

Abstract
Renal ischemia-reperfusion (IR) injury is one of the major causes of acute kidney injury and represents a significant risk factor for renal transplantation. The level of renal damage is influenced by the ischemic duration and is caused by excessive amounts of produced reactive oxygen species (ROS). Adaptor protein p66Shc is known to regulate cellular and organ’s sensitivity to oxidative stress and to contribute significantly to mitochondrial production of hydrogen peroxide in stress conditions. Studies carried out in cultured renal cells suggest that p66Shc-mediated mitochondrial dysfunction and ROS production are responsible for renal ischemic injury. We used our genetically modified rats, which either lack p66Shc expression, or express p66Shc variant, which constitutively generates increased quantities of hydrogen peroxide, to evaluate potential contribution of p66Shc signaling to renal damage in ischemia reperfusion rat model. Analysis of outer medulla tubule damage revealed the lack of contribution of either p66Shc expression or its constitutive signaling to IR injury in rat model.
Keywords: Ischemia-reperfusion, signaling, p66Shc, rat model, kidney injury, ROS
Introduction
Adaptor protein p66Shc contributes to production of hydrogen peroxide and is generally accepted to serve as an oxidative stress sensor [1,2]. Renal ischemia-reperfusion (IR) injury is a common cause of acute kidney injury (AKI) that results from multiple factors including alterations in renal blood flow which could increase oxidative stress [3]. Accordingly, the role of p66Shc expression and/or signaling in progression of IR injury was studied in multiple cellular systems. It was reported that p66Shc inhibits pro-survival signaling during severe oxidative stress in mouse renal proximal tubule cells [4]. Furthermore, p66Shc-mediated mitochondrial dysfunction in renal proximal tubule cells was observed during oxidative injury [5]. Additionally, the suppression of p66Shc seems to prevent hypoxia/reoxygenation-induced injury in cultured rat proximal tubular cells [6]. In summary, multiple studies carried out in cultured tubular epithelial cells support the idea that p66Shc signaling could be a principal factor in IR injury development. Nevertheless, studies carried out in animal models of IR injury are needed to establish the role of p66Shc in renal pathologies associated with IR injury. We have used our genetically modified rats with targeted editing of Shc1 gene in established model of IR injury to evaluate the role of p66Shc expression and signaling in progression of IR injury.
Materials and Methods
Renal ischemia and reperfusion
All animal procedures were conducted according to the National Institute of Health Guidelines for the Care and Use of Laboratory Animals and the Medical College of Wisconsin Institutional Animal Care and Use Committee. Transgenic p66Shc rats were established on Dahl Salt Sensitive MCW (SS) background using zinc-finger nucleases (ZFN) as described previously by our group [7]. Male rats 8–16 weeks of age were anesthetized with ketamine (25–40 mg/kg, i.m.) and sodium pentobarbital (25–50 mg/kg, i.p) and were placed on a heated surgical table to maintain body temperature at 37°C. Kidneys were exposed by a midline abdominal incision and the renal arteries and veins were subjected to bilateral occlusion for 30 minutes using microvascular clamps. The clamps were then removed, and the abdominal incision was closed. Post-surgery, rats were treated with Baytril (5 mg) and Buprenex (0.015 mg b.i.d.) and allowed to recover for 24 hours or seven days. Rats were then euthanized with sodium pentobarbital (100 mg/kg, i.p.), and blood was collected by cardiac puncture for measurement of plasma creatinine (Jaffe reaction, BioAssay Systems, USA) and blood urea nitrogen (improved Jung, BioAssay Systems, USA).
Tubular injury analysis
After euthanasia, kidneys were extracted, sectioned in half longitudinally, fixed in zinc formalin and embedded in paraffin blocks. 4-micron sections were prepared on slides and processed by hematoxylin-eosin staining. 10 random images were taken of each kidney in the outer medulla region where the most injury is expected to occur. A grid pattern was used to select up to 20 tubules per image, each being assessed for indications of damage, which resulted in 120–180 tubules counted per animal. A tubule was “damaged” if there were signs of apoptosis or necrosis, cell desquamation, intralumenal protein casts, or lumen dilation. Overall damage is tabulated as the percentage of tubules counted. No distinction was made to identify differences of the quality of damage or recovery.
