

Abstract
Duocarmycin natural products are promising anticancer cytotoxins but too potent for systemic use. Re-engineering of the duocarmycin scaffold has enabled the discovery of prodrugs designed for bioactivation by tissue-specific cytochrome P450 (P450) enzymes. Lead prodrugs bioactivated by both P450 isoforms CYP1A1 and CYP2W1 have shown promising results in xenograft studies; however, to fully understand the potential of these agents it is desirable to compare dual-targeting compounds with isoform-selective analogs. Such redesign requires insight into the molecular interactions with these P450 enzymes. Herein binding and metabolism of the individual stereoisomers of the indole-based duocarmycin prodrug ICT2700 and a nontoxic benzofuran analog ICT2726 were evaluated with CYP1A1 and CYP2W1, revealing differences exploitable for drug design. Although enantiomers of both compounds bound to and were metabolized by CYP1A1, the stereochemistry of the chloromethyl fragment was critical for CYP2W1 interactions. CYP2W1 differentially binds the S enantiomer of ICT2726, and its metabolite profile could potentially be used as a biomarker to identify CYP2W1 functional activity. In contrast to benzofuran-based ICT2726, CYP2W1 differentially binds the R isomer of the indole-based ICT2700 over the S stereoisomer. Thus the ICT2700 R configuration warrants further investigation as a scaffold to favor CYP2W1-selective bioactivation. Furthermore, structures of both duocarmycin S enantiomers with CYP1A1 reveal orientations correlating with nontoxic metabolites, and further drug design optimization could lead to a decrease of CYP1A1 bioactivation. Overall, distinctive structural features present in the two P450 active sites can be useful for improving P450—and thus tissue-selective—bioactivation.
SIGNIFICANCE STATEMENT
Prodrug versions of the natural product duocarmycin can be metabolized by human tissue-specific cytochrome P450 (P450) enzymes 1A1 and 2W1 to form an ultrapotent cytotoxin and/or high affinity 2W1 substrates to potentially probe functional activity in situ. The current work defines the binding and metabolism by both P450 enzymes to support the design of duocarmycins selectively activated by only one human P450 enzyme.
Introduction
Human cytochrome P450 (P450) enzymes are broadly recognized for their capacity to convert xenobiotics, including drugs, into metabolites more readily cleared from the body. However, P450 metabolism activates some foreign chemicals to reactive intermediates or toxins. Although these can have significant adverse effects and/or initiate disease, this capacity can be alternatively used advantageously to activate prodrugs in situ. Examples include many US Food and Drug Administration–approved anticancer compounds such as the DNA-alkylating cyclophosphamide/ifosfamide and dacarbazine/procarbazine compounds, the thymidylate synthase inhibitor tegafur, the nonsteroidal antiandrogen flutamide, the selective estrogen receptor modulator tamoxifen, the bioreductive topoisomerase II inhibitor AQ4N, and many others (see Sneha et al., 2021).
The ultrapotent cytotoxic duocarmycin natural products have significant anticancer potential, but their narrow therapeutic index and lack of tumor selectivity prevents systemic use (Jukes et al., 2021). Novel duocarmycin prodrugs based on the seco-duocarmycin scaffold have been developed (Pors et al., 2011; Sheldrake et al., 2013; Travica et al., 2013) that are bioactivated by two human cytochrome P450 enzymes: CYP1A1 and CYP2W1. CYP1A1 is an inducible extrahepatic enzyme present primarily in lung, placenta, and endothelial cells of the skin, intestine, and bladder with significant variation between tissues and individuals (Shimada et al., 1992; Lang et al., 2019). For tumors with high CYP1A1 levels, such as bladder tumors (Sutherland et al., 2013), CYP1A1-activated therapeutics could be clinically useful, but physicians would need to manage side effects due to bioactivation by low CYP1A1 levels in healthy tissues.
In contrast, CYP2W1, a poorly understood P450, is selectively expressed in colon cancer tissues (Karlgren et al., 2006; Gomez et al., 2007; Karlgren and Ingelman-Sundberg, 2007; Edler et al., 2009; Gomez et al., 2010; Stenstedt et al., 2014), where its ability to activate prodrugs could be a promising therapy for colorectal tumors. A duocarmycin-based prodrug was shown to arrest tumor growth in a CYP2W1-expressing xenograft model without host toxicity (Travica et al., 2013), suggesting such a prodrug approach exploiting tumor CYP2W1 holds promise for selective colon tumor targeting.
Both CYP1A1 and CYP2W1 hydroxylate several duocarmycin bioprecursors at the critical position on the chloromethyl indoline (Fig. 1) (Sheldrake et al., 2013). The hydroxylated seco-duocarmycin undergoes spontaneous spirocyclization, releasing the chlorine and generating a highly constrained cyclopropane (Pors et al., 2011). The indole at the opposite end of duocarmycins facilitates sequence-selective DNA binding, so that the newly formed cyclopropane is appropriately located to react with N3 of adenine. Such DNA alkylation is cytotoxic. Thus, site-specific hydroxylation is essential for P450 activation of duocarmycin-based bioprecursors.
Fig. 1.
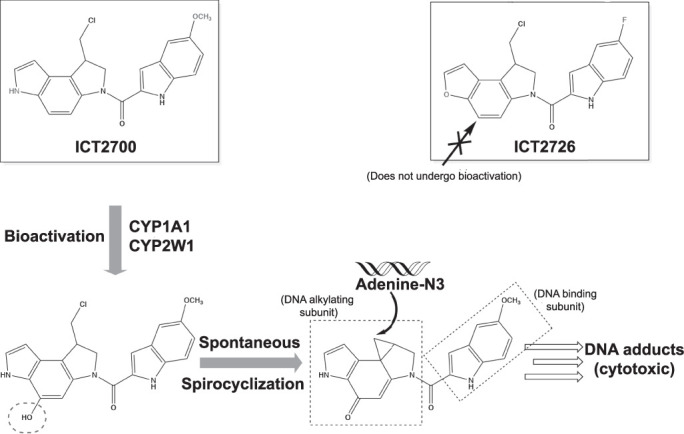
Duocarmycin analogs ICT2700 and ICT2726 (shown in boxes, differences colored in red). The ICT2700 analog has been reported to undergo bioactivation by CYP1A1 and CYP2W1 via a specific hydroxylation (red dotted oval) on the chloromethyl indoline substructure (Travica et al., 2013). Spontaneous spirocyclization occurs producing a reactive cyclopropane moiety. DNA adenine bases can be alkylated with this reactive group leading to cytotoxic DNA adducts. The ICT2726 analog does not undergo bioactivation. Instead, this analog can potentially serve as a biomarker for CYP2W1 activity due to the generation of a unique nontoxic metabolite specific to CYP2W1 (Sheldrake et al., 2013).
The specificity of hydroxylation depends on ligand orientation in the P450 active site with the desired site of metabolism adjacent to the heme iron. No structures are available for CYP2W1, but several exist for CYP1A1. The initial CYP1A1 structure with α-naphthoflavone revealed a highly planar enclosed active site (Walsh et al., 2013). Subsequent structures have been solved with various ligands, demonstrating how the CYP1A1 active site changes to accommodate larger and nonplanar compounds (Bart and Scott, 2018; Bart et al., 2020), but do not suggest how seco-duocarmycin compounds might be redesigned for increased selectivity for CYP1A1 or CYP2W1. The chemical scaffolds of ICT2700 and ICT2726 consist of an indole connected to another indole or a benzofuran, respectively (Fig. 1). Although each planar fused aromatic system would correlate well to a highly planar and narrow CYP1A1 active site individually, the linker between them does not constrain the two aromatic heterocyclic systems to the same plane. The chloromethyl arm also deviates from planarity and could sterically clash with a narrow active site. Although flexibility has been observed in certain structural elements in CYP1A1, it is not clear how these duocarmycin-type ligands would be accommodated.
