

Abstract
Alzheimer’s disease is characterized by amyloid plaques, neurofibrillary tangles, glial activation, and neurodegeneration. In mouse models, inflammatory activation of microglia accelerates tau pathology. The chemokine fractalkine serves as an endogenous neuronal modulator to quell microglial activation. Experiments with fractalkine receptor null mice suggest that fractalkine signaling diminishes tau pathology, but exacerbates amyloid pathology. Consistent with this outcome, we report here that soluble fractalkine overexpression using adeno-associated viral vectors significantly reduced tau pathology in the rTg4510 mouse model of tau deposition. Furthermore, this treatment reduced microglial activation and appeared to prevent neurodegeneration normally found in this model. However, in contrast to studies with fractalkine receptor null mice, parallel studies in an APP/PS1 model found no effect of increased fractalkine signaling on amyloid deposition. These data argue that agonism at fractalkine receptors might be an excellent target for therapeutic intervention in tauopathies, including those associated with amyloid deposition.
Keywords: Tau, Neurodegeneration, Fractalkine, Alzheimer
1. Introduction
There is evidence for microglial involvement in Alzheimer’s disease (AD) based on observations in human brain tissue, experimental animal models, and in vitro tissue culture data (Morales et al., 2010; Streit, 2004). Under normal conditions, microglia protect central nervous system (CNS) functions and remove cells damaged by acute injury. However, microglial neurotoxicity can occur after excessive and uncontrolled stimulation or when microglia function is impaired (Cardona et al., 2006; Streit, 2006; van Rossum and Hanisch, 2004). It has been suggested that a major difference between beneficial, resolving inflammation and detrimental, chronic inflammation is a failure to transition between classical inflammation and alternative activation states, leading to tissue destruction and organ failure (Duffield, 2003).
Under resting conditions, there are several signals produced by neurons that have an anti-inflammatory action on microglia, including fractalkine (FKN, CX3CL1). FKN signaling reduces the overproduction of inducible nitric oxide synthase, interleukin (IL)-1β, tumor necrosis factor-α, and IL-6 generated by microglia (Lyons et al., 2009; Zujovic et al., 2000). In the CNS, FKN is expressed by neurons, and only binds to a single receptor subtype (CX3CR1) located on microglia (Harrison et al., 1998; Ludwig and Weber, 2007; Mantovani et al., 2004). FKN is a transmembrane protein with a chemokine domain attached to a mucin-like stalk. The full-length, membrane-bound FKN is important for adhesion of monocytes to endothelial cells, and might also play a role in monocyte-induced endothelial cell death, at least in the periphery (Ludwig and Weber, 2007). However, cleavage by a disintegrin and metalloproteinase (ADAM)10/17 or cathepsin S produces a secreted or soluble form of fractalkine (sFKN) (Garton et al., 2001; Hundhausen et al., 2003). It has been proposed that dynamic proteolytic cleavage of FKN from neuronal membranes, in response to insults, is an early event in neuronal injury (Chapman et al., 2000). Evidence suggests that sFKN is important for chemotaxis and acts as a chemoattractant for lymphocytes and monocytes (Imai et al., 1997). Further, it has also been suggested that the membrane and soluble forms elicit different cytokine responses from immune cells (Kim et al., 2011; Yoneda et al., 2003). However, the exact roles of these subtypes of FKN are not completely established in the periphery or in the CNS.
There is some controversy about the effects of FKN in neurological disorders. sFKN has been shown to be neuroprotective (Morganti et al., 2012; Pabon et al., 2011) and also detrimental to neurons (Shan et al., 2011). CX3CR1 has been reported to be necessary for cell death in a mouse model of AD (Fuhrmann et al., 2010), but it has also been shown that loss of CX3CR1 exacerbates neurodegeneration (Cardona et al., 2006). A cross between CX3CR1−/− mice and amyloid precursor protein (APP)/presenilin 1 (PS1) transgenic mice resulted in a decrease in amyloid beta (Aβ) (Lee et al., 2010b), possibly by increasing the phagocytic capacity of the microglia (Lee et al., 2010b). Interestingly, a cross between CX3CR1−/− mice with a human tau line resulted in an increase in tau pathology (Bhaskar et al., 2010). Together, these data suggest that microglial activation (by reducing FKN ligation) can simultaneously affect Aβ and tau pathology in opposite directions. These opposing actions on the 2 sentinel pathologies of AD might cause this potentially important molecular target to be discarded as an AD therapeutic approach.
