interaction was found to sequester PKR and prevent its activation in sterile conditions107. Upon stimulation of cells with exogenous dsRNA or viral infection, RNase L degrades circRNAs globally, leading to the release and activation of PKR for innate immune response. In vitro binding assays between immune-sensing receptors and linear RNA or circRNA revealed that the dsRNA-binding domain of PKR is required for interaction and recognition of circRNA. Since these binding assays were conducted solely with the protein and the RNA, the presence of other cellular factors, including proteins that associate with endogenous circRNAs, may provide further insight into the recognition of circRNAs by PKR in physiological conditions and in disease. For example, sequestration of PKR by circRNAs has been suggested to prevent the initiation of autoimmune diseases such as systemic lupus erythematosus (SLE)107. Accordingly, overexpression of circRNAs with double-stranded secondary structure in peripheral blood mononuclear cells from patients with SLE reduced PKR autoactivation and expression of type I interferon. Whether other autoimmune diseases have a high aberrant activation of PKR due to lack of circRNA binding is also of interest.
Systemic lupus erythematosus.
(SLE). an autoimmune disease that causes widespread inflammation and tissue damage in the affected organs. it can affect the joints, skin, brain, lungs, kidneys and blood vessels.
Peripheral blood mononuclear cells.
Any peripheral blood cell having a round nucleus, which includes lymphocytes and monocytes.
Defects in RNA processing and degradation.
Multiple lines of evidence suggest that cells may accumulate immunostimulatory dsRNAs when there is a defect in RNA processing and degradation (FIG. 3c). While most ribonucleases in cells are inefficient at degrading dsRNAs, Dicer contains an RNase III domain, which specializes in cleaving dsRNAs. Dicer is best known for its role in processing microRNA precursors to generate mature microRNAs, but it can also process long dsRNAs from both endogenous and exogenous sources and generate small interfering RNAs109,110. Investigation of the sources of endogenous small interfering RNAs revealed that many of them are from sense–antisense hybrids of TEs or pseudogenes, or transcripts containing inverted repeats of retroelements. For example, LINE-1 has been shown to be subject to Dicer-mediated processing and to be the source of endogenous small interfering RNAs, which in turn mediate RNA-directed DNA methylation and silencing111,112. Therefore, defects in Dicer function can lead to accumulation of LINE-1 transcripts, presumably in the form of long dsRNAs112,113.
Dicer.
An endoribonuclease specialized in processing double-stranded RNA. it is typically involved in biogenesis of small regulatory RNAs such as microRNAs and small interfering RNAs.
Pseudogenes.
DNA sequences that resemble functional genes, but are inactive due to mutations.
Dicer deficiency was also shown to cause accumulation of Alu RNAs, and was proposed to cause age-related macular degeneration, a disease characterized by inflammation in the retinal pigment epithelium114,115. Accumulation of Alu RNAs in Dicer-deficient cells was reported to activate the NLRP3 inflammasome115 and cGAS116 through unclear mechanisms. Unlike NLRP1, NLRP3 has not been shown to directly recognize RNA, raising the question of whether NLRP3 is a direct sensor or whether there is another RNA sensor that directly detects Alu accumulation. Intriguingly, Alu upregulation induced by the Dicer deficiency was blocked by RNA polymerase III (Pol III) inhibition. Considering that Alu RNA synthesized by Pol III contains a single Alu (not Alu repeats) within individual transcripts, this observation raises the question as to what features of Pol III-transcribed Alu RNA are recognized by Dicer and cause inflammation.
Dicer was also proposed to regulate the level of RNAs with trinucleotide repeats and possibly contribute to the cause of triplet repeat expansion disorders. Triplet repeat expansion disorders, such as Huntington disease and spinocerebellar ataxia, are neurological and neuromuscular disorders that are associated with expansion of a trinucleotide repeat (in particular CNG) within either coding or non-coding regions of the causal genes. Expanded CNG repeats are partially self-complementary and thus can form a hairpin, a dimer and higher-order oligomers. It was found that Dicer can recognize and cleave RNAs containing a trinucleotide repeat expansion, producing small CNG-repeated RNAs117. Therefore, impaired function of Dicer may lead to accumulation of RNAs harbouring trinucleotide repeats, which may cause RNA-mediated toxicity that could potentially involve dsRNA sensor activation118.
SKIV2L is an RNA helicase and a component of the RNA exosome responsible for turnover of the bulk of cytosolic RNAs. SKIV2L assists the exosome by disrupting RNA secondary structure using the helicase activity, which is necessary for degrading RNA with secondary structures119. It was found that a defect in SKIV2L causes hyperactivation of RIG-I and is possibly involved in the pathogenesis of trichohepatoenteric syndrome, a disease associated with mutations in SKIV2L120. However, the identity of the RIG-I-stimulatory RNAs and the mechanism of RIG-I activation remain unclear.
RNA exosome.
A multisubunit protein complex that catalyses 3′-to-5′ processing or degradation of cellular RNAs
Trichohepatoenteric syndrome.
An inherited autosomal recessive condition that affects the hair, liver and intestines. Can be caused by mutations in SKIV2L or TTC37.
In addition to exosome-mediated degradation, cytosolic RNAs can be cleared through a recently described process termed ‘RNautophagy’, where RNA is directly taken up by lysosomes through the RNA transporter SIDT2 (REF.121) (FIG. 3c). SIDT2 is an orthologue of the dsRNA transporter SID-1, and was found to transport dsRNA in a bidirectional manner122. Whether deficiency of SIDT2 has a role in accumulation of endogenous dsRNAs would be an interesting area of future investigation.
Dysregulated RNA Pol III.
Unlike RNA Pol II transcripts, which are generally processed to contain a 7-methylguanosine cap at the 5′ end, many RNA Pol III transcripts, such as U6 small nuclear RNAs (snRNA), 5S ribosomal RNA, 7SK RNA and 7SL RNA, retain 5′ppp as in nascent transcripts123, although some acquire a monomethyl group on the γ-phosphate of 5′ppp124. Many Pol III transcripts also contain RNA secondary structures, which together with 5′ppp confers them with a potential to stimulate RIG-I (FIG. 3d). Consistent with this notion, several studies suggest that Pol III-transcribed RNAs can activate RIG-I in response to a variety of stimuli. For example, 7SL RNA, a component of the signal recognition particle (SRP) ribonucleoprotein complex, was found to activate RIG-I in breast cancer cells, causing tumorigenic inflammation125. Although 7SL RNA is abundant in cells, it does not normally activate RIG-I because it is bound by the protein partner SRP9/14, which shields 5′ppp and secondary structure. In breast cancer cells, ‘naked’ 7SL RNA accumulates as a result of hyperstimulation of Pol III through MYC activation, without the corresponding increase in the level of SRP9/14. Similarly, other Pol III transcripts were also reported to activate RIG-I in response to infection with several viruses, such as HSV-1, influenza A virus and Kaposi sarcoma-associated herpesvirus126–128. More recently, Pol III-mediated RIG-I activation was found to be regulated by the phosphatase DUSP11, which can remove 5′ppp from both endogenous and exogenous RNA128,129.
