

Abstract
The GABAA receptor is inhibited by the endogenous sulfated steroids pregnenolone sulfate (PS) and dehydroepiandrosterone sulfate (DHEAS). It has been proposed in previous work that these steroids act by enhancing desensitization of the receptor. Here, we have investigated the modulatory effects of the steroids on the human α1β3γ2L GABAA receptor. Using electrophysiology and quantitative model-based data analysis, we show that exposure to the steroid promotes occupancy of a nonconducting state that retains high affinity to the transmitter but whose properties differ from those of the classic, transmitter-induced desensitized state. From the analysis of the inhibitory actions of two combined steroids, we infer that PS and DHEAS act through shared or overlapping binding sites.
SIGNIFICANCE STATEMENT
Previous work has proposed that sulfated neurosteroids inhibit the GABAA receptor by enhancing the rate of entry into the desensitized state. This study shows that the inhibitory steroids pregnenolone sulfate and dehydroepiandrosterone sulfate act through a common interaction site by stabilizing a distinct nonconducting state.
Introduction
The sulfated steroids pregnenolone sulfate (PS) and dehydroepiandrosterone sulfate (DHEAS) are synthesized in the brain and are thus termed neurosteroids (Corpechot et al., 1981; Corpechot et al., 1983; Majewska and Schwartz, 1987). Both compounds have been linked to cognition, neuroprotection, stress response, and pathophysiology of schizophrenia, among others (Morgan et al., 2004; Charalampopoulos et al., 2006; Valenti et al., 2009; Ritsner, 2011), although it is not fully understood which targets are involved in the actions. Both steroids inhibit the GABAA receptor (Majewska et al., 1988; Majewska et al., 1990; Park-Chung et al., 1999). In addition, PS potentiates the N-methyl-D-aspartate receptor, whereas DHEAS is an agonist of the σ1 receptor (Wu et al., 1991; Monnet et al., 1995). The concentration of PS in brain regions and the concentration-dependence of some actions on brain receptors have been reviewed previously (Smith et al., 2014).
The inhibitory effect of PS on the function of the GABAA receptor manifests as an increase in the rate and extent of apparent desensitization in macroscopic recordings and a reduced mean cluster duration in single-channel recordings (Shen et al., 2000; Akk et al., 2001; Wang et al., 2002). In macroscopic recordings, the addition of PS in the absence of GABA does not significantly reduce the initial peak amplitude of the subsequent response to GABA, supporting the idea that PS selectively blocks active receptors. These effects have been interpreted as enhanced desensitization in the presence of steroid. At least qualitatively, the effect of DHEAS on apparent desensitization appears similar (Demirgören et al., 1991; Spivak, 1994) [but see: (Le Foll et al., 1997; Hansen et al., 1999; Sachidanandan and Bera, 2015)].
Direct examination of the effect of PS on muscimol binding has shown that the state(s) promoted by PS has high affinity to the transmitter (Akk et al., 2020), thereby rejecting the possible mechanism that PS stabilizes the resting (low-affinity) state. However, at present there is no direct evidence that the steroid increases occupancy of the classic, transmitter-induced desensitized state. The possibilities of fast or slow simple open-channel block have been excluded by lack of changes in single-channel kinetics or amplitude and by a nonzero current response in the presence of saturating PS or DHEAS (Mienville and Vicini, 1989; Akk et al., 2001; Sachidanandan and Bera, 2015; Germann et al., 2019a; Germann et al., 2019b).
The location of the binding sites for PS and DHEAS in the GABAA receptor is unknown. The α1(V257S) mutation, a Val-to-Ser substitution at the 2′ location in the second transmembrane domain (TM2), significantly reduces inhibition by PS, DHEAS, and several 3β-OH inhibitory steroids (Akk et al., 2001; Wang et al., 2002; Sachidanandan and Bera, 2015). This residue is located in the ion channel of the receptor, but a direct role in steroid binding was considered to be unlikely due to the observation that PS binding shows no voltage sensitivity (Akk et al., 2001; Eisenman et al., 2003). X-ray crystallographic analysis of a homomeric receptor of chimeric subunits containing the N-terminal sequence of Gloeobacter ligand-gated ion channel and the transmembrane sequence of the GABAA α1 subunit indicated a binding site for PS at the lipid-exposed face of TM3 and TM4 where the C3-sulfate group may interact with K390 at the cytoplasmic end of TM4 and the D-ring of steroid reaches F399 in the middle of TM4. Mutations to K390 and along the putative binding groove reduced the extent of inhibition at high PS concentrations (Laverty et al., 2017).
Here, we have investigated the mechanisms of inhibition of the α1β3γ2L GABAA receptor by the sulfated steroids PS and DHEAS. Our major conclusions are that the state promoted in the presence of the inhibitory steroids is distinct from the desensitized state promoted by the transmitter and that both steroids act by the same mechanism. We also show that the sulfated steroids PS and DHEAS inhibit the receptor by interacting with an overlapping binding site.
Materials and Methods
Receptor Expression and Electrophysiology
The experiments were conducted on human α1β3γ2L GABAA receptors expressed in oocytes from Xenopus laevis (African clawed frog). Frog quarter ovaries were purchased from Xenoocyte (Dexter, MI), and digested at 37°C with shaking at 250 RPM for 15–30 minutes in 2% w/v (mg/mL) collagenase A solution in ND96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES; pH 7.4) with 100 U/ml penicillin and 100 μg/ml streptomycin. After digestion, the oocytes were rinsed in ND96 and incubated in ND96 with supplements (2.5 mM Na pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml gentamycin) at 15°C for at least 4 hours before injection.
The cDNAs for human α1 (GenBank accession no NM_000806.5), β3 (NM_000814.5), and γ2L (NM_198904.2) subunits in the pcDNA3 vector were linearized with XbaI (NEB Laboratories, Ipswich, MA). The cRNAs were synthesized from linearized cDNA using mMessage mMachine (Ambion, Austin, TX).
The oocytes were injected with a total of 3.5 ng of cRNA per oocyte in the ratio of 1:1:5 (α1:β3:γ2L) to enhance the incorporation of the γ2L subunit and minimize the expression of α1β3 receptors. After injection, the oocytes were incubated in ND96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES; pH 7.4) with supplements (2.5 mM Na pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μg/ml gentamycin) at 15°C for 1–2 days prior to conducting electrophysiological recordings.
The electrophysiological recordings were done using a standard two-electrode voltage clamp. The oocytes were clamped at -60 mV. The solutions were gravity-applied from glass syringes with glass luer slips via Teflon tubing to the recording chamber (RC-1Z, Warner Instruments, Hamden, CT) at a flow rate of 6–8 ml/min (solution exchange time < 10 seconds). Solutions were switched manually using 4-port bulkhead switching valves and medium pressure 6-port bulkhead valves (IDEX Health and Science, Rohnert Park, CA). All experiments were conducted at room temperature. The current responses were amplified with an OC-725C amplifier (Warner Instruments, Hamden, CT), digitized with a Digidata 1200 series digitizer (Molecular Devices), and stored using pClamp (Molecular Devices). Records were low-pass filtered at 30 Hz and sampled at 100 Hz. The current traces were analyzed using Clampfit (Molecular Devices).
