

Abstract
Kratom (Mitragyna speciosa), a Southeast Asian tree, has been used for centuries in pain relief and mitigation of opium withdrawal symptoms. Mitragynine (MTG), the major kratom alkaloid, is being investigated for its potential to provide analgesia without the deleterious effects associated with typical opioids. Concerns have been raised regarding the active metabolite of MTG, 7-hydroxymitragynine (7HMG), which has higher affinity and efficacy at µ-opioid receptors than MTG. Here we investigated the hotplate antinociception, pharmacokinetics, and tissue distribution of MTG and 7HMG at equianalgesic oral doses in male and female C57BL/6 mice to determine the extent to which 7HMG metabolized from MTG accounts for the antinociceptive effects of MTG and investigate any sex differences. The mechanism of action was examined by performing studies with the opioid receptor antagonist naltrexone. A population pharmacokinetic/pharmacodynamic model was developed to predict the behavioral effects after administration of various doses of MTG and 7HMG. When administered alone, 7HMG was 2.8-fold more potent than MTG to produce antinociception. At equivalent effective doses of MTG and 7HMG, there was a marked difference in the maximum brain concentration of 7HMG achieved, i.e., 11-fold lower as a metabolite of MTG. The brain concentration of 7HMG observed 4 hours post administration, producing an analgesic effect <10%, was still 1.5-fold higher than the maximum concentration of 7HMG as a metabolite of MTG. These results provide strong evidence that 7HMG has a negligible role in the antinociceptive effects of MTG in mice.
SIGNIFICANCE STATEMENT
Mitragynine (MTG) is being investigated for its potential to aid in pain relief, opioid withdrawal syndrome, and opioid use disorder. The active metabolite of MTG, 7-hydroxymitragynine (7HMG), has been shown to have abuse potential and has been implicated in the opioid-like analgesic effect after MTG administration. The results of this study suggest a lack of involvement of 7HMG in the antinociceptive effects of MTG in mice.

Introduction
Mitragyna speciosa (Korth.) Havil., known in the United States as kratom, is a tree native to Southeast Asia (Sharma and McCurdy, 2021). Traditional use of kratom includes chewing the leaves to sustain energy while working in the hot and humid environment where it grows or brewing the leaves into a tea or decoction for analgesia. It has also been used as a substitution for opium and for the mitigation of opioid withdrawal symptoms (Veltri and Grundmann, 2019). In the Western world, kratom products are primarily used for pain, withdrawal from opioids, anxiety, and depression (Veltri and Grundmann, 2019). The most abundant kratom alkaloid in dried kratom leaves is mitragynine (MTG, Fig. 1 left) (Sharma et al., 2019). Although MTG has been reported to account for up to 66% of the total alkaloid content, more recent analyses demonstrate that MTG accounts for 0.7%–38.7% of total alkaloidal content in commercial products and traditional preparations. A metabolite of MTG, 7-hydroxymitragynine (7HMG, Fig. 1 right), is also found as < 0.01% in fresh samples and ∼2% in commercial products (Lydecker et al., 2016; Hemby et al., 2019; Singh et al., 2020; Chear et al., 2021). As a metabolite of MTG, 7HMG formation is primarily catalyzed by cytochrome P450 3A4 enzyme in humans (Kamble et al., 2019).
Fig. 1.

Chemical structures of MTG (left) and 7HMG (right).
Kratom alkaloids are active at a variety of receptor types, including opioidergic, adrenergic, and serotonergic (Ellis et al., 2020; Obeng et al., 2020; Obeng et al., 2021). Among the receptors, in vitro activities of MTG and 7HMG differ at µ-opioid receptors (MOR). 7HMG is a high affinity, partial agonist, whereas MTG is a low affinity, partial agonist with Ki values of 37 and 230 nM, respectively (Varadi et al., 2016). MTG differs from other standard MOR agonists as it does not activate the β-arrestin-2 signaling pathway, which has been hypothesized to be the cause of many of the adverse effects of MOR agonists, including respiratory depression and constipation (Eastlack et al., 2020). Study results have also indicated that MTG may produce analgesia through nonopioid mediated pathways (Matsumoto et al., 1996; Hiranita et al., 2019; Obeng et al., 2020).
Kratom has shown potential as a treatment of opioid dependence but has also produced adverse events, leading to scrutiny by public health and regulatory officials. In rats self-administering morphine, heroin, or methamphetamine, MTG was not self-administered above vehicle levels (Yue et al., 2018; Hemby et al., 2019). Additionally, pretreatment with MTG dose dependently decreased the rate of self-administration responding maintained by heroin but not methamphetamine (Yue et al., 2018). These results indicate MTG has limited abuse potential while also functionally antagonizing the reinforcing effects of opioids. Conversely, 7HMG substituted for morphine in assays of self-administration as well as being self-administered in naïve rats (Hemby et al., 2019). These results reveal that 7HMG has a high abuse potential (Hemby et al., 2019). Adverse effects in multiple organ systems have been reported from case studies of kratom users including acute liver failure, acute kidney injury, cardiotoxicity, acute respiratory distress syndrome, acute brain injury, seizures, coma, and even death (Eastlack et al., 2020). As of December 31, 2020, the Food and Drug Administration’s Adverse Events Reporting System public dashboard included 567 serious cases, and of those, 378 deaths associated with the use of kratom taken alone or concomitantly with other substances (FDA, 2021).
Due to the controversy surrounding the use of kratom products and the abuse potential of 7HMG, it is essential to compare the pharmacokinetic profile of 7HMG alone and converted from MTG to understand the respective contributions of each to the reported effects of MTG. The pharmacokinetic profile of active metabolites is essential for the drug development process, especially if the metabolites are suspected to be more potent and/or pose risks. Here we are investigating the systemic pharmacokinetics and tissue distribution of MTG and 7HMG after an equianalgesic oral dose of either MTG or 7HMG in male and female mice. To identify their equianalgesic doses, the time-effect functions of MTG and 7HMG were assessed using a hotplate assay. The opioid receptor antagonist naltrexone was used to determine the possible mechanism underlying the antinociceptive effects of MTG and 7HMG. In addition, each mouse concurrently underwent measurement of locomotor activity to assess if increases in hotplate response latency were due to nonspecific behavioral disruption. The pharmacokinetic-pharmacodynamic (PK/PD) relationship was then evaluated to determine how plasma concentrations of MTG and 7HMG (as both a metabolite and after individual administration) correlate to the measured hotplate latency. The results of this study will give insight into the magnitude of the contribution of 7HMG to the overall pharmacology of MTG.
Materials and Methods
Compounds.
MTG was isolated and purified from a kratom alkaloid enriched extract (OPMS, Choice Organics, Los Angeles, CA) as described in previous literature (Avery et al., 2019; Sharma et al., 2019) and was used as the hydrochloride salt with a purity ≥ 98%. 7HMG (purity ≥ 98%) was obtained through semisynthesis from MTG as previously reported (Maxwell et al., 2020) and used as the freebase. Purity and structural characterization of MTG and 7HMG were established using 1H proton and 13C NMR, ultra-high-performance liquid chromatography-photodiode array detection and liquid chromatography high-resolution quadrupole time of flight mass spectrometry. (-)-Naltrexone hydrochloride (purity ≥ 98%), midazolam (purity ≥ 98%), phenacetin [purity ≥ 98%, internal standard (IS)], and verapamil [purity ≥ 98%, (IS)] were purchased from Sigma-Aldrich Co. (St. Louis, MO). Tween-80, propylene glycol, and LC-MS grade acetonitrile, methanol, and formic acid were procured from Fisher Scientific (Waltham, MA). Blank pooled mouse plasma was obtained from Innovative Research, Inc. (Novi, MI). Nicotinamide adenine dinucleotide phosphate reduced tetrasodium salt (NADPH) was purchased from MP Biomedicals (Solon, OH). Male and female pooled mouse liver microsomes (MLM) were obtained from XenoTech, LLC (Lenexa, KS).
