

Abstract
Peroxisome proliferator activated receptor alpha (PPAR-α) deletion has been shown to increase blood pressure (BP). We hypothesized that the BP increase in PPAR-α KO mice was mediated by increased expression and activity of basolateral Na+/K+ ATPase (NKA) pump. To address this hypothesis, we treated wild-type (WT) and PPAR-α knockout (KO) mice with a slow-pressor dose of angiotensin II (400 ng/kg·min) for 12 days by osmotic minipump. Radiotelemetry showed no significant differences in baseline mean arterial pressure (MAP) between WT and PPAR-α KO mice; however, by day 12 of infusion, MAP was significantly higher in PPAR-α KO mice (156±16) compared to WT mice (138±11 mmHg). NKA activity and protein expression (α1 subunit) were significantly higher in PPAR-α KO mice compared to WT mice. There was no significant difference in NKA mRNA levels. Angiotensin II further increased the expression and activity of the NKA in both genotypes along with the water channel, aquaporin 1 (Aqp1). In contrast, angiotensin II decreased the expression (64-97% reduction in band density) of sodium-hydrogen exchanger-3 (NHE3), NHE regulatory factor-1 (NHERF1, Slc9a3r1), sodium-potassium-2-chloride cotransporter (NKCC2), and epithelial sodium channel (ENaC) β- and γ- subunits in the renal cortex of both WT and PPAR-α KO mice, with no difference between genotypes. The sodium-chloride cotransporter (NCC) was also decreased by angiotensin II, but significantly more in PPAR-α KO (59% WT versus 77% KO reduction from their respective vehicle-treated mice). Our results suggest that PPAR-α attenuates angiotensin II-mediated increased blood pressure potentially via reducing expression and activity of the NKA.
Keywords: PPAR-α, angiotensin II, hypertension, natriuresis, inflammation
Introduction
Peroxisomal Proliferator Activated Receptor-α (PPAR)- α, a nuclear transcription factor, is a member of a sub-family of nuclear receptors primarily involved in the regulation of lipid metabolism in peroxisomes of the cell [1]. PPAR-α are bound by omega-3-fatty acids, and the therapeutic class of drugs, fibrates [1,2]. Stimulation of this receptor leads to an increase in fatty acid β-oxidation and reduction in circulating triglycerides. PPAR-α is highly expressed in kidney, and its agonists have been shown to lower arterial blood pressure during angiotensin II-induced hypertension [3].
The dose of angiotensin II used during the infusion is a highly critical determinant of how the kidney reacts. The slow-pressor model of angiotensin II infusion involves a “relatively” lower infusion dose that results in changes in renal function that precede increases in conscious blood pressure, i.e., 10 – 13 days after subcutaneous infusion [4,5]. Therefore, during the “slow-pressor dose” infusion of angiotensin II, the blood pressure increase is thought to involve sodium retention, in addition to vascular constriction [6].
Previous results from our laboratory demonstrated that a slow-pressor dose of angiotensin II caused a greater rise in mean arterial pressure (MAP) in PPAR-α knockout (PPAR-α KO) mice versus wild type (WT) control mice. Fenofibrate, a PPAR-α agonist, attenuated angiotensin II-stimulated blood pressure rise in wild-type (WT) mice. It also increased renal expression of CYP4A and cytochrome P450 arachidonic acid epoxygenase (CYP2J2) expression, while reducing elevation in renal inflammatory mediators such as intracellular adhesion molecule-1 (ICAM-1), monocyte chemoattractant protein-1 (MCP-1), interleukin-6 (IL-6) and COX-2 in these mice. [7].
Although angiotensin II can bind to both natriuretic and anti-natriuretic receptors, it has stimulatory effects on sodium reabsorption primarily via the AT1 receptors [8]. In general, changes in sodium reabsorptive activity of the renal tubule are mediated by alterations in the activity or appearance of sodium reabsorption transporters, exchangers, and channels on the apical plasma membrane. In this regard, angiotensin II has been shown to increase abundance and/or activity of the distal convoluted tubule (DCT) sodium-chloride cotransporter (NCC; Slc12a3) and the cortical collecting duct (CD) epithelial sodium channel (ENaC; Scnn1b and Scnn1g) [9–14], as well as, decrease the abundance of the proximal tubule (PT) sodium-hydrogen exchanger-3 (NHE3; Slc9a3) and the thick ascending loop of Henle (TALH) Na-K-2Cl cotransporter 2 (NKCC2, Slc12a1) [11]. In rats, a 14-day infusion of angiotensin II caused a decrease in NHE3 expression in both cortex and medulla, whereas the more distal NKCC2 and NCC were increased in cortex. The phosphorylated form (active) of NKCC2 was also increased by angiotensin II in cortex [15]. In mice, an acute angiotensin II (in the absence of blood pressure changes) infusion stimulated the trafficking of NHE3 and sodium phosphate co-transporter 2 (NaPi2) into the proximal tubule (PT) brush-border microvilli [16].
However, recently the role of enhanced Na+/K+ ATPase (a basolateral enzyme) in mediating overall sodium-reabsorptive activity has received renewed attention. Many apical transporters are dependent upon the activity of Na+/K+ ATPase to maintain intracellular Na+. Abnormalities in regulation of Na+/K+ ATPase activity have been implicated in several forms of hypertension [17]. In renal tubules Na+/K+ ATPase is regulated by angiotensin II [18], parathyroid hormone and dopamine [19], PGE2 [20] and aldosterone [21]. However, regulation of Na+/K+ ATPase by PPAR-α is controversial. In one study, Jackson et al. showed increase in Na+/K+ ATPase α1 subunit expression by PPAR-α agonists in a chronic kidney perfusion model [22]. In another study, Newaz et al. showed an increase in Na+/K+ ATPase α1 subunit expression but a decrease in Na+/K+ ATPase activity by clofibrate, a PPAR-α agonist [23].
Regarding the major water channels that line renal epithelium, angiotensin II has been shown to increase the expression of the proximal tubular and thin-limb specific aquaporin-1 (Aqp1) in vitro, and in vivo. These studies implied a direct role of angiotensin II through the AT1 receptor, providing an important regulatory mechanism to link proximal tubular water reabsorption to body fluid homeostasis via the renin-angiotensin system [24].
Based on the above studies, the aim of the present study is to determine the effects of PPAR-α expression on angiotensin II mediated changes in major sodium transporters/channels/exchangers, and pumps in the kidney. We hypothesized that the BP increase in PPAR-α KO mice was mediated by increased sodium reabsorption, i.e., due to anti-natriuretic actions mediated by increased expression and activity of one or more of the sodium transporters/ channels/ exchangers that line the renal epithelium.
Methods
Animals:
Procedures involving animals were approved by the Howard University Institutional Animal Care and Use Committee. Age-matched (10–12 weeks, 25–28 grams) male PPAR-α knockout (B129S4/SvJae-Pparatm1Gonz/J) and wild type mice (B129S1/SvImJ) were obtained from Jackson Laboratories (Bar Harbor, ME, USA). For metabolic studies, mice were acclimatized in mouse metabolic cages for 3 days prior to measurements. Mice had access to water and food ad libitum while in these cages. Food intake (weight) was recorded, and urine was collected during baseline conditions and on days 10 - 12 of angiotensin II infusion. Sodium balance was calculated by using the following formula: sodium balance = sodium intake – urinary sodium excretion – insensitive sodium loss.