All experiments were conducted using a saturating concentration (1 mM) of GABA to activate receptors. This approach has several advantages over using a lower (e.g., EC50) concentration of agonist. First, it minimizes the fraction of receptors in the resting state and removes binding status from analysis. Second, it reduces cell-to-cell variability associated with differences in affinity to GABA. Lastly, it enables the use of lower concentrations of PS and DHEAS because the IC50 of inhibition is higher when measured at low GABA (Germann et al., 2019a).
The effects of steroids on apparent desensitization were determined by recording currents elicited by 1 mM GABA or 1 mM GABA coapplied with 30 µM PS or DHEAS. The time course of apparent desensitization decay was fitted to a single exponential or a sum of two exponentials. In the latter case, a weighted time constant was calculated.
Steroid concentration-response curves were measured by activating the receptors with 1 mM GABA and, upon reaching steady-state response, coapplying a steroid (PS or DHEAS) with GABA. Each oocyte was exposed to a single concentration of a single steroid. Experiments aimed at determining competition between steroids were conducted by consecutively applying a single steroid and a combination of two steroids during a steady-state response to 1 mM GABA.
All data are included in analysis. The results are presented as mean ± S.D. unless indicated otherwise. The study is exploratory by nature, and the findings are reported according to the guidelines detailed in (Michel et al., 2020).
Descriptive Characterization of Receptor Inhibition by Steroids
Descriptive characterization of steroid-induced inhibition of steady-state current was done by fitting a Hill equation to the normalized steroid concentration-response relationship:
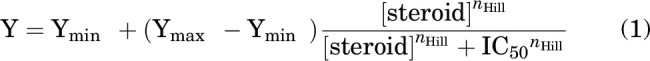 |
where Ymin and Ymax are the high- and low-concentration asymptotes, respectively; [steroid] is the concentration of steroid; IC50 is the concentration producing half-maximal effect; and nHill is the Hill coefficient. Fitting was done on data averaged from multiple oocytes (at least 5 per concentration of a steroid). Curve-fitting results are expressed as best fitting parameter ± S.E. of the fit.
Mechanistic Characterization of Receptor Inhibition by Steroids
Mechanistic characterization of steroid-induced inhibition was done in the framework of a four-state resting-active-desensitized/blocked (RADB) model (Fig. 1). The RADB model is based on the Monod-Wyman-Changeux concerted transition model originally used to describe enzyme function (Monod et al., 1965). The RADB model contains one state with lower affinity to the transmitter (resting, R, with a closed channel) than the other three. There are two higher affinity states that are also nonconducting (desensitized, D, and blocked, B). Finally, there is one state with high affinity that has an open channel (active, A). In this model, an inhibitory steroid stabilizes the blocked state.
Fig. 1.

The RADB model. The figure shows the kinetic scheme for the RADB model: a concerted transition, 4-state cyclic scheme with one active state. There are 2 identical sites for binding a hypothetical drug ×. Due to the difficulty of seeing the individual transition between states, the scheme is broken into 5 subschemes: three for the paths between states with the same number of bound molecules of X and two for the binding steps between degrees of ligation. The arrows indicate the numerator and denominator of the ratio: the start state (numerator) is at the circle and the end (denominator) is at arrow head. The constants give the ratio of the probability of being in the starting state to that of the ending state, so X2R/X2A = Lc2 and X2A/XA = 1/(cK/(2[X])). In the figure, the ratio K/[X] is given as K’. The highlighted ratios indicate the basic parameters of the scheme: L = R/A, Q = A/D, W = A/B, c = KX,A/KX,R, d = KX,D/KX,A, and e = KX,E/KX,A. Detailed balance sets the requirement that two paths leading between the same two states must result in the same overall ratios. The boxed scheme shows the full three-dimenstional kinetic scheme without equilibrium ratios.
Receptor properties in the RADB model are defined by three parameters: L = R/A (the ratio of resting to active receptors in the absence of any active compounds), Q = A/D (the ratio of active to desensitized receptors in the absence of active compounds), and W = A/B (the ratio of active to blocked receptors in the absence of active compounds). When referred to the R state, A = R/L, D = R/(LQ), and B = R/(LW).
Interaction of the hypothetical drug X with the receptor is described by five parameters: NX, the number of identical sites on a receptor for X; KR,X (KX in the equations), the affinity for the resting state; KA,X, the affinity for the active state (KA,X = cXKR,X); KD,X, the affinity for the desensitized state (KD,X = dXKA,X = cXdXKR,X); and KB,X, the affinity for the blocked state (KB,X = eXKA,X = cXeXKR,X). Fig. 1 shows an enlarged version of the RADB model with equilibrium ratios for prevalence between 2 states.
In the presence of drug X (and no other active compounds) the probability of being active (PA) is:
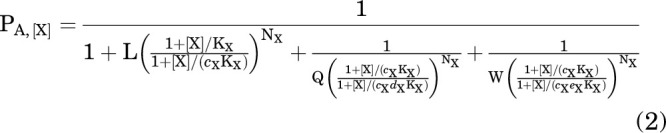 |
To simplify the notation, define:
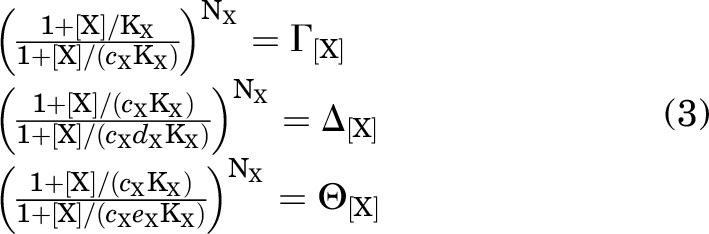 |
so:
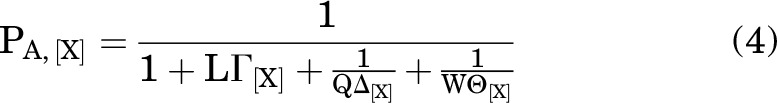 |
when [X] = 0, then ΓX = ΔX = ΘX = 1, and as [X] → ∞, then , and:
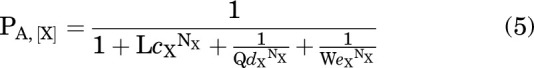 |
If 2 drugs (X and Y) that interact with nonoverlapping sites on the receptor are present, the values for Γ, Θ, and Δ multiply (Karlin, 1967; Steinbach and Akk, 2019), so:
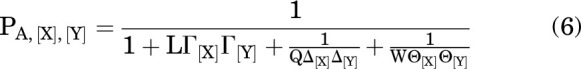 |
On the other hand, if X and Y bind to the same site(s) on the receptor, then a composite parameter replaces the product (Karlin, 1967; Steinbach and Akk, 2019). For example, ΔXΔY in Eq. 6 is replaced with:
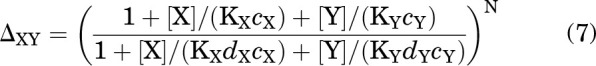 |
and analogously for ΓXY and ΘXY.
GABA is an agonist with cGABA less than 1, and it is assumed that it does not stabilize either the D or B states (dGABA = 1 and eGABA = 1). The steroids are not agonists (cPS = 1, cDHEAS = 1). For simplicity, it is assumed that neither stabilizes the D state (dPS = 1, dDHEAS = 1), whereas both stabilize the B state (ePS < 1, eDHEAS < 1).