Western blot analysis
After recovery from ischemia and reperfusion, kidneys were collected, the outer medulla separated from inner medulla and cortex, and snap frozen in liquid nitrogen and stored at −80°C until analysis. Protein extracts were prepared on ice in lysis buffer (in mM; 50 Tris-HCl, 150 NaCl, 5 EDTA, 25 beta-glycerophosphate, 50 NaF, 1 PMSF, 1% IGEPAL CA-650 and Complete-mini protease inhibitor (Roche, USA) from frozen tissue using 25 strokes of a Teflon Potter-Elvehjem homogenizer followed by a single 30-second sonication at setting 3.5 (Sonic Dismembrator 550, Fisher Scientific, USA). Samples were centrifuged at 12,000 × g for 10 minutes at 4°C to separate unbroken cells and debris. The supernatant was collected, and equal amounts of protein were loaded on SDS-PAGE gels and processed as a multi-strip western blot as described [8]. Briefly, after electrophoresis, each of the gels were cut in half at the 37 kDa marker and corresponding (top or bottom) strips were assembled together for simultaneous transfer onto the same PVDF membrane to allow statistically reliable comparisons of multiple experimental conditions. Protein expression levels were detected using primary antibodies against SHC (610081 BD Transduction Labs; 1:1000), NGAL/Lipocalin-2 (AF-3508 R&D Systems, USA; 1:2000), total Actin (A5060 Millipore-Sigma, USA; 1:10000), and near-infrared fluorescent IR-Dye secondary antibodies (LI-COR, USA; 1:15000) and data acquired by the Odyssey CLx imaging system (LI-COR). Total protein stain used for normalization of blot signals was obtained using Revert Total Protein Stain (LI-COR).
Results
Renal IR causes outer medulla tubule damage in rats [3]. Medullary blood flow in rats which undergo 30 minutes renal ischemia and 180 minutes of reperfusion fails to return to baseline levels exacerbating tubular epithelial cell injury [3]. Our panel of genetically modified rats which differ in p66Shc expression and/or signaling provided unique opportunity to evaluate the role of p66Shc in renal injury caused by IR. All our salt-sensitive rat strains were maintained at low salt diet to prevent the development of hypertension.
We have compared tubular injury in parental rat strain (p66Shc-WT), previously described p66Shc knockout rat strain (p66Shc-KO) [7] and previously described rat strain with introduced deletion in Shc1 gene which results in a presumably constitutive signaling by p66Shc (p66Shc-Del) [9].
Hematoxylin-eosin-stained histological images of the renal outer medulla of rats subjected to IR injury were evaluated for the loss of brush border, tubule dilation and presence of protein casts, necrosis and cellular desquamation (Figure 1A). The peak of tubular injury was observed 24 hours after IR injury with partial restoration of brush border, whereas an increased epithelial cell remodeling and proliferation occurred by the seventh day after IR. These characteristics were absent in control sham animals. Analysis and quantification of tubule damage failed to reveal any differences between WT, p66Shc-KO or p66Shc-Del rats subjected to IR (Figure 1B). Additional evidence of equal manifestation of kidney injury and recovery after IR in WT and our genetically modified rat strains with targeted editing of Shc1 gene was provided by measurements of Blood Urea Nitrogen (Figure 1C) and serum creatinine levels (Figure 1D).
Fig. 1. p66Shc does not protect against renal injury caused by ischemia and reperfusion.