Signal recognition particle (SRP) ribonucleoprotein complex.
A ribonucleoprotein complex that recognizes the signal sequence of a nascent peptide and targets it to the endoplasmic reticulum in eukaryotes and the plasma membrane in prokaryotes.
Genotoxic stress.
Ionizing radiation and other genotoxic stresses can potently activate antiviral signalling pathways, which have recently emerged as important part of cancer therapies. Multiple studies revealed that ionizing radiation-induced antiviral signalling is associated with activation of the cGAS pathway130–133. However, more recent studies suggested that both RIG-I and MDA5 also contribute to ionizing radiation-mediated immune activation in a cell type-dependent manner134,135 (FIG. 3d). Inhibition of the cell cycle checkpoint accelerates immune activation through both the cGAS pathway and the RLR pathway134,135. In an independent study, RIG-I, but not MDA5, was found to be activated upon ionizing radiation treatment136. Germline deletion of RIG-I, but not MDA5, protected mice from death following total body irradiation. LGP2, a third member of the RIG-I-like receptor family, was also found to be important for conferring radioresistance, consistent with its role in inhibiting RIG-I137.
Cell cycle checkpoint.
A checkpoint in the eukaryotic cell cycle at which the cell monitors the progression of cell division and decides whether or not to move forward.
Exactly which RNAs activate RIG-I or MDA5 upon ionizing radiation exposure remains unclear. The cell-type dependence of the activated receptor suggests that production of immunostimulatory ligands for each receptor differs depending on the cellular context. A study suggests that RNA components of the spliceosome, U1/U2 snRNAs136, are co-purified with RIG-I upon ionizing radiation treatment. U1/U2 snRNAs are synthesized by Pol II, and thus are co-transcriptionally processed to remove 5′ppp. It is unclear whether RIG-I activation is mediated by fully mature U1/U2 snRNAs or improperly processed RNA still retaining 5′ppp. In another study, it was proposed that RIG-I senses Pol III transcripts of certain cellular DNA fragments generated during ionizing radiation stress134. The identity of ionizing radiation-induced RNAs that activate RIG-I and MDA5 and their cell-type dependence need further investigation.
Mitochondrion-derived dsRNAs.
Mitochondria have traditionally been thought to be the major organelles for regulating cell metabolism and apoptosis. However, more recent studies have suggested their broader roles in cell stress response and innate immunity138. These include the role of mitochondrial DNA in activating cGAS116,139 and the role of the mitochondrial unfolded protein response in activating the cytosolic integrated stress response140. More recently, mitochondrial RNA has emerged as yet another ‘danger’ signal that indicates cell dysfunction and that can alert the innate immune system.
Unfolded protein response.
An evolutionarily conserved adaptive reaction that reduces unfolded protein load to maintain cell viability and function. Depending on the cell type involved and the nature of the stress stimuli, unfolded protein response signalling has different consequences and kinetics.
Integrated stress response.
An evolutionarily conserved cellular stress response that downregulates protein synthesis and upregulates specific genes in response to internal or environmental stresses.
Mitochondrial RNAs are transcribed from the circular mitochondrial genome in a bidirectional manner141 (FIG. 3e). Both RNA strands (H and L strands) are synthesized in long polycistronic precursor transcripts, which are processed into mature transcripts by endoribonucleolytic cleavage events. Despite these processing steps being in place, studies found that a detectable level of RNA exists as full-length RNA for both strands, and that sense–antisense hybrids are present142–145. The level of mitochondrial dsRNAs increases upon depletion of polynucleotide phosphorylase (PNPase) or SUV3, components of the mitochondrial RNA degradosome144. Intriguingly, deficiency in PNPase, but not SUV3, leads to both accumulation of mitochondrial dsRNAs and their leakage into the cytoplasm. This is in line with previous reports implicating PNPase in mitochondrial RNA transport as well as degradation146,147. As a result, knockdown of PNPase, but not SUV3, activates MDA5 (REF.144). A similar hyperinflammatory phenotype was observed in PNPase-deletion mice and cells derived from patients harbouring biallelic hypomorphic mutations in PNPT1 (the gene encoding PNPase)144.
Mitochondrial dsRNA can also activate PKR, but this does not require deficiency of PNPase or SUV3 (REF.145). Intriguingly, while the majority of PKR functions in the cytoplasm, a subset of PKR was found in the mitochondrial matrix and intermembrane space, explaining how PKR can be activated by mitochondrial dsRNAs without their erroneous leakage into the cytoplasm145,148. However, it remains unclear how PKR enters the mitochondria, and how activated PKR exits mitochondria to phosphorylate eIF2α.
Mitochondrial dsRNA was also proposed to mediate the innate immune response during ionizing radiation exposure149. Ionizing radiation was found to damage both nuclear and mitochondrial DNA, and the latter can lead to production of aberrant dsRNAs and their leakage into the cytoplasm through loss of mitochondrial membrane integrity. This observation raises the question as to whether other genotoxic stresses, such as those mediated by DNA-modifying chemotherapeutic agents, can also activate RLRs through similar mechanism involving mitochondria. The specific identity of the RIG-I ligand again requires further studies.
Consequences of endogenous dsRNAs
In the previous section, we described various physiological conditions activating dsRNA sensors where endogenous dsRNA is shown or suspected to mediate the virus-independent, ‘sterile’ immune activation. As discussed earlier herein, in many cases, the exact identity of the dsRNA remains speculative and requires further research. In this section, we focus on biological consequences of such sterile immune activation involving dsRNA sensors.
There are at least three categories of situations where sterile activation of dsRNA sensors has been observed (FIG. 4). First, sterile activation of dsRNA sensors can occur in a controlled fashion during normal physiological processes and can have important roles in maintaining cellular homeostasis. Second, sterile immune activation, when occurring in an uncontrolled and chronic fashion, can cause a broad range of disorders, including inflammatory disorders. Finally, the immune functions of dsRNA sensors can be leveraged for therapeutic purposes, where therapeutic agents induce endogenous dsRNA production to activate innate immune responses. Here, we describe examples of all three cases.
Fig. 4 |. Consequences of dsRNA recognition.
