

Abstract
G protein-coupled receptors (GPCRs) transduce a diverse variety of extracellular stimuli into intracellular signaling. These receptors are the most clinically productive drug targets at present. Despite decades of research on the signaling consequences of molecule-receptor interactions, conformational components of receptor-effector interactions remain incompletely described. The β2-adrenergic receptor (β2AR) is a prototypical and extensively studied GPCR that can provide insight into this aspect of GPCR signaling thanks to robust structural data and rich pharmacopeia. Using bioluminescence resonance energy transfer -based biosensors, second messenger assays, and biochemical techniques, we characterize the properties of β2AR-F193A. This single point mutation in extracellular loop 2 of the β2AR is sufficient to intrinsically bias the β2AR away from β-arrestin interaction and demonstrates altered regulatory outcomes downstream of this functional selectivity. This study highlights the importance of extracellular control of intracellular response to stimuli and suggests a previously undescribed role for the extracellular loops of the receptor and the extracellular pocket formed by transmembrane domains 2, 3, and 7 in GPCR regulation that may contribute to biased signaling at GPCRs.
SIGNIFICANCE STATEMENT
The role of extracellular G protein-coupled receptor (GPCR) domains in mediating intracellular interactions is poorly understood. We characterized the effects of extracellular loop mutations on agonist-promoted interactions of GPCRs with G protein and β-arrestin. Our studies reveal that F193 in extracellular loop 2 in the β2-adrenergic receptor mediates interactions with G protein and β-arrestin with a biased loss of β-arrestin binding. These results provide new insights on the role of the extracellular domain in differentially modulating intracellular interactions with GPCRs.
Introduction
G protein-coupled receptors (GPCRs) are well appreciated to be clinically important therapeutic targets and, as such, have been the subject of intense study for many decades (Jacobson, 2015). The β2-adrenergic receptor (β2AR) has served as an exemplar model for GPCR function, structure, and signaling paradigms as more was understood about these receptors’ complex pharmacology and interactome (Pierce et al., 2002). Canonically, when the β2AR is activated by an exogenous agonist or endogenous catecholamine, the receptor undergoes conformational changes that promote activation of the heterotrimeric Gs protein to initiate downstream signaling. The activated receptor is then phosphorylated by G protein-coupled receptor kinases (GRKs), which promote recruitment of β-arrestins to facilitate receptor unco-upling from Gs (desensitization), receptor endocytosis, and β-arrestin-mediated signaling (Krupnick and Benovic, 1998; Tan et al., 2018). β2AR ligands comprised of agonists, antagonists, inverse agonists, biased agonists, and other pharmacological agents have been discovered and are important tools in understanding the functional aspects of modulating β2AR signaling (Salon et al., 2011). Additionally, due to advances in biophysical characterization of GPCRs, the structural features of these classes of ligands have been elucidated to some extent (Manglik and Kobilka, 2014; Latorraca et al., 2017). While both the pharmacological tools and structural information around the β2AR is robust, it remains incomplete (Capper and Wacker, 2018), and the mechanism through which the receptor transitions from G protein interactions to GRKs to β-arrestins is unclear.
Previous studies have demonstrated the key role that the GPCR extracellular domains play in GPCR signaling outcomes, and several allosteric modulators of GPCRs have been discovered to occupy these regions (Strachan et al., 2014; Thal et al., 2018; Bermudez and Bock, 2019). In the β2AR, the extracellular vestibule, a region occupying space above the endogenous orthosteric ligand binding site, is a mediator of ligand entry and exit and activation dynamics (Dror et al., 2011). This pocket has been modeled to create a metastable energy barrier that must be overcome to achieve ligand binding in the orthosteric site (Dror et al., 2011). This occurs at a steric “gate” formed by phenylalanine 193 (F193) in extracellular loop 2 (ECL2) and tyrosine 308 (Y308) (Ballesteros-Weinstein number 7.35) in TM7 that caps this site (Ballesteros and Weinstein, 1995). The position of Y308 has been implicated in G protein interaction in corresponding residues in other GPCRs, and it has been suggested by molecular dynamics that F193 is a “functional hotspot” in the β2AR (Masureel et al., 2018; Nivedha et al., 2018). In this study, we examined the role of this entry barrier in receptor function by disrupting the steric gate in the extracellular vestibule through mutagenesis and subsequent characterization.
Understanding the molecular mechanisms of the transition from activation of G proteins to downstream regulation is paramount to developing the next generation of GPCR therapeutics. Here, we show that disruption of the steric gate at the extracellular vestibule substantially biases β2AR signaling toward G protein activation by inhibiting receptor phosphorylation and interaction with β-arrestin. This work provides insight into the molecular features of β2AR bias and subsequent cellular responses and provides insight into differential bias between adrenergic receptor family members.
Materials and Methods
Cell Culture
Human embryonic kidney 293 (HEK 293) cells were maintained in Dulbecco’s Modified Eagles Medium containing 10% FBS)and 12.5 mM of HEPES, pH 7.2. β2AR and β-arrestin1/2 triple-KO HEK 293 cells (hereafter simply referred to as β2AR KO) (Cahill et al., 2017) were maintained in Dulbecco’s modified Eagle’s medium containing 10% FBS, 12.5 mM of HEPES, pH 7.2, and penicillin-streptomycin. Cells were incubated at 37°C in a humidified incubator with 5% CO2.
cAMP Measurement
To eliminate endogenous adrenergic receptor-mediated responses, β2AR-HEK 293 KO cells were used. The cells were transiently transfected with wild-type (WT) β2AR or β2AR-F193A, seeded in poly-L-lysine-coated 24-well plates, incubated at 37°C and treated with indicated concentrations of ISO for 10 minutes. Cells were then lysed in 0.1 M HCl for 20 minutes at room temperature. cAMP levels were measured using the Cayman Chemical Cyclic AMP EIA kit following the manufacturer’s instructions.
Gs Recruitment by Bioluminescent Resonance Energy Transfer (BRET) Assay
HEK 293 cells were transiently co-transfected with wild-type (β2AR or β1-adrenergic receptor, β1AR) or mutated (β2AR-F193A or β1AR-F201A) receptor C-terminally fused with BRET donor RlucII and the mini-G protein BRET acceptor NES-Venus-mGs (Wan et al., 2018). Forty-eight hours post-transfection, medium was removed, and cells were incubated with the indicated concentration of isoproterenol (ISO) in the presence of 5 μM of Renilla luciferase substrate coelenterazine H for 15 minutes. BRET was measured using a Tecan Infinite F500 microplate reader. BRET ratios were calculated as the light emitted by the Venus acceptor divided by the total light emitted by the RLucII donor.