Formulations.
Each test compound and/or vehicle was administered orally by gavage at a dose volume of no more than 10 mL/kg. For the functional studies, each test compound was dissolved in a vehicle consisting of sterile water (HyClone Water for Injection, 1L, Fisher Scientific, Waltham, MA) containing 5% (v/v) Tween 80 and 5% (v/v) propylene glycol to reach behaviorally active doses of test compounds. The formulation or vehicle was filtered with a 0.2-µm pore size syringe filter (Millex-LG, Sigma Aldrich Co, St. Louis, MO) prior to administration. The dose ranges and pretreatment times were chosen based on preliminary data and published methods (Tanda, 2016; Hiranita et al., 2019; Behnood 2020; Obeng et al., 2020; Obeng et al., 2021). For the pharmacokinetic studies, MTG and 7HMG were prepared by dissolving MTG hydrochloride and 7HMG freebase in purified water with 3% (v/v) Tween 80 to get a freebase equivalent solution of 16.5 and 5.0 mg/mL, respectively.
Instrumentation and Analytical Conditions.
1H and 13C NMR spectra were obtained using Bruker Avance NEO 600 NMR and Bruker Avance II 600 MHz NMR spectrometers operating at 600 MHz in 1H and 151 MHz in 13C (Bruker Co., Billerica, MA). A previously reported ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) method validated for the quantification of MTG and 7HMG was used to analyze study samples (Maxwell et al., 2020). The UPLC-MS/MS was a Waters Acquity Class-I chromatograph coupled with a Xevo TQ-S Micro triple quadrupole mass spectrometer (Waters, Milford, MA).
In brief, a Waters Acquity UPLC BEH C18 column (1.7 µm, 2.1 × 50 mm) fitted with a Vanguard precolumn of the same chemistry was used with an aqueous mobile phase of 0.1% formic acid in water (A) and an organic mobile phase of acetonitrile (B). Gradient elution was used where 80% A was supplied for 0.5 minute and then decreased linearly to 68% up to 2.2 minutes and then 62% up to 3.5 minutes after which it was raised back up to 80% where it was maintained until 5.5 minutes at a flow rate of 0.35 mL/min. The method was updated to a quantitation range of 2–800 ng/mL with a 0.4 µL injection volume. In addition, the method was previously validated in dog plasma (Maxwell et al., 2020) so partial validations were performed for each of the additional matrices (mouse plasma, brain, lung, liver, kidney, and spleen).
For antinociception, a hard black anodized aluminum plate with a built-in digital thermometer (33.1 cm long, 28.8 cm wide, 9.9 cm high, Hot Plate Analgesia Meter, Columbus Instruments, Columbus, OH) was used. A clear acrylic cage (26.7 cm long, 26.7 cm wide, and 31.0 cm high) surrounded the square plate to confine the animal. The temperature was maintained at 52°C and measured to an accuracy of ± 0.1°C. For horizontal locomotor activity, a seamless open field system for mice was used (MED-OFAS-MSU, Med Associates Inc., Fairfax, VT). Med-PC software version 7 (SOF-812, Med Associates Inc., Fairfax, VT) controlled experimental events and provided a record of responses.
Preparation of Calibration Standards and Quality Control Samples.
MTG hydrochloride was first dissolved in acetonitrile to create a 1.0 mg/mL freebase equivalent of primary stock. 7HMG freebase was dissolved separately in acetonitrile to create an additional 1.0 mg/mL primary stock. Aliquots from these primary stocks were diluted in acetonitrile to create secondary stocks containing 10, 1.0, and 0.1 µg/mL of each compound. These secondary stocks were then used to create a set of eight working stocks ranging from 25–10,000 ng/mL for the preparation of the calibration standards, also in acetonitrile. Quality control (QC) working stocks ranging from 25–8750 ng/mL were created from a different primary stock than the calibration standards.
Sample Processing.
Samples were thawed to room temperature for one hour prior to analysis. To prepare blank or study samples for analysis, 25 µL aliquots of each sample were taken. Samples expected to fall outside the linearity range were diluted 5, 25, or 100 times. To prepare calibration standards, 2.0 µL of working stock was added to 23 µL of the appropriate blank matrix. This generated eight standards of 2.0, 10, 50, 100, 200, 400, 600, and 800 ng/mL. The same process was used to generate QCs: the lower limit of quantification (2.0 ng/mL), low (5.0 ng/mL), medium (350 ng/mL) and high (700 ng/mL) QCs. After spiking, the samples were vortex mixed for five minutes at 800 rpm. Simple protein precipitation using 100 µL methanol acidified with 0.05% formic acid containing 25 ng/mL IS was used to extract analytes. The samples were then vortex mixed for five minutes at 650 rpm before being transferred to a 96-well Millipore 0.45 µm filter plate and centrifuged at 850 x g for two minutes at 4°C. The filtrate was then subjected to UPLC-MS/MS analysis. The TargetLynx application of MassLynx 4.2 was used for data processing (Waters, Milford, MA).
Animals.
Adult male and female C57BL/6 mice (weight range 20–30 g) were purchased through Charles River (Wilmington, MA) and group housed in home cages (N = 5 per cage for pharmacokinetics studies; N = 4 per cage for behavioral studies). Each mouse was acclimated for at least three days to a temperature and humidity-controlled vivarium with a 12-hour light/dark cycle. The temperature, humidity, and light/dark cycle in the experimental rooms were equivalent to those in the vivarium. Food [2918 Teklad Global 18% protein rodent diet (Envigo, Frenchtown, NJ)] and reverse osmosis water was available in the home cage. Study protocols were approved by the Institutional Animal Care and Use Committee at the University of Florida, which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International and were written in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Pharmacodynamic Studies.
All functional experiments were conducted in the light cycle at the same time each day (0800 to 1200). The body weight of each mouse (N = 4 per sex, per dose) was measured before experiments. In each time course study, the hotplate latency and locomotor activity experiments were conducted sequentially under a within-subject design. A total of 72 mice were used. Each mouse was used only once.
Each mouse was placed on the heated plate and baseline latency was determined manually using a stopwatch (Martin Stopwatch, Martin Sports, Carlstadt, NJ) by trained and experimentally blinded raters. Hotplate latency was measured until the subject jumped, licked, and/or shook the back paws, any deemed toxicity (e.g., seizure) was observed, or up to 60 seconds to avoid tissue damage, whichever occurred first. After the measurement of hotplate latency, each mouse was placed singly into the clear acrylic chamber and activity counts were measured over a 5-minute period.
After determination of the baselines, each mouse orally received either a single dose of MTG or 7HMG, or vehicle and then was returned to the respective home cage. In each mouse, the hotplate response latency and activity counts were measured repeatedly every 30 minutes up to 240 minutes after dosing. A 1.0 mg/kg naltrexone dose was administered intraperitoneally five minutes prior to oral administration of the highest doses of MTG or 7HMG. Upon completion of each time-course test, each mouse was euthanized.
Pharmacokinetic and Tissue Distribution Studies.