To optimize the clinical utility, the seco-duocarmycins should be modified for more selective activation, preferably by CYP2W1 due to its exclusive expression in tumor tissue. Previous studies (Sheldrake et al., 2013) revealed functionalization of the DNA-binding motif that improved CYP1A1 bioactivation over CYP2W1, but structural changes favoring CYP2W1 activation are unknown. Such engineering has been impaired by the absence of information about how CYP1A1 and CYP2W1 interact with duocarmycin bioprecursors. Thus, the current study characterized binding and metabolism of duocarmycin-based compounds with both enzymes. Of specific interest are the prodrug duocarmycin ICT2700 and a potential biomarker version, ICT2726, that may be useful to identify CYP2W1-active cells prior to treatment. The current work examined both enantiomers of both compounds to CYP1A1 and CYP2W1, revealing key interactions that could be used to design second-generation duocarmycin bioprecursors with improved P450 selectivity and bioactivation.
Materials and Methods
Synthesis and Chromatographic Resolution of ICT2700 and ICT2726 Stereoisomers.
Racemic ICT2700 and ICT2726 were synthesized as previously described (Sheldrake et al., 2013; Travica et al., 2013). These compounds were dissolved in 99:1 dichloromethane/ethanol at the concentration of 4 mg/ml and 23.8 mg/ml, respectively. Enantiomers of ICT2700 and ICT2726 were resolved on a semipreparative Daicel ChiralPak ID column (10 mm × 250 mm; 5 μm) with 99:1:0.1 dichloromethane/ethanol/diethylamine elution and a flow of 2.5 ml/min.
Single crystal X-ray diffraction data on a single enantiomer of ICT2700 and of ICT2726 were collected using a Bruker X8 diffractometer with an APEX II detector and monochromatic Cu Kα radiation (λ = 1.5418 Å) at 173 K. The data were processed using Bruker SAINT. The structures were determined with SHELXT (Sheldrick, 2015) and subsequently refined with SHELXL (Sheldrick, 2008) within the program Olex2 (Dolomanov et al., 2009). Details of the crystallographic data are given in Supplemental Table 1. Crystal structures were visualized using Mercury (Macrae et al., 2008). The X-ray data for ICT2700 and ICT2726 have been deposited with the Cambridge Crystallographic Data Centre (numbers 2051115 and 2051117).
Protein Expression and Purification.
Human CYP1A1 and CYP2W1 enzymes were generated that had truncations of the N-terminal membrane-anchoring helix to increase solubility and addition of a C-terminal histidine tag to facilitate purification. The construct for CYP1A1 has been described previously (Walsh et al., 2013). For CYP2W1, a synthetic codon-optimized gene was generated in which the resulting amino acid sequence was almost the same as the 2W1 #3 construct described by Wu et al. (2006), in that residues 3–31 were replaced with KKTSSKGKL, but the C-terminal histidine tag consisted of only four histidines instead of five.
Expression and purification of CYP1A1 was performed without modification as described by Bart and Scott (2018). Expression of CYP2W1 was based on the method described for CYP1A1 (Bart and Scott, 2018) with some modifications. These included inoculation with 5 ml of starter culture per 1 L expression culture and growing in 2.8 L Fernbach flasks, a lower shaking speed of 225 rpm during cell growth and 200 rpm after induction, and addition of 5 mM imidazole to expression cultures to help stabilize CYP2W1. Purification of CYP2W1 was initially based on the CYP1A1 purification method; however, substantial modifications were made; therefore, for clarity a detailed protocol is provided below.
Purification of CYP2W1 began with thawing frozen cell pellets in a buffer consisting of 20 mM potassium phosphate, 20% (v/v) glycerol, 5 mM imidazole, with 1 mM of the serine protease inhibitor phenylmethanesulfonyl fluoride, pH 7.4. Thawed and resuspended cells were lysed by French press using a single pass with a pressure of 15,000 psi. The lysed cell suspension was then subjected to ultra-centrifugation for 30 minutes at 142,000g to isolate membranes. Pelleted membranes were gently washed twice with the lysis buffer, then resuspended by homogenization in an extraction buffer composed of 100 mM potassium phosphate, 500 mM NaCl, 20% (v/v) glycerol, 15 mM imidazole, 1% (w/v) 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (the detergent CHAPS), and 1 mM phenylmethylsulfonyl fluoride, pH 7.4. Resuspended membranes were stirred for 1 hour to promote P450 extraction from the membranes. Membranes were then pelleted by a second ultra-centrifugation using the above parameters. The clarified supernatant was loaded onto a 25 ml nickel–nitrilotriacetic acid (Ni-NTA) column (Qiagen, Germantown, MD) equilibrated with the extraction buffer. To remove unbound and nonspecifically bound proteins, four column volumes (CV) of extraction buffer followed by 10 CV of a Ni-NTA wash buffer [100 mM potassium phosphate, 100 mM NaCl, 20% (v/v) glycerol, 15 mM imidazole, 9 mM CHAPS, pH 7.4] were used. CYP2W1 was eluted using a linear gradient from Ni-NTA wash buffer to Ni-NTA elution buffer [10 mM potassium phosphate, 100 mM NaCl, 20% (v/v) glycerol, 250 mM imidazole, 9 mM CHAPS, pH 7.4] over 6 CV, with addition of 2 CV of Ni-NTA elution buffer to ensure complete elution. Ni-NTA fractions with A426/A280 > 0.8 were pooled and diluted fivefold with an ion-exchange wash buffer [10 mM potassium phosphate, 50 mM NaCl, 20% (v/v) glycerol, 1 mM EDTA, pH 7.4]. Diluted sample was then applied to two 5-ml Hi-Trap carboxymethyl-Sepharose fast-flow columns (GE Healthcare, Chicago, IL) connected in series (total 10 ml column volume). This was first washed with 10 CV of ion-exchange wash buffer; then a 6 CV linear gradient was applied transitioning from the ion-exchange wash buffer to an ion-exchange elution buffer [50 mM potassium phosphate, 500 mM NaCl, 20% (v/v) glycerol, 1 mM EDTA, pH 7.4] to elute CYP2W1. An additional 2 CV of ion-exchange elution buffer was run through the column to ensure complete elution. Eluted fractions from the CM column with A419/A280 > 1.0 were pooled and concentrated to ∼2 ml for application onto a Superdex 200 gel filtration column (GE Healthcare, Chicago, IL) run isocratically using the ion-exchange elution buffer. The major elution peak fractions with A419/A280 > 1.0 were pooled, flash-frozen in aliquots, and stored at −80°C.
The purified P450 proteins were also evaluated by SDS-PAGE to assess purity. The reduced carbon monoxide difference assay served as a method to evaluate P450 stability (Omura and Sato, 1964). Quantification of both CYP1A1 and CYP2W1 for use in ligand binding assays and crystallography was performed by UV-visible spectroscopy, using the Soret peak extinction coefficient of 100 mM−1cm−1 in the final buffer for the respective proteins plus 250 mM imidazole. The human NADPH–cytochrome P450 reductase construct hPORG3H6Δ27 (Sandee and Miller, 2011) was expressed and purified as described (Bart and Scott, 2017).
Ligand Binding Assays.
The affinities and binding modes of the duocarmycin compounds to CYP1A1 and CYP2W1 (1 µM) were measured in 2 cm tandem quartz cuvettes using the method previously described (Bart and Scott, 2018). Spectral binding data were determined in duplicate and fit to a quadratic equation used for tight binding (DeVore et al., 2009).