Analysis of β-Arrestin2 Binding to the β2AR and β1AR using BRET
HEK 293 cells were transfected with wild-type or mutant pcDNA-β-arrestin2-GFP10 and either pcDNA3-β2AR-RlucII, pcDNA3-β2AR-F193A-RlucII, pcDNA3-β1AR-RlucII, or pcDNA3-β1AR-F201A-RlucII using X-tremegene HP9 complexed in serum-free optiMEM. Twenty-four hours after transfection, cells were replated at 100,000 cells per well in an opaque, poly-L-lysine-coated 96-well plate and incubated overnight at 37°C. Coelenterazine 400a was added to cells prior to agonist stimulation. BRET was measured using a Tecan Infinite F500 microplate reader, and BRET ratios were calculated as the light emitted by the GFP10 acceptor divided by the total light emitted by the RLucII donor. To evaluate the role of receptor phosphorylation in β-arrestin binding, a constitutively active β-arrestin2 insensitive to the phosphorylation state of the β2AR was generated by mutation of Ile386, Val387, and Phe388 to alanine (AAA mutant) as previously described (Celver et al., 2002) and then analyzed for binding to WT and mutant β2AR as described above.
Cell Membrane Preparation
HEK 293 cells were transfected with β2AR WT or β2AR-F193A using X-tremegene HP9 complexed in serum-free optiMEM. After 48 hours, media was aspirated, and the cells were lysed on ice in 50 mM of Tris-HCl, pH 7.7, for 10 minutes. Cells were then scraped and pelleted by centrifugation at 1000 rpm for 5 minutes. The pellet was washed with 50 mM of Tris-HCl, pH 7.7, and passed through a 22-gauge syringe five times. Lysed cells were spun at 27,000 × g for 10 minutes, and the pellet was resuspended in buffer containing 15 mM of Tris-HCl, pH 7.4, 120 mM of sodium chloride, 5.4 mM of potassium chloride, 1.8 mM of calcium chloride, and 5 mM of glucose. Determination of total membrane protein concentration was determined by Bradford assay.
Competition Binding Assay
Sixty micrograms of cell membrane protein were incubated with 0.5 nM of [3H]dihydroxyalprenolol and the indicated concentrations of ISO in a volume of 1 ml. The mixture was incubated at 25°C for 1 hour and then filtered over GF/C glass fiber filters soaked in 0.05% polyethylenimine. The filters were washed three times with ice-cold Tris-buffered saline. Bound radioactivity was measured by liquid scintillation counting and expressed in counts per minute.
In-Cell β2AR Phosphorylation
HEK 293 cells seeded into poly-L-lysine coated 6-well plates were transiently transfected with FLAG-tagged β2AR WT or β2AR-F193A and incubated at 37°C. After 48 hours, cells were incubated with 0, 1, or 100 μM of ISO for 10 minutes and then rinsed with PBS, scraped in lysis buffer on ice, and sonicated. Lysates were immunoprecipitated using rabbit polyclonal anti-FLAG and Protein G agarose beads. Immunoprecipitated proteins were separated by SDS-PAGE and analyzed by western blot using a β2AR phospho-specific antibody or FLAG antibody.
Mutagenesis
β2AR-F193A and β1AR-F201A mutants were created with the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) according to manufacturer’s protocol.
Receptor Internalization
HEK 293 cells transiently transfected with FLAG-β2AR WT or FLAG-β2AR-F193A were seeded into poly-L-lysine-coated 24-well plates and incubated at 37°C. Cells were stimulated with 1 μM of ISO for 0–60 minutes. Cells were then fixed on ice and processed for cell surface ELISA with polyclonal anti-FLAG primary antibody, anti-rabbit HRP secondary antibody, and incubation with (2,2'-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt) (ABTS). Absorbance was then measured on a plate reader at 405 nm.
Functional Desensitization in HEK 293 Cells
β2AR KO HEK 293 cells were transiently transfected with pcDNA-β-arrestin2-GFP10 and either β2AR WT or β2AR-F193A and were seeded in poly-L-lysine-coated 24-well plates and incubated at 37°C. Cells were stimulated with 1 μM of ISO for 30 minutes. Cells were washed three times with PBS and incubated with various concentrations of ISO for 10 minutes at 37°C. Cells were then lysed in 0.1 M of HCl for 20 minutes at room temperature, and cAMP levels were measured using the Caymen Chemical Cyclic AMP EIA kit following the manufacturer’s instructions.
Extracellular Region Alignment
Alignment of the extracellular regions was done with Clustal Omega multiple sequence alignment tool (Madeira et al., 2019), and residue sequences of these regions were obtained from UniProt.
Quantification and Statistical Analysis
All statistical analyses were produced using Prism 8.0 (GraphPad Software). All data are expressed as the mean ± standard deviation (SD) from at least three independent experiments, unless otherwise stated in the figure legend. For normalizations, the values of all replicates were divided by the mean of isoproterenol-induced maximal responses and multiplied by 100 for any given read-out. Comparison of negative log of the half-maximal effective concentration (pEC50s), negative log of the half-maximal inhibitory concentration, and maximal efficacies were assessed by unpaired t test and considered statistically significant when P < 0.05. Stimulus inactivation was quantitated as previously described (Ostrom and Ehlert, 1997).
Results
Mutagenesis of the Extracellular Region of the β2AR Reveals F193 as a Key Determinant of β-Arrestin Interaction
Recent work in our laboratory has identified a potential role for the extracellular vestibule in coordinating response to biased ligands. This extracellular region located above the orthosteric binding site of the receptor, including extracellular loops 2 and 3, was used as a starting point to determine key residues for propagating a biased signaling profile (Fig. 1A). Generating point mutants around this site and subsequent evaluation by BRET revealed that isoproterenol (ISO)-promoted β2AR interaction with β-arrestin is robustly inhibited by the mutation of phenylalanine 193 to alanine (F193A), while mutation of other residues in ECL2 and ECL3 had modest effects (Fig. 1B). This suggests that F193 is a key residue involved in coordinating β-arrestin coupling to the β2AR.