According to the results of the hotplate test, the effective dose to reach 50% maximum possible effect (ED50) of each compound was tested in the pharmacokinetic study (165 and 50 mg/kg for MTG and 7HMG, respectively). The first pharmacokinetic study was performed in 40 males and 40 females using a single oral dose of 165 mg/kg MTG. The mice were randomly divided into 10 groups of N = 4 per sex, per time point with blood samples collected by cheek vein bleed and at the terminal time points for a total of 16 time points (predose, 0.083, 0.25, 0.50, 0.75, 1.0, 1.5, 2.0, 3.0, 4.0, 5.0, 6.0, 12, 18, 24, and 48 hours). Tissue samples were collected at the terminal time points (0.25, 0.50, 1.0, 2.0, 4.0, 6.0, 12, 18, 24, and 48 hours).
Another study was performed with a 50 mg/kg single oral dose of 7HMG in 24 males and 24 females (N = 4 per sex, per time point). Blood samples were collected at predose, 0.083, 0.25, 0.50, 0.75, 1.0, 1.5, 2.0, 3.0, 4.0, 6.0, and 8.0 hours for males and predose, 0.083, 0.25, 0.50, 0.75, 1.0, 1.5, 2.0, 3.0, 4.0, 5.0, and 6.0 hours for females, whereas tissues were collected at the terminal time points only (0.25, 0.5, 1.0, 2.0, 4.0, and 8.0 hours for males and 0.25, 0.5, 1.0, 2.0, 4.0, and 5.0 hours for females).
Blood samples were collected into heparinized tubes and plasma was separated by centrifugation for 10 minutes at 850 x g and 4°C. Organs were prepared for analysis by homogenization with aqueous buffer at a ratio of 1:4 organ to buffer. All samples were stored at -80°C until analysis.
Metabolic Stability of MTG and 7HMG in Mouse Liver Microsomes.
The in vitro metabolic stability of MTG and 7HMG was determined using male and female MLM. The incubation mixtures consisted of MLM (1 mg microsomal protein/mL), substrate (1 µM), and NADPH (1 mM) in a total volume of 0.2 mL potassium phosphate buffer (50 mM, pH 7.4). Midazolam was used as a positive control. Reactions were initiated with the addition of NADPH and kept in an incubator shaker at 37°C. For negative control, incubations without NADPH were also performed. All incubations were performed in triplicate. Aliquots of 25 µL each were withdrawn at 0, 5, 10, 15, 30, and 60 minutes and mixed with 125 µL of acetonitrile containing IS for the termination of the reaction. The samples were then vortex mixed and filtered through a 0.45 µm 96-well membrane filtration plate under centrifugation at speed of 850 × g for 5 minutes at 4°C. The filtrates were subjected to UPLC-MS/MS analysis.
Data Analysis.
Precision and accuracy were evaluated by calculating the percent relative standard deviation (RSD) and the percent bias (%Bias) at each concentration tested (N = 6, each concentration), respectively. RSD was calculated using the formula:
 |
While the %Bias was calculated using the equation:
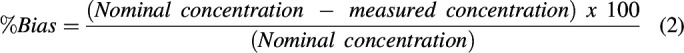 |
Organ concentrations were corrected for residual blood using the following formula (Khor and Mayersohn, 1991; Giudicelli et al., 2004):
 |
where Cmeasured was the concentration obtained after UPLC-MS/MS analysis, Cblood was calculated by multiplying Cmeasured times the blood to plasma partition coefficient (1.0 for MTG and 1.1 for 7HMG in females; 0.9 for both MTG and 7HMG in males, Supplemental Table 2), which was determined using previously published methods (Yu et al., 2005). Wblood represents the volume of residual blood in the organ relative to the volume of the organ. Wblood values for each organ are as follows: kidney = 0.24, liver = 0.31, lung = 0.50, spleen = 0.17, and brain = 0.03 (Brown et al., 1997). Once corrected, the concentration versus time profiles were generated using Graphpad Prism Version 8 (GraphPad Software, San Diego CA) and noncompartmental analysis was performed using the sparse sampling method and linear trapezoidal extrapolation to determine the pharmacokinetic parameters of each compound in plasma and tissues using Phoenix Version 6.4 (Certara, Princeton, NJ).
For the functional studies, all the experimental variables in each figure are shown as mean values (± SEM) per groups of subjects as a function of elapsed time after administration. Statistical analyses were conducted using GraphPad Prism version 8 (San Diego, CA) and SigmaPlot version 14.0 (Systat Software Inc., San Jose, CA). Statistical differences were considered significant when a p value was less than 0.05. A one- or two-way (repeated-measures) ANOVA followed by post hoc Bonferroni t tests was used as appropriate to analyze the effects of elapsed time after administration, dose, or their interactions.
Hotplate latency data in seconds was converted to percent maximum possible antinociceptive effect (MPE) with the following equation:
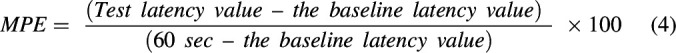 |
The locomotor activity counts of doses of test compounds were normalized to that of vehicle per corresponding time point. When the mean effect of a compound to produce an increase in percent MPE or decrease locomotor activity crossed 50% of vehicle level per time point, the ED50 and 95% confidence interval values in µmol/kg were calculated using linear regression (Snedecor and Cochran, 1967), where slopes were allowed to vary, according to Tallarida (Tallarida, 2002). The potency ratio was derived from the ED50 [(95% confidence intervals (CIs)] value for MTG divided by that for 7HMG. If the 95% CIs of the ED50 values did not overlap or the potency ratio of the compounds did not include one, the compounds were considered to have significantly different potencies. To compare the ED50 (95% CIs) values in µmol/kg, molecular weights for MTG (MTG HCl) and 7HMG used were 398.50 (434.96) and 414.50 g/mol, respectively.
The in vitro elimination half-life (t1/2) was determined using the following equation:
 |
where k is the slope of the line obtained by plotting natural logarithmic of the percentage of compound remaining versus time. The amount of 7HMG produced in MLM after MTG incubation was also quantified, and the metabolite to parent ratio was determined at 60 minutes by dividing the concentration of 7HMG formed by the concentration of MTG incubated (1 µM).
The PK/PD relationship for both MTG and 7HMG was examined by fitting the data to a population PK/PD model using Monolix (Monolix version 2020R1. Antony, France: Lixoft SAS, 2020). All parameters were fitted with the observed data, and simulations were performed at various doses to validate the models.
Results
Method Validation.
Supplemental Table 1 shows the results of all partial validations. The calibration curves for each matrix type were considered acceptable if the correlation coefficient was greater than 0.99. QC samples were prepared for each run and fell within the limits of ± 20% at the lower limit of quantification and ± 15% at the low quality control, medium quality control, and high quality control as required by the FDA Guidelines for Bioanalytical Method Validation (FDA, 2018).
Antinociception and Locomotor Activity.
Baseline values for hotplate response latency (approximately 11 seconds) and locomotor activity counts (approximately 825 counts) were statistically comparable across all groups regardless of sex, dose, or their interaction (F values ≤ 3.63; P values ≥ 0.062, Figs. 2–5 and Supplemental Table 3). Thus, data from females and males was combined and is shown in Figs. 2 and 4. After vehicle administration (filled circles), MPEs were stably low (Fig. 2, upper left); however, activity counts were gradually decreased over time and stabilized at less than 100 counts 120 minutes after vehicle administration (Fig. 2, lower left).
Fig. 2.

Effects of MTG and 7HMG on hotplate response latency and locomotor activity counts in mice. Abscissae: Baseline and elapsed time after oral administration in minutes; baseline values are shown for reference. Upper left ordinates: Baseline hotplate response latency in seconds. Upper right ordinates: Percentage of MPE. Lower left ordinates: Activity counts for 5 minutes immediately after each measurement of hotplate response latency. Left panels: Vehicle (filled circles), and 92, 163, and 293 mg/kg MTG (squares, upward triangles, and downward triangles, respectively). Right panels: Vehicle (filled circles), and 32, 56, and 100 mg/kg 7HMG (squares, upward triangles, and downward triangles, respectively). The data for vehicle were the same across left and right panels. Each point represents the mean ± SEM (4 mice per sex). Each gray symbol indicates a significant difference from vehicle per corresponding time point.