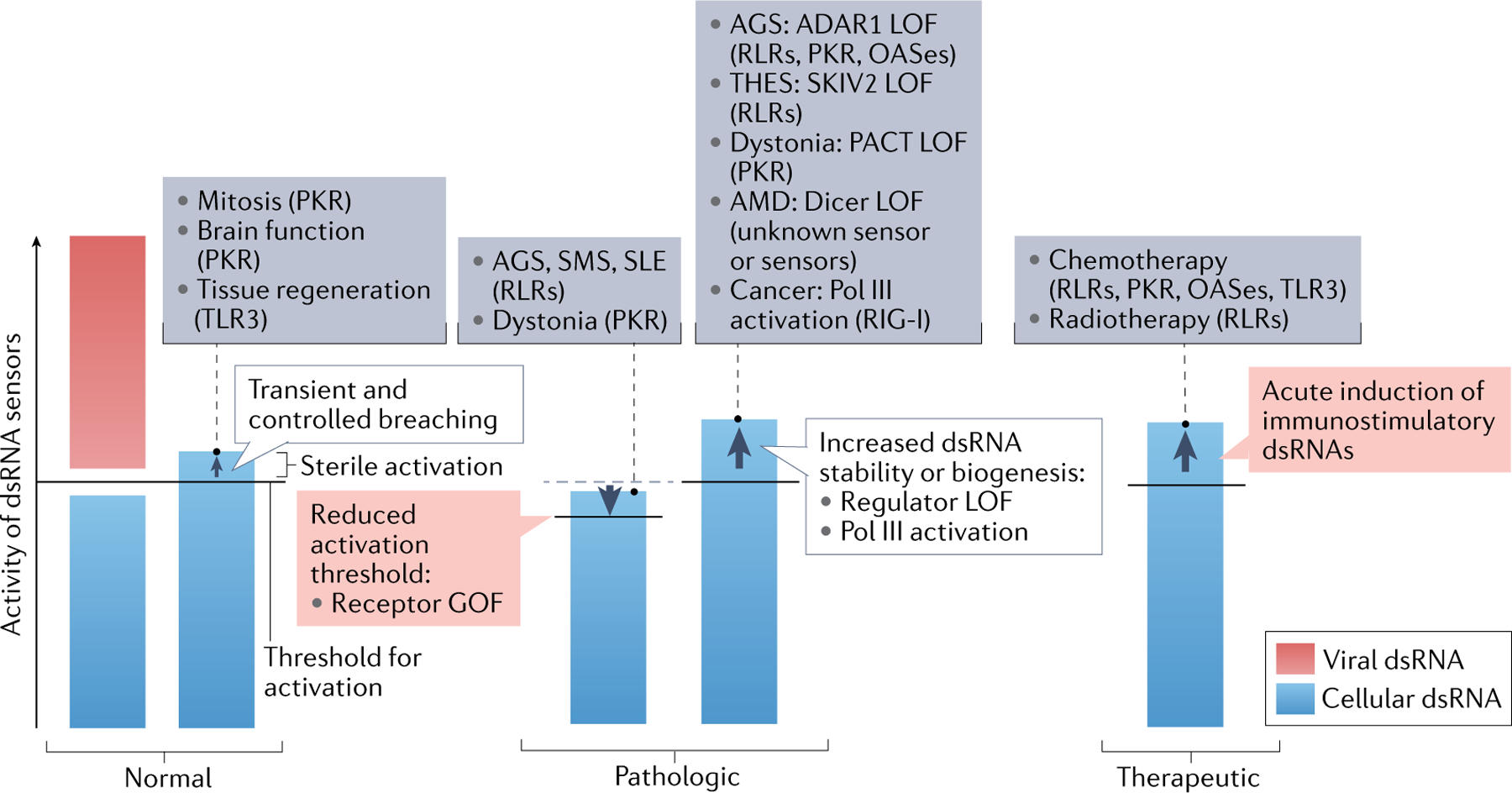
Sterile activation of double-stranded RNA (dsRNA) sensors can occur in normal, pathologic and therapeutic conditions. Here, we use an immunological threshold model to summarize examples in each category. dsRNA sensors have an evolutionarily optimized activation threshold that allows the receptors to tolerate a certain level and certain kinds of dsRNAs (for example, dsRNA with short and imperfect complementarity is normally tolerated by MDA5), while those RNAs beyond the threshold (for example, viral dsRNA) would activate the dsRNA sensors. In normal conditions, the levels of cellular dsRNAs are well below the activation threshold of dsRNA sensors. However, there are cases where a subset of cellular dsRNAs breach the threshold in a transient and controlled fashion, and innate immune functions of the dsRNA sensors are integrated into the normal biological processes. This includes protein kinase R (PKR) activation during mitosis, neuronal excitation in the brain and Toll-like receptor 3 (TLR3) activation during tissue regeneration in the skin. By contrast, constitutive and uncontrolled breaching of the tolerance threshold leads to pathogenesis of immune disorders and other diseases. This can occur through gain-of-function (GOF) mutations of the dsRNA sensor, which lowers the threshold, leading to misrecognition of otherwise inert cellular dsRNA. Such cases include Aicardi–Goutières syndrome (AGS), Singleton–Merten syndrome (SMS) and systemic lupus erythematosus (SLE) caused by GOF RIG-I-like receptors (RLRs) and dystonia caused by GOF PKR. Alternatively, similar diseases can be caused by loss-of-function (LOF) mutations in the regulators. For example, LOF in ADAR1 causes AGS through constitutive activation of RLRs, PKR or oligoadenylate synthases (OASes), LOF in SKIV2 causes trichohepatoenteric syndrome (THES), where constitutive activation of RIG-I is thought to contribute, LOF in PACT causes dystonia through constitutive activation of PKR and LOF in Dicer has been associated with age-related macular degeneration (AMD), although in the last case the exact sensor involved has not been determined. In addition, increased generation of immunostimulatory dsRNAs has been observed in cancer, where aberrant activation of RNA polymerase III (Pol III) downstream of the activity of the oncogenic protein MYC leads to increased generation of RNAs prone to forming secondary structures and RNAs containing 5′-triphosphate, which are recognized by RIG-I. Finally, immune functions of dsRNA sensors can be leveraged in cancer immunotherapy. Chemotherapy and radiotherapy were shown to confer anticancer efficacy partly by inducing biogenesis of immunostimulatory dsRNAs, which activate a broad range of dsRNA sensors.
Normal physiological process.
PKR, TLR3 and RLRs were found to be activated in several normal physiological conditions. During mitosis, PKR is activated and regulates the levels of multiple mitotic factors, likely through global suppression of protein synthesis and JNK phosphorylation, to ensure proper progression of mitosis145,148. PKR also regulates neuronal excitation during normal brain functions by suppressing the level of IFNγ and promoting GABAergic synaptic transmission150. Similarly, sterile activation of TLR3 by dsRNAs released from dead cells and consequent activities of downstream mediators, such as IL-6 and STAT3, are critical for skin regeneration following tissue damage151. Finally, activation of RLRs by TE RNA was found to occur during the formation of haematopoietic stem and progenitor cells, and the consequent inflammatory signalling is important for development of these cells152. These observations suggest dsRNA can serve as a cellular signalling molecule to coordinate innate immune response with various steps of normal physiological processes.
JNK.
JUN N-terminal kinase (JNK) that plays key roles in many cellular stress and inflammatory signalling pathways.
Disease process.
