

Abstract
Transcriptome profiling of tissues and single cells facilitates interrogation of gene expression changes within diverse biological contexts. However, spatial information is often lost during tissue homogenization or dissociation. Recent advances in transcriptome profiling preserve the in situ spatial context of RNA molecules and together comprise a group of techniques known as “spatial transcriptomics,” enabling localization of cell types and their associated gene expression within intact tissues. Here, we review spatial transcriptomics methods, summarize data analysis approaches including integration with single-cell transcriptomics data, and discuss their applications in dermatologic research. These tools offer a promising avenue towards improving our understanding of niche patterning and cell-cell interactions within heterogenous tissues that encompass skin homeostasis and disease.
Introduction
Rapid advancement in gene expression profiling methods has transformed our ability to understand tissue alterations associated with development, homeostasis, and disease. The advent of high-throughput next-generation sequencing (NGS) has prompted an “omics” revolution with exciting technological breakthroughs, such as single-cell RNA-sequencing (scRNA-seq), providing novel insights into the cellular heterogeneity of such tissues. ScRNA-seq has enabled identification of rare cell populations often masked within bulk profiling and pinpointing expression of genes of interest to specific cell types (Wu et al. 2018). However, scRNA-seq has significant limitations in that key spatial information is unobtainable, cells can be difficult to dissociate from archived samples, and dissociation can introduce artifacts in gene expression (Van Den Brink et al. 2017). Additionally, cells often coordinate their behaviors as part of niches consisting of multiple and diverse cell types, and lack of spatial information from single-cell assays has limited their biological interpretation in that respect. Addressing this key limitation, the latest omics techniques include a range of methods aimed to quantify gene expression within intact tissues, together known as “spatial transcriptomics” (ST), which preserve the in situ spatial location of transcripts expressed within a tissue of interest. ST has the potential to elucidate coordination of gene expression changes across cell types influenced by their proximity to one another, thereby informing intercellular communication. This article provides an overview of ST methods, principles of analysis for their assayed data, and their applications toward improving our understanding of cutaneous biology and disease.
Overview of Spatial Transcriptomics Techniques
ST encompasses in situ hybridization (ISH), in situ sequencing (ISS), and in situ capturing (ISC) technologies (Figure 1; abbreviations used throughout the text are included in Table 1). These techniques broadly divide into approaches that target specific genes a priori (ISH and ISS) or capture transcripts in an unbiased manner for transcriptome-wide profiling (ISC). A targeted approach can detect a higher percentage of existing transcripts (i.e., high efficiency) present within the tissue, but fewer unique genes can be assayed at once (typically a few hundred genes, although this number continues to increase). Conversely, unbiased profiling can capture thousands of genes at once, but at the cost of lower efficiency (i.e., a fraction of RNA copies for any gene is captured). Thus, each method comes with its own advantages and drawbacks as well as a range of achievable resolution (Table 2). The resolution of these technologies refers to how exact of a location is retained for any particular transcript and can range from subcellular localization to a 55μm-diameter “capture spot” with distinct x-y spatial coordinates. Subcellular approaches can pinpoint transcripts within individual cells and even within subcellular compartments such as the cytoplasm or nucleus, while capture spots that are greater than the diameter of a typical cell (~10μm) may encompass several cells, obfuscating which exact cell is expressing the transcript. In recent years, these technologies have increasingly demonstrated promise as a viable alternative to scRNA-seq because of their ability to both retrieve RNA information and provide spatial localization and visualization, but all ST technologies currently suffer from suboptimal depth and/or coverage of transcripts assayed from intact tissue when compared to scRNA-seq. Thus, investigators may find that combining ST with scRNA-seq from the same tissue samples could be synergistic for addressing certain biological questions (Longo et al. 2021).
Figure 1. Overview of ST Technologies.
