

Empagliflozin, a sodium-glucose cotransporter-2 (SGLT2) inhibitor has been recently shown to improve cardiovascular outcomes in patients with heart failure and preserved ejection fraction (HFpEF), independent of diabetes status.1 In addition to its advantageous renal and hemodynamic effects, empagliflozin can directly affect heart cells, and preclinical studies demonstrated empagliflozin effects in ex vivo perfused hearts and isolated cardiomyocytes.2 However, the exact mechanism by which empagliflozin affects the heart remains elusive. In cardiomyocytes, SGLT2 expression is virtually absent, suggesting off-target beneficial effects. While controversial, the sodium-hydrogen exchanger-1 has been suggested as a potential empagliflozin target in cardiomyocytes, especially during ischemia. Recently, Philippaert et al.3 showed that empagliflozin directly inhibited late Na+ current (INaL) in a transverse aortic constriction-induced heart failure murine model and in recombinant human NaV1.5 channels in HEK cells carrying long QT3 mutations or treated with H2O2. Moreover, empagliflozin reduces reactive oxygen species (ROS) production2 and Ca2+/calmodulin-dependent kinase II (CaMKII) activity,4 important known regulators of INaL in heart diseases. Here, we tested for empagliflozin effects on INaL and consequent action potential (AP) changes in a translational two-hit murine model of HFpEF.5
HFpEF was induced in C57BL/6J wild-type mice (male, 10-wk-old, Jackson Laboratory) by combining high-fat diet (D12492, Research Diets) and inhibition of nitric oxide synthases with L-NG-nitroarginine methyl-ester (L-NAME, 0.5 g/L, Sigma-Aldrich) in drinking water, while control mice were fed normal chow (5k52, LabDiet) and water. After 15 weeks of HFpEF treatment, consistent with previous report,5 morphometric and echocardiographic evaluations (Figure [A]) revealed significant obesity, concentric cardiac hypertrophy, preserved ejection fraction, and significant diastolic dysfunction, as demonstrated by increased heart mass, left ventricular remodeling index, and elevated E/e’. Empagliflozin effects were studied in freshly isolated left ventricular myocytes using patch-clamp at 37°C. In voltage-clamp INaL measurements, pipette solution contained (mmol/L): 110 CsCl, 20 tetraethylammonium chloride, 5 MgATP, 10 HEPES, 5 phosphocreatine-Na2, 0.0001 calmodulin, 10 EGTA, 4.1 CaCl2 (free [Ca2+]=100 nmol/L), pH=7.20. Bath solution contained (mmol/L): 140 NaCl, 4 CsCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, 5 Na-HEPES, 5.5 glucose, 5 4-aminopyridine, 0.01 nifedipine, pH=7.40.