Earlier studies showed that mice with a single point mutation (G821S) in MDA5 display multi-organ inflammatory symptoms153. In humans, gain-of-function mutations in MDA5 and RIG-I were shown to cause a broad spectrum of autoinflammatory diseases, such as Aicardi–Goutières syndrome, Singleton–Merten syndrome, neuroregression and spastic dystonia154–159. In many cases, the RNA-binding activity of the receptors is important, suggesting the role of endogenous dsRNAs in driving the pathogenesis17,154,160–162. A detailed mechanistic study showed that the mutations increase the ability of MDA5 to form filaments on cellular dsRNAs, which are normally inert due to their structural irregularities, such as mismatches and bulges17. For RIG-I, wild-type RIG-I uses a kinetic proofreading mechanism to prevent its oligomerization on self dsRNAs, and the disease mutations impair this regulatory function, allowing activation by self dsRNAs161,162. The diseases mentioned above and the mutations involved are rare. More common single-nucleotide polymorphisms in MDA5 have been linked to type 1 diabetes163,164 and SLE165,166, where the impact of an individual single-nucleotide polymorphism is probably subtler and context dependent.
Aicardi–Goutières syndrome.
An inherited encephalopathy that affects newborns and usually results in severe mental and physical handicap. it can be caused by gain-of-function mutations in IFIH1 (the gene encoding MDa5). Loss-of-function mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1 and ADAR1 were also shown to cause Aicardi–Goutières syndrome.
Singleton–Merten syndrome.
A rare autoimmune disorder characterized by tooth abnormalities, calcifications in the aorta and certain valves of the heart, and osteoporosis in the hands and feet. it can be caused by a gain-of-function mutation in IFIH1 (the gene encoding MDA5) or DDX58 (the gene encoding RIG-I).
Single-nucleotide polymorphisms.
Germline substitutions of a single nucleotide at a specific position in the genome, the most common type of genetic variation among people.
Aberrant activation of PKR has also been implicated in several diseases, including dystonia167,168, SLE107, Alzheimer disease169,170 and Huntington disease171. However, dystonia is one of the few cases with clear genetic evidence supporting the role of PKR in disease pathogenesis. In a subset of patients with dystonia, mutations were found in the genes encoding PKR and a PKR suppressor, PACT167,168, both of which lead to the constitutive activation of PKR and the consequent integrated stress response involving translational suppression. Studies of dietary and genetic obesity mouse models suggest that excessive nutrient leads to aberrant activation of PKR in adipose and liver tissues, and PKR inhibition ameliorates inflammation triggered by metabolic stress172.
Mutations in the coding and non-coding regions of NLRP1 have been linked to different types of disease, including metabolic disorders, cancer and autoimmune disorders173,174. NLRP1 has seemingly opposing effects by attenuating or augmenting certain cellular processes in a tissue-specific manner. For example, NLRP1 promotes inflammation and exacerbates neurological disorders, cardiopulmonary diseases and cancer, but NLRP1 also protects the gastrointestinal tract and decreases inflammation by modulating the microbiota composition173. Polymorphisms that lead to constitutive activation of NLRP1 have been associated with an increased risk of autoimmune diseases, including vitiligo, psoriasis and rheumatoid arthritis173. In these cases, however, the potential role of endogenous dsRNA remains unclear, given that NLRP1 can be activated by other PAMPs besides dsRNAs.
Vitiligo.
An autoimmune disease where the immune system attacks the melanocytes in the skin.
Psoriasis.
An autoimmune disease that speeds up the growth cycle of skin cells and causes red, itchy scaly patches over the body.
Unlike the situations described above, sterile immune activation can also occur without any alteration in dsRNA sensors, but through alteration in endogenous dsRNAs. For example, loss-of-function mutations in ADAR1 lead to an increase in the structural integrity of cellular dsRNAs and thus their stimulatory activity against MDA5, PKR and OASes17,67,80,82,83, causing Aicardi–Goutières syndrome175. Consistent with the importance of A-to-I editing, knock-in of catalytic-deficient ADAR1 in mice also results in MDA5 activation and a similar inflammatory phenotype as in ADAR1 depletion80. Although the mechanism is less clear, diseases caused by deficiency of SKIV2 and Dicer (trichohepatoenteric syndrome and age-related macular degeneration, respectively) can be considered in a similar disease category, where altered RNA leads to abnormal cellular response. Finally, tumorigenic inflammation caused by hyperactivation of Pol III and RIG-I is yet another case where altered cellular dsRNA population leads to disease pathogenesis, in this case breast cancer pathogenesis125.
Therapeutic applications.
Whereas chronic inflammation can promote tumour formation, it has become increasingly clear that acute inflammation can be an effective way to promote anticancer immunity. Studies suggest that some cancer cells are particularly susceptible to dsRNA sensor-mediated immune response176–179. Additionally, traditional chemotherapy and radiotherapy, which cause epigenetic dysregulation63–66 and genotoxic stress17,67,136,149, were also shown to activate multiple dsRNA receptors, including MDA5, RIG-I, PKR, OASes and TLR3 (as described in the section entitled Endogenous sources of dsRNA). Most importantly, the clinical efficacy of radiotherapy and chemotherapy correlates with the level of dsRNA sensor activation, and this innate immune activity synergizes with other immunotherapies63,64,136. Therefore, therapies that specifically leverage the acute activation of dsRNA sensors or acute induction of cellular dsRNAs, for example by developing agonists for RLRs180 or antagonists for ADAR1, would have the potential to contribute to existing cancer immunotherapies.
Conclusions and perspective
Over the last decade or so, our understanding of innate immune sensors for dsRNA and their signalling pathways has greatly improved at the level of structure and biochemical reconstitution. At the same time, there has been a rapid expansion of the list of human diseases and biological processes that involve sterile activation of dsRNA sensors. These studies identified new links connecting antiviral innate immune responses to diverse cellular processes, from the DNA damage response to neuronal excitation. Collectively, these observations support a model that dsRNAs are not simply PAMPs but are a new form of cellular signalling molecules that can alert cells to the presence of ‘danger’ or other biological processes that require involvement of antiviral immune response.
The diverse array of cellular sources for dsRNAs also implies that there are at least equally diverse and complex regulatory mechanisms at the level of both endogenous RNA and dsRNA sensors to prevent constitutive and uncontrolled activation of the dsRNA sensors. While multiple regulators of the dsRNA sensing pathways have been identified, it remains unclear exactly how these individual regulatory nodes work together and whether their dysregulation contributes to human diseases. Additionally, several dsRNA-binding proteins, such as ZBP1 (REFS181,182) and DHX9 (REF.183), have emerged as important regulators of cellular response to dsRNAs, but the detailed molecular mechanisms and their interactions with cellular dsRNAs need further investigation. Finally, it is also important to note that the precise identity of cellular dsRNAs that mediate sterile activation of the dsRNA sensors remain unclear in many cases. For instance, RNAs from repetitive elements have often been shown to associate with dsRNA sensors, but their functional connection has been difficult to probe, largely due to the lack of feasibility to genetically perturb the repetitive elements. Biochemical approaches to demonstrate the importance of such interactions beyond binding, for example in a biochemically reconstituted signalling pathway17, are necessary. Altogether, these future efforts will enable us to create a complete blueprint for cell biology of dsRNA.