Blood pressure measurements:
Mice were anesthetized using isoflurane and radiotelemetry transmitter devices (Data Science, PA-C10, St. Paul, MN, USA) were implanted using aseptic techniques. The catheter was implanted in the left carotid artery through an incision in the vessel wall made with a custom-shaped 27.5-gauge needle. The body of the transmitter was tunneled subcutaneously above the right shoulder and secured above the scapula. The incisions were infiltrated with 1% lidocaine, and mice were placed in warm cages to recover from surgery. All mice were individually housed in a shoebox cage and transferred to a light (with 12-hour light/dark cycles) and temperature-controlled room in the Howard University animal facility. Food and water were available ad libitum. Mice were given 7 days to recover from surgery before baseline mean arterial pressure (MAP), heart rate, and locomotor activity were recorded for at least 3 – 5 days. MAP data were collected at 500 Hz for 5s each minute, from 3 PM until 10 AM (i.e., 19 h)/day. The 3 PM to 6 PM period and the 6 AM to 10 AM period together (7 h) were analyzed as “day” MAP, and the 6 PM to 6 AM period (12 h) was “night.”
Osmotic minipump implantation:
Using isoflurane anesthesia, osmotic minipumps (Alzet, Durect, Cupertino, CA, USA) were implanted subcutaneously to deliver either vehicle (saline) or angiotensin II for 14 days at a rate of 400 ng/kg bw/min [25] a week after implantation of radiotelemetry transmitter following collection of baseline MAP.
Kidney sample preparation:
After the 12-days infusion, mice were euthanized under isoflurane anesthesia. Kidneys were decapsulated, flash frozen in liquid nitrogen and were transferred to −80°C until use. Crude membranes were prepared as described previously [26]. Briefly, kidney cortex was carefully separated from medulla. Whole cortex was homogenized in 50 mM mannitol, 5 mM Tris-HCl, pH 7.4. The homogenate was centrifuged at 2500xg, and the supernatant was again centrifuged at 30,000xg. The pellet (crude membrane) was resuspended in 300 mM mannitol, 5 mM Tris-HCl, pH 7.4. Protein was determined by bicinchoninic acid using bovine serum albumin, as standard.
Western blotting:
Blotting was performed as described previously [26]. Briefly, 25 μg crude membrane proteins were mixed with Laemmli buffer and boiled at 95°C for 5 min. The proteins were separated by 10% SDS PAGE using a BioRad Tris-Glycine-SDS gel electrophoresis system. The proteins were electrophoretically transferred to nitrocellulose membrane at 120 volts for 90 min. using the wet BioRad transfer system. The nitrocellulose membrane was incubated in 5% non-fat dry milk in Tris-buffered saline containing 0.05% Tween-20 (TTBS) to block non-specific proteins. The nitrocellulose membrane was then incubated overnight at 4°C with the respective primary antibodies (1:1000) in 5% non-fat dry milk in TTBS. The nitrocellulose membranes were washed 4 times (10 min each time) with TTBS, then incubated for 1 hr at room temperature with species appropriate horseradish peroxidase (HRP)-linked secondary antibodies (1:5000) together with HRP-linked anti-β actin antibodies (1:10,000; Santa Cruz Biotech, Santa Cruz, CA). Next, the nitrocellulose membranes were washed four times (10 min each time) in TTBS. The signal was detected using Super Signal (Thermo Fisher) in a BioRad Universal Hood II Gel documentation system. Rabbit polyclonal antibodies against sodium transporters and channel subunits and aquaporin-1 were provided by Dr. Mark Knepper (NHLBI, Bethesda, MD).
Plasma sodium:
On the day of euthanasia, blood was collected from the carotid artery and serum was collected after overnight coagulation. Urine was collected before the infusion of angiotensin II and a day before euthanizing the mice. Plasma sodium was measured directly in the plasma and the urine samples were diluted 1:10 with Medica diluent and measured with an EasyLyte Plus analyzer (Medica, Bedford, MA), following the manufacturer’s protocol.
Na+/K+ ATPase Activity:
Na+/K+ ATPase activity was measured as ouabain-sensitive ATPase as described previously [19]. Briefly, the kidney homogenates (50 μg protein) were incubated for 15 min at 37°C in medium containing in a final volume of 0.2 ml (final concentration in mM) 5 ATP, 120 NaCl, 10 KCl, 10 MgSO4, and 50 Tris-HCl, pH 7.6, with or without 5 mM ouabain. The reaction was terminated with 25μl 30% TCA. A calibration curve was prepared simultaneously with the test samples, using known concentrations of KH2PO4 (50–500 nmol Pi) and 5 mM ATP. The samples and standards were centrifuged at 14000xg for 5 min. The inorganic phosphate (Pi) released by conversion of ATP to ADP and Pi was determined according to the method of Taussky and Shorr [27]. Briefly, to 100 μl of the protein free supernatant, 100 μl of ferrous sulfate reagent [5 gm FeSO4 dissolved in 10% (wt/vol) ammonium molybdate in 10 N H2SO4] was added. The blue color obtained was read at 820 nm after a 20-min incubation at room temperature in a plate reader against a reagent blank. The difference in the ATPase activity (Pi released) assayed in the absence and presence of ouabain is taken as a measure of Na+-K+ ATPase activity. Na+-K+ ATPase activity is expressed as nanomoles Pi released per milligram protein per minute.
qRT-PCR: NKA α1 mRNA from wild-type (WT) and PPAR-α knockout (KO) mice kidneys was measured by quantitative real-time PCR (qRT-PCR) using the QuantStudio 3 real-time PCR (ThermoFisher). The kidneys were homogenized and total RNA from the WT, WT + Ang II, PPAR-α KO and PPAR-α KO + Ang II were extracted using Trizol (Thermo Fisher Scientific, Waltham, MA). RNA concentrations were measured on a NanoDrop 2000 (Thermo Fisher Scientific). First strand cDNA was made from 2 mg RNA using a cDNA kit from Thermo Fisher (Superscript, VILO cDNA Synthesis kit). The real-time PCR mixture consisted of RNase-free water, TaqMan Fast Advance Master Mix (Thermo Fisher Scientific), and the Na+/K+-ATPase α-1 (Atp1a1) (Mm00523255_m1, Thermo Fisher) specific primers (300 nM), β-actin (Mm00607939_s1, Thermo Fisher, 10 μM) or 18s (Mm03928990_g1, Thermo Fisher, 10 μM) RNA primers, and cDNA samples. PCRs without reverse transcription and no template were included to control for genomic DNA contamination. The data are expressed in ΔΔCt values relative to 18S or β-actin expression.
Statistics:
Radiotelemetry data were analyzed with a two-factor, repeated-measures ANOVA. Significant F-tests from the ANOVA at P < 0.05 was followed by post hoc comparisons using the Bonferroni test. Western blotting and metabolic data were analyzed by 2-way ANOVA (genotype X treatment) followed by Tukey’s multiple comparisons test (p < 0.05 considered significant).
Results
Effect of angiotensin II on blood pressure in WT and PPAR-α KO mice:
Baseline MAP did not differ between the genotypes; however, by days 11-12 of the angiotensin II infusion, MAP was significantly higher (p < 0.05 n = 6/group) in the PPAR-α KO mice as compared to WT mice (Figure 1). The plasma sodium concentrations and measured dietary sodium intakes were not different between the groups (Table 1). Likewise, our metabolic data demonstrate that sodium intake and urinary volumes were not different between groups during the control period and increased similarly in all groups after the implantation of the angiotensin II minipumps.