Fig. 5.

Effects of sex on the antinociceptive effects of 100 mg/kg 7HMG in the absence and presence of naltrexone. Abscissae: Baseline and elapsed time after oral administration in minutes. Left ordinates: Baseline hotplate response latency in seconds. Right ordinates: Percentage of MPE. Left and right panels: Females and males, respectively. Vehicle (filled circles), 100 mg/kg 7HMG in the absence and presence of naltrexone (downward triangles and diamonds, respectively). Each point represents the mean ± SEM (4 mice). Each gray symbol indicates a significant difference from vehicle per corresponding time point.
Fig. 4.

Effects of MTG and 7HMG on hotplate response latency and locomotor activity counts in the presence of the opioid receptor antagonist naltrexone. Abscissae: Baseline and elapsed time after oral administration in minutes. Upper left ordinates: Baseline hotplate response latency in seconds. Upper right ordinates: Percentage of MPE. Lower left ordinates: Activity counts for 5 minutes immediately after each measurement of hotplate response latency. Left panels: Vehicle (filled circles), and 293 mg/kg MTG in the absence and presence of naltrexone (downward triangles and diamonds, respectively). Right panels: Vehicle (filled circles), 100 mg/kg 7HMG in the absence and presence of naltrexone (downward triangles and diamonds, respectively). The data for vehicle were the same ones in Figure 2. Each symbol represents the mean ± SEM (4 mice/sex). Each gray symbol indicates a significant difference from vehicle per corresponding time point.
In addition to vehicle, effects of MTG and 7HMG on MPE and locomotor activity counts are shown in Fig. 2. MTG dose- and time-dependently produced significant increases in MPE (Fig. 2, top left; Supplemental Table 4). Each gray symbol in Fig. 2 indicates a significant difference from vehicle per corresponding time point (Supplemental Table 4). The MTG dose of 92 mg/kg (open squares) was inactive, whereas the increases in MPE and duration of action were greater in the 293 mg/kg MTG dose than the 163 mg/kg MTG dose (triangles downward and upward, respectively). There was no significant effect of sex on MPE (Supplemental Table 4). The ED50 (95% CIs) value of MTG to increase the percentage of MPE was 340 (262–400) µmol/kg at 90 minuted after administration (Table 1). In addition, activity counts (Fig. 2, lower left, Supplemental Table 4) were significantly increased by 163 mg/kg MTG at 60 and 90 minutes after administration (triangles upward) but decreased by 293 mg/kg MTG at 30 minutes after administration (triangles downward). There was no significant effect of sex on activity counts (Supplemental Table 4).
TABLE 1.
ED50 (95% CIs) values in µmol/kg for the antinociceptive and activity-decreasing effects of MTG and 7HMG in mice
| Compound | ED50 (95% CIs) | |
|---|---|---|
| MPE | Decrease in Locomotor Activity Counts | |
| MTG (Males and Females) | 340 (262–400) @ 90 min | 384 (255–487) @ 30 min |
| 7HMG (Males and Females) | 121 (114–152) @ 30 min | Not determined |
| 7HMG (Females) | 122 (111–158) @ 30 min | Not determined |
| 7HMG (Males) | 119 (108–154) @ 30 min | Not determined |
| Potency Ratio (males and females) | 2.81 (1.72–3.51) | Not applicable |
For the MPEs, the ED50 (95% CIs) value was calculated at the time point where the highest dose of MTG or 7HMG produced the 100% MPE as well as respective intermediate doses produced the highest MPEs (i.e., 90 and 30 minutes, respectively). For the activity-decreasing effects, only the 30-minute time point for MTG was eligible for the calculation of ED50 (95% CIs) value. Each sample size is four mice per sex per time points. The potency ratio was derived from the ED50 value for MTG divided by that for 7HMG. To obtain the ED50 (95% CIs) values, molecular weights for MTG and 7HMG used were 398.5 and 414.2 g/mol, respectively.
As with MTG, 7HMG dose- and time-dependently produced significant increases in MPE (Fig. 2, top right; Supplemental Table 4); however, the duration of action of 7HMG was shorter than that of MTG. For example, at 240 minutes after administration, the MPE was 94.1 ± 5.1% and 7.8 ± 4.8% for MTG and 7HMG, respectively. In contrast to MTG, there was a significant effect of sex on MPE (Supplemental Table 4). After the 30-minute time point, increases in MPE were significantly greater in males than females (Fig. 3, Supplemental Table 4). The duration of antinociceptive action of 7HMG was longer in males than females (Fig. 3). The ED50 (95% CIs) values of 7HMG to increase the MPE was 121 (114–152), 122 (111–158), and 119 (108–154) µmol/kg at 30 minutes after administration in a combination of females and males, females only, and males only (Table 1). The antinociceptive potency ratio (95% CIs) of 7HMG relative to MTG was 2.81 (1.72–3.51) (Table 1). Activity counts (Fig. 2, lower right) were only increased by 7HMG; the maximum increase in activity counts 60 minutes after administration was approximately 200 counts per minute higher in 100 mg/kg 7HMG than in 163 mg/kg MTG.
Fig. 3.

Effects of sex on the antinociceptive effects of 7HMG. Abscissae: Baseline and elapsed time after oral administration in minutes. Left ordinates: Baseline hotplate response latency in seconds. Right ordinates: Percentage of MPE. Left, middle, and right panels: 32, 56, and 100 mg/kg 7HMG, respectively. Females (open diamonds) and males (filled squares). Each point represents the mean ± SEM. The hotplate was maintained at 52 ± 0.1°C. After the baseline measurement, one of the 7HMG doses was administered PO. Each gray symbol indicates a significant difference from vehicle per corresponding time point. See Supplemental Table 3 for details of statistical analyses. Note that the antinociceptive effects of 7HMG lasted longer in males than females.
Naltrexone, a MOR antagonist, was used to assess a pharmacological mechanism underlying the effects of MTG and 7HMG (Fig. 4). Naltrexone fully antagonized the antinociceptive effects of 293 mg/kg MTG (Fig. 4, upper left; Supplemental Table 4). There were significant effects of sex alone and a combination of sex with dose on MPE (Supplemental Table 4). However, there was no significant interaction of dose with time (Supplemental Table 4). Thus, there is no gray symbol in percentage of MPEs (Supplemental Table 4). Naltrexone also fully antagonized the decrease in activity counts 30 minutes after administration of 100 mg/kg 7HMG (Fig. 4, lower left; Supplemental Table 4). There was a significant effect of sex on activity counts; however, there was no significant interaction of sex with dose or time (Supplemental Table 4). As with MTG, naltrexone fully antagonized the increases in MPE and activity counts after administration of 100 mg/kg 7HMG regardless of sex (Figs. 4 and 5, lower left; Supplemental Table 4).
Analysis of Pharmacokinetic Data.
The in vitro microsomal half-lives of MTG and 7HMG were determined after 60 minutes incubation with male and female MLM. The microsomal stability of MTG was found to be greater in females (0.52 ± 0.01 hour) than males (0.44 ± 0.01 hour). The metabolic stability of 7HMG was similar in males and females (0.94 ± 0.01 and 0.97 ± 0.03, respectively). There was no conversion of 7HMG to MTG observed after 60 minutes incubation of 7HMG with male or female MLM. The percent of 7HMG formed after incubation of MTG in MLM was 12 and 10 in males and females, respectively.