Acknowledgements
S.H. was supported by the NIH (R01AI154653, R01AI111784 and DP1AI152074). Y.G.C. was supported by the NIH (R35GM142687 and 5K12CA215110), the Rita Allen Foundation and the Yale Cancer Center.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Gesteland RF, Cech TR & Atkins JF The RNA World (Cold Springer Harbor Laboratory Press, 1999). [Google Scholar]
- 2.Hur S Double-stranded RNA sensors and modulators in innate immunity. Annu. Rev. Immunol 37, 349–375 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kato H, Takahasi K & Fujita T RIG-I-like receptors: cytoplasmic sensors for non-self. RNA. Immuno Rev 243, 91–98 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Cadena C & Hur S Filament-like assemblies of intracellular nucleic acid sensors: commonalities and differences. Mol. Cell 76, 243–254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venkataraman T et al. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol 178, 6444–6455 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Childs KS, Randall RE & Goodbourn S LGP2 plays a critical role in sensitizing mda-5 to activation by double-stranded RNA. PLoS ONE 8, e64202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Satoh T et al. LGP2 is a positive regulator of RIG-I– and MDA5-mediated antiviral responses. Proc Natl Acad Sci USA 107, 1512–1517 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cadena C et al. Ubiquitin-dependent and -independent roles of E3 ligase RIPLET in innate immunity. Cell 177, 1187–1200.e16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato K et al. Structural analysis of RIG-I-like receptors reveals ancient rules of engagement between diverse RNA helicases and TRIM ubiquitin ligases. Mol. Cell 81, 599–613.e8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang X et al. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 36, 959–973 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peisley A, Wu B, Xu H, Chen ZJ & Hur S Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509, 110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peisley A et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl Acad. Sci. USA 108, 21010–21015 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peisley A et al. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc. Natl Acad. Sci. USA 109, E3340–E3349 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu B et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152, 276–289 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Berke IC & Modis Y MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J 7, 1714–1726 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng Q et al. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep 29, 1187–1196 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmad S et al. Breaching self-tolerance to Alu duplex RNA underlies MDA5-mediated inflammation. Cell 172, 797–810 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals that constitutive activation of MDA5 under pathologic conditions is caused by MDA5 misrecognition of dsRNA formed by IR-Alus.
- 18.Mu X, Greenwald E, Ahmad S & Hur S An origin of the immunogenicity of in vitro transcribed RNA. Nucleic acids Res 46, 5239–5249 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peisley A, Wu B, Yao H, Walz T & Hur S RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol. Cell 51, 573–583 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Patel JR et al. ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep 14, 780–787 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goubau D et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514, 372 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myong S et al. Cytosolic viral sensor RIG-I is a 5′-triphosphate–dependent translocase on double-stranded RNA. Science 323, 1070–1074 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohlway A, Luo D, Rawling DC, Ding SC & Pyle AM Defining the functional determinants for RNA surveillance by RIG-I. EMBO Rep 14, 772–779 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinicke LA et al. RNA dimerization promotes PKR dimerization and activation. J. Mol. Biol 390, 319–338 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pohar J, Pirher N, Bencina M, Mancek-Keber M & Jerala R The role of UNC93B1 protein in surface localization of TLR3 receptor and in cell priming to nucleic acid agonists. J. Biol. Chem 288, 442–454 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leonard JN et al. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc. Natl Acad. Sci. USA 105, 258–263 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oshiumi H et al. The TLR3/TICAM-1 pathway is mandatory for innate immune responses to poliovirus infection. J. Immunol 187, 5320–5327 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Davey GM et al. Cutting edge: priming of CD8 T cell immunity to herpes simplex virus type 1 requires cognate TLR3 expression in vivo. J. Immunol 184, 2243–2246 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Zhang SY et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317, 1522–1527 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Liu L et al. Structural basis of Toll-like receptor 3 signaling with double-stranded RNA. Science 320, 379–381 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kariko K, Buckstein M, Ni H & Weissman D Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005). [DOI] [PubMed] [Google Scholar]; This study is the first to report that the modified nucleosides 5-methylcytidine, m6A, 5-methyluridine, 2-thiouridine and pseudouridine on RNAs suppress the stimulation of TLR3, TLR7 and TLR8 and decrease the potential of RNA to activate dendritic cells.
- 32.Tatematsu M, Nishikawa F, Seya T & Matsumoto M Toll-like receptor 3 recognizes incomplete stem structures in single-stranded viral RNA. Nat. Commun 4, 1833 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Liu Y et al. The role of host eIF2alpha in viral infection. Virol. J 17, 112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oh SW et al. Leader-containing uncapped viral transcript activates RIG-I in antiviral stress granules. PLoS Pathog 12, e1005444 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Onomoto K et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 7, e43031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cadena C et al. Stress granules are shock absorbers that prevent excessive innate immune responses to dsRNA. Preprint at bioRxiv 10.1101/2021.04.26.441141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schulz O et al. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 7, 354–361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McAllister CS, Taghavi N & Samuel CE Protein kinase PKR amplification of interferon beta induction occurs through initiation factor eIF-2alpha-mediated translational control. J. Biol. Chem 287, 36384–36392 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalet A et al. Protein synthesis inhibition and GADD34 control IFN-beta heterogeneous expression in response to dsRNA. EMBO J 36, 761–782 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nanduri S, Carpick BW, Yang Y, Williams BR & Qin J Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 17, 5458–5465 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dey M et al. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 122, 901–913 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Dar AC, Dever TE & Sicheri F Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 122, 887–900 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Mayo CB et al. Structural basis of protein kinase R autophosphorylation. Biochemistry 58, 2967–2977 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dey M, Mann BR, Anshu A & Mannan MA Activation of protein kinase PKR requires dimerization-induced cis-phosphorylation within the activation loop. J. Biol. Chem 289, 5747–5757 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Husain B, Mukerji I & Cole JL Analysis of high-affinity binding of protein kinase R to double-stranded RNA. Biochemistry 51, 8764–8770 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nallagatla SR et al. 5′-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 318, 1455–1458 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Launer-Felty K, Wong CJ & Cole JL Structural analysis of adenovirus VAI RNA defines the mechanism of inhibition of PKR. Biophys. J 108, 748–757 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kristiansen H, Gad HH, Eskildsen-Larsen S, Despres P & Hartmann R The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. J. Interferon Cytokine Res 31, 41–47 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Hovanessian AG & Justesen J The human 2′−5′ oligoadenylate synthetase family: unique interferon-inducible enzymes catalyzing 2′−5′ instead of 3′−5′ phosphodiester bond formation. Biochimie 89, 779–788 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Han Y et al. Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 343, 1244–1248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donovan J, Rath S, Kolet-Mandrikov D & Korennykh A Rapid RNase L-driven arrest of protein synthesis in the dsRNA response without degradation of translation machinery. RNA 23, 1660–1671 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hornung V, Hartmann R, Ablasser A & Hopfner KP OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat. Rev. Immunol 14, 521–528 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu J et al. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 40, 936–948 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee MS, Kim B, Oh GT & Kim Y-J OASL1 inhibits translation of the type I interferon–regulating transcription factor IRF7. Nat. Immunol 14, 346–355 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Donovan J, Whitney G, Rath S & Korennykh A Structural mechanism of sensing long dsRNA via a noncatalytic domain in human oligoadenylate synthetase 3. Proc. Natl Acad. Sci. USA 112, 3949–3954 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sarkar SN, Ghosh A, Wang HW, Sung SS & Sen GC The nature of the catalytic domain of 2′−5′-oligoadenylate synthetases. J. Biol. Chem 274, 25535–25542 (1999). [DOI] [PubMed] [Google Scholar]
- 57.Ilson DH, Torrence PF & Vilcek J Two molecular weight forms of human 2′,5′-oligoadenylate synthetase have different activation requirements. J. Interferon Res 6, 5–12 (1986). [DOI] [PubMed] [Google Scholar]
- 58.Ibsen MS et al. The 2′−5′-oligoadenylate synthetase 3 enzyme potently synthesizes the 2′−5′-oligoadenylates required for RNase L activation. J. Virol 88, 14222–14231 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y et al. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl Acad. Sci. USA 113, 2241–2246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martinon F, Burns K & Tschopp J The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE & Hornung V Human NLRP1 is a sensor for double-stranded RNA. Science 371, eabd0811 (2021). [DOI] [PubMed] [Google Scholar]; This article demonstrates that human NLRP1 directly binds dsRNA, which activates the inflammasome pathway; this is the first example of an inflammasome that directly senses foreign RNA.