Figure 1. Effect of angiotensin II on Blood Pressure in WT and PPAR-α KO mice:
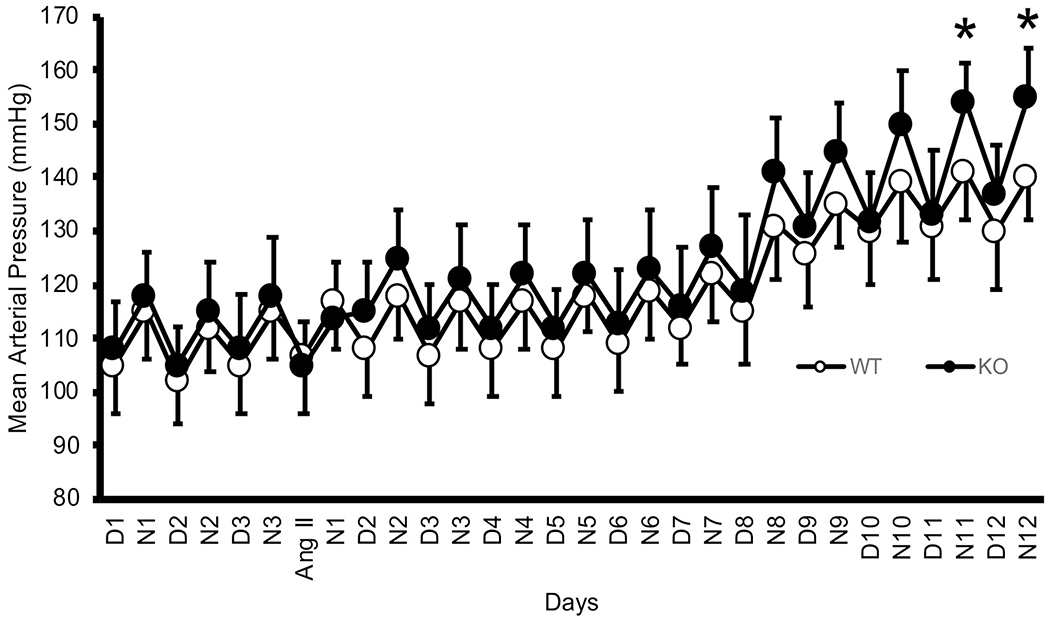
Mean arterial pressure in PPAR-α KO mice (open circles, n = 6) and WT mice (solid circles, n = 6) during 3 control days followed by infusion of angiotensin II 400 ng/kg·bw/min by osmotic minipump for 12 additional days, *indicates a significant difference (p < 0.05) by unpaired t-test.
Table 1:
Plasma Sodium and Urinary Volume in angiotensin II treated WT and PPAR-α KO mice
| Physiological Data* | |||||
|---|---|---|---|---|---|
| Groups | Sodium Intake (mEq/2 days) | Plasma Sodium (mmol/L) † | Baseline Urinary Volume (mL/2 days) ‡ | After (Days 10-12)‡ | Final Body weight (grams) |
| WT – Ctrl | 1.02 ± 0.16 | 134 ± 3.1 | 2.03 ± 0.05 | 2.15 ± 0.08 | 26.4 ± 0.5 |
| PPAR-α KO - Ctrl | 1.08 ± 0.14 | 136 ± 3.3 | 1.95 ± 0.07 | 1.98 ± 0.13 | 26.4 ± 0.5 |
| WT + angiotensin II | 1.23 ± 0.19 | 141 ± 3.4 | 2.10 ± 0.05 | 3.82 ± 0.17 | 27.5 ± 0.5 |
| PPAR-α KO + angiotensin II | 1.20 ± 0.22 | 137 ± 3.4 | 1.93 ± 0.07 | 3.98 ± 0.13 | 27.3 α 0.6 |
| Results of 2- or 3-way ANOVA (p-values) | |||||
| genotype | 0.92 | 0.80 | 0.50 | 0.81 | |
| treatment | 0.36 | 0.25 | <0.0001 | 0.076 | |
| time | - | - | <0.0001 | - | |
| treatment X genotype | 0.78 | 0.37 | 0.50 | 0.81 | |
| genotype x treatment x time | - | - | 0.065 | - | |
mean ± sem;
for plasma sodium (n = 11-13/group) for body weight and sodium intake (n = 6/group); 2-way ANOVA (treatment X genotype);
for urine volumes (n = 6/group/time point)-3-way repeated measures ANOVA (treatment x genotype x time)
Effect of angiotensin II on Na+/K+ ATPase (NKA) α1 expression and activity in WT and PPAR-α KO mice:
To determine the role of PPAR-α on angiotensin II-mediated changes in the basolateral NKA α1 expression and activity, we first determined by western blot the expression of NKA α1. As shown in Figure 2A, the NKA α1 subunit band density increased by angiotensin II in both genotypes (698 and 233% in WT and PPAR-α KO, respectively). The mean band densities were 211 and 47% higher in PPAR-α KO under vehicle and angiotensin II infusion, respectively, as compared to WT (Figure 2B). To test whether the increase in NKA α1 subunit expression is associated with an increase in the activity of the pump, we measured NKA activity as ouabain-sensitive ATP hydrolysis [19]. The liberated inorganic Pi was measured by the method of Taussky and Shorr [27]. The baseline NKA activity in the PPAR-α KO mice was about 270% higher than the WT animals (p = 0.015). Angiotensin II increased NKA activity by about 306% in WT mice and by about 154% in PPAR-α KO mice (p = 0.0029) as compared to their vehicle controls (Figure 2C).