All plasma and tissue samples after a single oral administration of MTG 165 mg/kg were analyzed for MTG and 7HMG content. The tissue concentrations were corrected for residual blood volume and once corrected, mean plasma/tissue concentration-time curves were constructed for each compound of interest as seen in Fig. 6 and Supplemental Fig. 1. The results of the noncompartmental analysis for brain and plasma MTG and 7HMG concentration after a 165 mg/kg dose of MTG are shown in Table 3. MTG was distributed in the following order: liver > kidney > lung > spleen > brain (Table 3, Supplemental Table 5). 7HMG after MTG administration was distributed in decreasing order of liver > kidney > spleen > lung > brain, similar to MTG distribution with a marked difference in lung concentrations (Table 2, Supplemental Table 5). The organ to plasma exposure ratios for MTG and 7HMG in the lung were 15.9 and 1.5, respectively (Supplemental Table 5).
Fig. 6.

Mean plasma (µg/mL) and brain (µg/g of wet tissue) concentration-time curves for MTG (A) and 7HMG (B) after a single oral dose of 165 mg/kg MTG in male and female mice. Each symbol represents the mean (N = 4), and error bars represent the SD.
TABLE 3.
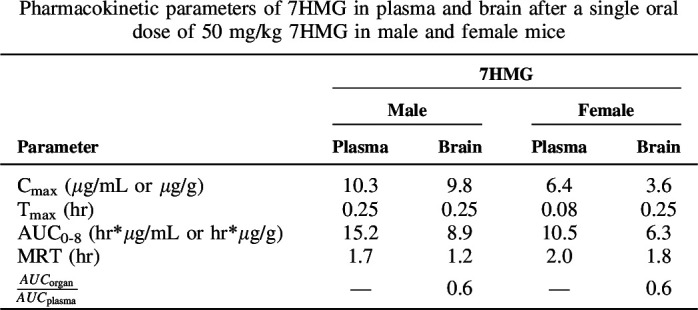
TABLE 2.

Figure 7 and Table 3 show the mean plasma and brain concentration-time curves and the results of the noncompartmental analysis for 7HMG after a single oral dose of 50 mg/kg. 7HMG was distributed in the following descending order: liver > kidney > spleen > lung > brain (Fig. 7, Supplemental Fig. 2). The tissue distribution followed the same order as that seen for 7HMG distribution after the MTG dose (Supplemental Table 6).
Fig. 7.

Mean plasma (µg/mL) and brain (µg/g of wet tissue) concentration-time curves for 7HMG after a single oral dose of 50 mg/kg MTG in male and female mice. Each symbol represents the mean (N = 4), and error bars represent the SD.
PK/PD Modeling.
For MTG, the PK/PD relationship was best characterized as a direct-link model where the effect (E) is determined using the Hill equation:
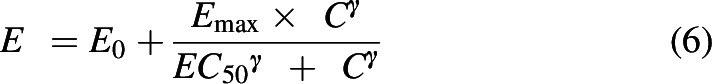 |
where E0 is the baseline effect after vehicle administration, Emax is the maximum effect, EC50 is the concentration at which 50% of the maximum effect is seen, and C is the concentration of compound (Derendorf and Meibohm, 1999). The factor gamma (γ) accounts for the sigmoid nature of the drug effect. The parameters of the models developed for MTG and 7HMG along with the likelihood distribution and error for both can be seen in Supplemental Table 7. The plots describing the goodness of fit of each model can be seen in Supplemental Fig. 3. The PK/PD model for MTG was a linear, two-compartment pharmacokinetic model with a direct pharmacodynamic effect. Sex was added as a covariate for clearance. The error model used was constant for MPE and proportional for plasma concentration. The predicted effect versus time were plotted for doses of 92, 163, and 293 mg/kg MTG, and this was compared with the observed effect values (Fig. 8A).
Fig. 8.
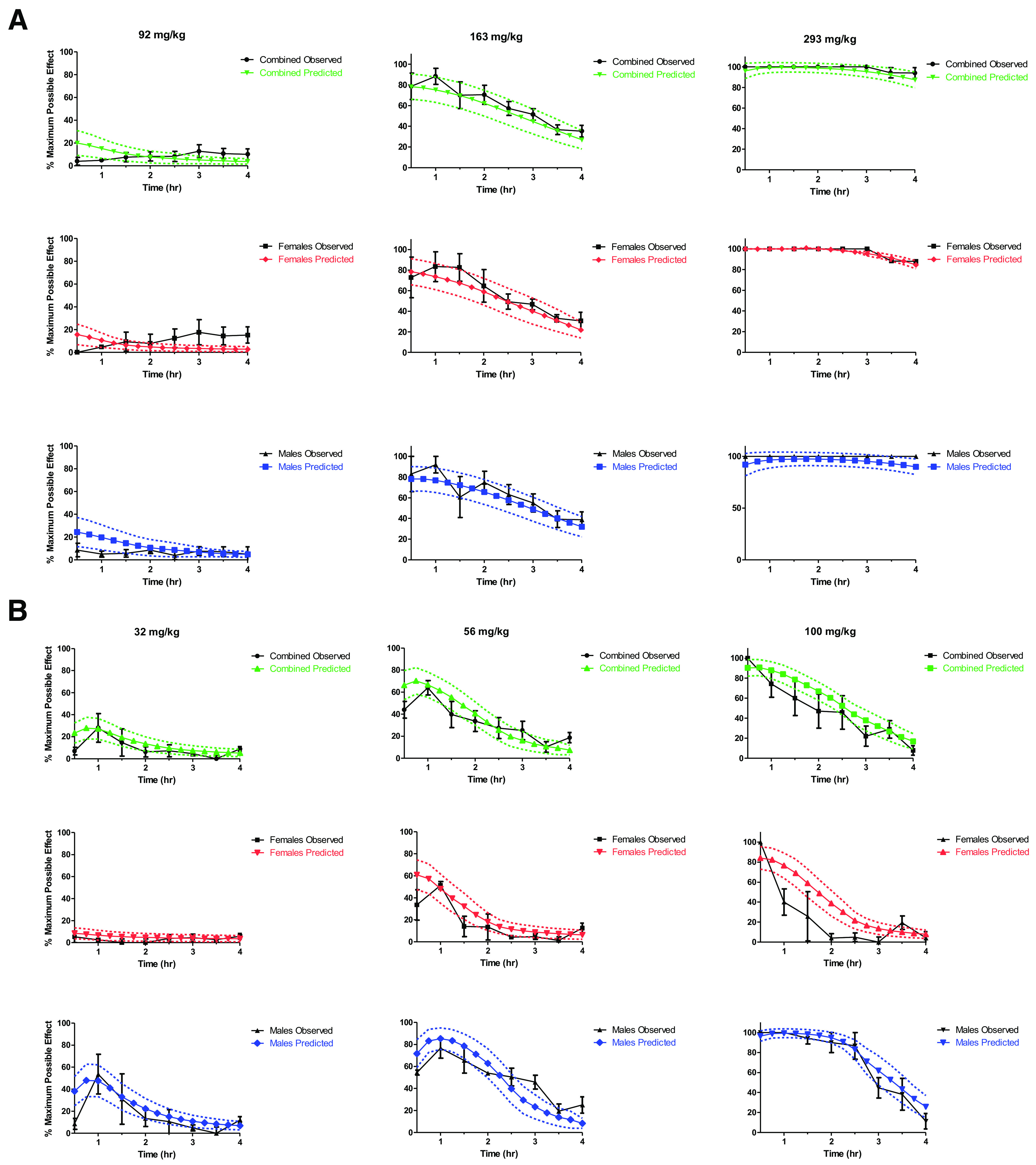
Simulation of effect versus time using the developed PK/PD model for doses of 92, 163, and 293 mg/kg MTG (A) and 32, 56, and 100 mg/kg 7HMG (B). Predicted error (dashed lines) represents the 95% CI (N = 25 subjects per sex, per simulation). Error bars on observations represent the SEM (N = 4 per time point).