Crystallization, Data Collection, and Structure Determination.
CYP1A1 was co-crystallized with the seco-duocarmycin compounds (S)-ICT2700 and (S)-ICT2726 as described previously (Bart and Scott, 2018), with modifications listed below. Purified CYP1A1 was saturated with the respective duocarmycin by three iterative rounds of dilution (in ion-exchange elution buffer with 0.4 M NH4NO3) and concentration via centrifugal ultrafiltration. Ligand was added at 20 μM for (S)-ICT2726 or 100 μM for (S)-ICT2700 during each dilution. The resulting complexes were then concentrated to a final protein concentration of 20 mg/ml. Crystals of both complexes were grown at 4°C using the sitting-drop vapor diffusion method. For CYP1A1/(S)-ICT2700, crystals were grown in a 96-well plate mixing 0.75 μl CYP1A1 with 0.75 μl of crystallization solution [0.18 M sodium phosphate dibasic, 18% (w/v) polyethylene glycol 3350, 9% (v/v) glycerol, 5 mM (S)-O-methyl-serine dodecylamide hydroxhloride] against a 50 μl reservoir of the same crystallization solution. Crystals of the CYP1A1/(S)-ICT2726 complex were grown in a 24-well plate by mixing 1 μl CYP1A1, 0.2 μl 18 mM n-decyl-β-d-maltoside, and 0.8 μl crystallization solution [0.2 M sodium phosphate dibasic, 20% (w/v) polyethylene glycol 3350], equilibrated with a 100 μl reservoir of the same crystallization solution. Crystals were harvested by transferring into the respective crystallization solution containing 20% glycerol and flash cooled in liquid nitrogen. Diffraction data for the CYP1A1/(S)-ICT2700 crystal were collected on beamline 21-ID-G at the Advanced Photon Source Life Sciences Collaborative Access Team, whereas data for CYP1A1/(S)-ICT2726 were collected on beamline 9-2 at the Stanford Synchrotron Radiation Lightsource.
Data processing for the CYP1A1/(S)-ICT2700 data set was performed using HKL2000 (Otwinowski and Minor, 1997), and a structure solution was obtained by molecular replacement via Phaser (McCoy et al., 2007) with the CYP1A1/α-naphthoflavone structure as the search model (Protein Data Bank code 4I8V, molecule A), resulting in a log likelihood of 667 and translation function Z score of 27.3. For the CYP1A1/(S)-ICT2726 complex, X-ray diffraction data were processed in XDS (Kabsch, 2010) and scaled in AIMLESS (Evans and Murshudov, 2013). A molecular replacement solution for this structure was obtained from MolRep (Vagin and Teplyakov, 1997), again using chain A of the CYP1A1/α-naphthoflavone structure as the search model (contrast score of 30.59). Iterative model building and refinement of both structures were performed using COOT (Emsley et al., 2010) and PHENIX (Adams et al., 2010), respectively. Ligand coordinates and restraints for the seco-duocarmycins were generated using PHENIX eLBOW (Moriarty et al., 2009) with AM1 geometry optimization. Data collection and refinement statistics are provided in Table 1. Crystal structure figures were prepared using PyMOL (Schrödinger 2017). The atomic coordinates and structure factors for human CYP1A1 structures have been deposited in the Protein Data Bank (http://wwpdb.org/). The Protein Data Bank code 6UDL is the complex with (S)-ICT2700 and 6UDM is the complex with (S)-ICT2726.
TABLE 1.
X-ray data collection and refinement statistics for P450/ligand structures.
| CYP1A1/(S)-ICT2700 (6UDL) | CYP1A1/(S)-ICT2726 (6UDM) | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P3121 |
| Cell dimensions (Å) | 65.37, 196.15, 237.22 | 241.28, 241.28, 125.30 |
| Molecules/asymmetric unit | 4 | 4 |
| Resolution (Å)a | 50.00–2.85 (2.90–2.85) | 39.64–3.07 (3.14–3.07) |
| Total reflectionsa | 510,182 (17,794) | 783,762 (35,174) |
| Unique reflectionsa | 72,081 (3,422) | 77,258 (3,944) |
| Redundancya | 7.1 (5.2) | 10.1 (8.9) |
| Rpima | 0.080 (0.942) | 0.054 (0.631) |
| <I/σ(I)> a | 28.0 (2.1) | 9.7 (1.6) |
| CC1/2a | 0.990 (0.322) | 0.997 (0.518) |
| Completeness (%)a | 99.8 (97.3) | 99.0 (85.6) |
| Refinement | ||
| Resolution (Å) | 48.799–2.850 | 39.64–3.10 |
| No. reflections | 71,709 | 76,831 |
| R/Rfree (%) | 23.8 / 28.7 | 20.1 / 22.1 |
| Ramachandran (%) favored/allowed/outliers | 96.19 / 3.81/ 0.00 | 97.15 / 2.85 / 0.00 |
| No. non-H atoms/B factors (Å2) | ||
| Protein | 15,015 / 68.60 | 15,026 / 80.62 |
| Ligand | 54 / 57.62 | 104 / 80.66 |
| Heme | 172 / 57.09 | 172 / 68.49 |
| CHAPS | —/— | 42 / 100.77 |
| Water | 1 / 49.31 | —/— |
| RMSD bond (Å) | 0.008 | 0.007 |
| RMSD angle (°) | 0.765 | 0.636 |
| Coordinate error (Å) | 0.37 | 0.37 |
CC1/2, Pearson correlation coefficient; <I/σ(I)>, average intensity over variation in intensity; R, measure of error between the observed intensities from the diffraction pattern and the predicted intensities calculated from the model; Rfree, measure of error between the observed diffraction pattern intensities from the diffraction pattern and the predicted intensities for a subset of data not used in model refinement; RMSD, root mean square deviation; Rpim, precision-indicating merging R factor
a Statistics for highest resolution shell shown in parentheses.
P450 Metabolism and Liquid Chromatography–Mass Spectrometry Analysis.
A reconstituted protein system was created by mixing 200 pmol P450 with 400 pmol human NADPH–cytochrome P450 reductase and incubating for 10 minutes at room temperature. Freshly prepared 1,2-dilauroyl-sn-glycero-3-phosphocholine (125 μM) in assay buffer (100 mM potassium phosphate, pH 7.4) was added, and incubation continued for an additional 10 minutes at room temperature. The reconstituted protein system was then added to an Eppendorf tube containing assay buffer and the ICT substrate (50 μM), at a total volume of 480 μl prior to the addition of NADPH. The mixture was incubated at 37°C for 3 minutes in a block heater (Grant Block Heater QBD4, UK). NADPH (20 μl of 25 mM stock in assay buffer) was added to the incubating mixture to a final concentration 1 mM to initiate the reaction. Aliquots (100 μl) were then removed over a 60-minute period, i.e., at 0, 15, 30, and 60 minutes, into labeled Eppendorf tubes containing dichloromethane (200 μl), gently mixed, and placed on ice. Tubes were centrifuged (4500g, 2 minutes) with 200 μl of the bottom organic layer carefully removed into separate tubes, and dried using a SP Genevac EZ-PLUS evaporator for 30 minutes. The dried reactions were each dissolved in 50 μl 90% acetonitrile, 10% H2O, 0.1% formic acid and transferred into a high-performance liquid chromatography (HPLC) vial for liquid chromatography–mass spectrometry (LC-MS) analysis.