Figure. Empagliflozin reduces late Na+ current and action potential prolongation in a preclinical model of heart failure with preserved ejection fraction (HFpEF).
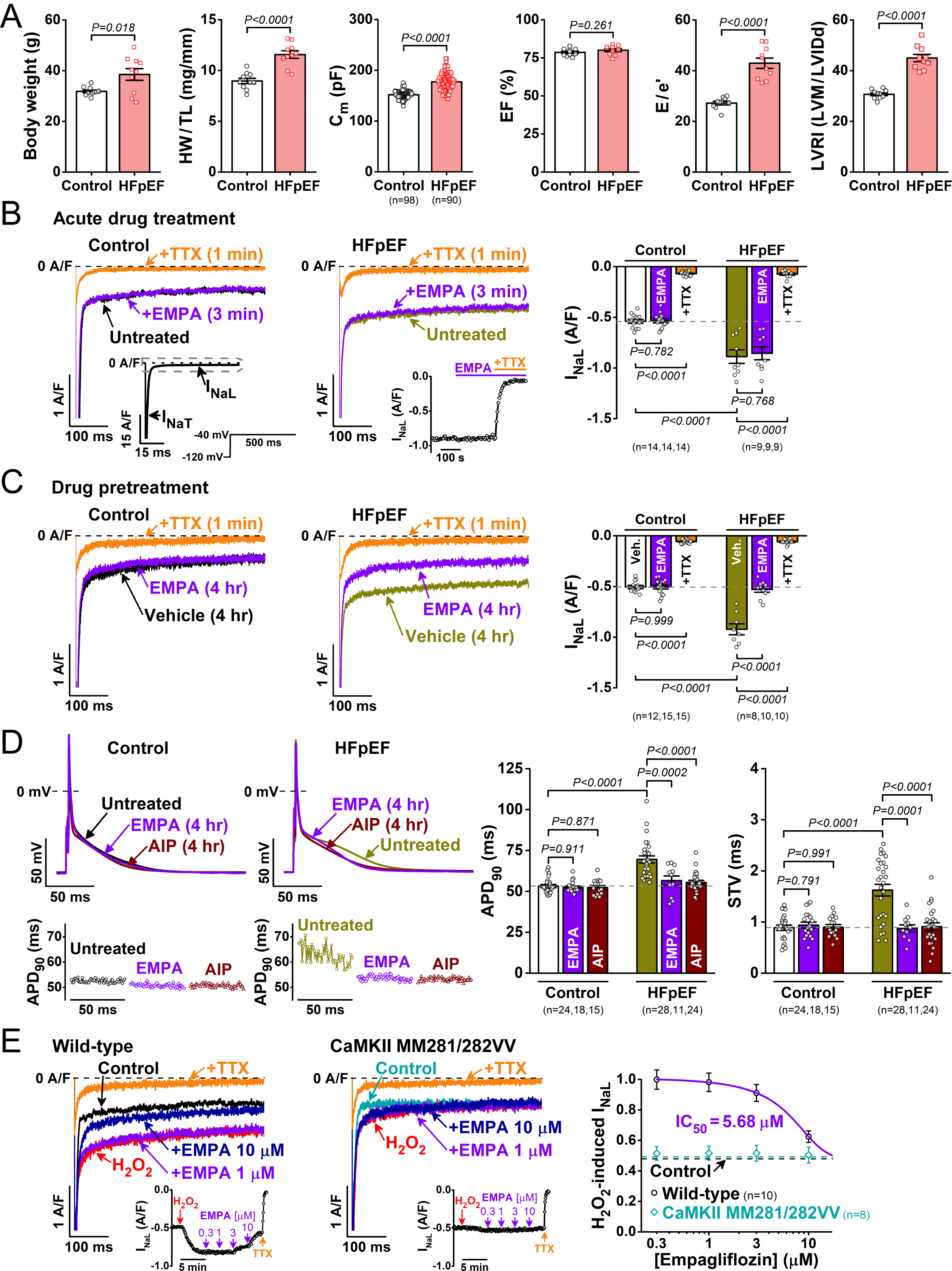
Wild-type mice (10-wk-old) on high-fat diet+L-NG-nitroarginine methyl ester (L-NAME) for 15 weeks were compared to those fed normal chow (N=10 per group). A, Body weight, heart weight to tibial length ratio (HW/TL), and membrane capacitance (Cm) of single ventricular cardiomyocytes. Left ventricular (LV) ejection fraction (EF), ratio between mitral E wave and e’ wave (E/e’), and LV remodeling index LVRI (LV mass/LV end-diastolic internal diameter, LVM/LVIDd). B, Representative late Na+ current (INaL) traces in control and HFpEF myocytes following acute empagliflozin (EMPA) and subsequent tetrodotoxin (TTX) applications. Insets shows voltage protocol, comparison of peak versus late INa, and the time course of EMPA and TTX effects on INaL. Peak INa was off-scale as shown. C, Four hour preincubation with empagliflozin reversed INaL upregulation in HFpEF. D, Action potential (AP) measurements in HFpEF myocytes paced at 1 Hz. Top left, Representative AP traces in control and HFpEF myocytes without or with preincubation with empagliflozin and autocamtide-2-related inhibitory peptide (AIP). Bottom left, Fifty consecutive AP durations at 90% repolarization (APD90) for control and HFpEF as above. Right, Average data on APD90 and short-term variability of APD90 (STV). E, Concentration-dependent acute effect of empagliflozin on H2O2 (100 μmol/L, 5 min)-induced INaL in wild-type and CaMKII MM281/282VV (N=4 per group). IC50, half-maximal inhibitory concentration. Data are presented as mean±SEM. Two-tailed Welch’s t-test was used for A. ANOVA followed by Tukey’s multiple-comparisons test was used for B, C, and D. All data, P values and n numbers are shown.
Acute empagliflozin treatment (1 μmol/L, 3 min), in contrast to a previous report,3 did not change INaL in control and HFpEF, whereas tetrodotoxin (TTX, 10 μmol/L) readily inhibited INaL in both cases (Figure [B]). Importantly, INaL density (current amplitude normalized to cell capacitance) was significantly increased in HFpEF. However, empagliflozin preincubation (4 hr) versus vehicle (0.01% dimethyl sulfoxide) reversed the TTX-sensitive INaL upregulation in HFpEF to the control level but had no effects in control myocytes (Figure [C]).