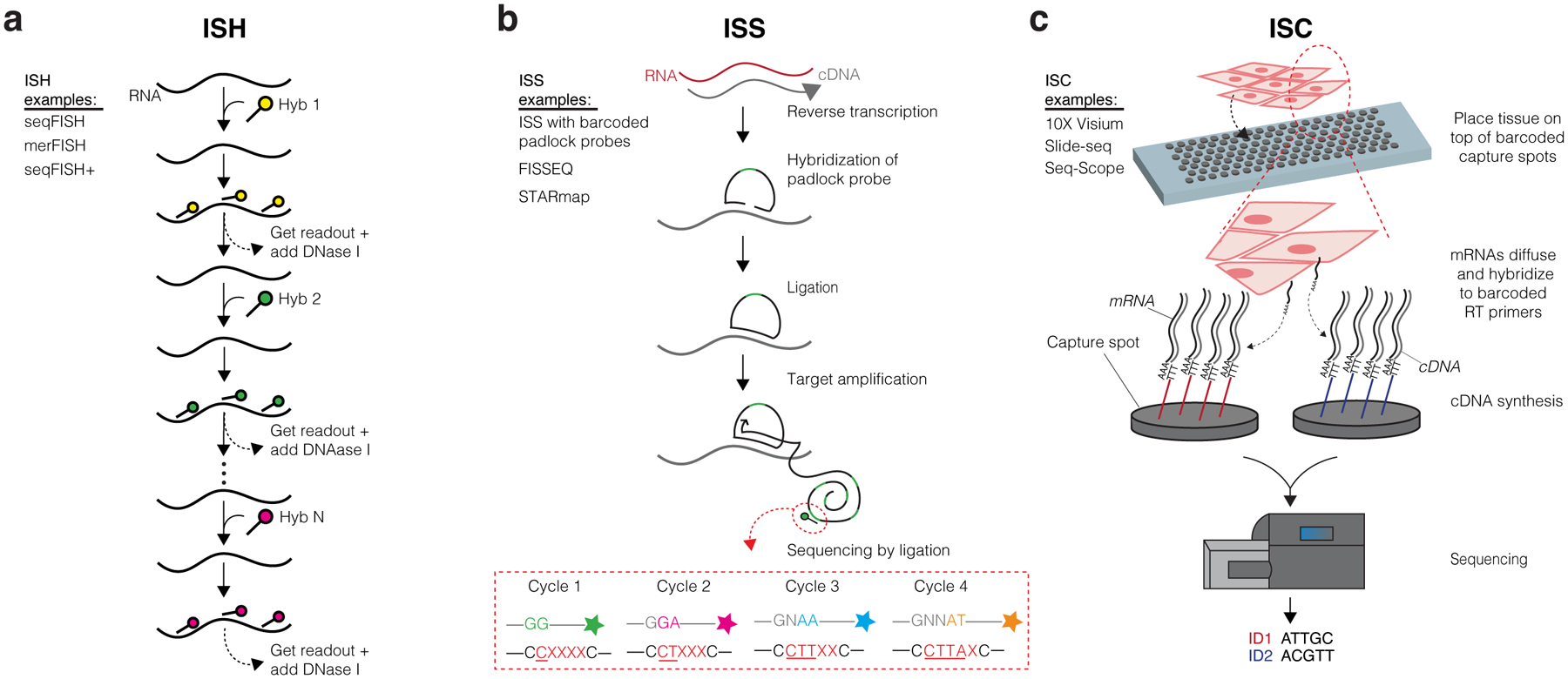
(a) In situ hybridization (ISH) methods detect specific target genes via use of fluorophore-labeled probes. Signals from probes targeting short sequences of transcripts are propagated by consecutive rounds of hybridization, imaging, and probe stripping. The steps depicted specifically follow seqFISH methods. (b) In situ sequencing (ISS) methods typically involve the hybridization and ligation of a barcoded padlock probe complementary to the RNA or cDNA of a target gene. Multiple rounds of target amplification and sequencing by ligation then allow for spatial resolution of the distinct target gene. The steps depicted specifically follow the ISS with barcoded padlock probes method. (c) In situ capturing (ISC) methods use capture spots containing an array of RT primers with distinct positional barcodes and poly-T sequences to capture mRNA transcripts. Reverse transcription produces cDNAs that are extracted and sequenced using next generation sequencing. Positional barcodes are mapped to specific locations on the tissue and enable spatial visualization of the transcriptome. The steps depicted specifically follow 10X Visium methods.
Table 1.
Abbreviations Summarized
| Abbreviation | Definition |
|---|---|
| NGS | Next-generation sequencing |
| scRNA-seq | Single-cell RNA-sequencing |
| ST | Spatial transcriptomics |
| ISH | In situ hybridization |
| ISS | In situ sequencing |
| ISC | In situ capturing |
| HPRI | High-plex RNA imaging |
| merFISH | Multiplexed error-robust fluorescence ISH |
| seqFISH | Sequential fluorescence ISH |
| FISSEQ | Fluorescent in situ sequencing |
| STARmap | Spatially Resolved transcript Amplicon Readout Mapping |
| CCI | Cell-cell interaction |
| RT | Reverse transcription |
| QC | Quality Control |
| PCA | Principal component analysis |
| t-SNE | t-distributed stochastic neighbor embedding |
| UMAP | Uniform manifold approximation and projection |
| TME | Tumor microenvironment |
| cSCC | Cutaneous squamous cell carcinoma |
| TSK | Tumor-specific keratinocyte |
| CAF | Cancer-associated fibroblast |
| T-lep | Tuberculoid leprosy |
| L-lep | Lepromatous leprosy |
| RR | Reversal reactions |
| ATAC-seq | Assay for transposase-accessible chromatin with sequencing |
Table 2.