Figure 2. Effect of angiotensin II on NKA α1 expression and activity in WT and PPAR-α KO mice:
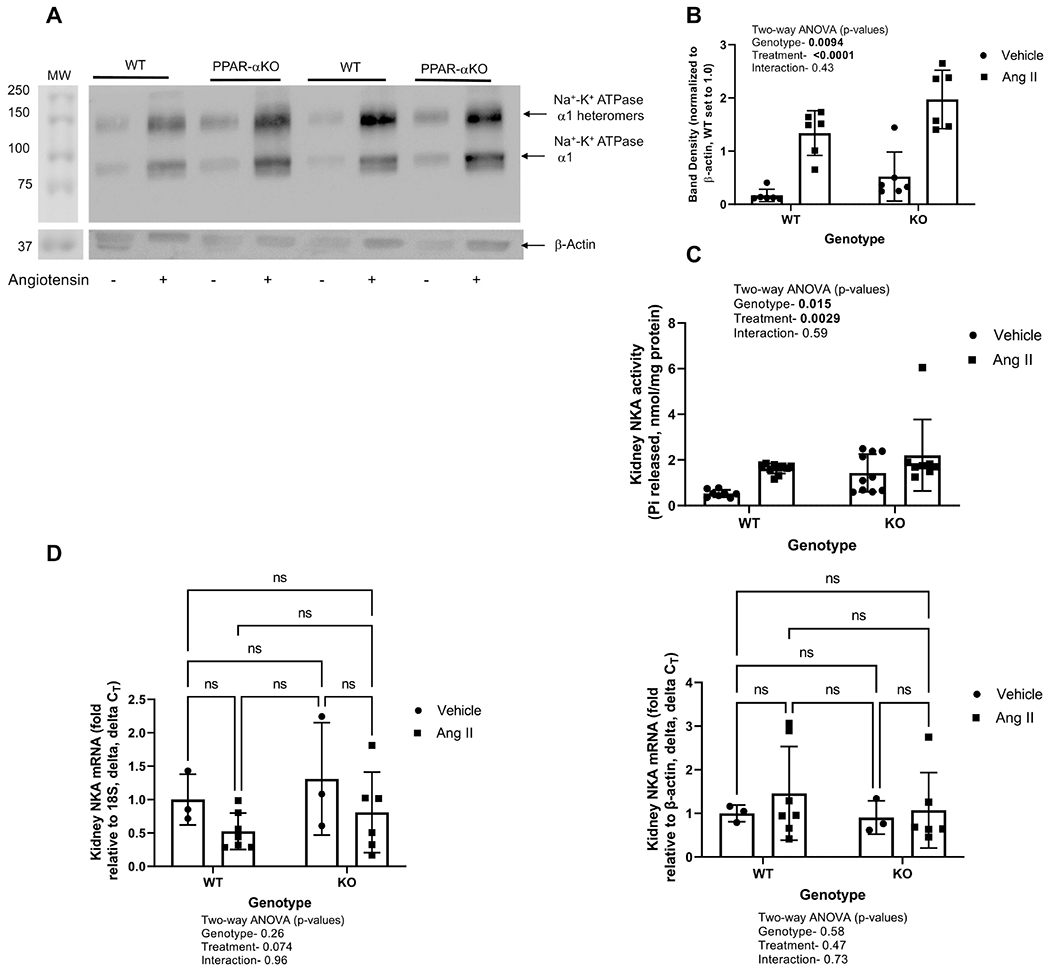
A, Representative western blot of NKA α1 (upper panel) and β-actin bottom panel is shown in membrane-enriched kidney cortex samples from WT and PPARα KO mice treated as vehicle (−) or with angiotensin II (+) infusion by osmotic minipump for 12 days. Gel was loaded with an equivalent amount of protein from each mouse kidney. B, density summary of NKA α1; and summary of NKA α1 densitometry (mean ± sem, n = 6/group); results of two-way ANOVA (genotype X treatment) displayed in graph insets. Significant effect (p < 0.05) shown in bold font. C, summary of Na+-K+ ATPase activity (mean ± sem, n = 6/group); results of two-way ANOVA (genotype X treatment). D, summary of NKA mRNA levels by qRT-PCR. Significant effect (p < 0.05) shown in bold font.
To determine if the increase in basal NKA α1 protein expression in PPAR-α KO mice and in response to angiotensin II in both WT and PPAR-α KO mice was due to an increase in the transcription, we measured the NKA mRNA expression by qRT-PCR. As shown in Figure 2D lack of PPAR-α expression or angiotensin treatment had no significant effect on NKA α1 mRNA expression.
Effect of angiotensin II on Aquaporin-1 expression in WT and PPAR-α KO mice:
The major water channel in the renal proximal tubule is aquaporin 1 (Aqp1) [24]. To determine if angiotensin II has an effect on Aqp1, we measured expression by western blot. Similar to NKA α1 subunit expression, Aqp1 was increased by angiotensin II (p < 0.0001; 211% in WT and 144% in PPAR-α KO), but not different between the genotypes (Figure 3).
Figure 3. Effect of angiotensin II on Aquaporin-1 (Aqp1) expression in WT and PPAR-α KO mice:
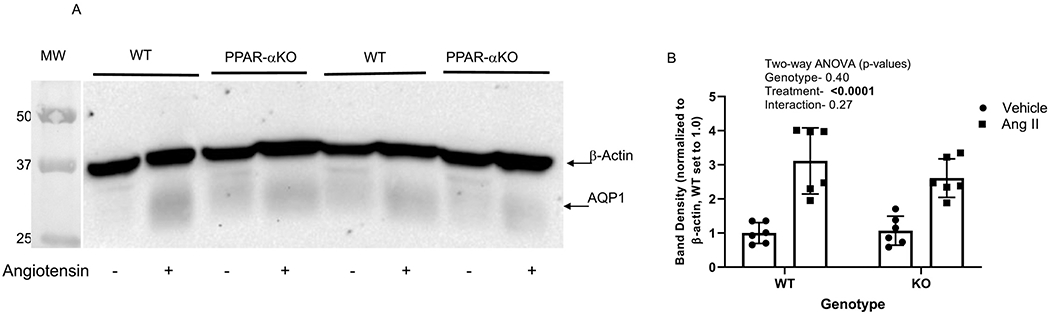
A, Representative western blot of Aqp1 and β-actin (normalized of membrane-enriched kidney cortex samples from WT and PPARα KO mice treated as vehicle (−) or with angiotensin II (+) infusion by osmotic minipump for 12 days. Gel was loaded with an equivalent amount of protein from each mouse kidney. B, density summary of Aqp1 (mean ± sem, n = 6/group); results of two-way ANOVA (genotype X treatment) displayed in graph inset. Significant effect (p < 0.05) shown in bold font.
Effect of angiotensin II on apical membrane sodium transporter expression and activity in WT and PPAR-α KO mice:
Unlike NKA α1 expression, the slow-pressor dose of angiotensin II markedly decreased the band densities of NHE3 in both PPAR-α knockout mice and their wild type littermates by 96-97% as compared to their respective vehicle-treated groups (n = 6/group, Figure 4). Two-way ANOVA (p-values shown in the inset of Figure 4B) revealed no significant difference between WT and PPAR-α KO animals.
Figure 4. Effect of angiotensin II on NHE3 expression in WT and PPAR-α KO mice:
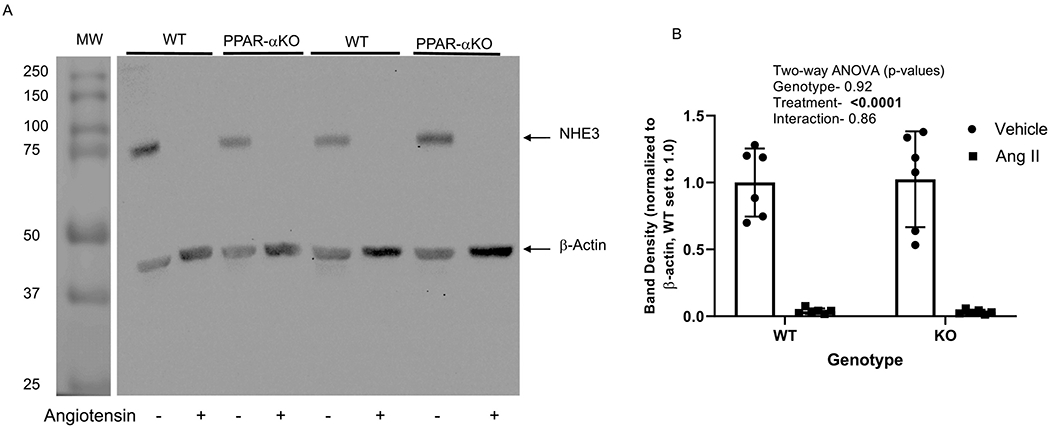
A. Representative western blot of NHE3 and β-actin (normalizer) of membrane-enriched kidney cortex samples from WT and PPARα KO mice treated as vehicle (−) or with angiotensin II (+) infusion by osmotic minipump for 12 days. Gel was loaded with an equivalent amount of protein from each mouse kidney. B, summary of western blotting densitometry (mean ± sem, n = 6/group); results of two-way ANOVA (genotype X treatment) displayed in graph inset. Significant effect (p < 0.05) shown in bold font.