In the presence of GABA and a single steroid (here, PS), the PA is:
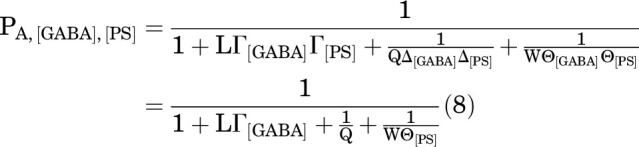 |
With both steroids (PS and DHEAS) binding to the same site, it is:
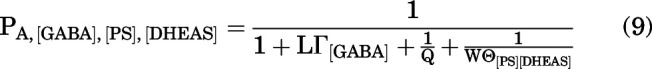 |
where
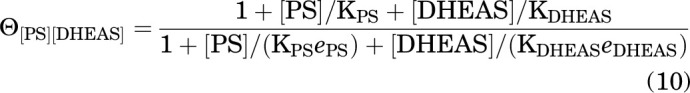 |
With the steroids PS and DHEAS binding to distinct sites, the probability of being active is:
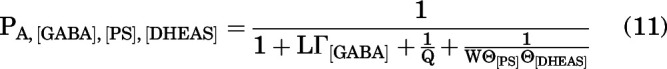 |
Materials
The stock solution of GABA was made in ND96 at 500 mM and stored in aliquots at -20°C. The steroids PS and DHEAS were dissolved in DMSO at 30–50 mM and stored at room temperature. Final dilutions were made on the day of experiment.
Results
Inhibition of GABAA Receptor Activity by PS and DHEAS
Exposure to PS or DHEAS inhibits the response of the α1β3γ2L GABAA receptor to the transmitter GABA. Coapplication of the steroid with GABA enhances the apparent rate of desensitization. In four cells exposed to 1 mM GABA and 1 mM GABA + 30 µM PS, the apparent desensitization time constants were 41 ± 12 seconds and 4.1 ± 0.4 seconds, respectively (P < 0.01; unequal variances t test). In a separate set of four cells, the apparent desensitization time constants were 43 ± 12 seconds in the presence of 1 mM GABA and 2.3 ± 0.8 seconds in the presence of 1 mM GABA + 30 µM DHEAS (P < 0.001; unequal variances t test). The peak amplitudes were reduced to 82 ± 7% of control in the presence of PS and to 80 ± 9% of control in the presence of DHEAS. Sample current traces are given in Fig. 2A. The data are summarized in Table 1.
Fig. 2.

Inhibition of the α1β3γ2L receptor by sulfated steroids. (A) Comparison of apparent desensitization in the presence of 1 mM GABA, 1 mM GABA coapplied with 30 µM PS (upper panel), or 30 µM DHEAS (lower panel). The paired traces are from the same cells and normalized to the peak response to GABA in the same cell. The fitted or weighted decay time constants are: 26 seconds (GABA) and 3.6 seconds (GABA + PS) in the upper panel, and 33 seconds (GABA) and 1.3 seconds (GABA + DHEAS) in the lower panel. (B) Sample traces showing PS-induced inhibition of steady-state current elicited by 1 mM GABA. The inset indicates the time course of inhibition with peak and sustained effects shown with arrows. (C) Concentration-response relationships for PS- and DHEAS-induced inhibition of steady-state current elicited by 1 mM GABA. The data points show mean ±S.D. from 5–8 cells per condition. Each cell was exposed to a single application of steroid. The curves were fitted with Eq. 1. The fitted IC50s, values of nHill, and high-concentration asymptotes are given in the text.
TABLE 1.
Summary of properties of PS and DHEAS.The table provides the desensitization time constants in the presence of 30 µM steroid (τdesensitization), results of fits of steroid concentration-response data to the Hill equation (IC50 and nH), and results of fits to Eq. 12 (K and e).
| Parameter | PS | DHEAS |
|---|---|---|
|
τdesensitization at 30 µM (s) (control in same cells) |
4.1 ± 0.4 (41 ± 12) |
2.3 ± 0.8 (43 ± 12) |
| IC50,peak (µM) | 0.26 ± 0.03 | 1.01 ± 0.12 |
| nH,peak | −0.73 ± 0.05 | −0.81 ± 0.06 |
| IC50,sustained (µM) | 1.45 ± 0.26 | 11.4 ± 0.8 |
| nH,sustained | −0.90 ± 0.11 | −1.51 ± 0.10 |
| K (µM) | 22.6 ± 3.6 | 336 ± 25 |
| e | 6.2 × 10−5 ± 3.3 × 10−5 | 6.2 × 10-5* |
*Constrained as described in Results. The numbers of cells are provided in text.
When coapplied during the steady-state response to saturating (1 mM) GABA, PS rapidly reduces the current amplitude. Particularly at lower steroid concentrations, the initial peak inhibitory effect is transient, followed by partial recovery of current response. For example, in the presence of 0.3 µM PS, the current level during the peak steroid effect was 50 ± 8% (n = 5 cells) of the control steady-state response to 1 mM GABA. Upon continued exposure to PS, the current response partially recovered and reached during the sustained effect 80 ± 14% of control (i.e., 20% inhibition). Sample current traces are shown in Fig. 2B.
The PS concentration-response relationship was established by measuring the effect of 0.01–50 µM PS on steady-state current elicited by 1 mM GABA. Each cell was exposed to a single concentration of PS to avoid measurement errors due to long-duration recordings and steroid exposure. Peak and sustained effects of the steroid were measured and analyzed separately. Fits of the mean data to the Hill equation (Eq. 1) yielded IC50s and nHill values of 0.26 ± 0.03 µM (best fitting value ± SE of the fit) and -0.73 ± 0.05 for peak effect, and 1.45 ± 0.26 µM and -0.90 ± 0.11 for sustained effect (Table 1). The fitted high-concentration asymptotes, indicating incomplete inhibition at saturating steroid concentrations, were 0.03 ± 0.02 and 0.01 ± 0.04 for peak and sustained effects, respectively. The concentration-response relationships are given in Fig. 2C.
DHEAS reduced the steady-state response to 1 mM GABA. Similar to PS, exposure to DHEAS resulted in a transient peak inhibitory effect followed by partial recovery to a sustained level of inhibition. Fits to the Hill equation (Eq. 1) gave an IC50 of 1.01 ± 0.12 µM (best fitting value ± SE of the fit) and nHill of -0.81 ± 0.06 for peak effect, and an IC50 of 11.4 ± 0.8 µM and nHill of -1.51 ± 0.10 for sustained effect (Fig. 2B). The fitted high-concentration asymptotes were 0.06 ± 0.03 and 0.21 ± 0.02 for the peak and sustained inhibition.
At up to 50 µM, neither steroid fully inhibited receptor function even during peak effect. The remaining current response was 4.3 ± 1.4% (n = 5 cells; P < 0.01 that the ratio is indistinguishable from 0; 1 sample t test) or 3.5 ± 1.3% (n = 5; P < 0.01; 1 sample t test) of the steady-state response to GABA alone in the presence of 30 or 50 µM PS, respectively. Exposure to 30 or 50 µM DHEAS reduced the current level to 11.2 ± 4.3% (n = 5; P < 0.01; 1 sample t test) or 8.7 ± 6.3% of control (n = 5; P < 0.05; 1 sample t test), respectively.