LC-MS investigations were carried out using a gradient method (Supplemental Table 2) on a HiChrom RPB column (25 cm × 2.1 mm inner diameter; HIRPB-250 AM; R6125) using a Waters Alliance 2695 HPLC (Micromass, Manchester, UK) with a photodiode array detector and connected in series with Waters Micromass ZQ quadrupole mass spectrometer in electrospray ionisation in positive moded (ESI+) mode. P450 substrates and their respective metabolites were detected using UV absorbance at 330 nm for ICT2700 and 312 nm for ICT2726, with their associated masses identified as singularly charged ions by mass spectrometry. Mass spectrometry electrospray ionisation in positive moded (ESI+) source parameters were as follows: desolvation gas, 650 l/h; cone gas, 50 l/h; capillary voltage, 3 kV; extraction voltage, 5 V; cone voltage, 20 V; radio frequency voltage, 0.2 V; source block temperature, 120°C; and desolvation temperature, 350°C.
Results
Resolution and Determination of ICT2700 and ICT2726 Enantiomers.
ICT2700 and ICT2726 are both chiral at the carbon adjacent to the chloromethyl group (Fig. 1). Previous reports (Sheldrake et al., 2013; Travica et al., 2013) have used racemic mixtures for pharmacological investigations. Although the racemic form of ICT2700 demonstrated potency in tumor growth inhibition and cell death in CYP1A1- and CYP2W1-transfected cancer cells (Sheldrake et al., 2013), it is unknown if differences in chirality of ICT2700 would affect binding or metabolism by these two P450 enzymes. In the current studies, the two enantiomers of ICT2700 and of ICT2726 were each successfully resolved by chiral phase (ChiralPak ID) HPLC with a 99:1:0.1 dichloromethane/ethanol/diethylamine eluent. A single stereoisomer from each ICT compound was successfully crystallized from ethanol in acetone, and crystallographic determination identified both as the R enantiomers of ICT2700 and ICT2726, respectively (Supplemental Fig. 1; Supplemental Table 1). For each compound the other HPLC peak was assigned as the S enantiomer. This separation and assignment allowed evaluation of the individual stereoisomers.
Seco-Duocarmycin Binding Modes and Affinities for Human CYP1A1 and CYP2W1.
Binding of ligands in a cytochrome P450 active site can usually be monitored by observing shifts in the absorbance maximum of the heme Soret peak. Substrates typically shift the Soret peak to shorter wavelengths as they bind in the active site close enough to the heme to displace water from its central iron. As a result, they shift the iron spin equilibrium from its six-coordinate, low-spin form to the five-coordinate, high-spin form. This is typically observed in the form of UV-visible difference spectra, where such “Type I” binding is associated with decreases in absorbance at ∼430 nm and increases at ∼393 nm. These were exactly the changes observed when CYP1A1 was progressively titrated with each ICT2726 enantiomer (Fig. 2A and 2B). Both (S)- and (R)-ICT2726 bound CYP1A1 with relatively high affinity, with Kd values of 0.23 ± 0.02 μM and 0.71 ± 0.05 μM (insets to Figs. 2, A and B), respectively. This suggests that both enantiomers of ICT2726, the potential CYP2W1-biomarker compound, bind in the CYP1A1 active site similar to substrates.
Fig. 2.

Spectral binding experiments of CYP1A1 and CYP2W1 with the seco-duocarmycin enantiomers of ICT2726. Representative difference binding spectra are shown of CYP1A1 (1A1) [(A) and (B)] and CYP2W1 (2W1) [(C) and (D)], with the respective ICT2726 enantiomer. Insets display the difference in peak-trough absorbance (average of two technical duplicates) versus compound concentration and are fit by nonlinear regression to a tight-binding equation. The fitted compound affinities (Kd values) are reported with ± S.E. (error bars).
CYP2W1 also bound (S)-ICT2726 (Fig. 1) with classic Type I, substrate-like spectral changes (Fig. 2C). The affinity of (S)-ICT2726 was also relatively high for CYP2W1, with a Kd of 0.41 ± 0.03 μM (Fig. 2C, inset). However, (R)-ICT2726 addition to CYP2W1 did not result in significant spectral shifts in the difference spectra at concentrations as high as ∼10 μM (Fig. 2D). This result suggests that either the R enantiomer does not bind CYP2W1 with significant affinity or that it binds in a different mode that does not disrupt water coordinated to the heme iron in the active site.
Similar titrations were used to evaluate CYP1A1 and CYP2W1 binding of ICT2700, which is bioactivated by both enzymes to generate a potent cell-killing metabolite (Fig. 1). Since ICT2700 is a validated substrate for both enzymes, it was expected that the titrations might show Type I spectral shifts similar to those observed for ICT2726 above. However, the binding modes observed with ICT2700 were markedly distinct. Binding to CYP1A1 was notable in three ways. First, the dominant feature of CYP1A1 binding of both ICT2700 enantiomers were increases in absorbance eventually emerging at 412–415 nm with a smaller trough forming at 432–434 nm (Fig. 3A and 3B). This primary maximum at ∼412–415 nm suggests that ligand binding likely promotes oxygen interaction with the heme iron, rather than displacing the iron-coordinated water. It is possible either that ligand orientation within the active site reinforces water coordination to the heme iron or that an oxygen of the ligand (e.g., the methoxy group) interacts with the heme iron. Second, the peak is broader throughout the titration, especially for the R enantiomer, with a secondary increase in absorbance at 398 nm (Fig. 3B). This broad peak shape is consistent with the presence of two different binding modes for ICT2700 across the population of CYP1A1 active sites, with the secondary one at 398 nm more similar to the substrate-like binding mode observed for ICT2726. Third, the affinities for (S)- and (R)-ICT2700 for CYP1A1 were 1.9 ± 0.1 μM (Fig. 3A, inset) and 1–2 µM (depending on the wavelength used; Fig. 3B, inset), respectively. Thus (S)-ICT2726 has about an eightfold higher affinity for CYP1A1 than (S)-ICT2700, whereas the affinities for the R enantiomers are more similar.
Fig. 3.
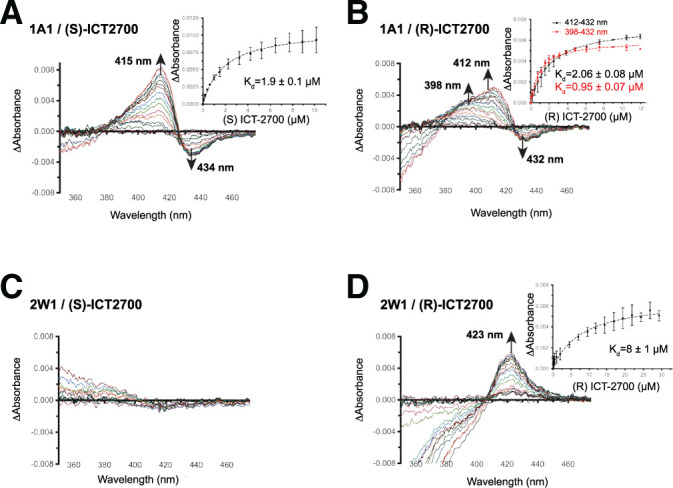
Spectral binding experiments of CYP1A1 and CYP2W1 with the seco-duocarmycin enantiomers of ICT2700. Difference binding spectra are shown of CYP1A1 (1A1) [(A) and (B)] and CYP2W1 (2W1) [(C) and (D)] binding the ICT2700 enantiomers. Insets display the difference in peak-trough absorbance (average of two technical duplicates) versus compound concentration and are fit by nonlinear regression to a tight-binding equation. The fitted compound affinities (Kd values) are reported with ± S.E. (error bars).