Comparison of ST Technologies
| Category | Resolution | Capture Approach | Number of Unique Genes Assayed (Multiplex Capacity) | Advantages | Limitations | |
|---|---|---|---|---|---|---|
| merFISH | ISH | Subcellular | Targeted | Up to 500 Up to 10,000* |
|
|
| seqFISH | ISH | Subcellular | Targeted | Up to 249 |
|
|
| seqFISH+ | ISH | Subcellular | Targeted | Up to 10,000 |
|
|
| Barcoded Padlock Probe ISS | ISS | Subcellular | Targeted | Up to 100 |
|
|
| FISSEQ | ISS | Subcellular | Unbiased | Whole transcriptome |
|
|
| STARmap | ISS | Subcellular | Targeted | Up to 1,000 |
|
|
| 10X Visium | ISC | 55 μm-diameter capture spots | Unbiased | Whole transcriptome |
|
|
| Slide-seq | ISC | 10 μm-diameter capture spots | Unbiased | Whole transcriptome |
|
|
| Seq-Scope | ISC | ~0.6 μm-diameter capture spots (subcellular) | Unbiased | Whole transcriptome |
|
|
Demonstrated exclusively in cultured cells; all values lacking an asterisk indicate validation in intact tissue samples
ISH technologies include subtypes of high-plex RNA imaging (HPRI), such as multiplexed error-robust fluorescence (merFISH), sequential fluorescence (seqFISH), and seqFISH+ (Chen et al. 2015; Eng et al. 2019; Lubeck et al. 2014; Shah et al. 2016), which differ in their multiplexing capacity (Table 2). ISH techniques allow researchers to directly visualize RNA molecules in their native environment instead of requiring cells to be extracted from tissue and analyzed ex situ. Direct visualization is achieved by hybridizing a fluorescent labeled probe complementary to a predetermined RNA target of interest (Figure 1a). The signals from the labeled probes are then used to determine quantitative measurements of transcripts in spatial context. The targeted nature of this approach enables high RNA capture efficiency and single-cell/subcellular resolution of transcripts, with ongoing improvements on the upper limits of the number of targets (Xia et al. 2019). There are also notable disadvantages to ISH technologies in that cost and labor significantly increase with the number of targeted readouts and they require specialized equipment (Asp et al. 2020). Currently, major drawbacks to ISH technologies are their relative inaccessibility, labor-intensive demands, and constraints on the number of probes (and therefore transcript targets) that can be simultaneously hybridized to the tissue.
ISS technologies, another form of HPRI, include the first ISS protocol using barcoded padlock probes and fluorescent in situ sequencing (FISSEQ), and implement direct base-pair fluorescence readout of cDNA amplicons containing barcodes assigned to known transcripts that are visualized throughout the tissue (Figure 1b) (Ke et al. 2013; Lee et al. 2014; Qian et al. 2020). These approaches also enable subcellular resolution and have the potential to enhance readout to a wider range of targets. The use of barcoded padlock probes further enabled the development of STARmap, which improved efficiency of ISS by bypassing the reverse transcription step and introduced three-dimensional (3D) localization of transcripts by immobilizing DNA amplicons in a 3D hydrogel (Wang et al. 2018). However, these methods are inherently limited by the need to target known genes and their small fields of view, and several have yet to be demonstrated outside of their originators’ labs, highlighting their relative inaccessibility.
ISC technologies, in contrast to ISH and ISS, capture transcripts in situ and sequencing is then completed ex situ, leveraging the massively parallel nature of NGS. This modification is additionally advantageous because it enables unbiased capture of the entire transcriptome. There are a variety of ISC approaches, including 10X Genomics Visium, Slide-seq, and Seq-Scope among others, which differ in their specific methods for capturing transcripts from tissue sections (Cho et al. 2021; Rodriques et al. 2019; Ståhl et al. 2016). However, the general approach of ISC involves placing an array of reverse transcription (RT) primers each containing distinct positional nucleotide barcodes (assigned to capture spots) and poly-T sequence for mRNA hybridization on slides (Figure 1c). Tissues are sectioned onto these slides, then either fixed, stained, imaged, and permeabilized (Visium and Seq-Scope) or directly hybridized to the barcoded RT primers (Slide-seq). In the Visium workflow, upon permeabilization, mRNA molecules from the tissue diffuse downward and hybridize to RT primers. After RT, cDNA is extracted and prepared into sequencing libraries. After sequencing, reads are superimposed back onto the tissue image using the positional barcodes for spatial visualization of the transcriptome (hence, ISC can also be referred to as “spatial barcoding”). Ståhl et al. validated their first ISC method (which they named “Spatial Transcriptomics,” a source of confusion when referencing the various technologies) with mouse brain and human breast cancer tissues. This initial method was later developed into the commercialized 10X Visium technology, which incorporates smaller distances between capture spots and spot diameters reduced to 55 μm for improved resolution. Spot diameter limits resolution because multiple cells are encompassed within the same barcoded regions. ISC methods also currently suffer from lower transcript capture efficiency (i.e., depth) than ISH and ISS in addition to scRNA-seq. While recent ISC advancements have increased their resolution down to 0.6 and 2μm-diameter capture spots, one major challenge to overcome will be the tradeoff between RNA capture efficiency and resolution, as decreasing spot size diameter typically hinders efficiency (Vickovic et al. 2019).
Data Analysis for Spatial Transcriptomics
Analysis of ST data follows similar principles for all high-dimensional data analysis. Here, we cover steps of ISC data analysis as a model workflow that is similar to scRNA-seq analysis and can be summarized into two main phases: pre-processing and downstream analysis (Figure 2). However, while ISH and ISS methods feature unique raw data processing steps associated with converting fluorescence signal into transcript quantification, similar pre-processing and downstream analytical methods can be applied thereafter. The goal of pre-processing is to ensure high-quality data can flow into downstream analysis, which subsequently seeks to unearth the biological implications of the data. The steps within pre-processing include but are not limited to quality control (QC), normalization, and dimensionality reduction (Figure 2A). QC metrics such as number of molecules per capture spot (counts per spot) and genes per spot indicate the depth of the data. Normalization accounts for differences in sequencing and capture depth across spots and is further complicated by variations in cellular density across the tissue. The choice of dimensionality reduction method may be based on priority of two main objectives: summarization (e.g., principal component analysis [PCA]) or visualization (e.g. t-distributed stochastic neighbor embedding [t-SNE] and uniform manifold approximation and projection [UMAP]) (Luecken and Theis 2019). Currently, limitations for ST methods, namely low transcriptome coverage for HPRI and low RNA capture efficiency for ISC, typically result in less heterogeneity captured within ST data than within scRNA-seq; thus, researchers may find that additional integration with scRNA-seq will yield more value.