Does PS Promote Receptor Desensitization?
Whole-cell and single-channel patch-clamp studies have suggested that PS inhibits the GABAA receptor by promoting entry into a desensitized state (Akk et al., 2001; Eisenman et al., 2003) (also, Fig. 2). Here, we compared the properties of the nonconducting states induced by prolonged exposure to the transmitter in the absence (i.e., desensitization) and presence of PS (i.e., PS-induced inhibition). The goal of these experiments was to determine whether the GABA- and PS-induced nonconducting states are functionally identical.
We compared the time courses of recovery from transmitter-induced desensitization and PS-induced inhibition. We reasoned that similar recovery rates upon the removal of GABA or GABA + PS would support a model where the steroid acts by enhancing transmitter-induced desensitization. In contrast, different recovery rates, particularly if recovery from PS-induced inhibition proceeds faster than recovery from GABA-induced desensitization, would be suggestive of the steroid promoting entry into a distinct nonconducting state with distinct properties.
Recovery from GABA-induced desensitization was measured by exposing the cells to 1 mM GABA until steady-state response was reached, followed by variable-duration wash in bath, and a second, test application of 1 mM GABA. A sample recording is shown in Fig. 3A. The ratio of the peak responses (test response over the initial control response) is plotted in Fig. 3B. Fitting the fractional recovery from GABA-induced desensitization over time to a single-exponential function gave a time constant of 81 ± 6 seconds. This is similar to recovery from desensitization reported previously (Chang et al., 2002; Xu et al., 2016).
Fig. 3.

Recovery from transmitter-induced desensitization and PS-induced inhibition. (A) The α1β3γ2L receptors were exposed to 1 mM GABA alone (top trace) or to a brief application of 1 mM GABA followed by a coapplication of GABA + 10 µM PS (bottom trace) for ∼5 minutes. This was followed by a 30- to 50-second washout in bath, and a test application of 1 mM GABA. (B) The graph shows fractional recovery from GABA-induced desensitization (open circles) or PS-induced inhibition. Fractional recovery was calculated as the ratio of peak responses to 1 mM GABA after and before prolonged exposure to GABA or GABA + PS, with appropriate subtraction of the steady-state current. The line is the predicted curve generated by the best-fitting exponential (81 ± 6 seconds) to the data. (C) The receptors were exposed to 1 mM GABA, followed by a coapplication of GABA + 3 µM PS (top) or GABA + 10 µM DHEAS (bottom), and subsequent return to GABA alone. The time courses of current response upon switch from GABA + steroid to GABA alone were fitted to sums of two exponentials (inset). For the trace in PS, the time constants were: 29 seconds (recovery) and 118 seconds (decay), whereas in DHEAS, the time constants were: 14 seconds and 29 seconds. (D) The graph compares recovery from steroid-induced inhibition and GABA-induced desensitization. The data points give mean ±S.D. from 5–8 cells per condition for recovery from PS- or DHEAS-induced inhibition at various steroid concentrations. The lines indicate mean (solid) ±S.D. (dashed) for recovery from GABA-induced desensitization from the fit shown in panel B.
Recovery from steroid-induced inhibition was measured by first activating the receptors with 1 mM GABA. Upon reaching the peak response, the solution was switched to GABA + 10 µM PS, which induced rapid current decay. This was followed by a ∼60- to 70-second wash in bath, and a second, test application of 1 mM GABA. The duration of the GABA + PS application was kept similar (∼5 minutes) to the duration of the GABA application in the experiments aimed at estimating recovery from GABA-induced desensitization (above). A sample current trace showing the order of applications is given in Fig. 3A. The ratio of the peak responses after and before exposure to PS is plotted in Fig. 3B. Data from 5 cells indicate that a ∼60- to 70-second wash is sufficient for full recovery (test/control = 1.08 ± 0.06). These observations indicate that the nonconducting states occupied in the presence of GABA or GABA + PS recover with different rates.
We then examined the time course of development of PS-induced inhibition and recovery in the continued presence of 1 mM GABA. Current traces in Fig. 3C show that the switch from GABA to GABA + PS or GABA + DHEAS generates an initial peak inhibitory effect that then partially recovers, and a transient rebound current upon the switch from GABA + steroid to GABA alone followed by slow decay to the original GABA steady-state current. Inspection of the traces in the figures indicates that inhibition develops and recovers rapidly. This suggests that the equilibrium between active and blocked states in the RADB model (Fig. 1) occurs rapidly. When this is the case, the RADB model predicts upon the application of steroid an initial rapid shift of receptors from the active to blocked state. This is followed by a slower re-equilibration among the desensitized, active, and blocked states. Similarly upon the removal of steroid, there is a rapid initial shift from blocked to active state, followed by the more slowly developing re-equilibration from active to desensitized state. We note that at this high [GABA] occupancy of the resting state is negligible.
Additional insight into the rate of recovery from steroid-induced inhibition was gained by examining the current rise times upon the removal of steroid in the continued presence of GABA. In the simplest interpretation, this time course will reflect the dissociation of steroid followed by transition from the blocked to active state. The cells were exposed to 1 mM GABA +0.1–50 µM PS (or 1 mM GABA +0.3–50 µM DHEAS). Upon reaching steady-state, sustained inhibition, the solution was switched to 1 mM GABA alone that produced a rapid rise in current response. Sample current traces are given in Fig. 3C. Fitting the rising phase of the current response to an exponential function yielded time constants that ranged from 12–25 seconds for 0.1 to 50 µM PS and 6–14 seconds for 0.3 to 50 µM DHEAS (Fig. 3D). The somewhat slower recovery at higher concentrations of steroids likely reflects slower removal of the steroid from the membrane (Eisenman et al., 2007; Chisari et al., 2019). Recovery was more rapid after inhibition by DHEAS compared with PS. This difference may arise from a more rapid dissociation of DHEAS from the receptor or a more rapid removal of DHEAS from the environment surrounding the receptor. Overall, the rates of recovery from steroid-induced inhibition were faster than the rate of recovery from desensitization, and we interpret these findings as indicating that the transmitter-induced desensitized and PS-induced inhibited states are distinct.
Finally, the rapid recovery after removal of steroid is postulated to reflect entry of receptors into the active state, followed by the re-establishment of the equilibrium between active and desensitized states. Accordingly, the time course for the decay from the rebound current should be the same as the initial development of desensitization. Due to the somewhat slower apparent washout of PS, this analysis was made of responses to DHEAS. As shown in the inset to Fig. 3C the rebound peak and subsequent decay were fit simultaneously with the sum of two exponentials of opposite sign. The initial desensitization in the presence of GABA in the same traces was fit with either a single exponential or the sum of two exponentials (in which case the weighted time constant was calculated). As we had noted previously with the recovery time constant, the time constant for the decay to steady-state was prolonged at higher concentrations of DHEAS. However, for 18 responses involving 0.3 to 3 μM DHEAS, the ratio of the recovery time constant to the desensitization time constant was 1.03 ± 0.28 (P = 0.68 that the ratio is indistinguishable from 1; 1 sample t test). In contrast, for 14 responses involving DHEAS concentrations of 10 to 50 μM, the ratio was 1.39 ± 0.46 (P = 0.006). The similarity of the two time constants is consistent with our interpretation that the decay of the rebound current results from desensitization.