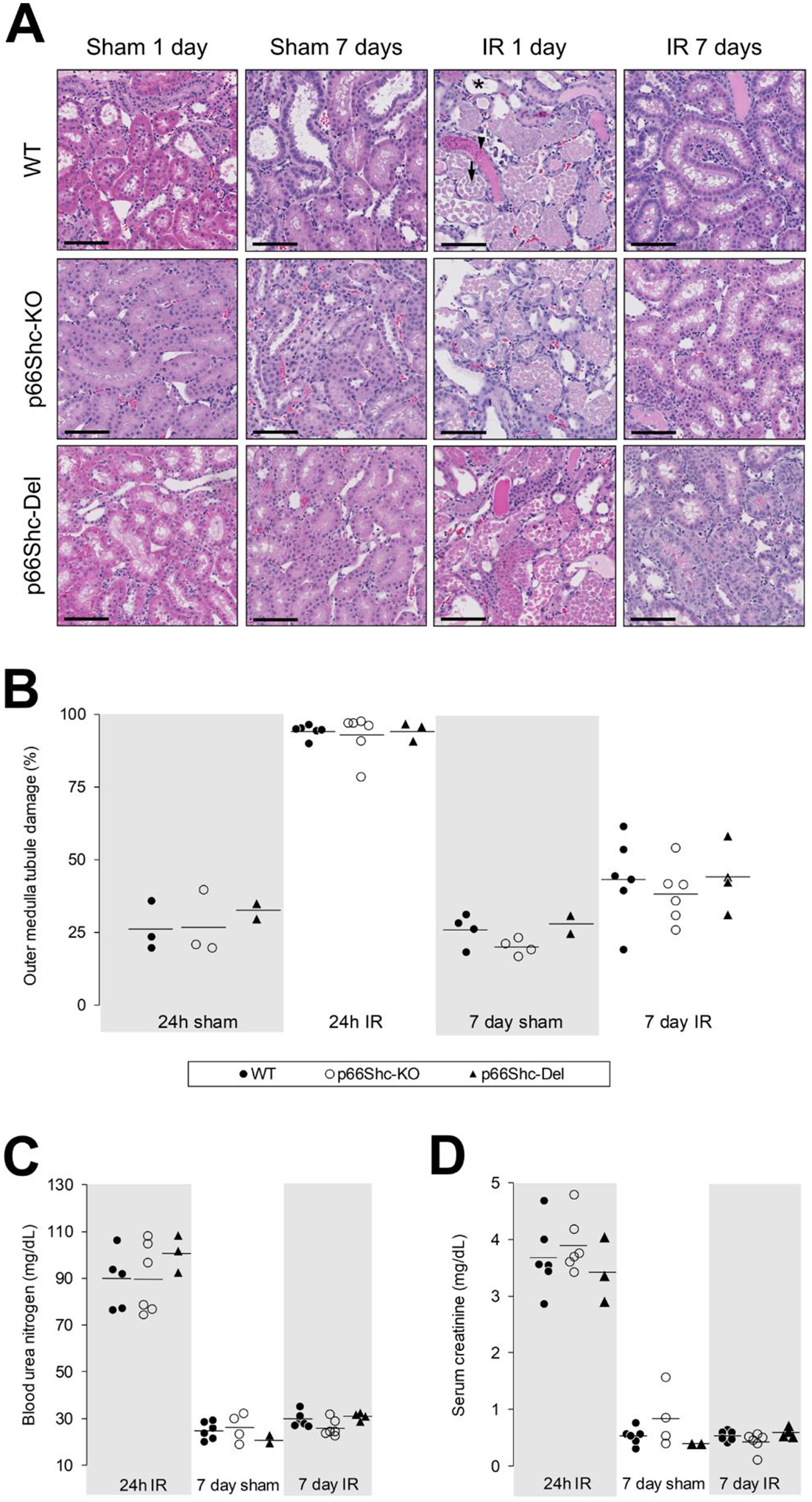
(A) Representative hematoxylin-eosin stained, histological images of the renal outer medulla during the course of IR injury in WT, p66Shc-KO and p66Shc-Del salt-sensitive rats. Peak injury (loss of brush border and tubule dilation indicated by asterisk; protein cast indicated by arrowhead; necrosis and cellular desquamation indicated by an arrow) was evident by 24 hours of reperfusion. Recovery was marked at 7 days by restoration of brush border and increased epithelial cell remodeling and proliferation. Scale bar is equal to 100 microns. (B) Summary analysis of ischemia-reperfusion injury in WT (black dot), p66Shc-KO (white dot) and p66Shc-Del (black triangle). There were no statistically significant differences in renal function among p66Shc-WT, p66Shc-KO and p66Shc-Del as measured by levels of (C) blood urea nitrogen and (D) serum creatinine.
Neutrophil Gelatinase-Associated Lipocalin (NGAL) is expressed in renal tubular epithelial cells in response to inflammation and serves as a marker of tubular damage [10]. The level of NGAL expression is associated with degree of kidney dysfunction and is used to monitor acute kidney injury [11,12]. Furthermore, NGAL could be used to differentiate the type of acute kidney injury [13]. Expression of NGAL is dramatically induced in the outer medulla in rats exposed to 30 minutes renal ischemia and after 24 hours reperfusion (Figure 2A) and expression returned to basal levels by the seventh day of recovery (Figure 2B). There was no significant difference in the level of NGAL expression following IR between WT, p66Shc-KO and p66Shc-Del rat strains.
Fig. 2. NGAL expression following IR is similar in WT, p66Shc-KO and p66Shc-Del rat strains.