- 62.Maharana J, Panda D & De S Deciphering the ATP-binding mechanism(s) in NLRP-NACHT 3D models using structural bioinformatics approaches. PLoS ONE 13, e0209420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chiappinelli KB et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roulois D et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mehdipour P et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 588, 169–173 (2020). [DOI] [PubMed] [Google Scholar]; This study is one of several studies reporting that chemotherapeutic agents (such as 5-aza-CdR) induce aberrant synthesis and accumulation of cellular dsRNAs, resulting in activation of MDA5.
- 66.Banerjee S et al. OAS-RNase L innate immune pathway mediates the cytotoxicity of a DNA-demethylating drug. Proc. Natl Acad. Sci. USA 116, 5071–5076 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chung H et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172, 811–824.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cuellar TL et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol 216, 3535–3549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tunbak H et al. The HUSH complex is a gatekeeper of type I interferon through epigenetic regulation of LINE-1s. Nat. Commun 11, 5387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sheng W et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174, 549–563.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Macfarlan TS et al. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev 25, 594–607 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kozub MM, Carr RM, Lomberk GL & Fernandez-Zapico ME LSD1, a double-edged sword, confers dynamic chromatin regulation but commonly promotes aberrant cell growth. F1000Res 6, 2016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walkley CR & Li JB Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol 18, 205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Slotkin W & Nishikura K Adenosine-to-inosine RNA editing and human disease. Genome Med 5, 105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.George CX, John L & Samuel CE An RNA editor, adenosine deaminase acting on double-stranded RNA (ADAR1). J. Interferon Cytokine Res 34, 437–446 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Samuel CE Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 411, 180–193 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tan MH et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bass BL & Weintraub H An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55, 1089–1098 (1988). [DOI] [PubMed] [Google Scholar]; This is the first study that revealed the presence of dsRNA-modifying activity, which partially unwinds dsRNA and increases the single-strandedness of the RNAs.
- 79.Wagner RW, Smith JE, Cooperman BS & Nishikura K A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc. Natl Acad. Sci. USA 86, 2647–2651 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liddicoat BJ et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mannion NM et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9, 1482–1494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Y et al. Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. eLife 6, e25687 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pestal K et al. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43, 933–944 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Athanasiadis A, Rich A & Maas S Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol 2, e391 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carmi S, Borukhov I & Levanon EY Identification of widespread ultra-edited human RNAs. PLoS Genet 7, e1002317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Meyer KD & Jaffrey SR The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol 15, 313–326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dominissini D et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012). [DOI] [PubMed] [Google Scholar]
- 88.Roundtree IA, Evans ME, Pan T & He C Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang X et al. m6A-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that the m6A modification on RNA is selectively recognized by the protein YTHDF2, which recruits the RNA to cellular RNA decay sites; this reveals that m6A can affect the lifetime of RNA.
- 90.Wang X et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals that YTHDF1 recognizes and promotes the translation of selective m6A-modified RNAs by interacting with protein synthesis machinery.
- 91.Liu N et al. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides evidence that the m6A modification can alter the structure of the modified RNA to change the binding affinity for proteins, which can affect splicing, gene expression and RNA maturation.
- 92.Han D et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature 566, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gao Y et al. m6A modification prevents formation of endogenous double-stranded RNAs and deleterious innate immune responses during hematopoietic development. Immunity 52, 1007–1021.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu J et al. N-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lu M et al. N6-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat. Microbiol 5, 584–598 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kane SE & Beemon K Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol. Cell. Biol 5, 2298–2306 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kennedy EdwardM. et al. Posttranscriptional m6A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe 19, 675–685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gokhale NS & Horner SM RNA modifications go viral. PLoS Pathog 13, e1006188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boccaletto P et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res 46, D303–D307 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Anderson BR et al. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res 38, 5884–5892 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bowling EA et al. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell 184, 384–403.e21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Blango MG & Bass BL Identification of the long, edited dsRNAome of LPS-stimulated immune cells. Genome Res 26, 852–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lybecker M, Zimmermann B, Bilusic I, Tukhtubaeva N & Schroeder R The double-stranded transcriptome of Escherichia coli. Proc. Natl Acad. Sci. USA 111, 3134–3139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen L-L The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol .Cell Biol 17, 205–211 (2016). [DOI] [PubMed] [Google Scholar]
- 105.Chen YG et al. N6-methyladenosine modification controls circular RNA immunity. Mol. Cell 76, 96–109.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that the m6A modification on human endogenous circRNAs binds the protein YTHDF2 to block innate immunity, whereas unmodified circRNAs activate RIG-I in the presence of a K63-linked polyubiquitin chain to cause MAVS filamentation, IRF3 dimerization and interferon production.
- 106.Chen YG et al. Sensing self and foreign circular RNAs by intron identity. Mol. Cell 67, 228–238.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu C-X et al. Structure and degradation of circular RNAs regulate PKR activation in innate immunity. Cell 177, 865–880.e21 (2019). [DOI] [PubMed] [Google Scholar]; This study finds that endogenous circRNAs have short dsRNA regions that bind and inhibit PKR to prevent its activity in sterile conditions; upon viral infection, RNase L degrades circRNAs to release PKR for innate immune responses.