Model-Based Analysis of Sustained Inhibition by PS and DHEAS
Given the overall support for the use of the RADB model we have just described, further characterization of steroid-induced inhibition was done in the framework of the RADB model (Fig. 1). In this model, a steroid has higher affinity to the blocked than active state, thereby stabilizing the blocked state and inhibiting current response. For simplicity, we assumed that the steroid is not an agonist (does not stabilize the active state relative to the resting state) and does not desensitize (does not stabilize the desensitized state).
Conversion of raw current responses to units of probability of being in the active state (PA units) was done by normalizing current amplitudes to the peak response to saturating (1 mM) GABA + 50 µM propofol, that was considered to have a peak PA indistinguishable from 1 (Shin et al., 2017). The steady-state PA in the presence of PS (or DHEAS), reflecting sustained inhibition by a steroid, was fitted to Eq. 12:
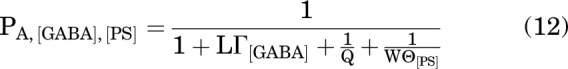 |
where the term LΓ[GABA] was calculated from the observed peak PA (0.88 ± 0.06; n = 9 cells) of the α1β3γ2L receptor in the presence of 1 mM GABA as:
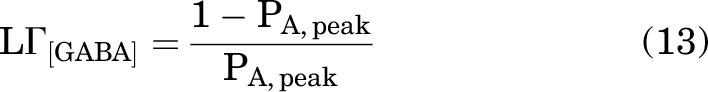 |
and
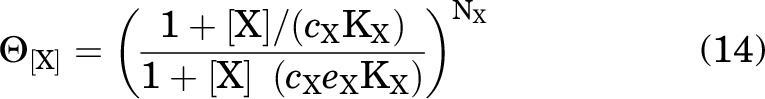 |
The value of Q in Eq. 12 was calculated from the response to GABA alone as:
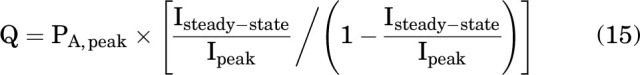 |
where Ipeak is the initial response to 1 mM GABA and Isteady-state is the steady response just before addition of steroid, with PA,peak constrained to 0.88. The value of W in eq. 12 was arbitrarily constrained to 100.
With the number of PS binding sites constrained to 1, fitting Eq. 12 to steady-state PA observed at various PS concentrations gave a KPS of 22.6 ± 3.6 µM (best fitting value ±SE of the fit) and an ePS of 6.2 × 10−5 ± 3.3 × 10−5. For fitting the PA data in the presence of DHEAS, we opted to constrain eDHEAS to the value estimated for PS while allowing KDHEAS vary freely. This was done due to lack of clear saturation in the PA curve at high concentrations of DHEAS. With this constraint, and NDHEAS fixed to 1, fitting Eq. 12 to steady-state PA in the presence of DHEAS gave a KDHEAS of 336 ± 25 µM (best fitting value ± SE of the fit). The fits are given as dashed lines in Fig. 4. The data are summarized in Table 1.
Fig. 4.

PA during peak and sustained inhibition by PS and DHEAS. The graphs show the probability of being in the active state (PA) during sustained (open symbols) and peak effect (filled symbols) of PS (A) or DHEAS (B). The data points give mean ±S.D. from 5–8 cells per steroid concentration. The dashed line shows a fit of sustained inhibition data to Eq. 12. The fitted parameters are given in the text. The solid line was calculated using the RADB model (Eq. 21) in which equilibration between the desensitized and active states is slow compared with the onset of inhibition in the presence of steroid, and in which the initial peak inhibitory effect of a steroid is due to rapid transition of receptors from active to blocked state.
Analysis of Peak Inhibition by PS and DHEAS
We predicted the concentration-dependence of peak inhibition from the parameters estimated from the steady-state responses. In the RADB model, the initial reduction in PA takes place due to rapid transition of receptors from the active into the blocked state, whereas the more slowly occurring equilibration between desensitized and other states increases PA, leading to reduced sustained inhibition. To predict the peak inhibition, we calculated the probability that a receptor is in either the active or blocked state at the steady-state response to 1 mM GABA. We then multiplied this by the probability that the receptor is in the active state in the presence of a given concentration of steroid, given that the receptor is in either the active or blocked state. The experiments were conducted using the steady-state response to a saturating concentration (1 mM) of GABA as the control condition. In this situation, a very small proportion of receptors is in the R state and its contribution can be neglected.
The steady-state probabilities of being in each of the 4 states are:
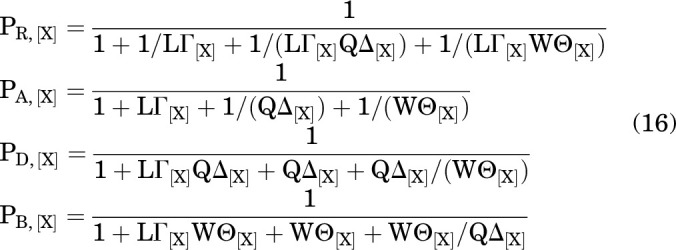 |
so
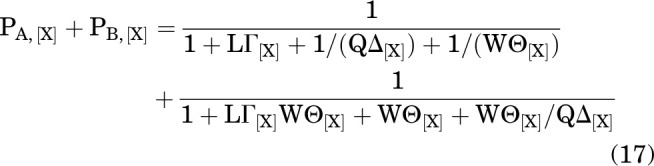 |
The ratio of A to B at equilibrium is:
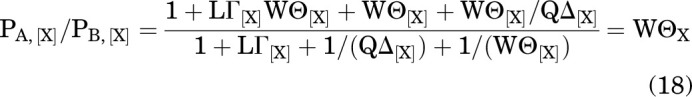 |
Accordingly, the fraction of the receptors in A, given that the receptors are in either A or B, is:
Under the assumptions that GABA stabilizes the A state (cGABA <1) but not the D or B states (dGABA = 1 and eGABA = 1), at the steady-state response to 1 mM GABA:
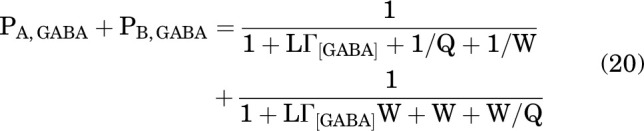 |
and during peak inhibition:
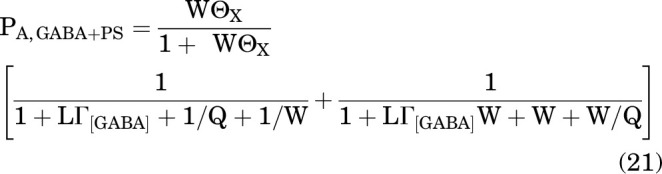 |
Using Eq. 21, we calculated the predicted PA during peak inhibition in the presence of GABA and PS. Rather than calculating all the terms in the equation, we used the global average estimate LΓ1 mM GABA = 0.136 (see above) and the experimentally determined value of Q = 0.135 for this group of cells (n = 58). For an example of the predicted PA at peak inhibition upon the addition of 0.3 μM PS, with KPS = 23 μM, cPS = 1, dPS = 1, ePS = 6.2 × 10−5, and NPS = 1 (all values from analysis of sustained inhibition; above), the calculated WΘPS = 0.48 and the predicted PA,GABA+PS at peak inhibition is 0.04. This is less than the observed value (0.056 ± 0.009) but comparable, given that it was calculated with no free parameters. The observed values and the predicted concentration-response curve for peak inhibition by PS are given in Fig. 4A.