To further investigate FKN signaling as a potential therapeutic target, we examined the effects of selective sFKN overexpression on histopathology in tau-depositing and amyloid-depositing transgenic mouse models.
2. Methods
2.1. Adeno-associated virus production
Recombinant adeno-associated virus (AAV) serotype (rAAV) 9-expressing fractalkine (geninfo identifier, GI: 114431260) were cloned using polymerase chain reaction from mouse brain cDNA. The sFKN was generated for expression as described previously (Morganti et al., 2012). sFKN was cloned into the pTR2-MCS vector at the Age I and Nhe I cloning sites. This vector contains the AAV2 terminal repeats and the hybrid cytomegalovirus-chicken β-actin (CBA) promoter for CX3CL1 messenger RNA transcription. An hemagglutinin (HA)-tag was added to the C-terminus of sFKN for protein detection. rAAV9 vectors were generated and purified as previously described (Carty et al., 2010). rAAV particles are expressed as vector genomes per mL. Vector genomes were quantitated using a modified version of the dot plot protocol described by Zolotukhin et al. (2002), using a nonradioactive biotinylated probe for fractalkine generated by polymerase chain reaction. Bound biotinylated probe was detected with IRDye 800CW (Li-Cor Biosciences, Lincoln, NE, USA) and quantitated on the Li-Cor Odyssey. UF11 plasmid was used to generate rAAV9 as described previously (Carty et al., 2010).
2.2. Transgenic mice
APP/PS1 mice (Holcomb et al., 1998) were acquired from the breeding colonies at the University of South Florida. Tg4510 mice and parental mutant tau and tetracycline-controlled transactivator protein lines were generated and maintained as described previously (Santacruz et al., 2005). Study animals were given water and food ad libitum and maintained on a 12-hour light/dark cycle and standard vivarium conditions.
Tg4510 mice aged 3 months were injected in the hippocampus with rAAV either expressing green fluorescent protein (GFP) (n = 8) or sFKN (n = 8). Animals were euthanized 3 months after administration of the viruses. For APP/PS1 mice, 2 cohorts were used; 13 months old (n = 16) and 6 months old (n = 16). Animals in each age group were randomly assigned to 1 of 2 groups. Group 1 received a control vector expressing GFP (n = 8), and group 2 received rAAV vector expressing sFKN (n = 8). Animals were euthanized 2 or 4 months after administration of the viral vectors for cohorts 1 and 2, respectively. Radial arm water maze was performed as described previously (Arendash et al., 2001).
2.3. Surgical procedure and tissue collection
Immediately before surgery, mice were weighed and anesthetized using isoflurane. Surgery was performed using a stereotaxic apparatus. The cranium was exposed using an incision through the skin along the midsagittal plane, and 2 holes were drilled through the cranium using a dental drill bit (SSW HP-3; SSWhite Burs Inc, Lakewood, NJ, USA). A Hamilton microsyringe was lowered, and injections of 2 μL of viral vector in sterile phosphate buffered saline (PBS) at a concentration of approximately 5 × 1012 vector genomes per mL were dispensed bilaterally into the hippocampus (coordinates from bregma: anteroposterior, ±2.7 mm; lateral −2.7 mm; vertical, −3.0 mm) using the convection enhanced method described previously (Carty et al., 2010). The incision was cleaned and closed with surgical sutures. Two to 4 months post surgery; mice were weighed, overdosed with pentobarbital (200 mg/kg) and perfused with 25 mL of 0.9% normal saline solution. Brains were collected from the animals immediately after perfusion. One hemisphere was dissected and frozen with dry ice for biochemical analysis. The second hemisphere was immersion-fixed in 4% paraformaldehyde for 24 hours. The fixed hemispheres were cryoprotected in successive incubations of 10%, 20%, and 30% solutions of sucrose for 24 hours in each solution. Subsequently, brains were frozen on a cold stage and sectioned in the horizontal plane (25 mm thickness) on a sliding microtome and stored in Dulbecco’s PBS with 10 mM sodium azide solution at 4 °C. For rTg4510 mice every 12th section was cut at 50 μm.