The model that was developed for 7HMG was a linear one-compartment model with an effect compartment equilibration rate constant to describe the time delay between the measured concentrations and the observed effect. This changes the Hill equation to:
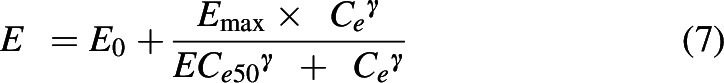 |
where Ce is the concentration in the effect compartment (Holford and Sheiner, 1981). Sex was added as a covariate for γ, the concentration to produce 50% of the maximum effect, the effect compartment equilibration rate constant, and the volume of distribution. The error model used for 7HMG was constant for MPE and proportional for plasma concentration. Meanwhile, the predicted effect versus time for 32, 56, and 100 mg/kg 7HMG was also compared with the observed values to validate the model (Fig. 8B). For each, the model with the lowest -2 log likelihood and least error was selected as the best fit to describe the observed data. The interindividual variability of both models was found to be high, but this can be attributed to the small sample sizes. Due to this, the population PK/PD model developed for MTG and 7HMG will be useful to provide a qualitative assessment of effect after different doses, but quantitative assessments will require more input parameters to further validate the model.
Discussion
The primary goal of this study was to assess the contribution of 7HMG to the antinociceptive effects of MTG after oral administration of MTG in mice. When individually administered orally based on ED50 values, both MTG and 7HMG significantly increased the hotplate response latency at doses of 163 mg/kg and 56 mg/kg, respectively. The apparent increases in hotplate response latency were not due to nonspecific behavioral disruption because the increases in hotplate response latency were present without decreases in locomotor activity counts. Based on the antinociceptive potency ratios, 7HMG was 2.8-fold more potent than MTG to produce the antinociceptive effects. Using the antinociceptive doses of MTG and 7HMG, the pharmacokinetic studies were conducted. The pharmacokinetic studies found that the concentration of 7HMG present in the brain as a metabolite of MTG (Cmax lower than concentrations found 4 hours post equianalgesic 7HMG dose) cannot account for the antinociception observed after MTG administration. Interestingly, the antinociceptive effects of MTG were fully reversed by the opioid receptor agonist naltrexone, indicating that the effect of MTG is in part mediated by MOR despite its lower affinity for the receptor.
The results of the tissue distribution analysis indicate that MTG is a perfusion limited compound as the calculated concentrations followed the blood flow to organs in mice (liver > kidney > brain > lung > spleen). The exception was the brain, which can be explained by the selectivity of the blood brain barrier (BBB). The organ to plasma exposure ratio for MTG in the brain was 2.2 and 1.8 in males and females, respectively, indicating that MTG has high BBB penetration. MTG was also rapidly distributed into the brain, as well as other organs, with time to the maximum concentration of 0.25 to 0.5 hour for all organs. The mean residence time (MRT), the average amount of time a molecule resides in the body, of MTG was 4.1–5.5 hours, indicating there is no accumulation of MTG after a single oral dose.
The metabolite to parent exposure after MTG administration was 2%–9% in all organs except the lungs where it was only 0.3%. The MRT of 7HMG after MTG administration was 2.4–3.1 hours. The Cmax of 7HMG achieved in the brain as a metabolite was 0.6–0.7 µg/g with an overall exposure of 1.7–2.2 hours*µg/g. Also, 7HMG as a metabolite showed adequate BBB penetration with a brain to plasma ratio of 0.6 in females and 1.5 in males. The brain to plasma ratios demonstrated in this study are higher than the previously reported values, but this difference may be explained by a single time point versus the entire time course of 7HMG as well as alternate routes of administration. The previously reported brain to plasma ratios of MTG and 7HMG after subcutaneous administration of MTG at two time points (15 and 60 minutes) were 1.0 and 0.2, respectively, in 129S1 mice (Kruegel et al., 2019).
It has been reported in the literature that 7HMG metabolizes to MTG (Manda et al., 2014), but no quantifiable MTG was found in any samples after oral administration of 50 mg/kg 7HMG, indicating that the quantifiable conversion of 7HMG to MTG did not occur in mice in this study. These results are supported by the in vitro microsomal data as well as the results of a PK study in female beagle dogs (Maxwell et al., 2021). The MRT of 7HMG after 7HMG oral dose was 1.2–2.0 hours in the various organs, which is less than the MRT of 7HMG after MTG administration. Independent of MTG, 7HMG has a shorter residence time in the body, but when present as a metabolite of MTG, it remains in the tissues longer, indicating that the rate of elimination of 7HMG as a metabolite is governed by its formation.
Compared with MTG, 7HMG is less perfused into tissues, which may be due to the differences in lipophilicity of the compounds, resulting in the lower ability of 7HMG than that of MTG to cross through lipid bilayers. The reported logPOct values for MTG and 7HMG were 4.1 and 1.7, respectively (Yusof et al., 2019). The organ to plasma ratios of 7HMG were 2.5- to 5.0-fold lower after a 50 mg/kg 7HMG dose as compared with a 165 mg/kg MTG dose, suggesting that MTG may have some influence on the ability of 7HMG to move freely into and out of tissues and/or MTG metabolism occurs in these organs. The MRT of each compound coincides with the functional results where the peak antinociceptive effects of MTG were present up to 4 hours post dose (MRT 4.1–5.5 hours), whereas 7HMG effects were only seen up to 1–2 hours post dose (MRT 1.2–2.0 hour). These results contrast with the in vitro microsomal data generated where 7HMG was found to be twice as stable as MTG (approximately 1 hour versus 0.5 hour, respectively) in both male and female MLM. This would suggest that 7HMG undergoes extrahepatic elimination and/or phase II metabolism. The Cmax of 7HMG achieved in the brain after administration of 50 mg/kg 7HMG was 9.8 µg/g in males and 3.6 µg/g in females, 16- and 5-fold higher than the Cmax as a metabolite of MTG.
Behavioral differences were noted in mice dosed either with MTG or 7HMG. In the MTG dosed group, no observable behaviors were noted, whereas in the 7HMG dosed group the Straub tail response was seen up to 4 hours post dose. This response has been reported to be related to MOR 2 activation, which is associated with the adverse effects associated with opioid abuse (Pasternak, 1988; Nath et al., 1994). The same behavior was not seen even after the 294 mg/kg MTG dose, indicating that the amount of 7HMG generated as a metabolite of MTG is either insufficient to produce the response or MTG may antagonize the 7HMG-induced Straub tail response. This is supported by in vitro evidence, as MTG antagonized activity of 7HMG at human MOR using a [35S] guanosine 5′-O-[γ-thio]triphosphate functional assay (Obeng et al., 2021).
Females had lower exposure of both MTG and 7HMG, which helps to explain their lower overall MPE results in behavioral assays. Sex differences in pharmacokinetics of animal models are common (Czerniak, 2001) but do not always lead to behavioral differences. For example, with MTG no statistically significant differences were observed in MPE after MTG administration despite a 2-fold difference in overall brain exposure. Meanwhile, a 1.4-fold difference in exposure (in both brain and plasma) of 7HMG after 7HMG administration led to statistically significant differences in MPE > 30 minutes after administration between males and females. Using the PK/PD model to simulate 250 subjects receiving the ED50, the EC50 of 7HMG was estimated to be 5.3 µg/mL for females, whereas for males it was only 3.2 µg/mL. This difference in sensitivity between sexes has been seen in rodent models of pain when researching typical opioid agonists like morphine (Craft et al., 2001). Specifically, in tests of hotplate antinociception in mice, males have been found to be more sensitive to morphine than females (Kavaliers and Innes, 1990; Kavaliers and Innes, 1992). Suggested mechanisms of sex variations include hormonal, genetic, and/or receptor density-based differences (Loyd et al., 2008; Lee and Ho, 2013; Liu et al., 2018). The differences in response to MTG and 7HMG by male versus female mice seen in this study further the assertion that 7HMG acts as a typical MOR agonist, whereas MTG acts on MOR atypically.