Similarly for inhibition by DHEAS, using the experimentally determined value of Q = 0.157 for this group of cells (n = 44), and KDHEAS = 336 μM, cDHEAS = 1, dDHEAS = 1, eDHEAS = 6.2 × 10−5, and NDHEAS = 1 (all values from analysis of sustained inhibition), the predicted PA at peak inhibition upon the addition of 3 μM DHEAS is 0.055. As shown in Fig. 4B, this is similar to the observed value (0.046 ± 0.008). Variations between observed data and the simulations likely arise from the underlying assumption in Eq. 21 that peak inhibition is fully resolved and not modified by the more slowly occurring equilibration from desensitized state. However, the generally good agreement between the predictions and observed data for the peak inhibition support the validity of the RADB model in describing the inhibitory effects of sulfated steroids.
Do PS and DHEAS Interact with a Common Site?
We next addressed whether the inhibitory effects of PS and DHEAS are mediated by common or distinct binding sites. The consequences of application of a mixture of two steroids will depend on whether the steroids bind to different (nonoverlapping) sites or to the same (overlapping) sites. In the first case, both steroids may be bound simultaneously and hence modulate receptor function at the same time, resulting in allosteric amplification of inhibition, whereas in the second case, they compete for binding. We employed an approach in which the observed current response in the simultaneous presence of the two steroids is compared with the responses predicted by the distinct and same site models (Shin et al., 2019; Germann et al., 2020).
PS and DHEAS are high-efficacy inhibitors of the GABAA receptor (Fig. 2), and both the common and distinct site models predict an increase in inhibition (i.e., smaller current) when the two steroids are coapplied. The extent of the predicted increase in inhibition is, however, remarkably different in the two models.
We measured the combined effects of the steroids by coapplying 10 µM DHEAS and 1 µM PS. In 5 cells, the mean steady-state PA in the presence of 1 mM GABA was 0.143 ± 0.031. Coapplication of 10 µM DHEAS with GABA reduced the PA to 0.081 ± 0.016. The PA was further reduced to 0.061 ± 0.014 in the presence of GABA + DHEAS + 1 µM PS. A sample current trace is shown in Fig. 5. Using the affinity and efficacy estimates for the two steroids (above) and the experimentally determined value of Q of 0.155 in this group of cells (n = 5), we estimated a probability of being in the active state of 0.000309 ± 0.000001 using the distinct site model (Eq. 11) and 0.053 ± 0.004 using the same site model (Eq. 9).
Fig. 5.

Coapplication of PS and DHEAS. The α1β3γ2L receptors were activated by 1 mM GABA. Upon reaching steady-state response, the cells were exposed to GABA + 10 µM DHEAS followed by a switch to GABA + DHEAS + 1 µM PS. The right panel shows a portion of the trace at the left (boxed in the left) at higher resolution. The dotted line shows holding current. The short solid lines marked as “distinct” or “same” indicate current levels predicted by models in which PS and DHEAS interact with distinct or same sites, respectively.
The abilities of the two models to describe the data were compared by calculating the difference in second-order Akaike information criterion scores of the two models (Wagenmakers and Farrell, 2004; Burnham et al., 2011):
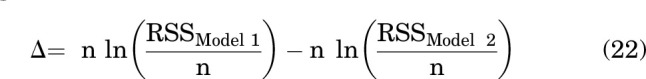 |
where n is the number of oocytes, RSS is the residual sum of squares, and Models 1 and 2 refer to models considering distinct sites and same sites, respectively. Akaike weights (w) for each model were calculated as:
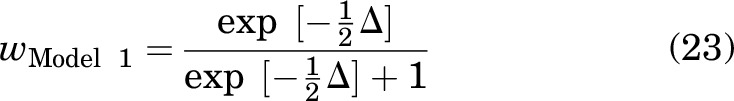 |
where wModel 1 is the probability that Model 1 is the best model describing data. The probability of Model 2 was then calculated as 1-wModel 1.
For the combination of DHEAS and PS, the wdistinct sites was 2 × 10−5 and the wcommon sites was 1 - (2 × 10−5). The same site model is clearly more likely than the distinct site model, although we note the caveat that the calculated w values rank the two chosen models but do not address the question of whether either is the globally best model.
Discussion
The major conclusion of this study is that the steroids PS and DHEAS inhibit the α1β3γ2L GABAA receptor by stabilizing a nonconducting state that is functionally distinct from the transmitter-induced desensitized state. Previous work using single-channel and whole-cell electrophysiology has proposed that PS speeds receptor desensitization (Akk et al., 2001; Eisenman et al., 2003; Wang et al., 2002). A recent study examining the effect of PS on binding of [3H]muscimol inferred that the PS-bound state has high affinity to the orthosteric agonist (Akk et al., 2020), similar to that of desensitized receptors (Chang et al., 2002). Our present data, in agreement with prior reports, indicate that coapplication of PS or DHEAS with GABA enhances the rate of current decay and inhibits steady-state current. The new interpretation regarding the nature of the nonconducting state is supported by the difference in rates of recovery from GABA-induced desensitization and steroid-induced inhibition. Specifically, the receptors recovered from PS- or DHEAS-induced inhibition more rapidly than from GABA-induced desensitization. This difference strongly supports the idea that the inactive states induced by prolonged exposure to GABA and to PS or DHEAS in the presence of GABA differ.
Additional support for the distinct nature of transmitter-induced desensitization and steroid-induced inhibition comes from the observation that the switch from GABA to GABA + steroid generates an initial peak effect that then partially recovers, and a transient rebound current upon the switch from GABA + steroid to GABA alone (e.g., Figs. 2A and, 3C). In the RADB model, the initial peak decrease in PA upon exposure to the steroid is due to rapid transition of receptors from the active into the blocked state. This is followed by the more slowly occurring equilibration between desensitized and other states during which PA partially recovers at the expense of desensitized receptors. The transient rebound current upon the removal of steroid is predicted to arise from transition of receptors from the blocked to the active state. The subsequent decline in current is due to re-equilibration between active and desensitized states, i.e., classic desensitization. In contrast, the Resting-Active-Desensitized model (Germann et al., 2019a), in which exposure to an inhibitory steroid enhances the rate of onset of desensitization, predicts a steady decrease in current upon exposure to steroid and a steady recovery to the original current level when the application of steroid is terminated. We caution, however, that while the RADB model predicts distinct peak and sustained inhibition at all steroid concentrations, this was not consistently observed at high (30–50 µM) PS concentrations (Fig. 2B; right trace). Thus, there may be additional complexities in how PS inhibits the GABAA receptor.
Our calculations indicate that the parameters derived for the steady-state responses by fitting the RADB model are able to satisfactorily describe the concentration dependence of the transient peak inhibition without any change in the parameters. The observation that PS and DHEAS share this qualitative pattern supports the idea that the steroids inhibit the α1β3γ2L GABAA receptor by the same mechanism. Experiments involving coapplications of PS and DHEAS indicate that the steroids interact with a common site or overlapping sites.