Fig. 1.

Mutagenesis of extracellular loops (ECLs) 2 and 3. (A) Top view of β2-adrenergic receptor (β2AR) showing the orthosteric binding site (yellow mesh) and allosteric region (blue mesh). Key residues forming the allosteric region are shown as sticks. (B) Isoproterenol (ISO) dose response curve for β-arrestin binding to wild-type and β2AR ECL2 and ECL3 mutants as measured by bioluminescence resonance energy transfer. Human embryonic kidney 293cells co-transfected with β-arrestin2-GFP10 and indicated β2AR-RlucII wild-type or mutant constructs were incubated with coelenterazine 400a for 2 minutes and then stimulated with indicated concentrations of ISO. Data for dose response curve was taken 12 minutes post ISO addition. n = 3 independent experiments.
Characterization of β2AR-F193A Demonstrates Intrinsic G Protein Bias
Fig. 2A shows the alteration of the steric gate in β2AR-F193A (WT, gray spheres, F193A, yellow sticks). Given the previously hypothesized role of this residue as a functional hotspot (Nivedha et al., 2018) and demonstrated inhibition of β-arrestin interaction by mutagenesis, we next characterized receptor function more broadly for this point mutant. Here, we verify that β2AR-F193A shows substantial inhibition of agonist-promoted β-arrestin interaction as assessed by BRET, with a 280-fold right-shift in the ISO dose response (mean pEC50 ± SD = 7.71 ± 0.29 versus 5.26 ± 0.11, Fig. 2B and Table 1). To evaluate whether this phenotype was caused by reduced agonist binding, we performed competition binding assays with membranes harvested from cells expressing either WT β2AR or β2AR-F193A. In these studies, various concentrations of ISO were incubated with the receptor preparations in the presence of the tritiated β2AR antagonist, [3H]dihydroxyalprenolol (Fig. 2C). It was found that both WT and mutant receptors show similar agonist binding profiles with a 2.8-fold right-shift in the IC50 for the mutant receptor (mean negative log of the half-maximal inhibitory concentration [pIC50] ± SD = 5.89 ± 0.02 versus 5.45 ± 0.03, unpaired t test, P < 0.05). Interestingly, this point mutation causes minimal ISO dose response shift for cAMP response (mean negative log of the half-maximal effective concentration (pEC50) ± SD = 7.92 ± 0.47 versus 7.86 ± 0.40), with no reduction in efficacy as measured by ELISA in β2AR-KO HEK 293 cells transiently transfected with wild-type or mutant receptors (Fig. 2D, Table 1). However, it is worth noting that the basal level of cAMP at very low levels of ISO is greatly reduced for the F193A mutant compared with WT β2AR (Fig. 2D). To control for the amplification of cellular cAMP, recruitment of mini-Gs by BRET was also performed. In these studies, a 20-fold right shift of mini-Gs recruitment was observed for the mutant receptor compared with wild-type (mean pEC50 ± SD = 7.98 ± 0.55 versus 6.67 ± 0.26, Fig. 2E, Table 1). It is possible that this effect is driven in part by intrinsic differences between endogenous WT Gs and the mini-Gs construct in addition to amplification effects in the cAMP measurements. Taken together, F193A is partially defective in mediating agonist-promoted interactions of the β2AR with both Gs and β-arrestin, although it is clearly more disrupted in β-arrestin binding. To further confirm this, we performed a quantitation of the stimulus inactivation for the pathways considered as previously described by Ostrom and Ehlert, 1997. Such analysis estimates the apparent fractional loss in the receptor population that could account for the loss in function. The stimulus inactivation analysis revealed a decrease in the apparent efficacy only for β-arrestin2 recruitment to F193A versus the WT β2AR (Table 1).
Fig. 2.
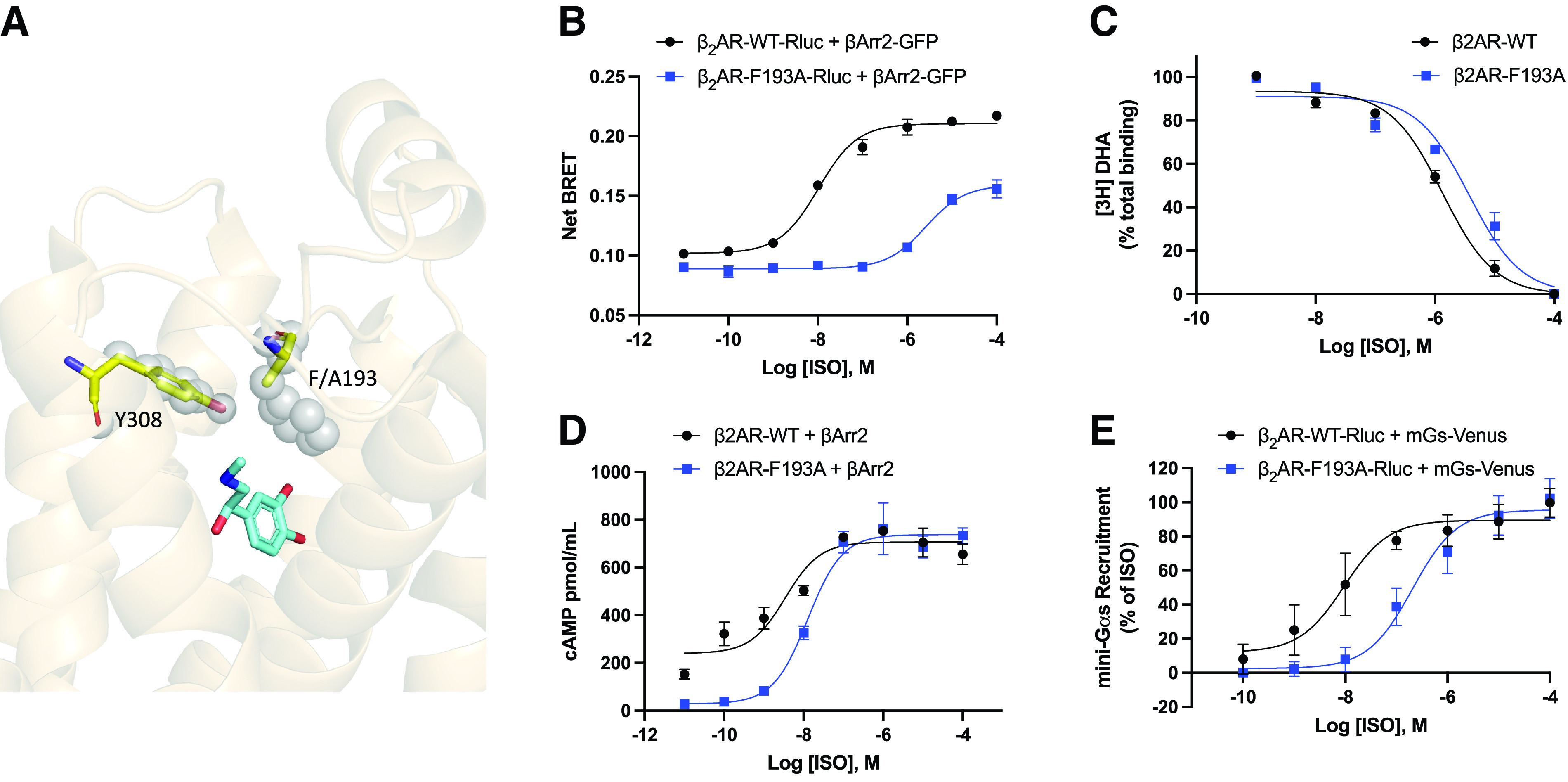