It is important to note that all the pharmacokinetic values in the present study are reported as the total concentration of the compound (fraction bound plus fraction unbound), whereas only the fraction unbound is available for pharmacological action. Previously, the unbound fraction of MTG and 7HMG in the rat brain has been reported as 0.027 ± 0.002 and 0.26 ± 0.04, respectively, whereas the unbound fraction in other tissues is not available in the literature (Yusof et al., 2019).
In summary, here we showed that the amount of 7HMG present in the brain after MTG administration alone cannot account for the functional activity of MTG, refuting an earlier study (Kruegel et al., 2019). In the present study, the exposure of 7HMG in the brain after an antinociceptive dose of MTG was 4.0-fold and 3.7-fold less in males and females, respectively, than that after an antinociceptive dose of 7HMG. The Cmax of 7HMG in the brain achieved as a metabolite of MTG was similar to concentrations seen 4 hours post dose of 7HMG where MPE was < 10%.
Conclusions
The results of this study give important information toward understanding the overall pharmacology of MTG. The 7HMG exposure generated from the metabolism of MTG administration cannot solely be responsible for the analgesic effects of MTG. Species differences in metabolism, brain penetration, and free fraction of drug must all be considered when translating these results to higher order species. Although these results provide valuable information about a preclinical rodent model, species differences in the fraction of MTG metabolized to 7HMG have been reported. In female beagle dogs, the AUC ratio of 7HMG to MTG was 13%, 1.4-fold greater than that found in female mice indicating that concentrations of free 7HMG may be greater in dogs and have more potential to influence the overall pharmacology of MTG (Maxwell et al., 2020). Combining the population PK/PD approach with physiologically based pharmacokinetic modeling that considers the aforementioned species and sex differences uncovered in this study will be helpful in translating preclinical results into first in human dose recommendations while also contributing to the continued evaluation of MTG.
Acknowledgments
The authors would like to acknowledge Dr. Bonnie A. Avery and her contributions to the success of this work group.
Abbreviations
- BBB
blood brain barrier
- %Bias
percent bias
- CI
confidence interval
- 7HMG
7-hydroxymitragynine
- IS
internal standard
- MLM
mouse liver microsome
- MOR
μ-opioid receptor
- MPE
maximum possible effect
- MRT
mean residence time
- MTG
mitragynine
- PK/PD
pharmacokinetic-pharmacodynamic
- QC
quality control
- RSD
relative standard deviation
- UPLC-MS/MS
ultra-performance liquid chromatography tandem mass spectrometry
Authorship Contributions
Participated in research design: Berthold, Kamble, Kanumuri, Sharma, Hiranita, McMahon, McCurdy.
Conducted experiments: Berthold, Kamble, Kanumuri, Kuntz, Senetra, Restrepo, Patel, Ho, Sharma.
Contributed new reagents or analytic tools: Mottinelli, León, McCurdy.
Performed data analysis: Berthold, Kamble, Kanumuri, Kuntz, Hiranita, Sharma.
Wrote or contributed to the writing of the manuscript: Berthold, Kamble, Kanumuri, Senetra, Sharma, Kuntz, Mottinelli, León, Hiranita, McMahon, McCurdy.
Footnotes
No author has an actual or perceived conflict of interest with the contents of this article.
This work was supported by National Institutes of Health National Institute of Drug Abuse [Grants UG3 DA048353, UH3 DA048353, R01 DA047855]; the University of Florida Clinical and Translational Science Institute, which is supported in part by National Institutes of Health National Center for Advancing Translational Sciences [UL1TR001427]; and the University of Florida Foundation and University of Florida Department of Pharmacodynamics Funding.
Current affiliation: Department of Drug Discovery and Biomedical Sciences, College of Pharmacy, University of South Carolina, Columbia, South Carolina
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Avery BA, Boddu SP, Sharma A, Furr EB, Leon F, Cutler SJ, McCurdy CR (2019) Comparative pharmacokinetics of mitragynine after oral administration of Mitragyna speciosa (kratom) leaf extracts in rats. Planta Med 85:340–346. [DOI] [PubMed] [Google Scholar]
- Behnood-Rod A, Chellian R, Wilson R, Hiranita T, Sharma A, Leon F, McCurdy CR, McMahon LR, Bruijnzeel AW (2020) Evaluation of the rewarding effects of mitragynine and 7- hydroxymitragynine in an intracranial self-stimulation procedure in male and female rats. Drug Alcohol Depend 215:108235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 13:407–484. [DOI] [PubMed] [Google Scholar]
- Chear NJLeón FSharma AKanumuri SRRZwolinski GAbboud KASingh DRestrepo LFPatel AHiranita T, et al. (2021) Exploring the chemistry of alkaloids from Malaysian Mitragyna speciosa (kratom) and the role of oxindoles on human opioid receptors. J Nat Prod 84:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft RM, Tseng AH, McNiel DM, Furness MS, Rice KC (2001) Receptor-selective antagonism of opioid antinociception in female versus male rats. Behav Pharmacol 12:591–602. [DOI] [PubMed] [Google Scholar]
- Czerniak R (2001) Gender-based differences in pharmacokinetics in laboratory animal models. Int J Toxicol 20:161–163. [DOI] [PubMed] [Google Scholar]
- Derendorf H, Meibohm B (1999) Modeling of pharmacokinetic/pharmacodynamic (PK/PD) relationships: concepts and perspectives. Pharm Res 16:176–185. [DOI] [PubMed] [Google Scholar]
- Eastlack SC, Cornett EM, Kaye AD (2020) Kratom-pharmacology, clinical implications, and outlook: a comprehensive review. Pain Ther 9:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis CR, Racz R, Kruhlak NL, Kim MT, Zakharov AV, Southall N, Hawkins EG, Burkhart K, Strauss DG, Stavitskaya L (2020) Evaluating kratom alkaloids using PHASE. PLoS One 15:e0229646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA (2018) Bioanalytical Method Validation Guidance for Industry. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research, Silver Spring, MD. [Google Scholar]
- FDA (2021) FDA Adverse Events Reporting System (FAERS). U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research, Silver Spring, MD. [Google Scholar]
- Giudicelli C, Dricot E, Moati F, Strolin-Benedetti M, Giudicelli JF (2004) Is it important to correct apparent drug tissue concentrations for blood contamination in the dog? Fundam Clin Pharmacol 18:281–286. [DOI] [PubMed] [Google Scholar]
- Hemby SE, McIntosh S, Leon F, Cutler SJ, McCurdy CR (2019) Abuse liability and therapeutic potential of the Mitragyna speciosa (kratom) alkaloids mitragynine and 7-hydroxymitragynine. Addict Biol 24:874–885. [DOI] [PubMed] [Google Scholar]
- Hiranita TLeon FFelix JSRestrepo LFReeves MEPennington AEObeng SAvery BAMcCurdy CRMcMahon LR, et al. (2019) The effects of mitragynine and morphine on schedule-controlled responding and antinociception in rats. Psychopharmacology (Berl) 236:2725–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holford NH, Sheiner LB (1981) Understanding the dose-effect relationship: clinical application of pharmacokinetic-pharmacodynamic models. Clin Pharmacokinet 6:429–453. [DOI] [PubMed] [Google Scholar]