The transient rebound peak of activity upon the removal of steroid in the continued presence of GABA also would not be seen in the model, postulating that steroids alter the rates for onset and/or recovery from desensitization. Instead, that model would predict a single exponential return to the steady-state level established in the presence of GABA alone. We note that the decline in activity from the transient rebound peak has a rate that is comparable to the onset of desensitization, as predicted by the RADB model.
It has been suggested previously that the mechanism of action of DHEAS may differ from that of PS because of observations that DHEAS inhibits the GABA-elicited current in pituitary cells without altering the kinetics of desensitization (Le Foll et al., 1997; Hansen et al., 1999). In contrast, we (Fig. 2A) and others (Demirgören et al., 1991; Spivak, 1994) have reported faster apparent desensitization in the presence of DHEAS. Overall, our data support the conclusions that DHEAS and PS bind to a common site and block GABAA receptor function by the same mechanism.
The value of the parameter W (= A/B) was arbitrarily constrained to 100 in our analysis. The occupancy of the blocked state in the absence of inhibitory steroid is likely very low although its precise value could not be determined. We note that a wide range of values for W could acceptably describe the effect of steroid. The PA at saturating steroid concentration is formally described by Eq. 5, which for a strong inhibitory steroid such as PS applied in the presence of a saturating concentration of a strong agonist reduces to:
 |
Accordingly, pairs of W and e can describe the data as long as We = W'e'. Indeed, fitting the PS concentration-response curve with N constrained to 1 gave a value of ePS of 1.11 × 10−4 when W was constrained to 50 (WePS = 0.0055) and a value of ePS of 5.61 × 10−6 with W constrained to 1000 (WePS = 0.0056). We note that this situation is also seen in the case of activation, in which the values for L and cX can be difficult to disentangle and essentially require measurements of constitutive activity in the absence of agonist and peak activity elicited by maximal agonist (Akk et al., 2018).
We note that an essential caveat exists, in that whereas the RADB model provides a description of block by PS and DHEAS that is better than that provided by the Resting-Active-Desensitized model, there is no evidence indicating that it is globally the best description. In particular, there is an alternative model involving selective block of the active receptor: a two-step open-channel block (Lingle et al., 2001). This model also postulates an additional inactive (blocked); this state does not exist in the absence of blocker and hence more closely resembles an “induced fit” mechanism. We have argued against a simple open channel block mechanism because at a high concentration of blocker, the inhibition is complete, whereas block by these inhibitory steroids is not complete at saturating concentrations. In the two-step process, however, the initial binding step does not result in block, which reflects a resultant conformation change either in the ligand, the receptor, or a change in orientation of the association. Accordingly, the block may not be complete even when binding is saturated. Modeling indicates that this model can also describe the data we have obtained, including the features of different peak and sustained levels of inhibition.
In sum, our data indicate that the inactive state induced by PS or DHEAS is a novel state that differs from the agonist-induced desensitized state. PS and DHEAS appear to share a mechanism and site of action, although it will require additional studies to determine whether other inhibitory steroids, particularly the uncharged 3β-OH steroids, act in this fashion.
Abbreviations
- DHEAS
dehydroepiandrosterone sulfate [(3aS, 3bR, 7S, 9aR, 9bS, 11aS)-7-hydroxy-9a, 11a-dimethyl-2, 3, 3a, 3b, 4, 6, 7, 8, 9, 9a, 9b, 10, 11, 11a-tetradecahydro-1H-cyclopenta[a]phenanthren-1-one sulfate]
- PA
probability of being in the active state
- PS
pregnenolone sulfate [(1S, 3aS, 3bS, 7S, 9aR, 9bS, 11aS)-1-acetyl-9a, 11a-dimethyl-2, 3, 3a, 3b, 4, 6, 7, 8, 9, 9a, 9b, 10, 11, 11a-tetradecahydro-1H-cyclopenta[a]phenanthren-7-yl hydrogen sulfate]
- RADB
resting-active-desensitized/blocked
Authorship Contributions
Participated in research design: Steinbach, Akk.
Conducted experiments: Pierce, Germann.
Performed data analysis: Pierce, Germann, Steinbach, Akk.
Wrote or contributed to the writing of the manuscript: Pierce, Germann, Steinbach, Akk.
Footnotes
This work was supported by National Institutes of Health National Institute of General Medical Sciences [Grants R01GM108580, R35GM140947] and funds from the Taylor Family Institute for Innovative Psychiatric Research.
No author has an actual or perceived conflict of interest with the contents of this article.
References
- Akk G, Bracamontes J, Steinbach JH (2001) Pregnenolone sulfate block of GABA(A) receptors: mechanism and involvement of a residue in the M2 region of the α subunit. J Physiol 532:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Germann AL, Sugasawa Y, Pierce SR, Evers AS, Steinbach JH (2020) Enhancement of muscimol binding and gating by allosteric modulators of the GABAA receptor: relating occupancy to state functions. Mol Pharmacol 98:303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Shin DJ, Germann AL, Steinbach JH (2018) GABA type A receptor activation in the allosteric coagonist model framework: relationship between EC50 and basal activity. Mol Pharmacol 93:90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham KP, Anderson DR, Huyvaert KP (2011) AIC model selection and multimodel inference in behavioral ecology: some background, observations, and comparisons. Behav Ecol Sociobiol 65:23–35. [Google Scholar]
- Chang Y, Ghansah E, Chen Y, Ye J, Weiss DS (2002) Desensitization mechanism of GABA receptors revealed by single oocyte binding and receptor function. J Neurosci 22:7982–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charalampopoulos IAlexaki VITsatsanis CMinas VDermitzaki ELasaridis IVardouli LStournaras CMargioris ANCastanas E, et al. (2006) Neurosteroids as endogenous inhibitors of neuronal cell apoptosis in aging. Ann N Y Acad Sci 1088:139–152. [DOI] [PubMed] [Google Scholar]
- Chisari M, Wilding TJ, Brunwasser S, Krishnan K, Qian M, Benz A, Huettner JE, Zorumski CF, Covey DF, Mennerick S (2019) Visualizing pregnenolone sulfate-like modulators of NMDA receptor function reveals intracellular and plasma-membrane localization. Neuropharmacology 144:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpéchot C, Robel P, Axelson M, Sjövall J, Baulieu EE (1981) Characterization and measurement of dehydroepiandrosterone sulfate in rat brain. Proc Natl Acad Sci USA 78:4704–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpéchot C, Synguelakis M, Talha S, Axelson M, Sjövall J, Vihko R, Baulieu EE, Robel P (1983) Pregnenolone and its sulfate ester in the rat brain. Brain Res 270:119–125. [DOI] [PubMed] [Google Scholar]
- Demirgören S, Majewska MD, Spivak CE, London ED (1991) Receptor binding and electrophysiological effects of dehydroepiandrosterone sulfate, an antagonist of the GABAA receptor. Neuroscience 45:127–135. [DOI] [PubMed] [Google Scholar]