Characterization of β2-adrenergic receptor (β2AR)-F193A. (A) Extracellular loop 3 facing side view of β2AR. The orthosteric site gate comprised of Y308 and F193 is represented by gray spheres. The mutated gate of the F193A mutant is represented by yellow sticks. Endogenous β2AR agonist adrenaline is shown in the orthosteric binding pocket (light blue, from PDB: 4LDO). (B) Isoproterenol (ISO) dose response curve for β-arrestin binding to β2AR wild-type and β2AR-F193A as measured by bioluminescence resonance energy transfer (BRET). Human embryonic kidney 293 (HEK 293) cells co-transfected with β-arrestin2-GFP10 and indicated β2AR-RlucII wild-type or β2AR-F193A-RlucII constructs were incubated with coelenterazine 400a for 2 minutes and then stimulated with indicated concentrations of ISO. Data for dose response curve was taken 12 minutes post ISO addition. n = 3 independent experiments. (C) [3H]dihydroxyalprenolol competition curves for ISO. n = 3 independent experiments. (D) Dose response curves for ISO promoted cAMP production by ELISA. β2AR-KO HEK 293 transiently transfected with β2AR-WT or β2AR-F193A were stimulated with the indicated concentrations of ISO for 10 minutes. Cells were lysed and cAMP production was measured. n = 3 independent experiments. (E) Dose response curves for ISO promoted mini-Gs recruitment by BRET. HEK 293 cells transiently transfected wild-type or mutated β2AR-RlucII and NES-Venus-mGs were stimulated with the indicated concentrations of ISO for 15 minutes, and BRET signal was measured. n = 3 independent experiments.
TABLE 1.
Comparison of β2AR-WT and β2AR-F193A
| pEC50 ± SD | Maximum | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Assay | WT | F193A | Shift | Significant | WT (%) | F193A (%WT) | Significant | SI (%) | |
| Arrestin BRET | 7.71 ± 0.29 | 5.26 ± 0.11 | 2.45 | Y | 100 | 56.4 ± 4.7 | Y | 60.8 | |
| cAMP | 7.92 ± 0.47 | 7.86 ± 0.41 | 0.06 | N | 100 | 111.9 ± 4.1 | N | 98.4 | |
| mGs BRET | 7.98 ± 0.55 | 6.67 ± 0.26 | 1.31 | Y | 100 | 102.2 ± 9.6 | N | 99.9 | |
Comparison of Wild-type β2-adrenergic receptor (β2AR-WT) and β2AR-F193A across assays. Negative log of the half-maximal effective concentration (pEC50) values and maximum response relative to β2AR-WT are listed. Shift indicates the difference in pEC50 values for β2AR-WT and β2AR-F193A. Significance for pEC50 and maximum response was determined by t test for n = 3 experiments. Stimulus inactivation indicates a comparison of agonist doses that promote equal response in WT and mutant receptor in each assay. This parameter was estimated as described previously (Ostrom and Ehlert, 1997). Briefly, equiactive isoproterenol concentrations were compared between β2AR-WT and β2AR-F193A by interpolating equivalent β2AR-F193A responses from β2AR-WT dose response curves. The stimulus inactivation (SI) parameter denotes the apparent efficacy of β2AR-F193A compared with wild-type.
Downstream Regulation of Agonist Activation is Impaired for β2AR-F193A
Typically, activation of the β2AR stimulates the Gs-adenylyl cyclase-cAMP-PKA pathway and is coupled to regulatory processes associated with phosphorylation of the receptor by GRKs and β-arrestin interaction (Hodavance et al., 2016). These regulatory mechanisms are critical to the inhibition of β2AR signaling via receptor desensitization and β-arrestin-mediated internalization (Ferguson et al., 1996; Goodman et al., 1996; Nobles et al., 2011). We evaluated these regulatory processes in the context of the intrinsically biased F193A mutant. Using an in-cell receptor phosphorylation assay, it was determined that GRK-mediated phosphorylation of Ser355/356 on the receptor C-tail is substantially inhibited for β2AR-F193A compared with wild-type even at very high concentrations of ISO (Fig. 3A and B). To determine whether the observed bias of β2AR-F193A was due to a difference in the conformational repertoire of the receptor or simply due to inefficient GRK coupling, we performed a BRET assay using a phosphorylation-insensitive β-arrestin mutant carrying a triple alanine substitution in the C-tail of β-arrestin2 (AAA) as previously described (Celver et al., 2002). ISO dose response curves were left-shifted 2- to 3-fold in the presence of AAA compared with WT β-arrestin2 for both WT and mutant receptor (Fig. 3C). This left shift is in line with previous studies showing that phosphorylation of the β2AR enhances the affinity of β-arrestin for the receptor by ∼10-fold (Lohse et al., 1992). It is also evident that the ISO promoted β-arrestin2 interaction was greatly reduced for β2AR-F193A compared with WT β2AR for both forms of β-arrestin examined (Fig. 3C). This indicates that the decreased capacity for β-arrestin interaction is at least partially due to a biased conformation of the receptor rather than simply reduced GRK phosphorylation or a general reduction in receptor efficacy.