We also measured APs to assess antiarrhythmic properties of empagliflozin. For physiological AP measurements, Cs+ was replaced with K+ in pipette and bath solutions without K+ and Ca2+ current inhibitors. Pipette EGTA was reduced to 10 μmol/L and CaCl2 was omitted. In line with upregulated INaL, myocyte AP duration (APD) was significantly prolonged, and the short-term APD-variability (STV) was markedly increased in HFpEF. These proarrhythmic APD changes were reversed in cells preincubated with empagliflozin (Figure [D]). Because CaMKII is an important regulator of INaL in heart diseases, we also tested the effect of cell-pretreatment with the selective CaMKII inhibitor autocamtide-2-related inhibitory peptide (AIP, myristoylated, 1 μmol/L). Importantly, CaMKII inhibition also reversed APD prolongation and temporal variability in HFpEF (Figure [D]).
Finally, we measured concentration-response effects of empagliflozin on ROS-promoted INaL. Empagliflozin acutely inhibited the H2O2-induced INaL, but significantly only above 3 µmol/L with a half-maximal inhibition (IC50) at 5.68 μmol/L (Figure [E]). Critically, myocytes with oxidation-resistant mutations in CaMKII (MM281/282VV) prevented both the H2O2-induced increase in INaL and the empagliflozin effect (Figure [E]).
Our data show that INaL is elevated in a preclinical murine model of HFpEF and that empagliflozin at a clinically-relevant 1 μmol/L concentration reverts INaL upregulation and arrhythmogenic AP changes. However, this empagliflozin effect on the Na+ channel in HFpEF is not acute inhibition (unlike the direct acute selective INaL blocker TTX) but requires drug preincubation, and this effect is mimicked by CaMKII inhibition and absent when CaMKII lacks two critical oxidation sites. Moreover, empagliflozin acutely inhibits H2O2-induced INaL with a 7 times higher IC50 in cardiomyocytes than in HEK cells3 (5.68 μmol/L versus 0.79 μmol/L). Thus, we suggest that empagliflozin likely regulates INaL in HFpEF by an indirect mechanism, which may involve CaMKII. That agrees with results that empagliflozin treatment for 24 hr (but not 30 min) suppresses CaMKII activity and sarcoplasmic reticulum Ca2+ leak, and enhanced Ca2+ transients.4 Mechanisms could include reduced ROS and its ability to activate CaMKII. While the key molecular targets of empagliflozin in cardiomyocytes remain unclear, INaL and CaMKII suppression may be important mediators of its beneficial effects that merit further investigation. Our study is limited in that we studied empagliflozin effects in isolated murine myocytes and focused on cell electrophysiology. Further studies are required to confirm these findings in human cardiomyocytes and on other phenotypical aspects of HFpEF.
All procedures involving animals were approved by institutional and national authorities. All supporting data are available within the article.
Acknowledgments
We thank Mark E. Anderson (Johns Hopkins University) for providing CaMKIIδ-MM281/282VV knock-in mice, Daria Smoliarchuk, Avery Mandel, Emily Spencer, and Adam Wilder for their help in animal care, cell isolation, and laboratory tasks.
Sources of Funding
This work was supported by grants from the National Institutes of Health: P01-HL141084 (Bers) and R01-HL142282 (Bers and Bossuyt), and Minciencias – Fulbright Colombia Scholarship (Mira Hernandez).
Footnotes
Disclosures
None.
REFERENCES
- 1.Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Bohm M, Brunner-La Rocca HP, Choi DJ, Chopra V, Chuquiure-Valenzuela E, et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N Engl J Med 2021;385:1451–1461. doi: 10.1056/NEJMoa2107038. [DOI] [PubMed] [Google Scholar]
- 2.Cowie MR and Fisher M. SGLT2 inhibitors: mechanisms of cardiovascular benefit beyond glycaemic control. Nat Rev Cardiol 2020;17:761–772. doi: 10.1038/s41569-020-0406-8. [DOI] [PubMed] [Google Scholar]
- 3.Philippaert K, Kalyaanamoorthy S, Fatehi M, Long W, Soni S, Byrne NJ, Barr A, Singh J, Wong J, Palechuk T, et al. Cardiac Late Sodium Channel Current Is a Molecular Target for the Sodium/Glucose Cotransporter 2 Inhibitor Empagliflozin. Circulation 2021;143:2188–2204. doi: 10.1161/CIRCULATIONAHA.121.053350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mustroph J, Wagemann O, Lucht CM, Trum M, Hammer KP, Sag CM, Lebek S, Tarnowski D, Reinders J, Perbellini F, et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail 2018;5:642–648. doi: 10.1002/ehf2.12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019;568:351–356. doi: 10.1038/s41586-019-1100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]