- Eisenman LN, He Y, Fields C, Zorumski CF, Mennerick S (2003) Activation-dependent properties of pregnenolone sulfate inhibition of GABAA receptor-mediated current. J Physiol 550:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman LNShu HJAkk GWang CManion BDKress GJEvers ASSteinbach JHCovey DFZorumski CF, et al. (2007) Anticonvulsant and anesthetic effects of a fluorescent neurosteroid analog activated by visible light. Nat Neurosci 10:523–530. [DOI] [PubMed] [Google Scholar]
- Germann AL, Pierce SR, Burbridge AB, Steinbach JH, Akk G (2019a) Steady-state activation and modulation of the concatemeric α1β2γ2L GABAA receptor. Mol Pharmacol 96:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germann AL, Pierce SR, Senneff TC, Burbridge AB, Steinbach JH, Akk G (2019b) Steady-state activation and modulation of the synaptic-type α1β2γ2L GABAA receptor by combinations of physiological and clinical ligands. Physiol Rep 7:e14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germann AL, Reichert DE, Burbridge AB, Pierce SR, Evers AS, Steinbach JH, Akk G (2020) Analysis of modulation of the ρ1 GABAA receptor by combinations of inhibitory and potentiating neurosteroids reveals shared and distinct binding sites. Mol Pharmacol 98:280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SL, Fjalland B, Jackson MB (1999) Differential blockade of γ-aminobutyric acid type A receptors by the neuroactive steroid dehydroepiandrosterone sulfate in posterior and intermediate pituitary. Mol Pharmacol 55:489–496. [PubMed] [Google Scholar]
- Karlin A (1967) On the application of “a plausible model” of allosteric proteins to the receptor for acetylcholine. J Theor Biol 16:306–320. [DOI] [PubMed] [Google Scholar]
- Laverty D, Thomas P, Field M, Andersen OJ, Gold MG, Biggin PC, Gielen M, Smart TG (2017) Crystal structures of a GABAA-receptor chimera reveal new endogenous neurosteroid-binding sites. Nat Struct Mol Biol 24:977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll F, Louiset E, Castel H, Vaudry H, Cazin L (1997) Electrophysiological effects of various neuroactive steroids on the GABA(A) receptor in pituitary melanotrope cells. Eur J Pharmacol 331:303–311. [DOI] [PubMed] [Google Scholar]
- Lingle CJ, Zeng XH, Ding JP, Xia XM (2001) Inactivation of BK channels mediated by the NH(2) terminus of the β3b auxiliary subunit involves a two-step mechanism: possible separation of binding and blockade. J Gen Physiol 117:583–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska MD, Demirgören S, Spivak CE, London ED (1990) The neurosteroid dehydroepiandrosterone sulfate is an allosteric antagonist of the GABAA receptor. Brain Res 526:143–146. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Mienville JM, Vicini S (1988) Neurosteroid pregnenolone sulfate antagonizes electrophysiological responses to GABA in neurons. Neurosci Lett 90:279–284. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Schwartz RD (1987) Pregnenolone-sulfate: an endogenous antagonist of the γ-aminobutyric acid receptor complex in brain? Brain Res 404:355–360. [DOI] [PubMed] [Google Scholar]
- Michel MC, Murphy TJ, Motulsky HJ(2020) New author guidelines for displaying data and reporting data analysis and statistical methods in experimental biology. Mol Pharmacol 97:49–60. [DOI] [PubMed] [Google Scholar]
- Mienville JM, Vicini S (1989) Pregnenolone sulfate antagonizes GABAA receptor-mediated currents via a reduction of channel opening frequency. Brain Res 489:190–194. [DOI] [PubMed] [Google Scholar]
- Monnet FP, Mahé V, Robel P, Baulieu EE (1995) Neurosteroids, via sigma receptors, modulate the [3H]norepinephrine release evoked by N-methyl-D-aspartate in the rat hippocampus. Proc Natl Acad Sci USA 92:3774–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12:88–118. [DOI] [PubMed] [Google Scholar]
- Morgan CA III, Southwick S, Hazlett G, Rasmusson A, Hoyt G, Zimolo Z, Charney D (2004) Relationships among plasma dehydroepiandrosterone sulfate and cortisol levels, symptoms of dissociation, and objective performance in humans exposed to acute stress. Arch Gen Psychiatry 61:819–825. [DOI] [PubMed] [Google Scholar]
- Park-Chung M, Malayev A, Purdy RH, Gibbs TT, Farb DH (1999) Sulfated and unsulfated steroids modulate γ-aminobutyric acidA receptor function through distinct sites. Brain Res 830:72–87. [DOI] [PubMed] [Google Scholar]
- Ritsner MS (2011) The clinical and therapeutic potentials of dehydroepiandrosterone and pregnenolone in schizophrenia. Neuroscience 191:91–100. [DOI] [PubMed] [Google Scholar]
- Sachidanandan D, Bera AK (2015) Inhibition of the GABAA receptor by sulfated neurosteroids: a mechanistic comparison study between pregnenolone sulfate and dehydroepiandrosterone sulfate. J Mol Neurosci 56:868–877. [DOI] [PubMed] [Google Scholar]
- Shen W, Mennerick S, Covey DF, Zorumski CF (2000) Pregnenolone sulfate modulates inhibitory synaptic transmission by enhancing GABA(A) receptor desensitization. J Neurosci 20:3571–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DJ, Germann AL, Covey DF, Steinbach JH, Akk G (2019) Analysis of GABAA receptor activation by combinations of agonists acting at the same or distinct binding sites. Mol Pharmacol 95:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DJ, Germann AL, Steinbach JH, Akk G (2017) The actions of drug combinations on the GABAA receptor manifest as curvilinear isoboles of additivity. Mol Pharmacol 92:556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Gibbs TT, Farb DH (2014) Pregnenolone sulfate as a modulator of synaptic plasticity. Psychopharmacology (Berl) 231:3537–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak CE (1994) Desensitization and noncompetitive blockade of GABAA receptors in ventral midbrain neurons by a neurosteroid dehydroepiandrosterone sulfate. Synapse 16:113–122. [DOI] [PubMed] [Google Scholar]
- Steinbach JH, Akk G (2019) Applying the Monod-Wyman-Changeux allosteric activation model to pseudo-steady-state responses from GABAA receptors. Mol Pharmacol 95:106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti G, Ferrucci L, Lauretani F, Ceresini G, Bandinelli S, Luci M, Ceda G, Maggio M, Schwartz RS (2009) Dehydroepiandrosterone sulfate and cognitive function in the elderly: the InCHIANTI Study. J Endocrinol Invest 32:766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenmakers EJ, Farrell S (2004) AIC model selection using Akaike weights. Psychon Bull Rev 11:192–196. [DOI] [PubMed] [Google Scholar]
- Wang MHe YEisenman LNFields CZeng CMMathews JBenz AFu TZorumski ESteinbach JH, et al. (2002) 3β-hydroxypregnane steroids are pregnenolone sulfate-like GABA(A) receptor antagonists. J Neurosci 22:3366–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FS, Gibbs TT, Farb DH (1991) Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-D-aspartate receptor. Mol Pharmacol 40:333–336. [PubMed] [Google Scholar]
- Xu XJ, Roberts D, Zhu GN, Chang YC (2016) Competitive antagonists facilitate the recovery from desensitization of α1β2γ2 GABAA receptors expressed in Xenopus oocytes. Acta Pharmacol Sin 37:1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]