Fig. 3.

G protein-coupled receptor kinase phosphorylation, receptor internalization and desensitization for wild-type (WT) and mutant β2AR. (A) Representative blot for agonist promoted phosphorylation of the β2-adrenergic receptor (β2AR). Human embryonic kidney 293 (HEK 293) cells expressing FLAG-β2AR-WT or FLAG-β2AR-F193A were stimulated with 0, 1, or 100 μM of isoproterenol (ISO) for 10 minutes. Cells were lysed, and the FLAG-β2AR was immunoprecipitated. Phosphorylation at serines 355 and 356 was analyzed by western blot using a pSer355/356 antibody, while total β2AR was measured using an anti-FLAG antibody. The western blot is representative of at least 3 independent experiments and is quantified in panel (B). (C) ISO dose response curve for WT or AAA β-arrestin binding to β2AR wild-type and β2AR-F193A as measured by bioluminescence resonance energy transfer. HEK 293 cells co-transfected with β-arrestin2-GFP10 or β-arrestin2-AAA-GFP10 and indicated β2AR-RlucII WT or β2AR-F193A-RlucII constructs were incubated with coelenterazine 400a for 2 minutes and then stimulated with indicated concentrations of ISO. Data for dose response curve was taken 12 minutes post ISO addition. n = 3 independent experiments. (D) Effect of F193A mutation on agonist-promoted internalization of the β2AR. HEK 293 cells expressing FLAG-β2AR-WT or FLAG-β2AR-F193A were stimulated with 1 μM of ISO for up to 60 minutes. Cells were fixed, and receptor surface expression was measured by ELISA. Loss of cell surface expression at 60 minutes is quantified in (E). n = 3 independent experiments. HEK 293 cells expressing β2AR-WT (F) or β2AR-F193A (G) were desensitized by incubating with 1 μM of ISO, washing with PBS, and then re-stimulating with ISO at the indicated concentrations for 10 minutes. Cells were lysed, and cAMP production was measured by ELISA. n = 3 independent experiments.
We also examined agonist-promoted receptor internalization using a cell-surface ELISA. We found that β2AR-F193A was unresponsive to agonist-promoted internalization relative to wild-type, with cell surface levels of β2AR-F193A unchanged up to 60 minutes post agonist addition (Fig. 3D and E). These results support the loss of β-arrestin binding in this point mutant and demonstrate an altered regulatory phenotype when compared with WT β2AR.
Agonist promoted desensitization of cellular response to β-agonists is a known consequence of prolonged treatment that manifests downstream of GRK phosphorylation and β-arrestin binding (Gagnon et al., 1998; Deshpande and Penn, 2006; Deshpande et al., 2008). To evaluate whether β2AR-F193A was sensitive to this phenomenon, β2AR-KO cells transiently transfected with wild-type or F193A mutant were stimulated with ISO for 30 minutes, washed, and then re-stimulated with various doses of ISO to generate dose response curves for cAMP production. Relative to a control generated from cells that were not pre-treated with ISO, we observe a right-shift in the EC50 of ISO and decreased response in wild-type β2AR cells that is greatly diminished in cells expressing β2AR-F193A (Fig. 3F and G). These results suggest that the point mutant is insensitive to acute desensitization promoted by agonist treatment, and is consistent with the observation that receptor phosphorylation, β-arrestin binding, and receptor internalization are inhibited for β2AR-F193A.
F193 Represents a Highly Conserved ECL2 Residue in Class A GPCRs
Class A GPCRs like the β2AR represent the largest and most diverse GPCR subfamily (Venkatakrishnan et al., 2019). This class of receptors, despite regulating varied physiologic functions, share many evolutionarily conserved sequence motifs and activation/regulatory processes (Hanyaloglu and von Zastrow, 2008; Filipek, 2019). An alignment of the extracellular regions of several β-adrenergic, adenosine, muscarinic acetylcholine, and dopamine receptors is shown in Fig. 4A. In the alignment, it is apparent that a phenylalanine residue corresponding to F193 in the β2AR (or tyrosine in the dopamine D2R) is conserved across several GPCR families and within GPCR family members.
Fig. 4.