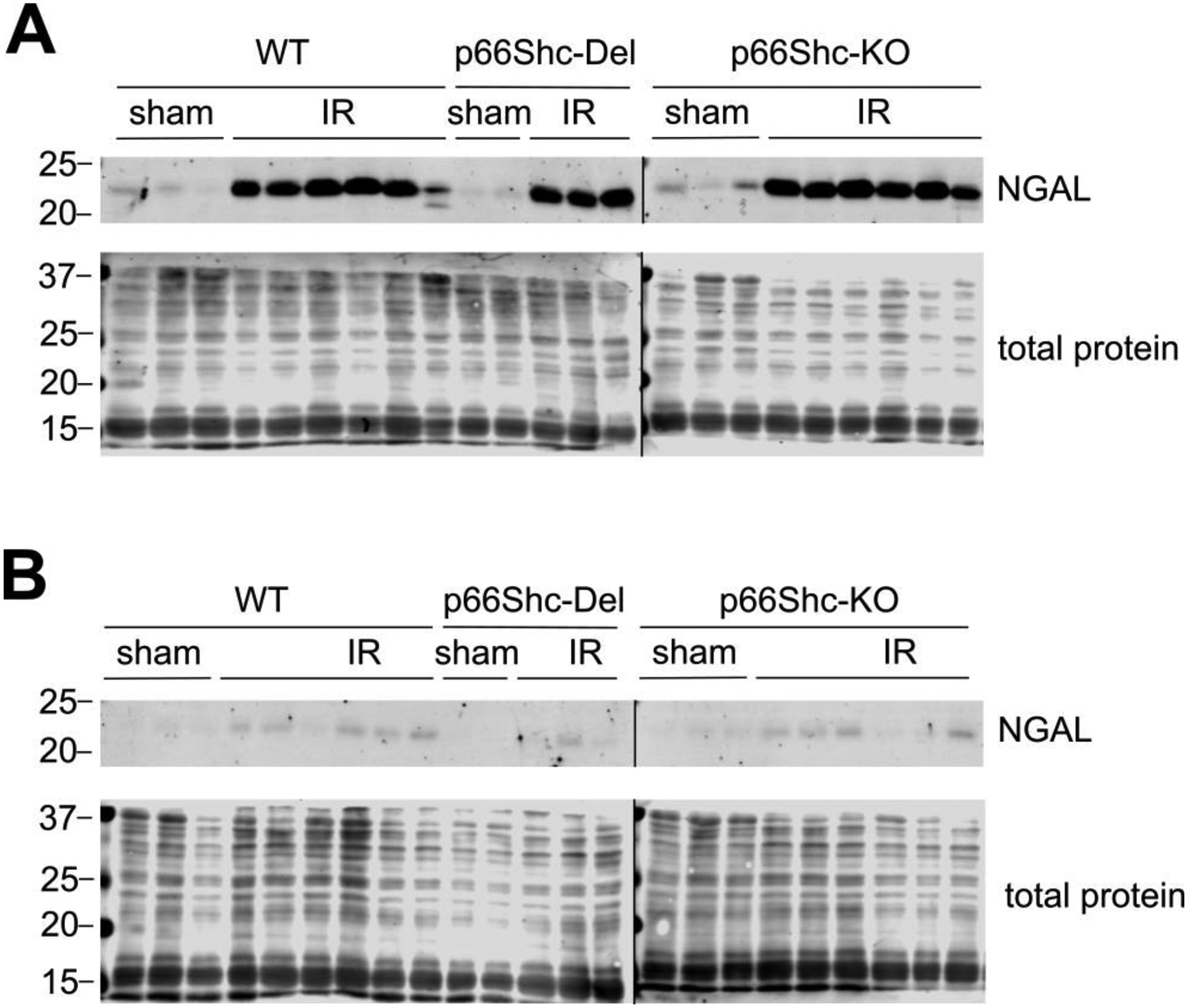
Western blot analysis of outer medullar proteins at 24 hours (A) and seven days (B) post-ischemia. Each lane represents a unique replicate animal. Vertical black lines indicate image splices from the same blot used in both panels. Total protein stain was used to normalize NGAL signals and densitometry revealed no significant statistical difference in NGAL expression among rats undergoing IR.