- 108.Salzman J, Gawad C, Wang PL, Lacayo N & Brown PO Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 7, e30733 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tam OH et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 453, 534–538 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Watanabe T et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 453, 539–543 (2008). [DOI] [PubMed] [Google Scholar]
- 111.Chen L, Dahlstrom JE, Lee SH & Rangasamy D Naturally occurring endo-siRNA silences LINE-1 retrotransposons in human cells through DNA methylation. Epigenetics 7, 758–771 (2012). [DOI] [PubMed] [Google Scholar]
- 112.Yang N & Kazazian HH Jr. L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat. Struct. Mol. Biol 13, 763–771 (2006). [DOI] [PubMed] [Google Scholar]
- 113.Heras SR et al. The Microprocessor controls the activity of mammalian retrotransposons. Nat. Struct. Mol. Biol 20, 1173–1181 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kaneko H et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 471, 325–330 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tarallo V et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149, 847–859 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kerur N et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med 24, 50–61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Krol J et al. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol. Cell 25, 575–586 (2007). [DOI] [PubMed] [Google Scholar]
- 118.Zhang N & Ashizawa T RNA toxicity and foci formation in microsatellite expansion diseases. Curr. Opin. Genet. Dev 44, 17–29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Halbach F, Reichelt P, Rode M & Conti E The yeast Ski complex: crystal structure and RNA channeling to the exosome complex. Cell 154, 814–826 (2013). [DOI] [PubMed] [Google Scholar]
- 120.Eckard SC et al. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat. Immunol 15, 839–845 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Aizawa S et al. Lysosomal putative RNA transporter SIDT2 mediates direct uptake of RNA by lysosomes. Autophagy 12, 565–578 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nguyen TA et al. SIDT2 transports extracellular dsRNA into the cytoplasm for innate immune recognition. Immunity 47, 498–509.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dieci G, Conti A, Pagano A & Carnevali D Identification of RNA polymerase III-transcribed genes in eukaryotic genomes. Biochim. Biophys. Acta 1829, 296–305 (2013). [DOI] [PubMed] [Google Scholar]
- 124.Singh R & Reddy R Gamma-monomethyl phosphate: a cap structure in spliceosomal U6 small nuclear RNA. Proc. Natl Acad. Sci. USA 86, 8280–8283 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nabet BY et al. Exosome RNA unshielding couples stromal activation to pattern recognition receptor signaling in cancer. Cell 170, 352–366.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that the imbalanced upregulation of the RNA component (without the corresponding upregulation of the protein component) of the SRP leads to activation of RIG-I, which then promotes breast cancer progression.
- 126.Chiang JJ et al. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol 19, 53–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Karijolich J, Abernathy E & Glaunsinger BA Infection-induced retrotransposon-derived noncoding RNAs enhance herpesviral gene expression via the NF-kappaB pathway. PLoS Pathog 11, e1005260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhao Y, Ye X, Dunker W, Song Y & Karijolich J RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat. Commun 9, 4841 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Choi JH et al. DUSP11-mediated control of 5′-triphosphate RNA regulates RIG-I sensitivity. Genes Dev 34, 1697–1712 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bakhoum SF et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mackenzie KJ et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Santaguida S et al. Chromosome mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev. Cell 41, 638–651.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Harding SM et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Feng X et al. ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J. 39, e104036 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chen J et al. Cell cycle checkpoints cooperate to suppress DNA-and RNA-associated molecular pattern recognition and anti-tumor immune responses. Cell Rep 32, 108080 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ranoa DR et al. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 7, 26496–26515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Widau RC et al. RIG-I-like receptor LGP2 protects tumor cells from ionizing radiation. Proc. Natl Acad. Sci. USA 111, E484–E491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Grazioli S & Pugin J Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front. Immunol 9, 832 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Huang LS et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 52, 475–486.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Guo X et al. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 579, 427–432 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.D’Souza AR & Minczuk M Mitochondrial transcription and translation: overview. Essays Biochem 62, 309–320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Murphy WI, Attardi B, Tu C & Attardi G Evidence for complete symmetrical transcription in vivo of mitochondrial DNA in HeLa cells. J. Mol. Biol 99, 809–814 (1975). [DOI] [PubMed] [Google Scholar]
- 143.Harel L, Riou G & Montagnier L Nuclear and mitochondrial origin of rat liver double-stranded RNA. Biochimie 57, 227–233 (1975). [DOI] [PubMed] [Google Scholar]
- 144.Dhir A et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals that mitochondria can be a potent source of immunostimulatory dsRNA and that deficiency of the mitochondrial protein PNPase can lead to both accumulation of mitochondrial dsRNAs and their leakage into the cytoplasm, leading to the pathologic activation of MDA5.
- 145.Kim Y et al. PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. Mol. Cell 71, 1051–1063.e6 (2018). [DOI] [PubMed] [Google Scholar]; This study shows that both nuclear transcripts (IR-Alus) and mitochondrial transcripts (mitochondrial sense–antisense hybrids) can activate PKR in homeostatic conditions.
- 146.Chen HW et al. Mammalian polynucleotide phosphorylase is an intermembrane space RNase that maintains mitochondrial homeostasis. Mol. Cell Biol 26, 8475–8487 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang G et al. PNPASE regulates RNA import into mitochondria. Cell 142, 456–467 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kim Y et al. PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Genes Dev 28, 1310–1322 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tigano M, Vargas DC, Tremblay-Belzile S, Fu Y & Sfeir A Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 591, 477–481 (2021). [DOI] [PubMed] [Google Scholar]
- 150.Zhu PJ et al. Suppression of PKR promotes network excitability and enhanced cognition by interferon-gamma-mediated disinhibition. Cell 147, 1384–1396 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nelson AM et al. dsRNA released by tissue damage activates TLR3 to drive skin regeneration. Cell Stem Cell 17, 139–151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lefkopoulos S et al. Repetitive elements trigger RIG-I-like receptor signaling that regulates the emergence of hematopoietic stem and progenitor cells. Immunity 53, 934–951.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 153.Funabiki M et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 40, 199–212 (2014). [DOI] [PubMed] [Google Scholar]
- 154.Rice GI et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet 46, 503–509 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies mutations in IFIH1 (the gene encoding MDA5) as a cause of the inflammatory disorder Aicardi-Goutières syndrome.