Figure 2. Data Analysis Workflows.
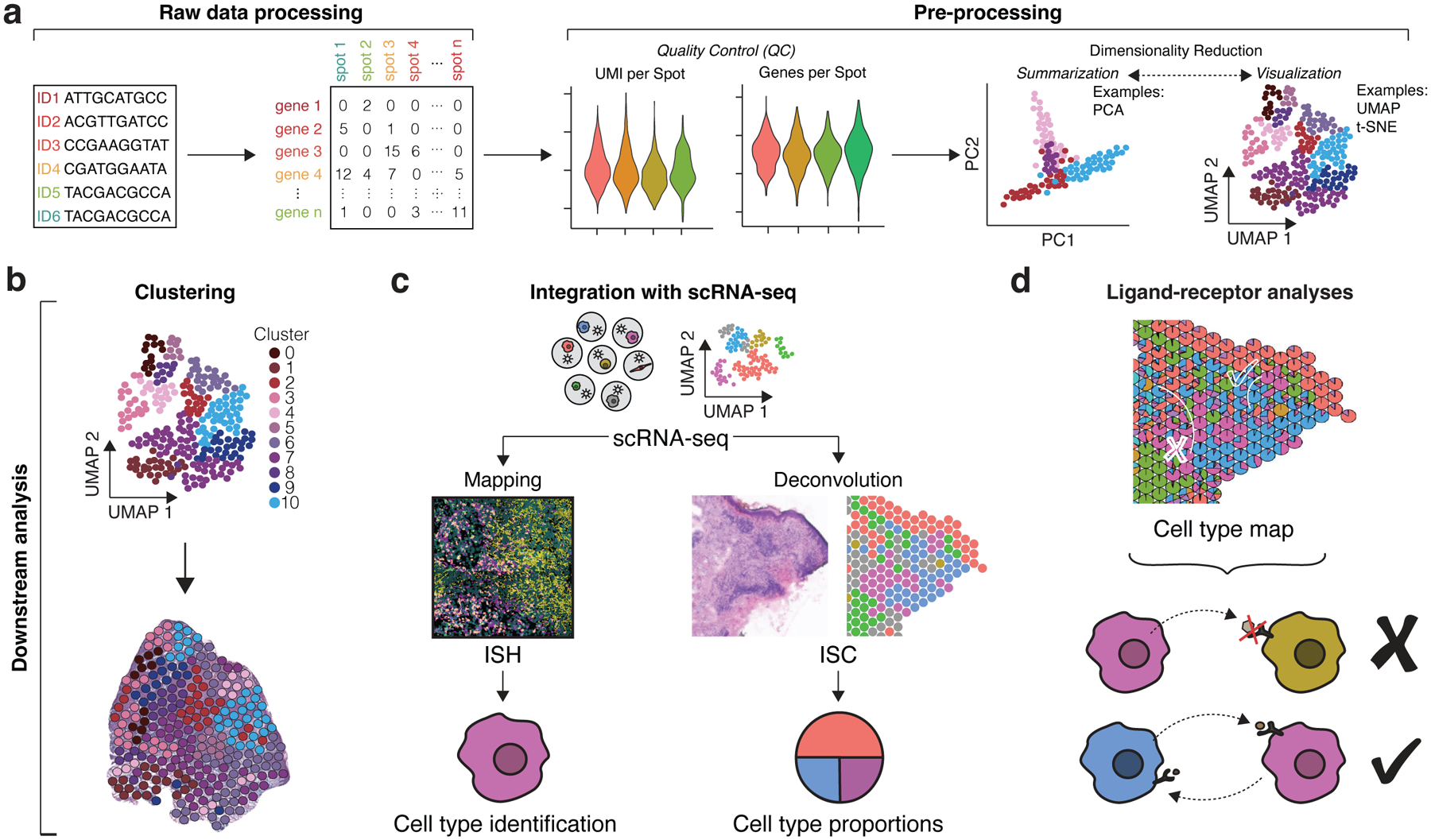
(a) For ISC, raw sequencing data is processed into count matrices consisting of genes and capture spots (this step varies depending on ST technique as ISH/ISS involve converting fluorescent imaging signal into similar count matrices). Pre-processing of matrices involves obtaining quality control metrics such as distributions of UMIs per spot (counts per spot) and genes per spot as well as data normalization. Dimensionality reduction methods include summarization methods (PCA) and visualization methods (UMAP and t-SNE), which both seek to reduce gene expression data into fewer dimensions for more informative analysis. (b) Clustering groups similar spot transcriptomes, which can be transposed over the original tissue images for general interpretation. (c) Mapping and deconvolution combine scRNA-seq data with spatial transcriptomics data to localize cell subpopulations. Mapping typically uses ISH data to localize scRNA-seq profiles and predict specific cell types within the tissue. Deconvolution typically uses ISC data to infer cell type proportions per capture spot. (d) Cell type maps generated from mapping and deconvolution can be applied for ligand-receptor analyses. The proximity of cell types can help infer cell-cell communication events. (c) and (d) were adapted with permission from Ma et al., Nature Immunology 2021.