Since p66Shc expression is known to be a subject of regulation by stress factors, it was necessary to verify that p66Shc and p66Shc-Del mutant are present in corresponding rat strains after IR. As demonstrated in Figure 3 wild type p66Shc expression is indeed induced in parental rats, whereas p66Shc-Del mutant expression is observed in genetically modified rats, albeit stimulation of expression is less prominent. As expected, p66Shc is not expressed and is not detected in p66Shc-KO rats. There is no difference in expression of p52Shc and p46Shc isoforms between WT, p66Shc-KO and p66Shc-Del rats. Some changes in expression of p52Shc and p46Shc during recovery phase could reflect the increase of the number of tubular cells due to remodeling/proliferation.
Fig.3. Expression of p66Shc isoforms after IR in wild type and genetically modified rat strains.
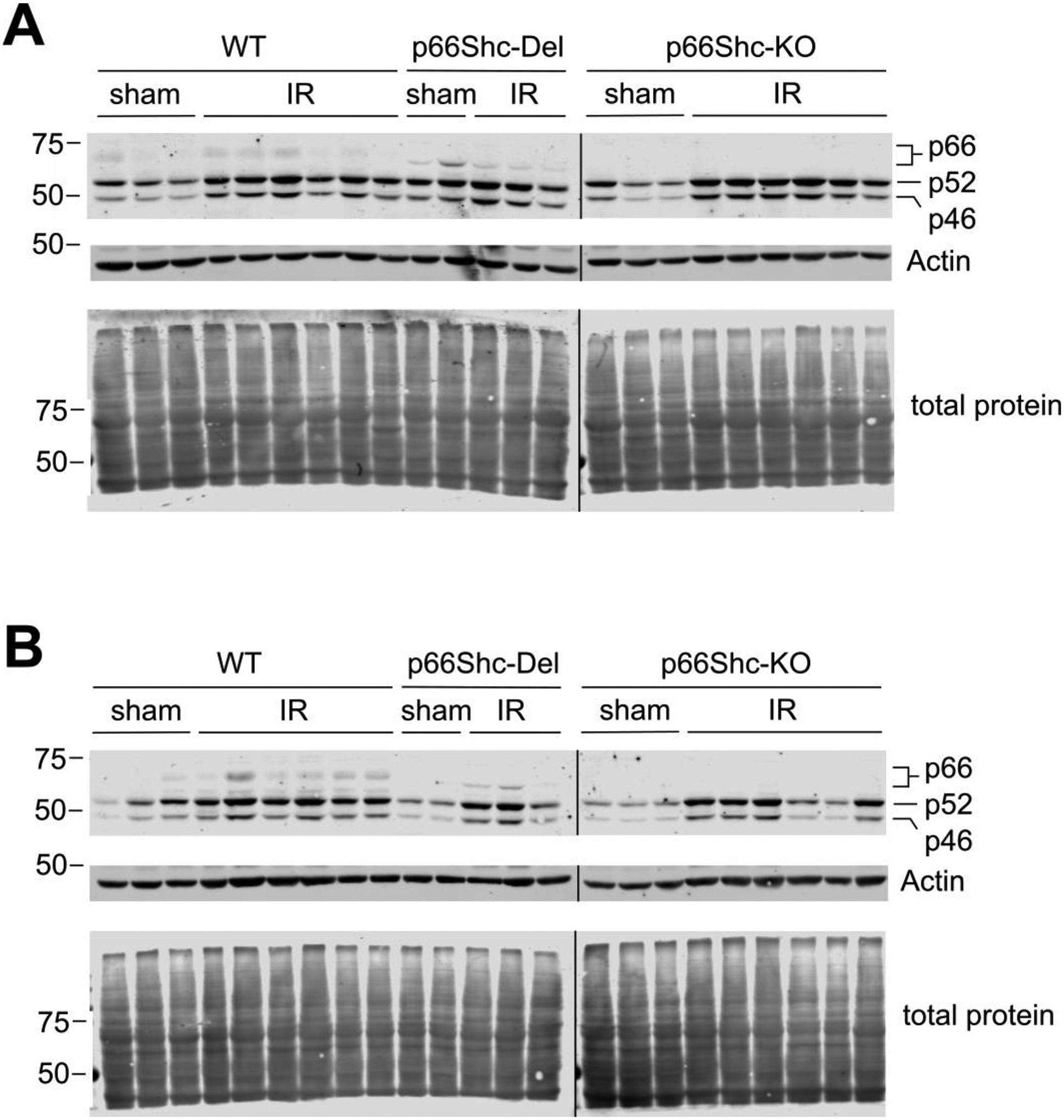
Western blot analysis of outer medullar proteins at 24 hours (A) and seven days (B) post-ischemia. Each lane represents a unique replicate animal. The faster migration of p66Shc in p66Shc-Del is indicative of the nine amino acid deletion. Vertical black lines indicate image splices from the same blot used in (A) and (B). Total protein stain was used to normalize SHC signals for densitometry analysis. No significant statistical difference in the induction of p66-, p52- and p46Shc expression was seen among rats undergoing IR.