As was the case for ICT2726, titrations of the ICT2700 enantiomers into CYP2W1 revealed a distinct preference for one enantiomer. Addition of up to ∼15 μM (S)-ICT2700 into 1 µM CYP2W1 had minimal effects on the P450 spectrum (Fig. 3C). This could indicate that the S enantiomer either has weak or no affinity toward CYP2W1 or does not bind in the active site close enough to disrupt the water on the heme iron. In contrast, the R enantiomer produced a unique binding mode characterized primarily by a peak around 423 nm (Fig. 3D). Although there does not appear to be a clear corresponding decrease in absorbance, this could be partially obscured by changes in background absorbance caused by this compound itself in the UV range (350–380 nm). This red-shifting of the Soret peak position is generally consistent with spectral changes that occur when a ligand nitrogen replaces the water interacting with the heme iron. ICT2700 has two nitrogen-containing indole ring systems that are possibilities for direct iron coordination, but the most likely position would be the indole on the alkylation subunit as it may be sterically less hindered, as well as being close to the known site of metabolism (Fig. 1). The affinity for this interaction is relatively weak, with a Kd = 8 ± 1 μM (Fig. 3D, inset). Since the N-Fe coordination appears to be weak, it is possible that this N-Fe feature helps to orient the molecule in the active site for bioactivation but also allows for the dissociation from the iron so that hydroxylation can occur at the adjacent C5 as required for bioactivation. Notably, replacement of the indole with a naphthyl moiety omitting the nitrogen drastically reduced duocarmycin activation by CYP2W1 (and CYP1A1), which would be consistent with a propensity of this nitrogen directing the nearby critical C5 toward the heme iron (Sheldrake et al., 2013).
Metabolism of ICT2700 and ICT2726 Enantiomers by CYP1A1 and CYP2W1.
A previous study reported that racemic ICT2700 is metabolized by CYP1A1 bactosomes to generate four major metabolites, two of which were identified as the hydroxylated seco-duocarmycin (Fig. 1, bottom left), which undergoes spontaneous spirocyclization, and the resulting cyclopropane product responsible for DNA damage and potent cell killing (Fig. 1, bottom right) (Pors et al., 2011). Another study showed that the racemic ICT2726 analog did not cause DNA damage or kill cells in CYP2W1-transfected cells despite high metabolism by CYP2W1 (Travica et al., 2013). The current study determined whether the R and S enantiomers were differentially metabolized using a reconstituted protein system composed of either purified CYP1A1 or CYP2W1, human cytochrome P450 reductase, NADPH, and 1,2-dilauroyl-sn-glycero-3-phosphocholine liposome. The R and S enantiomers were each incubated with this reconstituted protein system at 37°C, and aliquots were taken at four time points (0, 15, 30, and 60 minutes). These were analyzed by LC-MS and revealed several metabolic differences.
CYP1A1 metabolizes (S)-ICT2700 to at least 12 different metabolites (Fig. 4A, red). These include the initial hydroxylated seco-duocarmycin critical for spirocyclization (Fig. 4A, M13 or ICT2740) and the post-spirocyclization cytotoxic product, which is a major metabolite (Fig. 4A, red, M6). CYP1A1 also metabolizes (R)-ICT2700 but with lower yield and to fewer metabolites. In this case, the hydroxylated seco-duocarmycin metabolite (M13) is not observed at significant levels but must be transiently generated because the spirocyclized metabolite is also detected, albeit at much lower levels (Fig. 4A, green, M6). Significantly, the hydroxylated seco-duocarmycin ICT2740 (eluting at t = 25.4 minutes, m/z 396.1) and the spirocyclized metabolite necessary for DNA binding and cytotoxicity were generated primarily from the S enantiomer. In contrast, CYP2W1 metabolizes ICT2700 stereoisomers more specifically, to only one (R) or two (S) products (Fig. 4B). These are generated in quite low yield (1 and 4%, respectively) and are not related to prodrug activation because they contain an intact chloromethyl fragment. Metabolite M1 is supportive of a single hydroxylation (m/z 396.1), but it was not possible to analyze M2 due to the low yield (Fig. 4B).
Fig. 4.

LC-MS chromatograms of ICT compounds incubated with CYP1A1 (1A1) or CYP2W1 (2W1) for 30 minutes; M6 = spirocyclized product. (A) ICT2700 R/S CYP1A1 metabolism. (B) ICT2700 R/S CYP2W1 metabolism. (C) ICT2726 R/S CYP1A1 metabolism (D) ICT2726 R/S CYP2W1 metabolism. AU, absorbance units; ESI+, electrospray ionization in positive mode; MS, mass; M+H, singly protonated mass; RT, retention time.
Metabolism of the stereoisomers of the noncytotoxic analog ICT2726 also differed between the two P450 enzymes. CYP1A1 converted both stereoisomers to multiple products, with similar overall metabolite profiles, although the R enantiomer is the better substrate, whereas the S enantiomer was a little more selective in generating the major singly hydroxylated product (Fig. 4C, M6, parent m/z 368.9 + 16). However, CYP2W1 had very distinct metabolism of the ICT2726 isomers. CYP2W1 converted (R)-ICT2726 to very few metabolites and at exceedingly low yield (Fig. 4D, red), whereas (S)-ICT2726 was highly metabolized with >14 identifiable peaks accounting for ∼38% metabolism (measured by the area under the curve; Fig. 4D, green). Comparison of the metabolite m/z profiles (Fig. 4C versus Fig. 4D) reveals that most of these metabolites are unique for CYP2W1 versus CYP1A1 and thus have the potential to serve as biomarker of CYP2W1 enzymatic activity. The major (S)-ICT2726 metabolites generated by CYP2W1 (M1 and M2) produced m/z (M+1) of 356.1 and 372.1 with an intact Cl isotope pattern. This is consistent with a single or double hydroxylation, corresponding to the addition of 16 or 32 Da, rather than the hydroxymethyl metabolite dominant in colon cancer cell studies (Travica et al., 2013). These two major metabolites had a UV-visible spectrum with a maximum at 330 nm, indicating that oxidation had not altered the duocarmycin pharmacophore to generate the spirocyclized duocarmycin cytotoxin. Thus these major metabolites are expected to be nontoxic.
Co-Crystal Structures of CYP1A1 with (S)-ICT2700 and (S)-ICT2726.
Co-crystallization was attempted for both enantiomers of ICT2700 and ICT2726 with both CYP1A1 and CYP2W1 enzymes. No structures have been reported for CYP2W1 with any ligand, and similarly our efforts did not yield crystals with these ICT compounds. However, diffraction quality crystals were obtained for CYP1A1 with (S)-ICT2700 and (S)-ICT2726, yielding structures at 2.9 Å and 3.1 Å, respectively. The (S)-ICT2700 complex was solved in the orthorhombic P212121 crystal form that other CYP1A1 structures have displayed; however, the co-crystal structure with (S)-ICT2726 solved in a new crystal system, with a trigonal P3121 space group. Even with different symmetry, the crystal contacts between molecules were similar, and each structure contained four copies of the CYP1A1/ICT ligand complex in the asymmetric unit. In both complexes the overall fold of CYP1A1 was as seen in previous solved structures of the enzyme (Walsh et al., 2013; Bart and Scott, 2018; Bart et al., 2020). The one region of CYP1A1 that has been observed in varied conformations when different ligands are bound consists of helices F through G that form the roof of the active site (Bart and Scott, 2018). Despite the significantly more elongated structures of the duocarmycins, this region displayed a conformation of this region most similar to that reported when smaller, more compact α-naphthoflavone is bound (Walsh et al., 2013). This includes a five-residue break in the F helix as it passes over the active site, a distinctive common feature for human CYP1 structures (Sansen et al., 2007; Wang et al., 2011).