After pre-processing, downstream analysis for ST data seeks to identify spatial domains with coherent gene expression, such as tissue niches and cell-cell interactions (CCIs) within, or pathogenic domains in diseased tissues where aberrant interactions may occur. This can be accomplished by methods such as clustering as well as those involving integration with scRNA-seq data such as mapping, deconvolution, and ligand-receptor analyses, which informs cell-to-cell communication (Longo et al. 2021) (Figure 2b–2d; Table 3). Clustering compares gene expression profiles to group and identify similar cell types (if the data are single-cell resolution such as HPRI) or patterns of cell type organization such as tissue layers or niches, which cannot be reconstructed using scRNA-seq alone. While non-spatial methods of clustering can be applied to ST data, clustering methods directly incorporating spatial information are rapidly advancing. These include methods that account for neighboring spot information, tissue histology, and cellular morphology to improve the recovery of tissue structure from the data (Biancalani et al. 2021; Dries et al. 2021; Hu et al. 2021; Zhao et al. 2021). Mapping and deconvolution integrate scRNA-seq data with spatial data from either HPRI data or ISC data, respectively. Mapping seeks to assign a cell type resolved by scRNA-seq to its spatial counterpart in HPRI data. Deconvolution seeks to predict the proportion of scRNA-seq cell types present within the mixture of transcripts recovered from each capture spot in ISC data, which can convert multi-cell resolution ISC data to single-cell resolution. Both mapping and deconvolution computational methods are extensively reviewed elsewhere (Longo et al. 2021). A common goal of mapping and deconvolution is to generate cell type maps that can be used for spatially informed ligand-receptor analyses. These analyses look for statistically significant co-expression of ligands and receptors at proximal locations (such as neighboring cell types) and predict the likelihood of cell communication events by accounting for the effect of distance on gene expression (Cang and Nie 2020; Dries et al. 2021). Given the spatial restriction of most juxtracrine and paracrine signaling events, spatially-informed ligand-receptor analyses can maximize predictions of CCIs by eliminating CCIs derived from scRNA-seq analyses alone that are spatially implausible. Computational methods for these tasks are quickly evolving, with different methods better suited for different tasks. Thus, it is important to keep in mind that several methods may need to be applied to accomplish one’s analysis goals.
Table 3.
Analysis Packages for ST Data
| Programming Language | Clustering | Incorporates Histology | Integration with scRNA-seq | CCI Analysis | Additional Features | |
|---|---|---|---|---|---|---|
| Seurat | R | Yes | No | Yes | No |
|
| Giotto | R | Yes | No | Yes | Yes |
|
| BayesSpace | R | Yes | No | No | No |
|
| Tangram | Python | No | Yes | Yes | No |
|
| SpaOTsc | Python | No | No | Yes | Yes |
|
| SpaGCN | Python | Yes | Yes | No | No |
|
For comprehensive overview of data analysis and packages, see Lewis et al. 2021; Longo et al. 2021.
Applications of Spatial Transcriptomics in Dermatologic Research
Given the diversity of cell types present in normal and diseased skin, ST is poised to become a powerful tool for dissecting the relationships among these various cells. Areas in which ST has proven to be valuable thus far are skin cancer and immune responses in infectious skin diseases (Figure 3). Elucidating ligand-receptor and CCIs in these settings offers the potential for biomarker discovery and therapeutic opportunities. Thus far, ISC technologies have emerged as the most frequently used tool in cutaneous research.
Figure 3. ST Applications in Dermatologic Research.