NHERF1 is a PDZ binding chaperone protein that regulates trafficking of ion transporters specifically NHE3, and GPCRs in renal tubules [28]. To determine the effects of angiotensin II on NHERF1 expression, we determined the expression by western blot. Similar to the expression of ion transporters, NHERF1 band density was markedly decreased by angiotensin II in both PPAR-α KO (89%) and WT (93%) mice (Figure 5 A & B). There was no effect of genotype. Similar to NHE3, angiotensin II decreased NKCC2 by an average of 87% and 81% in the PPAR-α KO and WT, respectively (relative to their own vehicle, Figure 6), with no significant effect of genotype.
Figure 5. Effect of angiotensin II on NHERF1 expression in WT and PPAR-α KO mice:
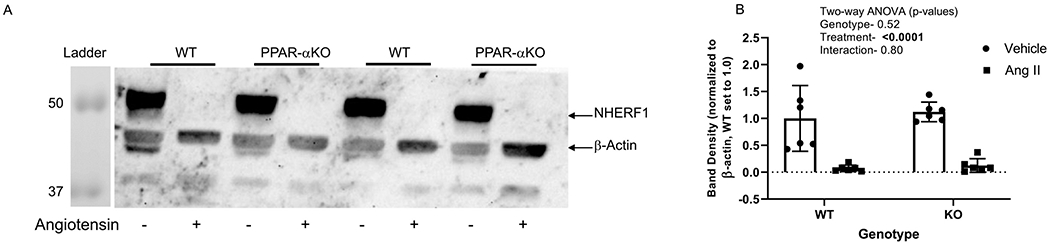
A, Representative western blot of NHERF1 and β-actin (normalized of membrane-enriched kidney cortex samples from WT and PPARa KO mice treated as vehicle (−) or with angiotensin II (+) infusion by osmotic minipump for 12 days; Gel was loaded with an equivalent amount of protein from each mouse kidney. B. density summary of NHERF1 densitometry (mean ± sem, n = 6/group); results of two-way ANOVA (genotype X treatment) displayed in graph insets. Significant effect (p < 0.05) shown in bold font.
Figure 6. Effect of angiotensin II on NHERF1 expression in WT and PPAR-α KO mice:
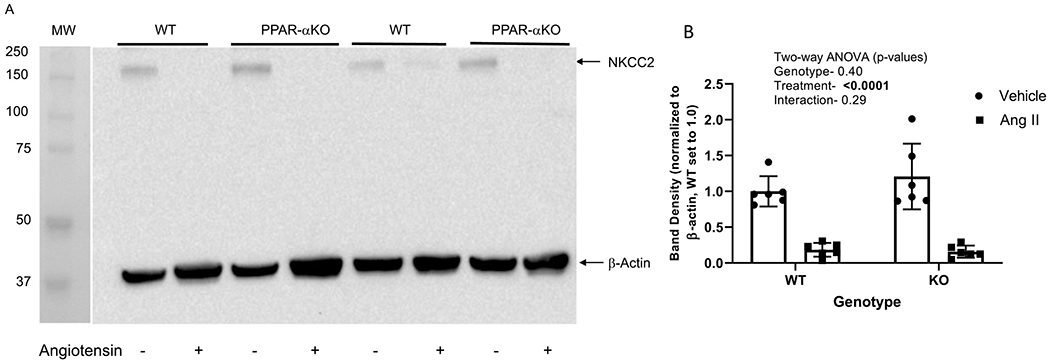
A, Representative western blot of NKCC2 and β-actin (normalizer) of membrane-enriched kidney cortex samples from WT and PPARα KO mice treated as vehicle (−) or with angiotensin II (+) infusion by osmotic minipump for 12 days. Gel was loaded with an equivalent amount of protein from each mouse kidney. B, summary of western blotting densitometry (mean ± sem, n = 6/group); results of two-way ANOVA (genotype X treatment) displayed in graph inset. Significant effect (p < 0.05) shown in bold font.
The slow-pressor dose of angiotensin II also decreased the band densities of NCC in both PPAR-α knockout (77%) and WT (59%) mice (Figure 7). While there was no significant effect of genotype, there was a significant interaction between genotype and treatment, in that the PPAR-α KO mice showed a significantly larger fall in band density with angiotensin II than did WT mice (slope of the change indicated by dashed lines in the figure).
Figure 7. Effect of angiotensin II on NCC expression in WT and PPAR-α KO mice:
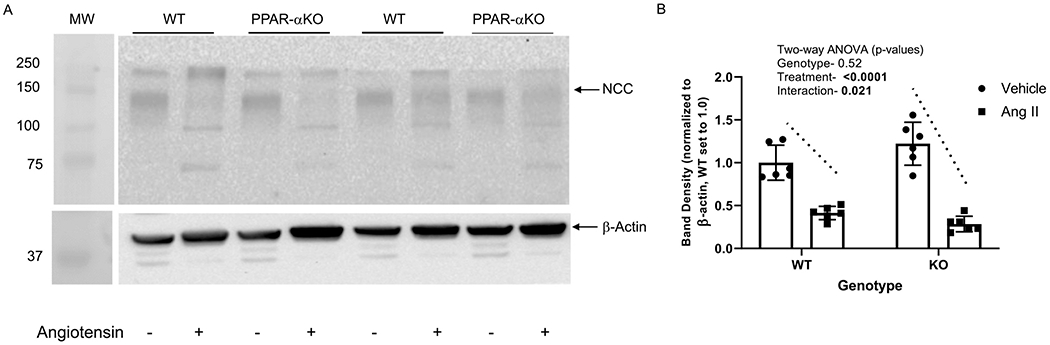
A, Representative western blot of NCC and β-actin (normalizer) of membrane-enriched kidney cortex samples from WT and PPARα KO mice treated as vehicle (−) or with angiotensin II (+) infusion by osmotic minipump for 12 days. Gel was loaded with an equivalent amount of protein from each mouse kidney. B, summary of western blotting densitometry (mean ± sem, n = 6/group); results of two-way ANOVA (genotype X treatment) displayed in graph inset. Significant effect (p < 0.05) shown in bold font. Significant interactive term shown by dotted lines indicating genotype differences in response to angiotensin II.
With regard to the 3 ENaC subunits, β-ENaC (90-kDa) was reduced 72% (WT) and 64% (PPAR-α KO) by angiotensin II (Figure 8A, top panel). Two-way ANOVA showed (similar to NHE3 and NKCC2) a large effect of treatment, with no effect of genotype (Fig 8B). Our antibody for γ-ENaC produced two bands as reported by Beutler et al [9], a higher molecular weight full-length protein and a cleaved active low molecular weight form (Fig 8A, middle panel). Similar to other proteins, both bands were markedly suppressed by angiotensin II. Band density for the full-length (85-kDa γ-ENaC) was reduced 93% in both WT and PPAR-α KO (Fig 5C). The cleaved (active) band was reduced 79 and 76% in WT and PPAR-α KO, respectively (Fig 5D). The effect of treatment was highly significant for each band, with no effect of genotype.