Taken together, our data suggest that tubular injury in rat model of renal IR injury proceeds independently of p66Shc expression and signaling. Therefore, IR injury is not caused by p66Shc.
Discussion
Ability of p66Shc to promote oxidative stress and contribute to oxidant injury has been established in different systems [14–16]. In renal proximal tubular cells chronic nicotine exposure-induced ROS production was mediated by p66Shc expression [17]. Furthermore, p66Shc was shown to play an essential role in injury of cultured mouse renal proximal tubule cells during oxidative stress [4]. Accordingly, it seems logical to hypothesize that proximal tubular cells injury that occurs in IR and is accompanied by oxidative stress should be mediated by p66Shc. However, our studies carried out with genetically modified rats do not provide data in support of this hypothesis and, on the contrary, suggest that expression and signaling by p66Shc play no significant role in renal IR injury. One possible reason of this discrepancy is that studies demonstrating p66Shc principal role in oxidative stress-related injury of renal proximal tubular cells were carried out in cell cultures [4,6], whereas we have employed in vivo system with rats genetically modified to differ in p66Shc expression and signaling. It seems important that in a study carried out with p66Shc knockout mice authors also reached conclusion that p66Shc-derived ROS do not contribute either to IR injury or to cardioprotection by ischemic preconditioning [18]. p66Shc is also not involved in the development and progression of right ventricular hypertrophy, the process in which ROS have been suggested to play a role [19].
We have previously reported that p66Shc-Del mutant is characterized by increased hydrogen peroxide production in rat renal tissues and could be considered to represent a constitutively signaling p66Shc variant [9]. Thus, inability of p66Shc-Del to exacerbate proximal tubular epithelial cell injury at 24 hours after IR, or to mitigate restoration of brush border after 7 days is another evidence of lack of contribution of p66Shc signaling to these processes.
As kidney is sensitive to increases in oxidative stress, p66Shc plays a critical role in the progression and aggravation of hypertension induced nephrology, diabetic-induced hyperglycemia, renal disease induced by the glycoxidation product Nε-(carboxymethyl) lysine CML and age-related pathologies [2,20,21]. Apparently, it is not however a crucial player in IR injury of renal tissues and particularly renal epithelial proximal tubular cells in vivo. It may be argued, that p66Shc contribution becomes important, when the damage is somewhat related to vascular pathologies. Adaptor protein p66Shc is a regulator of renal vascular tone and in hypertension induced nephropathy it impairs renal vascular reactivity [7]. Inactivation of p66Shc restores renal vascular reactivity and decreases renal damage in diabetic rats [20]. Protection of p66Shc knockout mice from CML-induced glomerulopathy [21] could be also related to prevention of Advanced Glycation End products-induced vascular dysfunction [22]. Beside kidney, p66Shc mediates oxidative stress-linked cerebrovascular events, which can be responsible for diabetes-related cognitive decline [23]. p66Shc was shown to be involved in regulation of the pulmonary vascular tone with effect of p66Shc knockout being specific for the pulmonary vasculature [24]. Recently, we have shown that modulation of p66Shc signaling impairs cerebral artery myogenic tone in a low renin model of hypertension [25].