2.4. Immunohistochemistry and staining
Six to 8 sections 200 μm apart spanning the site of injection were chosen for analysis. Immunohistochemical procedural methods were described by Gordon et al. (2002). For each marker, sections from all animals were placed in a multisample staining tray, and endogenous peroxidase was blocked (10% methanol, 10% H202 in PBS; 30 minutes). Tissue samples were permeabilized (with 0.2% lysine, 1% Triton X-100 in PBS solution), and incubated overnight in appropriate primary antibody. Anti-NeuN (Millipore); anti-CD45 (Thermo Scientific), and anti-pS396 tau (Anaspec, Fremont, CA, USA) antibodies were used in this study. An anti-HA biotinylated rabbit polyclonal antibody was used to determine transgene expression (Roche, Indianapolis, IN, USA). Sections were washed in PBS, then incubated in corresponding biotinylated secondary antibody (Vector Laboratories, Burlingame, CA, USA), if necessary. The tissue was again washed after 2 hours, and incubated with Vectastain Elite ABC kit (Vector Laboratories) for enzyme conjugation. Finally, sections were stained using 0.05% diaminobenzidine and 0.03% H202. Tissue sections were mounted onto slides, dehydrated, and coverslipped. Each immunochemical assay omitted some sections from primary antibody incubation period to evaluate nonspecific reaction of the secondary antibody.
Congo red and Gallyas histology were performed using sections that were premounted on slides, then air-dried for a minimum of 24 hours. The sections were rehydrated for 30 seconds before beginning the staining protocol. For Congo red, 2.5 mM NaOH was added to a saturated sodium chloride—ethanol solution, and slides were incubated for 20 minutes. Subsequently, slides were incubated in 0.2% Congo red in alkaline alcoholic saturated sodium chloride solution for 30 minutes. Slides were rinsed through 3 changes of 100% ethanol, cleared through 3 changes of xylene, and cover-slipped with Di-N-butyle phthalate in xylene (DPX). Gallyas staining was performed as described in Lee et al. (2010a). Slides were treated with 5% periodic acid for 5 minutes, washed with water, and incubated sequentially in silver iodide (1 minute) and 0.5% acetic acid (10 minutes) solutions before being placed in developer solution (2.5% sodium carbonate, 0.1% ammonium nitrate, 0.1% silver nitrate, 1% tungstosilicic acid, 0.7% formaldehyde). Slides were treated with 0.5% acetic acid to stop the reaction, incubated with 0.1% gold chloride, placed in 1% sodium thiosulphate, and counterstained with 0.1% nuclear fast red in 2.5% aqueous aluminum sulfate, with each step separated by washes in water. After a final wash, slides were dehydrated and coverslipped.
Stained sections were imaged using a Zeiss Mirax-scan 150 microscope and Image Analysis software v1.0 (created by Andrew Lesniak). Area of positive stain in each hippocampal section was analyzed. The software used hue, saturation, and intensity to segment the image fields. Thresholds for object segmentation were established with images of high and low levels of staining to identify positive staining over any background levels. These limits were held constant for the analysis of every section in each study (Gordon et al., 2002). Stereologer software (Stereology Resource Center) with a Leica DM4000B microscope and a Prior Optiscan II stage were used for stereological counts. Nissl-stained 50 μm sections every 300 μm were used for stereological counting. Analysis of variance statistical analysis was performed using StatView version 5.0.1 (SAS Institute, Raleigh, NC, USA).
2.5. Biochemical analysis
Tissues for Western blot analysis were prepared as described in Carroll et al. (2011). Briefly, the dissected hippocampal tissue was weighed and resuspended in radio-immunoprecipitation assay (RIPA) buffer (50 mM Tris pH 7.6, 140 mM NaCl, 1% NP40, 0.5% Na deoxycholate, 0.1% sodium dodecyl sulfate), with protease inhibitor cocktail (Sigma) and phosphatase inhibitor cocktails I and II (Sigma) at 10 vol/wt of tissue. Tissue was homogenized with a pestle followed by a brief sonication pulse. The samples were centrifuged at 40,000 g for 30 minutes at 4 °C. The soluble fraction was taken for Western analysis. The pellet fraction was resuspended with 70% formic acid (2 μL/mg of tissue) and incubated for 30 minutes at room temperature. Equal volume of 1 M Tris pH 7.5 was added. The pH was adjusted to 7.5 with NaOH if required (pH paper used to measure pH). Pierce BCA protein assay (Thermo Scientific) was used to determine protein concentrations. For Western analysis, 1 μg of protein was loaded for each sample. H150, anti-pS199/S202 tau, anti-pS356 tau, and anti-pS262 tau were obtained from Anaspec. Anti-GSK3 α216/β279 was obtained from Abcam. AT8, AT180, AT270, and HT7 anti-bodies were obtained from Thermo Scientific. CX3CR1 antibody was obtained from Abcam. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Meridian Life Science, Inc, Memphis, TN, USA) was used as a loading control. For the formic acid fraction, which does not contain GAPDH, the amount of protein loaded was based on the BCA of the crude extract. Fractalkine enzyme-linked immunosorbent assay kit was obtained from RayBiotech.