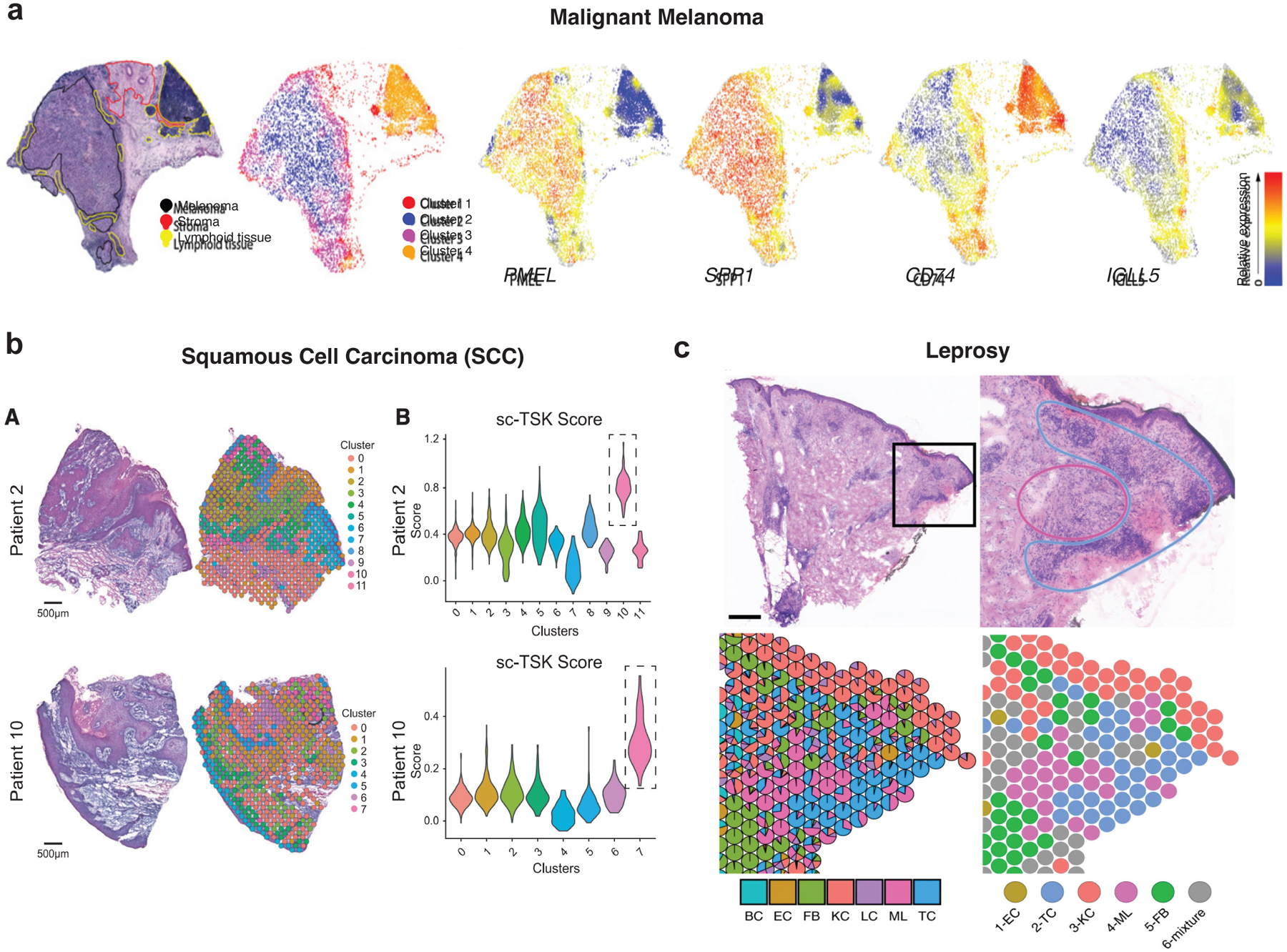
(a) ISC from lymph node biopsy of melanoma. Left, H&E staining with pathologic annotation and clustering. Lymphoid tissue occupies two clusters. Right, Spatial heatmaps of select highly-expressed and variable genes. Adapted with permission from Thrane et al., Cancer Research 2018. (b) Left, H&E staining and clustering of ISC spots in squamous cell carcinoma (SCC). Right, Violin plots of TSK scores of individual spots derived from scRNA-seq data (sc-TSK score) for each cluster. One spatial cluster within each sample demonstrates highest sc-TSK score (dotted boxes), highlighting areas occupied by this subpopulation. Reprinted from Ji et al., Cell 2020 under the CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/legalcode). (c) Leprosy granuloma architecture and antimicrobial ecosystem. Top, H&E staining of a T-lep biopsy; bottom, cell type composition map showing myeloid cells in the granuloma center, while T cells and fibroblasts occupy the periphery. Scale bar: 0.5 mm. BC, B cell; EC, endothelial cell; FB, fibroblast; KC, keratinocyte; LC, Langerhans cell; ML, myeloid cell; TC, T cell. Reprinted with permission from Ma et al., Nature Immunology 2021.
With respect to melanoma research, ST (specifically ISC) was utilized to dissect spatial heterogeneity within lymph node metastases in stage III cutaneous melanoma (Thrane et al. 2018). The authors obtained 2,200 spatial transcriptomes from four patient lymph node biopsies and used PCA and factor analysis to demonstrate significant intertumor heterogeneity of spatial expression patterns across patients. Within an individual biopsy, they observed different expression profiles overlapping regions of lymphoid tissue that were either proximal or distant to tumor cells, suggesting possible influence of the immune composition by tumor cells (Figure 3a). Moreover, this was the very first application of an ISC technology to skin disease.
Another group utilized ST to dissect the tumor microenvironment (TME) of cutaneous squamous cell carcinoma (cSCC). They combined ST (ISC, including Visium) and scRNA-seqon a common cohort of patient tumor samples to identify cSCC tumor and stromal cell populations and the spatial niches in which they resided (Figure 3b). They further mapped ligand-receptor networks potentially operating at the leading edge of tumors (Ji et al. 2020). This multimodal approach discovered a tumor-specific keratinocyte (TSK) population localized in a heterogenous manner at the leading edge of tumors, surrounded by a fibrovascular niche, and identified the most likely ligand-receptor pairs engaged in crosstalk among TSKs, cancer-associated fibroblasts (CAFs), and endothelial cells. The integration of scRNA-seq data with ISC data was essential for prioritizing specific ligand-receptor pairs, some of which were further functionally assessed through in vivo CRISPR screening that identified tumorigenic genes enriched in TSKs and other tumor subpopulations.