Expression or enhanced signaling by adaptor protein p66Shc does not contribute to tubular damage induced by renal ischemia-reperfusion injury in rat
p66Shc expression or signaling does not mitigate restoration of brush border in rat model of ischemia-reperfusion injury
Ischemia-reperfusion injury is not caused by p66Shc
Acknowledgements
This work was supported by the National Institutes of Health grant number R01 HL147976 to Sorokin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Fadini GP, Albiero M, Menegazzo L, Boscaro E, Pagnin E, Iori E, Cosma C, Lapolla A, Pengo V, Stendardo M, Agostini C, Pelicci PG, Giorgio M, Avogaro A, The redox enzyme p66Shc contributes to diabetes and ischemia-induced delay in cutaneous wound healing, Diabetes 59 (2010) 2306–2314. 10.2337/db09-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wright KD, Staruschenko A, Sorokin A, Role of adaptor protein p66Shc in renal pathologies, Am J Physiol Renal Physiol 314 (2018) F143–F153. 10.1152/ajprenal.00414.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Regner KR, Roman RJ, Role of medullary blood flow in the pathogenesis of renal ischemia-reperfusion injury, Curr Opin Nephrol Hypertens 21 (2012) 33–38. 10.1097/MNH.0b013e32834d085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Arany I, Faisal A, Nagamine Y, Safirstein RL, p66shc inhibits pro-survival epidermal growth factor receptor/ERK signaling during severe oxidative stress in mouse renal proximal tubule cells, J Biol Chem 283 (2008) 6110–6117. 10.1074/jbc.M708799200. [DOI] [PubMed] [Google Scholar]
- [5].Arany I, Faisal A, Clark JS, Vera T, Baliga R, Nagamine Y, p66SHC-mediated mitochondrial dysfunction in renal proximal tubule cells during oxidative injury, Am J Physiol Renal Physiol 298 (2010) F1214–1221. 10.1152/ajprenal.00639.2009. [DOI] [PubMed] [Google Scholar]
- [6].Zhao WY, Han S, Zhang L, Zhu YH, Wang LM, Zeng L, Mitochondria-targeted antioxidant peptide SS31 prevents hypoxia/reoxygenation-induced apoptosis by down-regulating p66Shc in renal tubular epithelial cells, Cell Physiol Biochem 32 (2013) 591–600. 10.1159/000354463. [DOI] [PubMed] [Google Scholar]
- [7].Miller B, Palygin O, Rufanova VA, Chong A, Lazar J, Jacob HJ, Mattson D, Roman RJ, Williams JM, Cowley AW Jr., Geurts AM, Staruschenko A, Imig JD, Sorokin A, p66Shc regulates renal vascular tone in hypertension-induced nephropathy, J Clin Invest 126 (2016) 2533–2546. 10.1172/JCI75079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Aksamitiene E, Hoek JB, Kholodenko B, Kiyatkin A, Multistrip Western blotting to increase quantitative data output, Electrophoresis 28 (2007) 3163–3173. 10.1002/elps.200700002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Miller B, Palygin O, El-Meanawy A, Mattson DL, Geurts AM, Staruschenko A, Sorokin A, p66Shc-mediated hydrogen peroxide production impairs nephrogenesis causing reduction of number of glomeruli, Life Sci 279 (2021) 119661. 10.1016/j.lfs.2021.119661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cassidy H, Slyne J, Higgins M, Radford R, Conlon PJ, Watson AJ, Ryan MP, McMorrow T, Slattery C, Neutrophil gelatinase-associated lipocalin (NGAL) is localised to the primary cilium in renal tubular epithelial cells - A novel source of urinary biomarkers of renal injury, Biochim Biophys Acta Mol Basis Dis 1865 (2019) 165532. 10.1016/j.bbadis.2019.165532. [DOI] [PubMed] [Google Scholar]
- [11].Tejchman K, Nowacki A, Kotfis K, Skwirczynska E, Kotowski M, Zair L, Ostrowski M, Sienko J, The Role of Endothelins, IL-18, and NGAL in Kidney Hypothermic Machine Perfusion, Biomedicines 9 (2021). 10.3390/biomedicines9040417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lisowska-Myjak B, Serum and urinary biomarkers of acute kidney injury, Blood Purif 29 (2010) 357–365. 10.1159/000309421. [DOI] [PubMed] [Google Scholar]