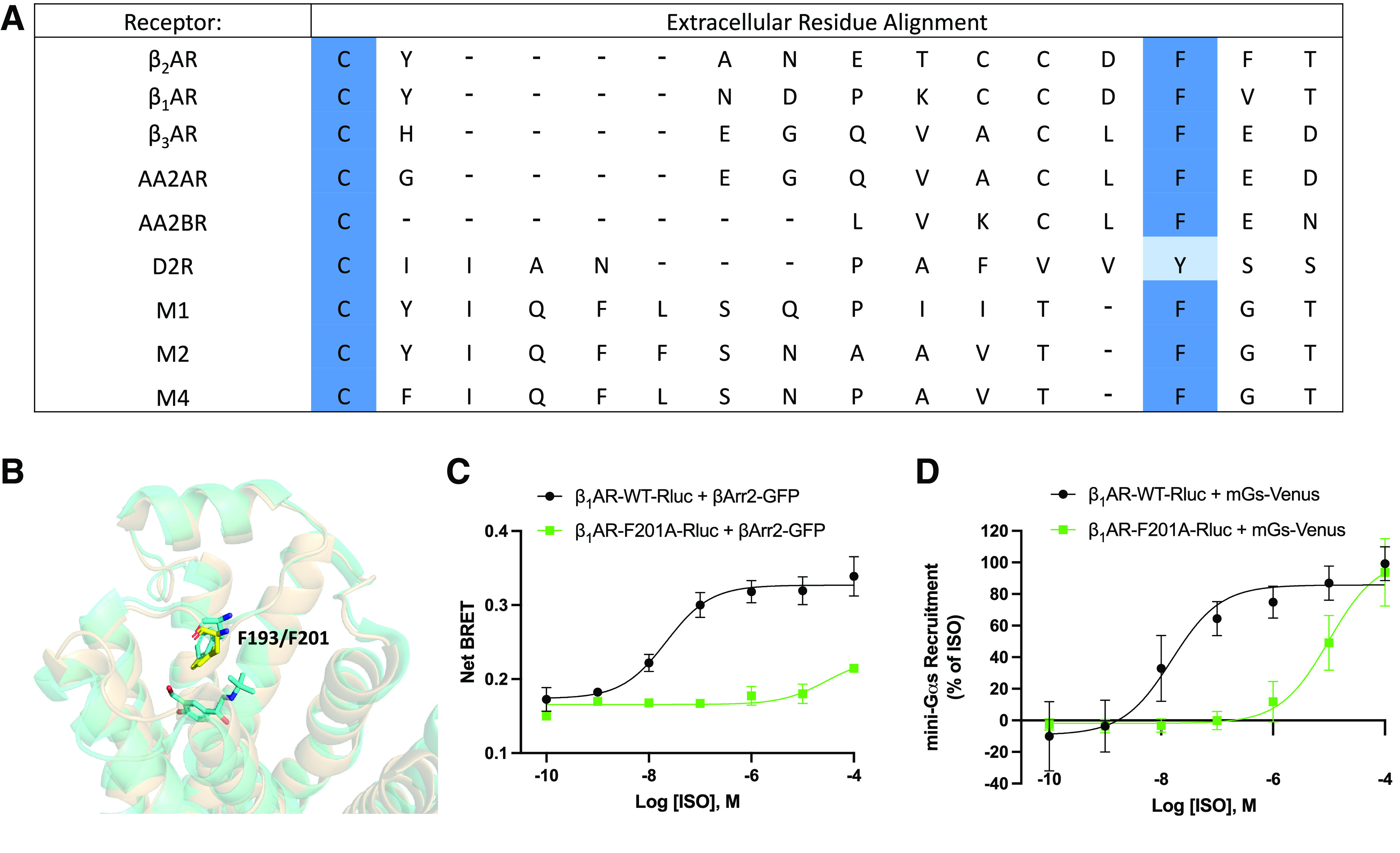
Analysis of extracellular residues of class A G protein-coupled receptors. (A) Multiple sequence alignment of several class A G protein-coupled receptors. βAR=β-adrenergic receptor, AAR=adenosine receptor, DR=dopamine receptor, MR=muscarinic acetylcholine receptor. (B) Structural alignment of β2-adrenergic receptor (β2AR, tan) and β1-adrenergic receptor (β1AR, yellow, PDB: 7BTS). β2AR residue F193 and β1AR residue F201 are indicated by sticks. (C) β-arrestin recruitment to wild-type β1AR (β1AR-WT) and β1AR-F201A as measured by bioluminescence resonance energy transfer (BRET). Human embryonic kidney 293cells co-transfected with β-arrestin2-GFP10 and indicated β1AR-WT-RlucII or β1AR-F201A-RlucII constructs were incubated with coelenterazine 400a for 2 minutes and then stimulated with indicated concentrations of isoproterenol (ISO). Data were collected at 6 minutes post agonist addition. n = 3 independent experiments. (E) Dose response curves for ISO promoted mini-Gs recruitment to β1AR by BRET. Human embryonic kidney 293cells transiently transfected wild-type or mutated β1AR-Rluc and NES-Venus-mGs were stimulated with the indicated concentrations of ISO for 15 minutes and BRET signal was measured. n = 3 independent experiments.
To initially evaluate whether the observed extracellular control of receptor regulation was functionally conserved for other GPCRs, we evaluated Gs and β-arrestin interaction with the β1-adrenergic receptor (β1AR) using BRET. To do this, we generated the point mutant β1AR-F201A, the position that is analogous to β2AR-F193A (Fig. 4B). Comparisons between ISO-stimulated wild-type β1AR and β1AR-F201A show an even greater reduction in β-arrestin interaction as compared with the β2AR-F193A with a ∼10,000-fold shift in EC50 (Fig. 4C). Interestingly, this mutation also causes a substantial right shift in mini-Gs recruitment to the β1AR with a ∼500-fold shift in EC50 (Fig. 4D). Due to the conservation of this residue in multiple receptor extracellular regions and functional consequences observed in the β1AR, it seems likely that this extracellular region coordinates control over intracellular signaling outcomes for other GPCRs.
Discussion
Biased signaling of GPCRs remains a promising avenue for clinical application that has yet unmet applications in areas of clinical need (Kenakin, 2018; Michel and Charlton, 2018). These span pathologies ranging from psychiatric disorders, airway disease, metabolic disorders, cardiovascular disease, and cancer. Untangling physiologic processes from specific signaling pathways may provide a path to new generations of therapeutics with enhanced clinical benefit and fewer side effects. The molecular determinants of biased signaling through various class A GPCRs has been a subject of increased investigation in recent studies (Rahmeh et al., 2012; Choi et al., 2018; Lamichhane et al., 2020; Suomivuori et al., 2020). Many laboratories have provided insight into this signaling paradigm in different systems, including the dopamine D2 receptor, the angiotensin II receptor type 1, and the β2AR (Carr III et al., 2014; Sanchez-Soto et al., 2020; Wingler et al., 2020). Functional selectivity in activation of these receptors and others is now widely accepted as an intrinsic feature of GPCR signaling, but there is less agreement about how these signaling paradigms are achieved.
Previous work has shown the importance of the extracellular loops of β2AR in mediating ligand specificity and dynamic changes, but this hasn’t been extended to a comprehensive analysis of signaling profiles (Ahuja et al., 2009; Bokoch et al., 2010; Baker et al., 2015). A very recent study examined the role of transmembrane domain 7 (TM7) of the β2AR using an in vitro single molecule fluorescence system to examine the role of conformational exchange kinetics on β-arrestin bias (Lamichhane et al., 2020). In this study, TM7 conformers were demonstrated to diverge for balanced and arrestin-biased agonists of β2AR, indicating a role for TM7 in arrestin coupling. Additionally, molecular dynamics simulations of β2AR have implicated F193 as an allosteric propagator of conformational changes through TM2 and TM7 and suggested a role for this residue in coordinating an allosteric pipeline during active state transitions (Bhattacharya and Vaidehi, 2014). Consistent with previous studies, we demonstrate that this region is able to stabilize a biased conformation of the receptor and could act as a switch propagating conformational changes to facilitate either G protein, GRK, and/or β-arrestin interaction. By mutating residues in the extracellular domains of β2AR, we were able to identify phenylalanine 193 on extracellular loop 2 as a powerful modulator of downstream protein-protein interaction.