The (S)-ICT2726 ligand is present in all four CYP1A1 copies of the crystal structure, and the binding mode appears to be consistent among them. The best fit to the observed electron density positions the 5-fluoroindole ring of (S)-ICT2726 above the heme (Fig. 5A) with the closest carbon atoms of the six-membered ring each 4.3 Å from the iron (Fig. 5A, red dashed lines). The fluorine projects toward a cluster of hydrophobic residues forming the bottom portion of the CYP1A1 active site. On the opposite half of the molecule the pyrrole ring pi-pi stacks with Phe224 in the F helix break. The linker between the two rings is co-planar with the two ring systems and packs against the peptide bond between Gly316 and Ala317 in the I helix. These and other interactions constrain the overall molecule with the two halves of the small molecule in a co-planar conformation. The only exception is the chloromethyl arm of the ligand, which is directed into the break in the F helix and is in close contact (2.7 Å) to the side-chain oxygen of Asn222 where it could potentially form a halogen-oxygen interaction (Fig. 5A, yellow dashed line). In some copies of the complex there is evidence for an active-site water forming a hydrogen bond network bridging from the side chain of Ser122 (B'/C loop) to the ligand’s linker carbonyl oxygen.
Fig. 5.
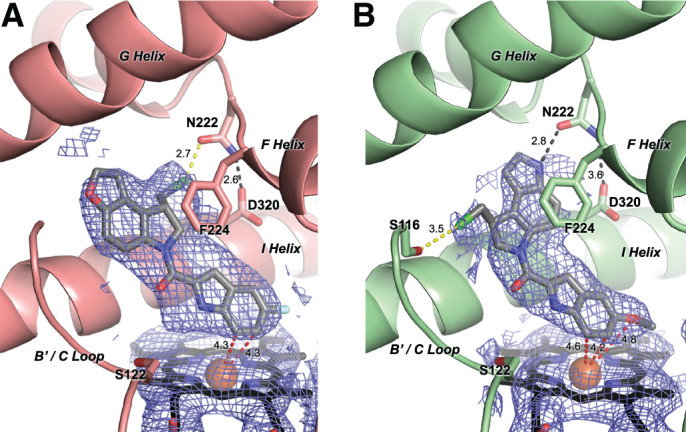
Co-crystallization structures of CYP1A1 (ribbons) bound to (S)-ICT2726 (A) and (S)-ICT2700 (B) (gray sticks). Electron density is shown as 2Fo – Fc composite omit map contoured at 1.0 σ (blue mesh) around the ligand and heme (black sticks with orange Fe sphere). Potential hydrogen bonds (black dashes), halogen bonds (yellow dashes), and ligand atoms in closest proximity to the heme iron (red dashes) are shown with their corresponding distances.
The second CYP1A1 structure was determined with (S)-ICT2700, which is bioactivated by CYP1A1 to form the DNA-alkylating metabolite (vide supra). Due to ligand disorder and/or low occupancy, this molecule could only be placed into the observed electron density for two of four CYP1A1 molecules in the asymmetric unit. In these two CYP1A1 molecules, the overall conformation of (S)-ICT2700 also has the two halves of the molecule co-planar (Fig. 5B). The overall orientation is similar to (S)-ICT2726 in that the DNA recognition motif is directly above the heme and the DNA-alkylating subunit is more distant. However, the DNA-alkylating subunit of (S)-ICT2700 appears to be flipped by 180° with respect to the DNA-binding subunit compared with (S)-ICT2726 (Fig. 5A versus Fig. 5B). This reorientation of the DNA-alkylating ring system for (S)-ICT2700 directs the chloromethyl arm toward the B' helix and the fused pyrrole into the F helix break. This still permits stacking between Phe224 and the aromatic pyrroloindole system of (S)-ICT2700. The indole nitrogen of this portion of the ligand makes a hydrogen bond interaction with the oxygen of Asn222 (2.8 Å). Similar to (S)-ICT2726, the chlorine of the chloromethyl substituent also makes a potential halogen-oxygen interaction, but in this case, it is a longer interaction with the oxygen of Ser116 in the B' helix (3.5 Å; Fig. 5B, yellow dashed line). In one molecule, electron density suggested water-mediated hydrogen bonding interactions between Ser122, and the ligand linker carbonyl oxygen and nitrogen of the methoxy-indole ring, similar to the CYP1A1/(S)-ICT2726 complex. One of the differences between ICT2700 and ICT2726 is the substitution of a methoxy group on the DNA-recognition motif of ICT2700 versus a fluorine substituent at this position for ICT2726. The positioning of these two groups is quite similar in a hydrophobic pocket over the heme, with the oxygen in ICT2700 only 4.8 Å from the iron.
Discussion
Comparisons and Applications of CYP1A1 Versus CYP2W1 Interactions with Seco-Duocarmycins.
CYP2W1 is still classified as an orphan enzyme. Little is known about its substrate scope or endogenous role. One possible endogenous role that has been suggested is oxidation of lysophospholipids, which could have importance in inflammation and tumor development (Xiao and Guengerich, 2012). Based on sequence identity, CYP2W1 groups with the xenobiotic-metabolizing P450 enzymes and does have examples of drug substrates, some of which suggest a partially overlapping substrate profile with CYP1A1. In addition to the re-engineered duocarmycin molecules such as those examined herein, CYP1A1 and CYP2W1 both metabolize substrates such as the benzothiazole-based anticancer compounds 5F 203 and GW 610 (Wang and Guengerich, 2012), AQ4N (Yakkundi et al., 2006; Nishida et al., 2010), all-trans-retinoic acid (Marill et al., 2000; Zhao et al., 2016), and the polycyclic aromatic hydrocarbon benzo[a]pyrene (Wu et al., 2006). This indicates that shared active-site elements are present between the two enzymes that would need to be elucidated to design more selective duocarmycin prodrugs.
The clinical utility of duocarmycin prodrugs activated by in situ cytochrome P450 activation is dependent on 1) the selectivity of the duocarmycin for an individual P450 and 2) the selective expression of that P450 in the targeted tissue. Duocarmycin prodrugs designed for selective in situ bioactivation by CYP1A1 are of potential clinical utility in tissues such as the lung and bladder where this enzyme is expressed at higher levels. However, this enzyme is also normally expressed at low levels in other tissues, which could result in undesirable side effects. Duocarmycin prodrugs designed to be selectively activated by CYP2W1 would not suffer from the latter issue because CYP2W1 is selectively expressed in colon cancer tissues and not in normal adult tissues (Choong et al., 2015). Modulating the P450—and thus tissue—selectivity of these prodrugs relies on understanding the molecular-level interactions of these compounds with both P450 enzymes. Such insight could result in duocarmycin analogs with increased CYP2W1 bioactivation and aid us to evaluate whether single CYP1A1- or CYP2W1-targeting prodrugs or dual-targeting CYP1A1 and CYP2W1 prodrugs are better therapeutics.