Figure 8. Effect of angiotensin II on ENaC subunit expression in WT and PPAR-α KO mice:
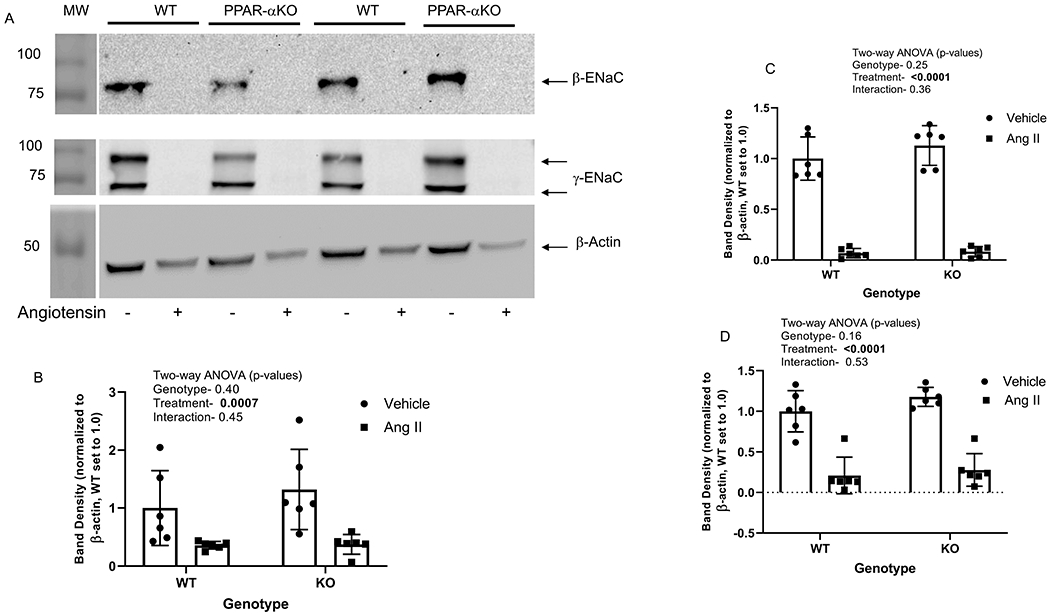
A, Representative western blots of β-ENaC, γ-ENaC and β-actin (normalizer) of membrane-enriched kidney cortex samples from WT and PPARα KO mice treated as vehicle (−) or with angiotensin II (+) infusion by osmotic minipump for 12 days. Gel was loaded with an equivalent amount of protein from each mouse kidney. B-D, summary of western blotting densitometry (mean ± sem, n = 6/group) for β- and two bands of γ-ENaC, respectively, as indicated; results of two-way ANOVA (genotype X treatment) displayed in graph inset. Significant effect (p < 0.05) shown in bold font.
Discussion
Up or down “tuning” of apical transport protein abundance or localization in the membrane is considered the major means by which the kidney regulates sodium reabsorption/excretion activity. This regulation is related to blood pressure by expansion or contraction of extracellular volume. In this study, we demonstrated that the elevation in blood pressure observed in PPAR-α KO mice in response to sustained, slow-pressor dose angiotensin II infusion was associated with relatively higher renal expression of the α-1 subunit of NKA, as well as the activity of the NKA enzyme. Similar to the NKA α1 expression, Aqp1, the major water channel of the proximal tubule and thin descending limbs [24] was increased by angiotensin II, but to a similar degree in both genotypes. All other sodium-transport related proteins we assessed, with the exception of NCC, were similarly, markedly decreased in abundance by angiotensin II infusion. NCC was, in fact, downregulated to a greater extent in the PPAR-α KO.
In the current study, we found both activity of NKA, as well as protein expression of the α1 subunit of the basolateral NKA pump was significantly higher in the PPAR-α KO under control and angiotensin II conditions, and its abundance was increased by angiotensin II infusion. Although NKA is an oligomer composed of α-, β- and γ-subunits, the α subunit (of which α1 is most common in kidney) is thought to be most sensitive to expression changes. The increase in NKA expression and activity together with an increase in Aqp1 expression may have a role in BP differences between the genotypes. Jackson et al demonstrated that chronic perfusion of kidneys results in decreased NKA activity and decrease renal preservation. Addition of PPARα agonist gemfibrozil and mannitol caused improved renal preservation by increasing the activity of NKA activity [22]. Our findings are in partial agreement with those of Newaz et al., who showed reduced activity but increased expression of the NKA pump with the PPAR-α agonist Clofibrate [23]. Their studies also postulated a link between PPAR-α expression together with increased nitric oxide (NO) production and reduced NKA activity in proximal tubule. In general, it is expected that the activity should match the expression of the NKA. However, the differences in the activity and western blot data in Newaz study may be due to the methodological differences. The authors should have used a higher concentration of ouabain (around 5 mM vs. 1 mM) in their method as the rodent NKA is highly resistant to cardiac glycosides like ouabain.
PPARs are nuclear receptor family members and are activated by fatty acids and naturally occurring fatty acid-derived molecules. They are known to regulate energy homeostasis, lipid and lipoprotein metabolism, glucose homeostasis, and are key regulators of inflammatory and immune responses. They regulate gene expression by ligand-dependent transactivation or transrepression and by ligand-independent repression [29]. Our studies demonstrate that knockout of PPAR-α results in an increase in NKA α1 subunit protein expression without evidence of increased mRNA levels, suggesting that PPAR-α decreases the NKA α1 subunit protein abundance by post-translational changes in the protein, perhaps by shortening half-life or increased degradation. We [19,30] and others [31] have demonstrated that the localization of NKA α1 protein in the basolateral membrane can be enhanced by increasing phosphorylation of a tyrosine at position 10 in the protein or decreased by serine phosphorylation at position 11 and 23. Low pressor dose of Ang II-mediated increase in NKA α1 subunit expression may be occurring through inflammatory mechanisms. Further studies are required to confirm these mechanisms.
The dose and chronicity are majorly important in determining the sodium transporter expression protein in kidneys. Acute or low-dose angiotensin II infusions have been shown to upregulate both proximal and distal tubule sodium transport and transporters. In agreement with our study, a previous report in rats demonstrated that a slow-pressor dose of angiotensin II decreased NHE3 and NKCC2 abundance in homogenates from the cortex and medulla [15]. A decrease in mouse whole kidney NKCC2 expression during angiotensin II infusion has also been previously observed [6]. In the current study, we observed a similar slow pressor dose of angiotensin II-mediated decrease in NHE3 and NKCC2 expression in the cortex. The role of NHE3 during angiotensin II-induced pressure natriuresis has been previously shown [32]. The slow-pressor dose of angiotensin II gradually increases blood pressure by sodium retention. Once BP has reached a trigger threshold, anti-natriuresis ensues as part of an escape mechanism.
It is interesting to note that nitric oxide (NO) has also been shown to have a role in the increase in NKCC2 and NHE3 in models of more modest angiotensin II infusion [33,34]; as inhibition of NO production by L-NAME reduced the upregulation of these proteins by angiotensin II. Because we saw no differences in the downregulation of NHE3 and NKCC2 by our sustained slow-pressor dose of angiotensin II between the genotypes, we suspect NO levels were relatively low in both genotypes.
Our studies clearly show that the relative higher MAP in the PPAR-α KO mice with angiotensin II infusion was not due to relatively higher abundance of any major apical sodium transport protein. While there are other sodium transport-related pathways in the apical epithelium, which we did not address, e.g., sodium phosphate and sodium glucose transporters, these proteins are thought to be responsible for much less total sodium uptake and are unlikely to be major contributors to this phenotype. NHE3 alone is thought to be responsible for about 50% of sodium reabsorption along the entire nephron. The proximal tubule accounts for about 60-70% of total reabsorptive activity with another 25-30 reabsorbed primarily through NKCC2 in the thick ascending limb.