- Kamble SH, Sharma A, King TI, León F, McCurdy CR, Avery BA (2019) Metabolite profiling and identification of enzymes responsible for the metabolism of mitragynine, the major alkaloid of Mitragyna speciosa (kratom). Xenobiotica 49:1279–1288. [DOI] [PubMed] [Google Scholar]
- Kavaliers M, Innes D (1992) Sex differences in the effects of neuropeptide FF and IgG from neuropeptide FF on morphine- and stress-induced analgesia. Peptides 13:603–607. [DOI] [PubMed] [Google Scholar]
- Kavaliers M, Innes DG (1990) Developmental changes in opiate-induced analgesia in deer mice: sex and population differences. Brain Res 516:326–331. [DOI] [PubMed] [Google Scholar]
- Khor SP, Mayersohn M (1991) Potential error in the measurement of tissue to blood distribution coefficients in physiological pharmacokinetic modeling. Residual tissue blood. I. Theoretical considerations. Drug Metab Dispos 19:478–485. [PubMed] [Google Scholar]
- Kruegel ACUprety RGrinnell SGLangreck CPekarskaya EALe Rouzic VAnsonoff MGassaway MMPintar JEPasternak GW, et al. (2019) 7-Hydroxymitragynine is an active metabolite of mitragynine and a key mediator of its analgesic effects. ACS Cent Sci 5:992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Ho IK (2013) Sex differences in opioid analgesia and addiction: interactions among opioid receptors and estrogen receptors. Mol Pain 9:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Zhang H, Qin F, Wang Q, Sun Q, Xie S, Wang Q, Tang Z, Lu Z (2018) Sex associated differential expressions of the alternatively spliced variants mRNA of OPRM1 in brain regions of C57BL/6 mouse. Cell Physiol Biochem 50:1441–1459. [DOI] [PubMed] [Google Scholar]
- Loyd DR, Wang X, Murphy AZ (2008) Sex differences in micro-opioid receptor expression in the rat midbrain periaqueductal gray are essential for eliciting sex differences in morphine analgesia. J Neurosci 28:14007–14017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydecker AG, Sharma A, McCurdy CR, Avery BA, Babu KM, Boyer EW (2016) Suspected adulteration of commercial kratom products with 7-hydroxymitragynine. J Med Toxicol 12:341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manda VK, Avula B, Ali Z, Khan IA, Walker LA, Khan SI (2014) Evaluation of in vitro absorption, distribution, metabolism, and excretion (ADME) properties of mitragynine, 7-hydroxymitragynine, and mitraphylline. Planta Med 80:568–576. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Mizowaki M, Suchitra T, Murakami Y, Takayama H, Sakai S, Aimi N, Watanabe H (1996) Central antinociceptive effects of mitragynine in mice: contribution of descending noradrenergic and serotonergic systems. Eur J Pharmacol 317:75–81. [DOI] [PubMed] [Google Scholar]
- Maxwell EA, King TI, Kamble SH, Raju KSR, Berthold EC, León F, Avery BA, McMahon LR, McCurdy CR, Sharma A (2020) Pharmacokinetics and safety of mitragynine in beagle dogs. Planta Med 86:1278–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell EA, King TI, Kamble SH, Raju KSR, Berthold EC, León F, Hampson A, McMahon LR, McCurdy CR, Sharma A (2021) Oral pharmacokinetics in beagle dogs of the mitragynine metabolite, 7-hydroxymitragynine. Eur J Drug Metab Pharmacokinet 46:459–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath C, Gupta MB, Patnaik GK, Dhawan KN (1994) Morphine-induced straub tail response: mediated by central mu2-opioid receptor. Eur J Pharmacol 263:203–205. [DOI] [PubMed] [Google Scholar]
- Obeng SKamble SHReeves MERestrepo LFPatel ABehnke MChear NJRamanathan SSharma ALeón F, et al. (2020) Investigation of the adrenergic and opioid binding affinities, metabolic stability, plasma protein binding properties, and functional effects of selected indole-based kratom alkaloids. J Med Chem 63:433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng SWilkerson JLLeón FReeves MERestrepo LFGamez-Jimenez LRPatel APennington AETaylor VAHo NP, et al. (2021) Pharmacological comparison of mitragynine and 7-hydroxymitragynine: in vitro affinity and efficacy for μ-opioid receptor and opioid-like behavioral effects in rats. J Pharmacol Exp Ther 376:410–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak GW (1988) Multiple morphine and enkephalin receptors and the relief of pain. JAMA 259:1362–1367. [PubMed] [Google Scholar]
- Sharma A, Kamble SH, León F, Chear NJ, King TI, Berthold EC, Ramanathan S, McCurdy CR, Avery BA (2019) Simultaneous quantification of ten key kratom alkaloids in Mitragyna speciosa leaf extracts and commercial products by ultra-performance liquid chromatography-tandem mass spectrometry. Drug Test Anal 11:1162–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, McCurdy CR (2021) Assessing the therapeutic potential and toxicity of Mitragyna speciosa in opioid use disorder. Expert Opin Drug Metab Toxicol 17:255–257. [DOI] [PubMed] [Google Scholar]
- Singh D, Yeou Chear NJ, Narayanan S, Leon F, Sharma A, McCurdy CR, Avery BA, Balasingam V (2020) Patterns and reasons for kratom (Mitragyna speciosa) use among current and former opioid poly-drug users. J Ethnopharmacol 249:112462. [DOI] [PubMed] [Google Scholar]
- Snedecor GW, Cochran WG (1967) Statistical Methods, Iowa State University Press, Ames. [Google Scholar]
- Tallarida RJ (2002) The interaction index: a measure of drug synergism. Pain 98:163–168. [DOI] [PubMed] [Google Scholar]
- Tanda G, Mereu M, Hiranita T, Quarterman JC, Coggiano M, Katz JL (2016) Lack of specific unvolvement of (+)-naloxone and (+)-naltrexone on the reinforcing and neurochemical effects of cocaine and opioids. Neuropsychopharmacology 41:2772–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Váradi AMarrone GFPalmer TCNarayan ASzabó MRLe Rouzic VGrinnell SGSubrath JJWarner EKalra S, et al. (2016) Mitragynine/corynantheidine pseudoindoxyls as opioid analgesics with mu agonism and delta antagonism, which do not recruit β-arrestin-2. J Med Chem 59:8381–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltri C, Grundmann O (2019) Current perspectives on the impact of kratom use. Subst Abuse Rehabil 10:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Li S, Yang H, Lee F, Wu JT, Qian MG (2005) A novel liquid chromatography/tandem mass spectrometry based depletion method for measuring red blood cell partitioning of pharmaceutical compounds in drug discovery. Rapid Commun Mass Spectrom 19:250–254. [DOI] [PubMed] [Google Scholar]
- Yue K, Kopajtic TA, Katz JL (2018) Abuse liability of mitragynine assessed with a self-administration procedure in rats. Psychopharmacology (Berl) 235:2823–2829. [DOI] [PubMed] [Google Scholar]
- Yusof SR, Mohd Uzid M, Teh EH, Hanapi NA, Mohideen M, Mohamad Arshad AS, Mordi MN, Loryan I, Hammarlund-Udenaes M (2019) Rate and extent of mitragynine and 7-hydroxymitragynine blood-brain barrier transport and their intra-brain distribution: the missing link in pharmacodynamic studies. Addict Biol 24:935–945. [DOI] [PubMed] [Google Scholar]