CYP1A1 binds both enantiomers of both ICT compounds examined herein. Spectral changes for the nontoxic ICT2726 analogs were similar to those commonly observed for substrates (Fig. 2A, 2B), and multiple metabolites (Fig. 4C) were indeed detected. The R enantiomer is more extensively metabolized, but the S enantiomer appears to bind about twofold more tightly. The structure of the CYP1A1/(S)-ICT2726 complex (Fig. 5A) reveals that the ligand scaffold is constrained to a planar conformation, with the chloromethyl fragment accommodated by a break in the F helix that is common in CYP1 structures. This overall orientation is consistent with oxidation on the DNA-binding half of the molecule to generate the nontoxic metabolites observed. By comparison, the bioactivated ICT2700 compounds demonstrated evidence for two binding modes, especially for the R enantiomer (Fig. 3A and 3B), and were both metabolized to multiple metabolites (Fig. 4A). Metabolites of ICT2700 included the cytotoxic product, with substantially less from (R)-ICT2700 compared with the S enantiomer. However, the structure of the CYP1A1/(S)-ICT2700 complex revealed the orientation of (S)-ICT2700 with the DNA-alkylating subunit distant from the heme and the DNA-binding subunit closer to the heme for metabolism (Fig. 5B). This orientation places the carbon oxidized to generate the cytotoxic metabolite 9.6 Å away from the heme iron. This orientation is consistent with the multiple nontoxic metabolites observed, but not the spirocyclized toxic metabolite. Thus, a second orientation with the (S)-ICT2700 DNA-alkylating domain toward the heme must also occur to generate the observed cytotoxic metabolite (Fig. 4A, M13→M6). A possible explanation for this could be that the orientation observed in the crystal structure may be the dominant binding pose, with a minor pose being responsible for bioactivation. This would be consistent with the reported observation that the CYP2W1-activated species undergoing spirocyclization is a minor product rather than the major product (Pors et al., 2011). The ICT2700 molecule has two planar fused-ring halves, which are constrained to be coplanar by the active site, so it is not difficult to imagine that an orientation with the molecule with the ligand flipped 180° lengthwise, swapping the DNA-alkylating and DNA-binding domains, might also be accommodated by the CYP1A1 active site. Modifying (S)-ICT2700 to further favor the orientation observed in this structure should decrease CYP1A1 bioactivation.
In contrast to CYP1A1, CYP2W1 appears to be more selective in the binding and metabolism of these duocarmycins. The spectral binding data suggest that this enzyme binds the nontoxic analog (S)-ICT2726 over the R enantiomer and correspondingly generates more total turnover of the S stereoisomer, as well as more different metabolites. Many of these seem to be distinct from those generated by CYP1A1, further supporting the idea that the (S)-ICT2726 metabolite profile could be used to identify CYP2W1-expressing cells. Again, demonstrating the importance of steric factors for CYP2W1, spectral binding changes suggested that compared with the S enantiomer, the (R)-ICT2700 stereoisomer bound and was also more extensively metabolized.
Although there are no structures of CYP2W1 with duocarmycins or any other ligands, the binding information herein suggests differences between the two active sites could potentially be exploited. It is interesting that CYP2W1 appears to be distinctively affected by the chirality of the chloromethyl moiety for both compounds. CYP2W1 appears to bind (R)-ICT2700, but the S enantiomer of ICT2726. CYP1A1 binds both enantiomers of both compounds, although there are differences in affinity and homogeneity. These observations may indicate that the CYP2W1 active site could be less flexible than that of CYP1A1. Regardless, employing only the R isomer as a scaffold should help direct CYP2W1 bioactivation. This enantiomeric preference could therefore be useful in the design of duocarmycin prodrugs with improved P450 isoform selectivity.
Another potentially important observation became apparent with the current studies. Herein it was observed that recombinant, purified CYP2W1 incubated with recombinant, purified NADPH–cytochrome P450 reductase, the typical redox partner for microsomal human P450 enzymes, resulted in CYP2W1-mediated metabolism of both ICT compounds (Fig. 4B and 4D), although metabolism was low for some compounds and the spirocyclized toxic metabolite was not generated from ICT2700 at detectable levels. This result is not specific to the ICT substrates herein, as in our hands this same enzyme system metabolizes several proluciferin substrates sold by Promega to probe the function of other cytochrome P450 enzymes (unpublished data). However, this is in contradiction with two prior reports. First, Sheldrake et al. (2013) reported that recombinant CYP2W1 did generate the toxic spirocyclized ICT2700 metabolite in experiments where CYP2W1 was reduced by not NADPH–cytochrome P450 reductase but rather by cumene hydroperoxide, a small molecule artificially used to generate the active intermediate of P450 enzymes. Second, Guo et al. used in vivo knockdown and inhibitor studies to suggest that CYP2W1 activity may not be supported by the typical NADPH–cytochrome P450 reductase—or several possible alternative redox partners—in human embryonic kidney cells stably transformed to express CYP2W1. However, xenograft experiments using colon cancer cells expressing CYP2W1 did successfully convert a similar seco-duocarmycin prodrug to a cytotoxic product (Travica et al., 2013). This latter result indicates there must be a cellular redox partner, either NADPH–cytochrome P450 reductase or an unknown molecule. Thus, a critical unresolved aspect that will affect the success of drug development and the clinical utility of this approach is to determine the appropriate redox partner(s) to appropriately compare in vitro and in vivo studies and to determine if the redox partner influences which metabolite(s) are produced.
Overall, this information indicates that NADPH–cytochrome P450 reductase can support CYP2W1 metabolism in vitro and that distinctive structural features present in the CYP1A1 and CYP2W1 P450 active sites control differential in binding and/or positioning, which bodes well for improving compound selectivity. Despite the unclear role of CYP2W1 in normal human physiology, its tumor localization and ability to bioactivate duocarmycin prodrugs make CYP2W1 an attractive enzyme for selective cancer therapeutics.
Acknowledgments
Crystallization screening employed instrumentation in the University of Michigan (U-M) Center for Structural Biology (CSB). The CSB is grateful for support from the U-M Life Sciences Institute, the U-M Rogel Cancer Center, the U-M Medical School Endowment for the Basic Sciences, and grants from the National Institutes of Health and Yorkshire Cancer Research Program Grant (B381PA). X-ray diffraction data were collected at both Advanced Photon Source and the Stanford Synchrotron Radiation Lightsource. This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences. We thank Dr. Colin C. Seaton for support with single crystal small molecule X-ray data determinations and Dr. Francis Mprah Barnier for assistance with LC-MS experiments discussed in this study.
Abbreviations
- CHAPS
3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate
- CV
column volume(s)
- HPLC
high-performance liquid chromatography
- LC-MS
liquid chromatography–mass spectrometry
- Ni-NTA
nickel–nitrilotriacetic acid
- P450
cytochrome P450
Authorship Contributions
Participated in research design: Bart, Scott, Pors.
Conducted experiments: Bart, Morais, Vangala, Pors.
Performed data analysis: Bart, Loadman, Pors, Scott.
Wrote or contributed to the writing of the manuscript: Bart, Pors, Scott.
Footnotes
Current affiliation: Merck & Co., Boston, Massachusetts.