Our findings are in agreement with the strong effect that mutation of F193 has on the ability of the β2AR to interact with GRKs and β-arrestins. A plausible mechanism for this is that transmembrane domain shifts are coordinated and propagated by residues at the extracellular loops of the receptor, and that these transitions are disrupted in the mutant receptor. Here, we find that mutation of F193 to an alanine is sufficient to right shift agonist promoted β-arrestin recruitment to β2AR over 200-fold while having a lesser detrimental effect on Gs binding and activation. This residue has also been characterized by molecular dynamics as forming a steric gate that represents the final major energy barrier to agonist binding at the orthosteric site (Dror et al., 2011). In this work, we demonstrate that a point mutation at this energy barrier has little effect on agonist binding affinity, but greatly inhibits downstream regulation of the β2AR. This may suggest a role for dynamic features of ligand binding as mediators of ligand bias.
Despite large numbers of GPCR structures elucidated by crystallography and cryo-electron microscopy in recent years with a variety of pharmacological agents, a complete understanding of the structural transitions between G protein coupling, GRK interaction, and arrestin interaction has not been established. Understanding these transitions will provide a framework for improved drug design for targeting specific GPCR pathways. This work helps to elucidate the molecular mechanism through which biased signaling is propagated at adrenergic receptors. Our data are primarily focused around the β2AR, although our work also demonstrates applicability to the β1AR and suggests that the role of the extracellular regions in signaling bias may be applicable to many class A GPCRs. Previous studies have used similar approaches for other GPCRs, including the dopamine D2 receptor and allosteric modulation of the M2 muscarinic acetylcholine receptor (Croy et al., 2014; Sanchez-Soto et al., 2020).
In summary, this study demonstrates that extracellular control over signaling transitions from G protein pathways to regulatory pathways is a feature of the β2AR, and it may have broader implications for other class A GPCRs. F193 in extracellular loop 2 is a key determinant of β2AR signaling activity, and mutation of this residue is sufficient to generate a receptor that is intrinsically biased away from β-arrestin. Our data implicate transducer stoichiometry-independent achievement of signaling bias. Additionally, the pocket formed by TMs 2, 3, and 7 and ECL2 and 3 functions as a regulatory domain that can either positively or negatively regulate β-arrestin interaction with the β2AR. Ultimately, this will provide a broader context through which to evaluate the pharmacological properties of novel β2AR ligands and inform drug design for biased ligands.
Acknowledgments
The authors thank Dr. Michel Bouvier for providing pcDNA-β-arrestin2-GFP10 and pcDNA3-β2AR-RlucII and Dr. Nevin A. Lambert for the NES-Venus-mGs construct.
Abbreviations
- β1AR
β1-adrenergic receptor
- β2AR
β2-adrenergic receptor
- BRET
bioluminescence resonance energy transfer
- ECL
extracellular loop
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- HEK 293
human embryonic kidney 293
- ISO
isoproterenol
- pEC50
negative log of the half-maximal effective concentration
- TM
transmembrane domain
- WT
wild-type
Authorship Contributions
Participated in research design: Ippolito, De Pascali, Benovic.
Conducted experiments: Ippolito, De Pascali.
Contributed new reagents or analytic tools: Inoue.
Performed data analysis: Ippolito, De Pascali.
Wrote or contributed to the writing of the manuscript: Ippolito, De Pascali, Inoue, Benovic.
Footnotes
Research reported in this publication was supported by the National Institutes of Health awards R35GM122541 (J.L.B.), R01HL136219 (J.L.B.), P01HL114471 (J.L.B.), T32GM100836 (M.I.), and F31HL139104 (M.I.), and used the MetaOmics Shared Resource at Sidney Kimmel Cancer Center at Jefferson Health supported by National Institutes of Health award P30CA056036. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. A.I. was funded by KAKENHI 21H04791 from The Japan Society for the Promotion of Science (JSPS); the PRIME 20gm5910013, the LEAP JP20gm0010004 and the BINDS JP20am0101095 from the Japan Agency for Medical Research and Development (AMED), and the Uehara Memorial Foundation.
No author has a conflict with the contents of this article.
References
- Ahuja SHornak VYan ECYSyrett NGoncalves JAHirshfeld AZiliox MSakmar TPSheves MReeves PJ, et al. (2009) Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat Struct Mol Biol 16:168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Proudman RGW, Hill SJ (2015) Salmeterol’s extreme β2 selectivity is due to residues in both extracellular loops and transmembrane domains. Mol Pharmacol 87:103–120. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci 25:366–428. [Google Scholar]
- Bermudez M, Bock A (2019) Does divergent binding pocket closure drive ligand bias for class A GPCRs? Trends Pharmacol Sci 40:236–239. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Vaidehi N (2014) Differences in allosteric communication pipelines in the inactive and active states of a GPCR. Biophys J 107:422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch MPZou YRasmussen SGFLiu CWNygaard RRosenbaum DMFung JJChoi H-JThian FSKobilka TS, et al. (2010) Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 463:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill TJ 3rdThomsen ARBTarrasch JTPlouffe BNguyen AHYang FHuang LYKahsai AWBassoni DLGavino BJ, et al. (2017) Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci USA 114:2562–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capper MJ, Wacker D (2018) How the ubiquitous GPCR receptor family selectively activates signalling pathways. Nature 558:529–530. [DOI] [PubMed] [Google Scholar]
- Carr R 3rd, Du Y, Quoyer J, Panettieri RA Jr, Janz JM, Bouvier M, Kobilka BK, Benovic JL, (2014) Development and characterization of pepducins as Gs-biased allosteric agonists. J Biol Chem 289:35668–35684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV (2002) Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem 277:9043–9048. [DOI] [PubMed] [Google Scholar]
- Choi M, Staus DP, Wingler LM, Ahn S, Pani B, Capel WD, Lefkowitz RJ (2018) G protein-coupled receptor kinases (GRKs) orchestrate biased agonism at the β2-adrenergic receptor. Sci Signal 11:eaar7084. [DOI] [PubMed] [Google Scholar]