- 155.Van Eyck L et al. IFIH1 mutation causes systemic lupus erythematosus with selective IgA deficiency. Arthritis Rheumtol 67, 1592–1597 (2015). [DOI] [PubMed] [Google Scholar]
- 156.Bursztejn AC et al. Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: overlap between Aicardi-Goutieres and Singleton-Merten syndromes. Br. J. Dermatol 173, 1505–1513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jang M-A et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet 96, 266–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ferreira CR et al. DDX58 and classic Singleton-Merten syndrome. J. Clin. Immunol 39, 75–80 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rice GI et al. Genetic and phenotypic spectrum associated with IFIH1 gain-of-function. Hum. Mutat 41, 837–849 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Louber J, Brunel J, Uchikawa E, Cusack S & Gerlier D Kinetic discrimination of self/non-self RNA by the ATPase activity of RIG-I and MDA5. BMC Biol 13, 54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lassig C et al. Unified mechanisms for self-RNA recognition by RIG-I Singleton-Merten syndrome variants. eLife 7, e38958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Devarkar SC, Schweibenz B, Wang C, Marcotrigiano J & Patel SS RIG-I uses an ATPase-powered translocation-throttling mechanism for kinetic proofreading of RNAs and pligomerization. Mol. Cell 72, 355–368.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chistiakov DA, Voronova NV, Savost’Anov KV & Turakulov RI Loss-of-function mutations E627X and I923V of IFIH1 are associated with lower poly(I:C)–induced interferon-a production in peripheral blood mononuclear cells of type 1 diabetes patients. Hum. Immunol 71, 1128–1134 (2010). [DOI] [PubMed] [Google Scholar]
- 164.Smyth DJ et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet 38, 617–619 (2006). [DOI] [PubMed] [Google Scholar]
- 165.Molineros JE et al. Admixture mapping in lupus identifies multiple functional variants within IFIH1 associated with apoptosis, inflammation, and autoantibody production. PLoS Genet 9, e1003222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Robinson T et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-alpha and serologic autoimmunity in lupus patients. J. Immunol 187, 1298–1303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Burnett SB, Vaughn LS, Sharma N, Kulkarni R & Patel RC Dystonia 16 (DYT16) mutations in PACT cause dysregulated PKR activation and eIF2alpha signaling leading to a compromised stress response. Neurobiol. Dis 146, 105135 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kuipers DJS et al. EIF2AK2 missense variants associated with early onset generalized dystonia. Ann. Neurol 89, 485–497 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Reimer L et al. PKR kinase directly regulates tau expression and Alzheimer’s disease-related tau phosphorylation. Brain Pathol 31, 103–119 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hugon J, Mouton-Liger F, Dumurgier J & Paquet C PKR involvement in Alzheimer’s disease. Alzheimers Res. Ther 9, 83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lee H et al. Cell type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron 107, 891–908.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lu B et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yu C-H, Moecking J, Geyer M & Masters SL Mechanisms of NLRP1-mediated autoinflammatory disease in humans and mice. J. Mol. Biol 430, 142–152 (2018). [DOI] [PubMed] [Google Scholar]
- 174.Drutman SB et al. Homozygous NLRP1 gain-of-function mutation in siblings with a syndromic form of recurrent respiratory papillomatosis. Proc. Natl Acad. Sci. USA 116, 19055–19063 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rice GI et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet 44, 1243–1248 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies mutations in ADAR1 as a cause of Aicardi–Goutières syndrome, thereby linking for the first time misregulated cellular RNA to immunological disorders.
- 176.van den Boorn JG & Hartmann G Turning tumors into vaccines: co-opting the innate immune system. Immunity 39, 27–37 (2013). [DOI] [PubMed] [Google Scholar]
- 177.Ishizuka JJ et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gannon HS et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun 9, 5450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Liu H et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med 25, 95–102 (2019). [DOI] [PubMed] [Google Scholar]
- 180.Iurescia S, Fioretti D & Rinaldi M The innate immune signalling pathways: turning RIG-I sensor activation against cancer. Cancers 12, 3158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Maelfait J et al. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 36, 2529–2543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jiao H et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 580, 391–395 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Aktas T et al. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature 544, 115–119 (2017). [DOI] [PubMed] [Google Scholar]
- 184.Lemay G Synthesis and translation of viral mRNA in reovirus-infected cells: progress and remaining questions. Viruses 10, 671 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Abad AT & Danthi P Recognition of reovirus RNAs by the innate immune system. Viruses 12, 667 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Goody RJ, Beckham JD, Rubtsova K & Tyler KL JAK-STAT signaling pathways are activated in the brain following reovirus infection. J. Neurovirol 13, 373–383 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Connolly JL et al. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J. Virol 74, 2981–2989 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kowal KJ & Youngner JS Induction of interferon by temperature-sensitive mutants of Newcastle disease virus. Virology 90, 90–102 (1978). [DOI] [PubMed] [Google Scholar]
- 189.Long WF & Burke DC Interferon production by double-stranded RNA: a comparison of induction by reovirus to that by a synthetic double-stranded polynucleotide. J. Gen. Virol 12, 1–11 (1971). [DOI] [PubMed] [Google Scholar]
- 190.Nagy PD, Strating JR & van Kuppeveld FJ Building viral replication organelles: close encounters of the membrane types. PLoS Pathog 12, e1005912 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Romero-Brey I & Bartenschlager R Membranous replication factories induced by plus-strand RNA viruses. Viruses 6, 2826–2857 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Triantafilou K et al. Visualisation of direct interaction of MDA5 and the dsRNA replicative intermediate form of positive strand RNA viruses. J. Cell Sci 125, 4761–4769 (2012). [DOI] [PubMed] [Google Scholar]
- 193.Weber F, Wagner V, Rasmussen SB, Hartmann R & Paludan SR Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol 80, 5059–5064 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Son KN, Liang Z & Lipton HL Double-stranded RNA is detected by immunofluorescence analysis in RNA and DNA virus infections, including those by negative-stranded RNA viruses. J. Virol 89, 9383–9392 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ziegler CM & Botten JW Defective interfering particles of negative-strand RNA viruses. Trends Microbiol 28, 554–565 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Vignuzzi M & Lopez CB Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol 4, 1075–1087 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Strahle L, Garcin D & Kolakofsky D Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351, 101–111 (2006). [DOI] [PubMed] [Google Scholar]
- 198.Tapia K et al. Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog 9, e1003703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Xu J et al. Identification of a natural viral RNA motif that optimizes sensing of viral RNA by RIG-I. mBio 6, e01265–01215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Weber M et al. Incoming RNA virus nucleocapsids containing a 5′-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe 13, 336–346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rehwinkel J et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140, 397–408 (2010). [DOI] [PubMed] [Google Scholar]
- 202.Davis WG et al. The 3′ untranslated regions of influenza genomic sequences are 5′PPP-independent ligands for RIG-I. PLoS ONE 7, e32661 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Te Velthuis AJW et al. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat. Microbiol 3, 1234–1242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Baum A, Sachidanandam R & Garcia-Sastre A Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl Acad. Sci. USA 107, 16303–16308 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Vasilijevic J et al. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog 13, e1006650 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Boone RF, Parr RP & Moss B Intermolecular duplexes formed from polyadenylylated vaccinia virus RNA. J. Virol 30, 365–374 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Colby C, Jurale C & Kates JR Mechanism of synthesis of vaccinia virus double-stranded ribonucleic acid in vivo and in vitro. J. Virol 7, 71–76 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Samanta M, Iwakiri D, Kanda T, Imaizumi T & Takada K EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J 25, 4207–4214 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Samanta M, Iwakiri D & Takada K Epstein-Barr virus-encoded small RNA induces IL-10 through RIG-I-mediated IRF-3 signaling. Oncogene 27, 4150–4160 (2008). [DOI] [PubMed] [Google Scholar]