With regard to the post-macula densa region of the tubule, in the current study, we found that NCC protein levels were suppressed by angiotensin II to a greater extent in the PPAR-α KO. NCC, a distal convoluted tubule sodium transporter, has been reported to remain elevated in some studies with angiotensin II infusion [15]. Furthermore, its downregulation has been demonstrated to underlie both the aldosterone- and angiotensin II-escape phenomenon, which has been shown by infusing aldosterone and offering a high-NaCl diet [34]. Our results suggest that PPAR-α mice were attempting to mount a greater escape (relative to the WT), in line with their elevated BP. Thus, we may conclude that PPAR-α does not contribute to the classic escape process, as mediated by the distal tubule; however, it may have a role in pressure natriuresis as mediated by the proximal tubule.
In the current study, contrary to the previous study [15], we also observed a decrease in major bands for β-ENaC and γ-ENaC. The difference between our study and the Nguyen et al study is unclear, but maybe due to differences in the background strain or in western blotting sample preparation. While we used crude membrane preparations from whole cortex, the Nguyen et al. study used cortex and medulla homogenates [15]. It may very well be that whole-cell levels were not affected, but the amount in the membrane was reduced. Moreove, Barrick et al [35] demonstrated that C57BL/6J mice and B129S1/SvImJ mice respond differently to overload in a time-dependent manner. The C57BL/6J mice had earlier onset and more pronounced impairment in cardiac contractile function as compared to B129S1/SvimJ mice. These differences may account for the differences in expression of sodium transporters (NCC and ENaC) in our studies as compared to studies in mice on C57BL/6J background.
The regulation of sodium channel activity and expression by peroxisome proliferator activated receptors is not clearly understood at this time. In some respects, PPAR-α and PPAR-γ have opposing effects, for example, a recent investigation revealed a suppressive influence of PPAR-α on amiloride-sensitive current density when co-expressed with ENaC subunits in CHO cells [36]. Furthermore, it is important to note that PPAR deletions of the other subclasses may not result in the same phenotype. Activation of PPAR-γ, e.g., has been associated with expansion of extracellular volume and increased sodium retention and increased whole-kidney expression of several sodium carriers including α1 subunit of Na+/K+ ATPase, NHE3, NpT2a, and NKCC2 in rats [37]. In addition, PPAR-γ activation has been proposed to stimulate renal Na+ reabsorption by stimulating serum- and glucocorticoid-Regulated Kinase 1 (SGK1) and ENaC [38].
We also found that angiotensin II infusion increased Aqp1 to an equivalent level in the WT and KO mice. Previously, Bouley et al. demonstrated a dose-responsive increase in Aqp1 mRNA in PT cells and in rats treated with 10−12-10−8 M angiotensin II, which was ablatable by AT1R antagonism [24]. The fact that we found no difference in this increase between the genotypes would suggest that the mechanisms for this increase does not involve PPAR-α.
In summary, the data presented in this study suggest that the relatively higher level of activity and protein expression of NKA and Aqp1 may play a role in angiotensin II-mediated increase in blood pressure. The attenuated decreases in apically expressed sodium transporters, exchangers, and channels and resultant natriuresis in the PPAR-α versus WT mice to angiotensin II infusion using the slow pressor dose, may perhaps be a classical pressure natriuresis response to the elevated BP. The regulation of NKA and Aqp1 by PPAR-α may be through post-translational mechanisms. Additional studies will be required to determine the post-translational mechanisms and whether impaired vasodilatory responses that accompany pressure natriuresis may also contribute to the genotype differences.
Acknowledgments
The authors acknowledge the summer students David Hardin and Sarah Stewart for their help in western blotting experiments. We thank Dr. Xie Wu for help with the qRT-PCR experiments. The authors thank Dr. Mark Knepper (NHLBI, NIH) for providing antibodies against NKCC2, NCC, ENaC.
Funding
This project was also supported (in part) by the National Institute on Minority Health and Health Disparities of the National Institutes of Health (2U54MD007597) and by the National Center for Advancing Translational Sciences Program (TL1TR001431). Funding was also provided by National Institute of Aging (5R21AG047474, NIH) and AHA (GIA, 16GRNT31030019) to SJK and R25AG047843 to Howard University. Additional support was provided by the Georgetown University Biomedical Graduate Research Organization (to CME).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References:
- [1].Han L, Shen W-J, Bittner S, Kraemer FB, Azhar S, PPARs: regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-α., Future Cardiology. 13 (2017) 259–278. 10.2217/fca-2016-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Okopień B, Buldak L, Bołdys A, Fibrates in the management of atherogenic dyslipidemia., Expert Review of Cardiovascular Therapy. 15 (2017) 913–921. 10.1080/14779072.2017.1408410. [DOI] [PubMed] [Google Scholar]
- [3].Banks T, Oyekan A, Peroxisome proliferator-activated receptor alpha activation attenuated angiotensin type 1-mediated but enhanced angiotensin type 2-mediated hemodynamic effects to angiotensin II in the rat., Journal of Hypertension. 26 (2008) 468–77. 10.1097/HJH.0b013e3282f2f0f3. [DOI] [PubMed] [Google Scholar]
- [4].Li P, Jackson EK, Enhanced slow-pressor response to angiotensin II in spontaneously hypertensive rats, Journal of Pharmacology and Experimental Therapeutics. 251 (1989). [PubMed] [Google Scholar]
- [5].Kawada N, Imai E, Karber A, Welch WJ, Wilcox CS, A mouse model of angiotensin II slow pressor response: Role of oxidative stress, Journal of the American Society of Nephrology. 13 (2002) 2860–2868. 10.1097/01.ASN.0000035087.11758.ED. [DOI] [PubMed] [Google Scholar]
- [6].Lever AF, Slow pressor mechanisms in hypertension: a role for hypertrophy of resistance vessels? Journal of Hypertension. 4 (1986) 515–24. 10.1097/00004872-198610000-00001. [DOI] [PubMed] [Google Scholar]
- [7].Lee DL, Wilson JL, Duan R, Hudson T, El-Marakby A, Peroxisome Proliferator-Activated Receptor-α Activation Decreases Mean Arterial Pressure, Plasma Interleukin-6, and COX-2 While Increasing Renal CYP4A Expression in an Acute Model of DOCA-Salt Hypertension., PPAR Research. 2011 (2011)502631. 10.1155/2011/502631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhou Y, Luo P, Chang H-H, Huang H, Yang T, Dong Z, Wang C-Y, Wang M-H, Colfibrate attenuates blood pressure and sodium retention in DOCA-salt hypertension., Kidney International. 74 (2008) 1040–8. 10.1038/ki.2008.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Beutler KT, Masilamani S, Turban S, Nielsen J, Brooks HL, Ageloff S, Fenton RA, Packer RK, Knepper MA, Long-term regulation of ENaC expression in kidney by angiotensin II., Hypertension (Dallas, Tex.: 1979). 41 (2003) 1143–50. 10.1161/01.HYP.0000066129.12106.E2. [DOI] [PubMed] [Google Scholar]
- [10].Zhao D, Seth DM, Navar LG, Enhanced distal nephron sodium reabsorption in chronic angiotensin II-infused mice., Hypertension (Dallas, TX: 1979). 54 (2009) 120–6. 10.1161/HYPERTENSIONAHA.109.133785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gurley SB, Riquier-Brison ADM, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM, AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure., Cell Metabolism. 13 (2011) 469–475. 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].van der Lubbe N, Lim CH, Fenton RA, Meima ME, Jan Danser AH, Zietse R, Hoorn EJ, Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone., Kidney International. 79 (2011)66–76. 10.1038/ki.2010.290. [DOI] [PubMed] [Google Scholar]