- Croy CH, Schober DA, Xiao H, Quets A, Christopoulos A, Felder CC (2014) Characterization of the novel positive allosteric modulator, LY2119620, at the muscarinic M(2) and M(4) receptors. Mol Pharmacol 86:106–115. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Penn RB (2006) Targeting G protein-coupled receptor signaling in asthma. Cell Signal 18:2105–2120. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Theriot BS, Penn RB, Walker JKL (2008) β-arrestins specifically constrain β2-adrenergic receptor signaling and function in airway smooth muscle. FASEB J 22:2134–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci USA 108:13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SSG, Downey WE 3rd, Colapietro A-M, Barak LS, Ménard L, Caron MG (1996) Role of β-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271:363–366. [DOI] [PubMed] [Google Scholar]
- Filipek S (2019) Molecular switches in GPCRs. Curr Opin Struct Biol 55:114–120. [DOI] [PubMed] [Google Scholar]
- Gagnon AW, Kallal L, Benovic JL (1998) Role of clathrin-mediated endocytosis in agonist-induced down-regulation of the β2-adrenergic receptor. J Biol Chem 273:6976–6981. [DOI] [PubMed] [Google Scholar]
- Goodman OB Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL (1996) β-arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature 383:447–450. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, von Zastrow M (2008) Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol 48:537–568. [DOI] [PubMed] [Google Scholar]
- Hodavance SY, Gareri C, Torok RD, Rockman HA (2016) G protein-coupled receptor biased agonism. J Cardiovasc Pharmacol 67:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA (2015) New paradigms in GPCR drug discovery. Biochem Pharmacol 98:541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T (2018) Is the quest for signaling bias worth the effort? Mol Pharmacol 93:266–269. [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Benovic JL (1998) The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol 38:289–319. [DOI] [PubMed] [Google Scholar]
- Lamichhane R, Liu JJ, White KL, Katritch V, Stevens RC, Wüthrich K, Millar DP (2020) Biased signaling of the G-protein-coupled receptor β2AR is governed by conformational exchange kinetics. Structure 28:371–377.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorraca NR, Venkatakrishnan AJ, Dror RO (2017) GPCR dynamics: Structures in motion. Chem Rev 117:139–155. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Andexinger S, Pitcher J, Trukawinski S, Codina J, Faure JP, Caron MG, Lefkowitz RJ(1992) Receptor-specific desensitization with purified proteins. Kinase dependence and receptor specificity of beta-arrestin and arrestin in the beta 2-adrenergic receptor and rhodopsin systems. Journal of Biological Chemistry 267:8558–8564. [PubMed] [Google Scholar]
- Madeira FPark YMLee JBuso NGur TMadhusoodanan NBasutkar PTivey ARNPotter SCFinn RD, et al. (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47 (W1):W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kobilka B (2014) The role of protein dynamics in GPCR function: insights from the β2AR and rhodopsin. Curr Opin Cell Biol 27:136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masureel MZou YPicard L-Pvan der Westhuizen EMahoney JPRodrigues JPGLMMildorf TJDror ROShaw DEBouvier M, et al. (2018) Structural insights into binding specificity, efficacy and bias of a β2AR partial agonist. Nat Chem Biol 14:1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MC, Charlton SJ (2018) Biased agonism in drug discovery - is it too soon to choose a path? Mol Pharmacol 93:259–265. [DOI] [PubMed] [Google Scholar]
- Nivedha AK, Tautermann CS, Bhattacharya S, Lee S, Casarosa P, Kollak I, Kiechle T, Vaidehi N (2018) Identifying functional hotspot residues for biased ligand design in G-protein-coupled receptors. Mol Pharmacol 93:288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles KNXiao KAhn SShukla AKLam CMRajagopal SStrachan RTHuang T-YBressler EAHara MR, et al. (2011) Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal 4:ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom RS, Ehlert FJ (1997) M2 muscarinic receptor inhibition of agonist-induced cyclic adenosine monophosphate accumulation and relaxation in the guinea pig ileum. J Pharmacol Exp Ther 280:189–199. [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ (2002) Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3:639–650. [DOI] [PubMed] [Google Scholar]
- Rahmeh RDamian MCottet MOrcel HMendre CDurroux TSharma KSDurand GPucci BTrinquet E, et al. (2012) Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci USA 109:6733–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salon JA, Lodowski DT, Palczewski K (2011) The significance of G protein-coupled receptor crystallography for drug discovery. Pharmacol Rev 63:901–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Soto MVerma RKWillette BKAGonye ECMoore AMMoritz AEBoateng CAYano HFree RBShi L, et al. (2020) A structural basis for how ligand binding site changes can allosterically regulate GPCR signaling and engender functional selectivity. Sci Signal 13:eaaw5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan RT, Sun JP, Rominger DH, Violin JD, Ahn S, Rojas Bie Thomsen A, Zhu X, Kleist A, Costa T, Lefkowitz RJ (2014) Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J Biol Chem 289:14211–14224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomivuori CMLatorraca NRWingler LMEismann SKing MCKleinhenz ALWSkiba MAStaus DPKruse ACLefkowitz RJ, et al. (2020) Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science 367:881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Yan W, McCorvy JD, Cheng J (2018) Biased ligands of G protein-coupled receptors (GPCRs): structure-functional selectivity relationships (SFSRs) and therapeutic potential. J Med Chem 61:9841–9878. [DOI] [PubMed] [Google Scholar]
- Thal DM, Glukhova A, Sexton PM, Christopoulos A (2018) Structural insights into G-protein-coupled receptor allostery. Nature 559:45–53. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Ma AK, Fonseca R, Latorraca NR, Kelly B, Betz RM, Asawa C, Kobilka BK, Dror RO (2019) Diverse GPCRs exhibit conserved water networks for stabilization and activation. Proc Natl Acad Sci USA 116:3288–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Okashah N, Inoue A, Nehmé R, Carpenter B, Tate CG, Lambert NA (2018) Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells. J Biol Chem 293:7466–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler LM, Skiba MA, McMahon C, Staus DP, Kleinhenz ALW, Suomivuori CM, Latorraca NR, Dror RO, Lefkowitz RJ, Kruse AC (2020) Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 367:888–892. [DOI] [PMC free article] [PubMed] [Google Scholar]