This work was supported by National Institutes of Health National Institute of General Medical Sciences [Grant R37-GM076343 to E.E.S.]. A.G.B. was previously a trainee on National Institutes of Health National Institute of General Medical Sciences [Grant T32-GM008545]. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This content was previously presented as part of the first author’s dissertation and a poster at the 2019 Experimental Biology in Orlando, FL.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bart AG, Scott EE (2017) Structural and functional effects of cytochrome b5 interactions with human cytochrome P450 enzymes. J Biol Chem 292:20818–20833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bart AG, Scott EE (2018) Structures of human cytochrome P450 1A1 with bergamottin and erlotinib reveal active-site modifications for binding of diverse ligands. J Biol Chem 293:19201–19210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bart AG, Takahashi RH, Wang X, Scott EE (2020) Human cytochrome P450 1A1 adapts active site for atypical nonplanar substrate. Drug Metab Dispos 48:86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choong E, Guo J, Persson A, Virding S, Johansson I, Mkrtchian S, Ingelman-Sundberg M (2015) Developmental regulation and induction of cytochrome P450 2W1, an enzyme expressed in colon tumors. PLoS One 10:e0122820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVore NM, Smith BD, Wang JL, Lushington GH, Scott EE (2009) Key residues controlling binding of diverse ligands to human cytochrome P450 2A enzymes. Drug Metab Dispos 37:1319–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) OLEX2: a complete structure solution, refinement and analysis program. J Appl Crystallogr 42:339–341. [Google Scholar]
- Edler D, Stenstedt K, Ohrling K, Hallström M, Karlgren M, Ingelman-Sundberg M, Ragnhammar P (2009) The expression of the novel CYP2W1 enzyme is an independent prognostic factor in colorectal cancer - a pilot study. Eur J Cancer 45:705–712. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR, Murshudov GN (2013) How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez A, Karlgren M, Edler D, Bernal ML, Mkrtchian S, Ingelman-Sundberg M (2007) Expression of CYP2W1 in colon tumors: regulation by gene methylation. Pharmacogenomics 8:1315–1325. [DOI] [PubMed] [Google Scholar]
- Gomez A, Nekvindova J, Travica S, Lee MY, Johansson I, Edler D, Mkrtchian S, Ingelman-Sundberg M (2010) Colorectal cancer-specific cytochrome P450 2W1: intracellular localization, glycosylation, and catalytic activity. Mol Pharmacol 78:1004–1011. [DOI] [PubMed] [Google Scholar]
- Guo J, Thiess S, Johansson I, Mkrtchian S, Ingelman-Sundberg M (2016) Membrane topology and search for potential redox partners of colon cancer-specific cytochrome P450 2W1. FEBS Lett 590:330–339. [DOI] [PubMed] [Google Scholar]
- Jukes Z, Morais GR, Loadman PM, Pors K (2021) How can the potential of the duocarmycins be unlocked for cancer therapy? Drug Discov Today 26:577–584. [DOI] [PubMed] [Google Scholar]
- Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlgren M, Gomez A, Stark K, Svärd J, Rodriguez-Antona C, Oliw E, Bernal ML, Ramón y Cajal S, Johansson I, Ingelman-Sundberg M (2006) Tumor-specific expression of the novel cytochrome P450 enzyme, CYP2W1. Biochem Biophys Res Commun 341:451–458. [DOI] [PubMed] [Google Scholar]
- Karlgren M, Ingelman-Sundberg M (2007) Tumour-specific expression of CYP2W1: its potential as a drug target in cancer therapy. Expert Opin Ther Targets 11:61–67. [DOI] [PubMed] [Google Scholar]
- Lang D, Radtke M, Bairlein M (2019) Highly variable expression of CYP1A1 in human liver and impact on pharmacokinetics of riociguat and granisetron in humans. Chem Res Toxicol 32:1115–1122. [DOI] [PubMed] [Google Scholar]
- Macrae CF, Bruno IJ, Chisholm JA, Edgington PR, McCabe P, Pidcock E, Rodriguez-Monge L, Taylor R, van de Streek J, Wood PA (2008) Mercury: visualization and analysis of crystal structures. J Appl Crystallogr 41:466–470. [Google Scholar]
- Marill J, Cresteil T, Lanotte M, Chabot GG (2000) Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Mol Pharmacol 58:1341–1348. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty NW, Grosse-Kunstleve RW, Adams PD (2009) Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr 65:1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida CR, Lee M, de Montellano PRO (2010) Efficient hypoxic activation of the anticancer agent AQ4N by CYP2S1 and CYP2W1. Mol Pharmacol 78:497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Sato R (1964) The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem 239:2370–2378. [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- Pors K, Loadman PM, Shnyder SD, Sutherland M, Sheldrake HM, Guino M, Kiakos K, Hartley JA, Searcey M, Patterson LH (2011) Modification of the duocarmycin pharmacophore enables CYP1A1 targeting for biological activity. Chem Commun (Camb) 47:12062–12064. [DOI] [PubMed] [Google Scholar]
- Sandee D, Miller WL (2011) High-yield expression of a catalytically active membrane-bound protein: human P450 oxidoreductase. Endocrinology 152:2904–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansen S, Yano JK, Reynald RL, Schoch GA, Griffin KJ, Stout CD, Johnson EF (2007) Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J Biol Chem 282:14348–14355. [DOI] [PubMed] [Google Scholar]
- Schrödinger (2017) The PyMOL Molecular Graphics System, Version 1.8.6.2. Schrödinger, New York. [Google Scholar]
- Sheldrake HM, Travica S, Johansson I, Loadman PM, Sutherland M, Elsalem L, Illingworth N, Cresswell AJ, Reuillon T, Shnyder SD, et al. (2013) Re-engineering of the duocarmycin structural architecture enables bioprecursor development targeting CYP1A1 and CYP2W1 for biological activity. J Med Chem 56:6273–6277. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr A 64:112–122. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2015) SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr A Found Adv 71:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Yun CH, Yamazaki H, Gautier JC, Beaune PH, Guengerich FP (1992) Characterization of human lung microsomal cytochrome P-450 1A1 and its role in the oxidation of chemical carcinogens. Mol Pharmacol 41:856–864. [PubMed] [Google Scholar]
- Sneha S, Baker SC, Green A, Storr S, Aiyappa R, Martin S, Pors K (2021) Intratumoural cytochrome P450 expression in breast cancer: impact on standard of care treatment and new efforts to develop tumour-selective therapies. Biomedicines 9:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenstedt K, Hallstrom M, Lédel F, Ragnhammar P, Ingelman-Sundberg M, Johansson I, Edler D (2014) The expression of CYP2W1 in colorectal primary tumors, corresponding lymph node metastases and liver metastases. Acta Oncol 53:885–891. [DOI] [PubMed] [Google Scholar]
- Sutherland M, Gill JH, Loadman PM, Laye JP, Sheldrake HM, Illingworth NA, Alandas MN, Cooper PA, Searcey M, Pors K, et al. (2013) Antitumor activity of a duocarmycin analogue rationalized to be metabolically activated by cytochrome P450 1A1 in human transitional cell carcinoma of the bladder. Mol Cancer Ther 12:27–37. [DOI] [PubMed] [Google Scholar]
- Travica S, Pors K, Loadman PM, Shnyder SD, Johansson I, Alandas MN, Sheldrake HM, Mkrtchian S, Patterson LH, Ingelman-Sundberg M (2013) Colon cancer-specific cytochrome P450 2W1 converts duocarmycin analogues into potent tumor cytotoxins. Clin Cancer Res 19:2952–2961. [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (1997) MOLREP: an automated program for molecular replacement. J Appl Crystallogr 30:1022–1025. [Google Scholar]
- Walsh AA, Szklarz GD, Scott EE (2013) Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J Biol Chem 288:12932–12943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Savas U, Stout CD, Johnson EF (2011) Structural characterization of the complex between alpha-naphthoflavone and human cytochrome P450 1B1. J Biol Chem 286:5736–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Guengerich FP (2012) Bioactivation of fluorinated 2-aryl-benzothiazole antitumor molecules by human cytochrome P450s 1A1 and 2W1 and deactivation by cytochrome P450 2S1. Chem Res Toxicol 25:1740–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZL, Sohl CD, Shimada T, Guengerich FP (2006) Recombinant enzymes overexpressed in bacteria show broad catalytic specificity of human cytochrome P450 2W1 and limited activity of human cytochrome P450 2S1. Mol Pharmacol 69:2007–2014. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Guengerich FP (2012) Metabolomic analysis and identification of a role for the orphan human cytochrome P450 2W1 in selective oxidation of lysophospholipids. J Lipid Res 53:1610–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakkundi A, McErlane V, Murray M, McCarthy HO, Ward C, Hughes CM, Patterson LH, Hirst DG, McKeown SR, Robson T (2006) Tumor-selective drug activation: a GDEPT approach utilizing cytochrome P450 1A1 and AQ4N. Cancer Gene Ther 13:598–605. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Wan D, Yang J, Hammock BD, Ortiz De Montellano PR (2016) Catalytic activities of tumor-specific human cytochrome P450 CYP2W1 toward endogenous substrates. Drug Metab Dispos 44:771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]