ST was also recently utilized to understand the immune response associated with leprosy. Leprosy is histologically characterized by granulomas, organized structures of myeloid cells and lymphocytes surrounding and killing the causative Mycobacterium leprae pathogen, and traverses a clinical spectrum from limited (tuberculoid leprosy, or T-lep) to disseminated (lepromatous leprosy, or L-lep) disease. A recent study examined patient samples obtained from L-lep, T-lep, and reversal reactions (RR), during which patients transition from L-lep toward lesions resembling T-lep either via chemotherapy or spontaneously (Figure 3c) (Ma et al. 2021). Utilizing both scRNA-seq and Visium, the authors found that at least two subpopulations of macrophages occupied the central zone of RR granulomas, surrounded by T cells and dendritic cells in the mantle zone periphery. Multiple cell types appeared to contribute to the antimicrobial response in RR, which was driven by interferon-γ and interleukin-1β produced by lymphocytes (TH17 and cytotoxic T cells) and dendritic cells (including Langerhans cells), respectively. Thus, ISC enabled reconstruction of a spatially accurate map of the architecture within leprosy granulomas, allowing for deeper analysis of the contributing factors to the immune response associated with this particular skin disease.
While several aforementioned studies utilized the combination of scRNA-seq and ISC on matched tissues, additional molecular and genomics assays can be integrated to yield further insights. By combining ISC along with lineage tracing and bulk/single-cell RNA-seq and assay for transposase-accessible chromatin with sequencing (ATAC-seq), a recent study characterized wound-associated fibroblast populations in a mouse model of cutaneous wound healing (Foster et al. 2021). They identified four subpopulations of fibroblasts: “activated-responder”, “mechano-fibrotic”, “proliferator”, and “remodeling,” and localized them within the wound by applying Visium across various timepoints for 2 weeks after wounding. Integration of single cell genomics data with Visium data enabled imputation of chromatin accessibility changes within fibroblasts through space and time within the healing wound, providing deeper resolution and nuance to the three “classical” wound-healing stages of inflammation, proliferation, and remodeling. Notably, Visium also identified macrophages in the wound center after one week of healing, highlighting additional insights gained from unbiased transcript capture associated with ISC. Thus, the integration of ISC with scRNA-seq and/or other assays can compensate for the current limited capture efficiency of ISC and facilitate detailed spatiotemporal characterization of a coordinated multicellular and multi-state process such as skin wound repair.
Conclusions and Future Directions
ST can provide a high-dimensional and high-resolution approach to investigating in situ tissue dynamics. Each ST technology has its own tradeoffs in terms of accessibility, transcriptome coverage and depth, field of view, and spatial resolution. These factors, along with the specific biological or clinical question, are all considerations when selecting from the various ST assays currently available. Further synergy can be achieved by integrating techniques with deeper transcriptome coverage, such as scRNA-seq, on cells dissociated from the same tissue. Similar to the diversity of ST assays available, a variety of computational methods also exist to perform different tasks for ST data analysis, including clustering, integration with scRNA-seq data, and CCI predictions. In assays where visual information is concurrently obtained, such as H&E staining during the Visium workflow, efforts to incorporate information such as the density or morphology of cells using deep learning approaches continue to emerge (Bergenstråhle et al. 2020; Pham et al. 2020). These methods may help overcome current normalization challenges and extract additional insights from these data. Despite the shortcomings of current ST technologies, we envision their broad utility in dermatologic research by elucidating the organization of various cell types present in skin homeostasis and disease and aiding in characterization of CCIs.
Supplementary Material
Summary Points.
Spatial transcriptomics (ST) encompass a variety of methods aimed to quantify RNA expression directly from intact tissues.
ST methods include in situ hybridization (ISH), in situ sequencing (ISS), and in situ capturing (ISC) technologies, each with their own strengths and limitations.
ST methods can be combined with single-cell RNA-sequencing (scRNA-seq) to maximize resolution and depth of data.
Analysis of ST can localize cell types of interest, identify organizational patterns, i.e., niches of cell types within tissues, and reveal potential cell-cell interactions.
Advantages.
ST offers high-throughput profiling of hundreds to thousands of genes simultaneously within intact tissue in their native context, preserving the spatial positioning of transcripts
Ability to assess spatial organization of cell types within tissue niches and cell-cell interactions (CCIs)
Availability of a wide range of computational analysis tools that can also facilitate integration with scRNA-seq data
Disadvantages.
Each ST method has its own limitations in resolution, capture efficiency, and/or transcriptome coverage, and the burden of choice falls on the investigator
Most ST methods are optimized for fresh frozen tissue, while adaptation of protocols to formalin-fixed paraffin-embedded (FFPE) tissue has lagged
Each tissue of interest may require additional optimization of assay parameters
High cost, labor, and/or need for specialized equipment has thus far limited widespread adoption
Wide range of analysis methods can be difficult to navigate for finding the method best suited for the investigator’s needs
Acknowledgements
This work was supported by the National Institutes of Health K08CA263187 to A.L.J.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflicts of interest.