- [13].Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G, Activation of the renal Na+:Cl-cotransporter by angiotensin II is a WNK4-dependent process., Proceedings of the National Academy of Sciences of the United States of America. 109 (2012) 7929–34. 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MTX, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA, The absence of intrarenal ACE protects against hypertension., The Journal of Clinical Investigation. 123 (2013) 2011–23. 10.1172/JCI65460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nguyen MTX, Lee DH, Delpire E, McDonough AA, Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: distal stimulation counteracted by proximal inhibition., American Journal of Physiology. Renal Physiology. 305 (2013) F510–9. 10.1152/ajprenal.00183.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Riquier-Brison ADM, Leong PKK, Pihakaski-Maunsbach K, McDonough AA, Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli., American Journal of Physiology. Renal Physiology. 298 (2010) F177–86. 10.1152/ajprenal.00464.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hussain T, Lokhandwala MF, Renal dopamine receptors and hypertension., Experimental Biology and Medicine (Maywood, N.J.). 228 (2003) 134–42. [DOI] [PubMed] [Google Scholar]
- [18].Yingst DR, Massey KJ, Rossi NF, Mohanty MJ, Mattingly RR, Angiotensin II directly stimulates activity and alters the phosphorylation of Na-K-ATPase in rat proximal tubule with a rapid time course., American Journal of Physiology. Renal Physiology. 287 (2004) F713–21. 10.1152/ajprenal.00065.2004. [DOI] [PubMed] [Google Scholar]
- [19].Khundmiri SJ, Lederer E, PTH and DA regulate Na-K ATPase through divergent pathways, American Journal of Physiology - Renal Physiology. 282 (2002). 10.1152/AJPRENAL.00111.2000. [DOI] [PubMed] [Google Scholar]
- [20].Herman MB, Rajkhowa T, Cutuli F, Springate JE, Taub M, Regulation of renal proximal tubule Na-K-ATPase by prostaglandins, American Journal of Physiology - Renal Physiology. 298 (2010). 10.1152/ajprenal.00467.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Feraille E, Dizin E, Coordinated control of ENaC and Na+, K+-ATPase in renal collecting duct, Journal of the American Society of Nephrology. 27 (2016) 2554–2563. 10.1681/ASN.2016020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jackson TC, Mi Z, Bastacky SI, McHale T, Melhem MF, Sonalker PA, Tofovic SP, Jackson EK, PPARα agonists improve renal preservation in kidneys subjected to chronic in vitro perfusion: Interaction with mannitol, Transplant International. 20 (2007) 277–290. 10.1111/j.1432-2277.2006.00431.x. [DOI] [PubMed] [Google Scholar]
- [23].Newaz MA, Ranganna K, Oyekan AO, Relationship between PPARalpha activation and NO on proximal tubular Na+ transport in the rat., BMC Pharmacology. 4 (2004) 1. 10.1186/1471-2210-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bouley R, Palomino Z, Tang S-S, Nunes P, Kobori H, Lu HA, Shum WW, Sabolic I, Brown D, Ingelfinger JR, Jung FF, Angiotensin II and hypertonicity modulate proximal tubular aquaporin 1 expression., American Journal of Physiology. Renal Physiology. 297 (2009) F1575–86. 10.1152/ajprenal.90762.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee DL, Sturgis LC, Labazi H, Osborne JB, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW, Angiotensin II hypertension is attenuated in interleukin-6 knockout mice., American Journal of Physiology. Heart and Circulatory Physiology. 290 (2006) H935–40. 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
- [26].Pushpakumar S, Ahmad A, Ketchem CJ, Jose PA, Weinman EJ, Sen U, Lederer ED, Khundmiri SJ, Sodium-hydrogen exchanger regulatory factor-1 (NHERF1) confers salt sensitivity in both male and female models of hypertension in aging, Life Sciences. 243 (2020) 117226. 10.1016/j.lfs.2019.117226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].TAUSSKY HH, SHORR E, A microcolorimetric method for the determination of inorganic phosphorus., The Journal of Biological Chemistry. 202 (1953) 675–85. [PubMed] [Google Scholar]
- [28].Weinman EJ, Lederer ED, NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones., Am J Physiol Renal Physiol. 303 (2012) F321–7. 10.1152/ajprenal.00093.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lee JW, Chou C-L, Knepper MA, Deep Sequencing in Microdissected Renal Tubules Identifies Nephron Segment-Specific Transcriptomes., Journal of the American Society of Nephrology: JASN. 26 (2015) 2669–77. 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].RICOTE M, GLASS C, PPARs and molecular mechanisms of transrepression, Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1771 (2007) 926–935. 10.1016/j.bbalip.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Khundmiri SJ, Advances in understanding the role of cardiac glycosides in control of sodium transport in renal tubules, Journal of Endocrinology. 222 (2014). 10.1530/JOE-13-0613. [DOI] [PubMed] [Google Scholar]
- [32].Blanco G, Mercer RW, Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function, Am J Physiol. 275 (1998) F633–50. [DOI] [PubMed] [Google Scholar]
- [33].Li XC, Soleimani M, Zhu D, Rubera I, Tauc M, Zheng X, Zhang J, Chen X, Zhuo JL, Proximal Tubule-Specific Deletion of the NHE3 (Na+/H+ Exchanger 3) Promotes the Pressure-Natriuresis Response and Lowers Blood Pressure in Mice., Hypertension (Dallas, Tex.: 1979). 72 (2018) 1328–1336. 10.1161/HYPERTENSIONAHA.118.10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kwon T-H, Nielsen J, Kim Y-H, Knepper MA, Fr∅kiaer J, Nielsen S, Regulation of sodium transporters in the thick ascending limb of rat kidney: response to angiotensin II., American Journal of Physiology. Renal Physiology. 285 (2003) F152–65. 10.1152/ajprenal.00307.2002. [DOI] [PubMed] [Google Scholar]
- [35].Turban S, Wang X-Y, Knepper MA, Regulation of NHE3, NKCC2, and NCC abundance in kidney during aldosterone escape phenomenon: role of NO., American Journal of Physiology. Renal Physiology. 285 (2003) F843–51. 10.1152/ajprenal.00110.2003. [DOI] [PubMed] [Google Scholar]
- [36].Barrick CJ, Rojas M, Schoonhoven R, Smyth SS, Threadgill DW, Cardiac response to pressure overload in 129S1/SvImJ and C57BL/6J mice: temporal- and background-dependent development of concentric left ventricular hypertrophy., American Journal of Physiology. Heart and Circulatory Physiology. 292 (2007) H2119–30. 10.1152/ajpheart.00816.2006. [DOI] [PubMed] [Google Scholar]
- [37].Pavlov TS, Imig JD, Staruschenko A, Regulation of ENaC-Mediated Sodium Reabsorption by Peroxisome Proliferator-Activated Receptors., PPAR Research. 2010 (2010) 703735. 10.1155/2010/703735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Song J, Knepper MA, Hu X, Verbalis JG, Ecelbarger CA, Rosiglitazone activates renal sodium- and water-reabsorptive pathways and lowers blood pressure in normal rats., The Journal of Pharmacology and Experimental Therapeutics. 308 (2004) 426–33. 10.1124/jpet.103.058008. [DOI] [PubMed] [Google Scholar]
- [39].Vallon V, Lang F, New insights into the role of serum- and glucocorticoid-inducible kinase SGK1 in the regulation of renal function and blood pressure, Current Opinion in Nephrology and Hypertension. 14 (2005) 59–66. 10.1097/00041552-200501000-00010. [DOI] [PubMed] [Google Scholar]