2.6. Radial arm water maze
A detailed description of the general procedure has been published, complete with goal arm assignments and scoring sheets (Alamed et al., 2006). Briefly, entry into an incorrect arm (all 4 limbs within the arm) was scored as an error. If a mouse failed to make an arm entry within 20 seconds, this also was scored as an error. The errors for blocks of 3 consecutive trials were averaged for data analysis. Each cohort was given 3 trials (1 block) sequentially then returned to their home cages while a second cohort was tested. Next, the first cohort is tested for the second block, alternating with a second cohort until 15 trials (5 blocks) were completed. The start arm is varied for each trial so that mice rely upon spatial cues to solve the task instead of learning motor rules (i.e., second arm on the right). The goal arm for each successive mouse was different to avoid use of odor cues to locate the goal arm. Group averages of less than 1 error indicate learning of platform location (Arendash et al., 2001).
3. Results
We examined the levels of fractalkine ligand and receptor in nontransgenic mice compared with transgenic (APP/PS1 and Tg4510). With respect to CX3CR1, we observed no significant differences between APP/PS1 mice and their nontransgenic littermates, however Tg4510 mice showed approximately a 5-fold increase compared with nontransgenic littermates (Fig. 1A). Fractalkine was slightly reduced in APP/PS1 mice compared with littermates yet slightly increased in Tg4510 mice compared with littermate controls (Fig. 1B).
Fig. 1.
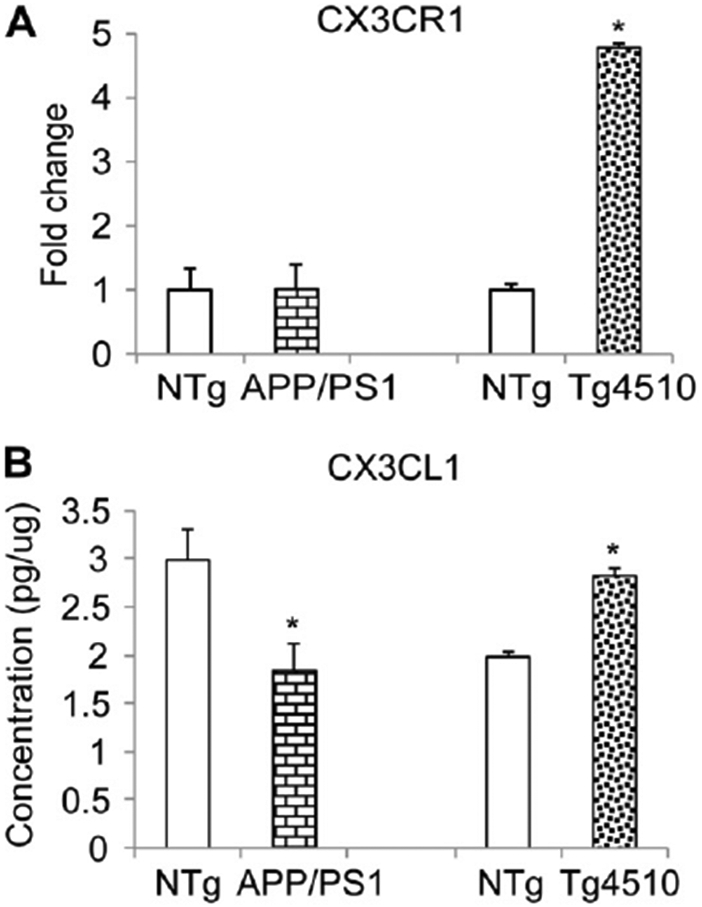
Fractalkine ligand (CX3CL1) and receptor(CX3CR1) levels in transgenic mice. (A) Western blot with anti-CX3CR1 antibody using hippocampal cell lysates of APP/PS1, Tg4510, or nontransgenic (NTg) littermate controls and normalizing to GAPDH staining; (B) Enzyme-linked immunosorbent assay for CX3CL1 of hippocampal cell lysates of APP/PS1, Tg4510, or nontransgenic littermate controls.