CRediT Author Statement
Writing – Original draft preparation: A.J.P, A.E.H; Writing – reviewing and editing: A.L.J; Supervision: A.L.J.
References
- Asp M, Bergenstråhle J, Lundeberg J. Spatially Resolved Transcriptomes—Next Generation Tools for Tissue Exploration. BioEssays. 2020;42(10):1–16 [DOI] [PubMed] [Google Scholar]
- Bergenstråhle L, He B, Bergenstråhle J, Andersson A, Lundeberg J, Zou J, et al. Super-resolved spatial transcriptomics by deep data fusion. bioRxiv. 2020;2020.02.28.963413 Available from: https://www.biorxiv.org/content/10.1101/2020.02.28.963413v1.full.pdf+html [DOI] [PubMed] [Google Scholar]
- Biancalani T, Scalia G, Buffoni L, Avasthi R, Lu Z, Sanger A, et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson-Maduro J, Baron CS, et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 2017;14(10):935–6 [DOI] [PubMed] [Google Scholar]
- Cang Z, Nie Q. Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat. Commun Springer US; 2020;11(1):1–13 Available from: 10.1038/s41467-020-15968-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science (80-. ) 2015;348(6233):1360–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C, Xi J, Si Y, Jun G, Kang HM, Lee JH, et al. Microscopic examination of spatial transcriptome using Seq-Scope. Cell. Elsevier Inc; 2021;184:1–14 Available from: 10.1016/j.cell.2021.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dries R, Zhu Q, Dong R, Eng CHL, Li H, Liu K, et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. Genome Biology; 2021;22(1):1–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CHL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature. Springer US; 2019;568(7751):235–9 Available from: 10.1038/s41586-019-1049-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DS, Januszyk M, Yost KE, Chinta MS, Gulati GS, Nguyen AT, et al. Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proc. Natl. Acad. Sci. U. S. A 2021;118(41):1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Li X, Coleman K, Schroeder A, Ma N, Irwin DJ, et al. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods Springer US; 2021; Available from: 10.1038/s41592-021-01255-8 [DOI] [PubMed] [Google Scholar]
- Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell. 2020;497–514 Available from: 10.1101/827071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke R, Mignardi M, Pacureanu A, Svedlund J, Botling J, Wählby C, et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 2013;10(9):857–60 [DOI] [PubMed] [Google Scholar]
- Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, et al. Highly Multiplexed Subcellular RNA Sequencing in Situ. Science (80-. ) 2014;343(6177):1360–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SM, Asselin-Labat M-L, Nguyen Q, Berthelet J, Tan X, Wimmer VC, et al. Spatial omics and multiplexed imaging to explore cancer biology. Nat. Methods. Springer US; 2021; Available from: 10.1038/s41592-021-01203-6 [DOI] [PubMed] [Google Scholar]
- Longo SK, Guo MG, Ji AL, Khavari PA. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet Springer US; 2021;0123456789 Available from: 10.1038/s41576-021-00370-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods Nature Publishing Group; 2014;11(4):360–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol 2019;15(6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F, Hughes TK, Teles RMB, Andrade PR, de Andrade Silva BJ, Plazyo O, et al. The cellular architecture of the antimicrobial response network in human leprosy granulomas. Nat. Immunol Springer US; 2021;22(7):839–50 Available from: 10.1038/s41590-021-00956-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham D, Tan X, Xu J, Grice LF, Lam PY, Raghubar A, et al. stLearn: Integrating spatial location, tissue morphology and gene expression to find cell types, cell-cell interactions and spatial trajectories within undissociated tissues. bioRxiv. 2020; [Google Scholar]
- Qian X, Harris KD, Hauling T, Nicoloutsopoulos D, Muñoz-Manchado AB, Skene N, et al. Probabilistic cell typing enables fine mapping of closely related cell types in situ. Nat. Methods Springer US; 2020;17(1):101–6 Available from: 10.1038/s41592-019-0631-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science (80-. ) 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Lubeck E, Zhou W, Cai L. In Situ Transcription Profiling of Single Cells Reveals Spatial Organization of Cells in the Mouse Hippocampus. Neuron. Elsevier Inc; 2016;92(2):342–57 Available from: 10.1016/j.neuron.2016.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science (80-. ) 2016;353(6294):78–82 [DOI] [PubMed] [Google Scholar]
- Thrane K, Eriksson H, Maaskola J, Hansson J, Lundeberg J. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res. 2018;78(20):5970–9 [DOI] [PubMed] [Google Scholar]
- Xia C, Fan J, Emanuel G, Hao J, Zhuang X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl. Acad. Sci. U. S. A 2019;116(39):19490–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickovic S, Eraslan G, Salmén F, Klughammer J, Stenbeck L, Schapiro D, et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods Springer US; 2019;16(10):987–90 Available from: 10.1038/s41592-019-0548-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. 2018;361(eaat5691) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Yang B, Udo-Inyang I, Ji S, Ozog D, Zhou L, et al. Research Techniques Made Simple: Single-Cell RNA Sequencing and its Applications in Dermatology. J. Invest. Dermatol 2018;138(5):1004–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao E, Stone MR, Ren X, Guenthoer J, Smythe KS, Pulliam T, et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol Springer US; 2021; Available from: 10.1038/s41587-021-00935-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.