- [13].Allegretti AS, Parada XV, Endres P, Zhao S, Krinsky S, St Hillien SA, Kalim S, Nigwekar SU, Flood JG, Nixon A, Simonetto DA, Juncos LA, Karakala N, Wadei HM, Regner KR, Belcher JM, Nadim MK, Garcia-Tsao G, Velez JCQ, Parikh SM, Chung RT, H.-H.s. investigators, Urinary NGAL as a Diagnostic and Prognostic Marker for Acute Kidney Injury in Cirrhosis: A Prospective Study, Clin Transl Gastroenterol 12 (2021) e00359. 10.14309/ctg.0000000000000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lebiedzinska-Arciszewska M, Oparka M, Vega-Naredo I, Karkucinska-Wieckowska A, Pinton P, Duszynski J, Wieckowski MR, The interplay between p66Shc, reactive oxygen species and cancer cell metabolism, Eur J Clin Invest 45 Suppl 1 (2015) 25–31. 10.1111/eci.12364. [DOI] [PubMed] [Google Scholar]
- [15].Natalicchio A, Tortosa F, Perrini S, Laviola L, Giorgino F, p66Shc, a multifaceted protein linking Erk signalling, glucose metabolism, and oxidative stress, Arch Physiol Biochem 117 (2011) 116–124. 10.3109/13813455.2011.562513. [DOI] [PubMed] [Google Scholar]
- [16].Pellegrini M, Baldari CT, Apoptosis and oxidative stress-related diseases: the p66Shc connection, Curr Mol Med 9 (2009) 392–398. [DOI] [PubMed] [Google Scholar]
- [17].Arany I, Clark J, Reed DK, Juncos LA, Chronic nicotine exposure augments renal oxidative stress and injury through transcriptional activation of p66shc, Nephrol Dial Transplant 28 (2013) 1417–1425. 10.1093/ndt/gfs596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Boengler K, Bencsik P, Paloczi J, Kiss K, Pipicz M, Pipis J, Ferdinandy P, Schluter KD, Schulz R, Lack of Contribution of p66shc and Its Mitochondrial Translocation to Ischemia-Reperfusion Injury and Cardioprotection by Ischemic Preconditioning, Front Physiol 8 (2017) 733. 10.3389/fphys.2017.00733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hirschhauser C, Sydykov A, Wolf A, Esfandiary A, Bornbaum J, Kutsche HS, Boengler K, Sommer N, Schreckenberg R, Schluter KD, Weissmann N, Schermuly R, Schulz R, Lack of Contribution of p66shc to Pressure Overload-Induced Right Heart Hypertrophy, Int J Mol Sci 21 (2020). 10.3390/ijms21249339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Miller BS, Blumenthal SR, Shalygin A, Wright KD, Staruschenko A, Imig JD, Sorokin A, Inactivation of p66Shc Decreases Afferent Arteriolar KATP Channel Activity and Decreases Renal Damage in Diabetic Dahl SS Rats, Diabetes 67 (2018) 2206–2212. 10.2337/db18-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Menini S, Iacobini C, Ricci C, Oddi G, Pesce C, Pugliese F, Block K, Abboud HE, Giorgio M, Migliaccio E, Pelicci PG, Pugliese G, Ablation of the gene encoding p66Shc protects mice against AGE-induced glomerulopathy by preventing oxidant-dependent tissue injury and further AGE accumulation, Diabetologia 50 (2007) 1997–2007. 10.1007/s00125-007-0728-7. [DOI] [PubMed] [Google Scholar]
- [22].Wautier JL, Wautier MP, Schmidt AM, Anderson GM, Hori O, Zoukourian C, Capron L, Chappey O, Yan SD, Brett J, et al. , Advanced glycation end products (AGEs) on the surface of diabetic erythrocytes bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: a link between surface-associated AGEs and diabetic complications, Proc Natl Acad Sci U S A 91 (1994) 7742–7746. 10.1073/pnas.91.16.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Minami Y, Sonoda N, Hayashida E, Makimura H, Ide M, Ikeda N, Ohgidani M, Kato TA, Seki Y, Maeda Y, Kanba S, Takayanagi R, Ogawa Y, Inoguchi T, p66Shc Signaling Mediates Diabetes-Related Cognitive Decline, Sci Rep 8 (2018) 3213. 10.1038/s41598-018-21426-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gierhardt M, Pak O, Sydykov A, Kraut S, Schaffer J, Garcia C, Veith C, Zeidan EM, Brosien M, Quanz K, Esfandiary A, Saraji A, Hadzic S, Kojonazarov B, Wilhelm J, Ghofrani HA, Schermuly RT, Seeger W, Grimminger F, Herden C, Schulz R, Weissmann N, Heger J, Sommer N, Genetic Deletion of p66shc and/or Cyclophilin D Results in Decreased Pulmonary Vascular Tone, Cardiovasc Res (2020). 10.1093/cvr/cvaa310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hughes WE, Hockenberry J, Miller B, Sorokin A, Beyer AM, Modulation of p66Shc impairs cerebrovascular myogenic tone in low renin but not low nitric oxide models of systemic hypertension, Am J Physiol Heart Circ Physiol 321 (2021) H1096–H1102. 10.1152/ajpheart.00542.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]