We have constructed an rAAV that overexpresses the soluble domain of the mouse fractalkine protein (sFKN) with an HA-tag on the C-terminus for protein detection (Morganti et al., 2012). The sFKN was placed under the CBA promoter for overexpression. This promoter system has previously been shown to express almost exclusively in neurons, by our lab and others (Burger et al., 2004; Carty et al., 2010; Klein et al., 2008). Vector expression was tested in HEK293 cells. As expected, we observed expression and secretion of the sFKN into the cell culture media (Fig. 2A and B). Viral vector expression in vivo was also confirmed by immunohistochemical staining of injected mice with an anti-HA antibody (Fig. 2C and D).
Fig. 2.
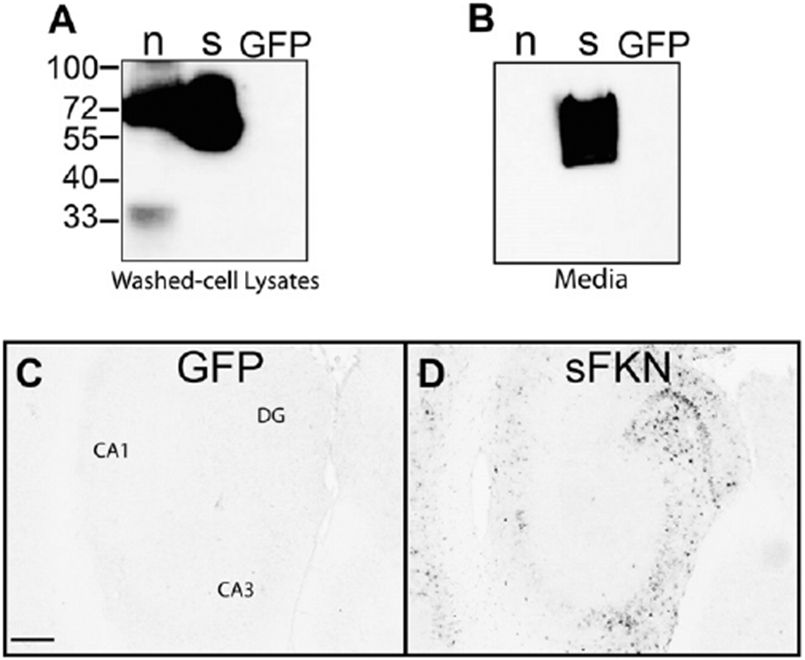
Expression of FKN constructs. (A) Western blot with anti-HA tag antibody using cell lysates of plasmid transfected HEK293 cells. (B) Anti-HA Western blot of the media from transfected HEK293 cells, only sFKN is observed in the media. (C and D) Anti-HA immunohistochemistry of hippocampus of GFP-injected and sFKN-injected mice. Scale = 200 μm. Abbreviations: DG, dentate gyrus; FKN, fractalkine; GFP, green fluorescent protein (control); HA, hemagglutinin; n, native (membrane associated) FKN; sFKN, secreted FKN.
Tg4510 mice overexpress human tau with a P301L mutation, and start exhibiting significant neuron loss by 6 months of age (Dickey et al., 2009; Santacruz et al., 2005). We injected 3-month-old Tg4510 mice with either a GFP- (control) or sFKN-expressing virus. Treatment with sFKN significantly decreased the levels of tau pathology in these mice 3 months later (Fig. 3). We observed reductions in both Gallyas staining and anti-pS396 tau immuno-staining by 40% and 35%, respectively (Fig. 3E and F). In addition, neuronal staining using antibody against NeuN revealed a higher density of staining in the sFKN-treated group compared with the control mice treated with rAAV-expressing GFP (Fig. 4). This was most apparent in the dentate region of the hippocampus (Fig. 4D), especially in the lateral blade granule neurons. Stereological analysis of the hippocampal volume in these groups shows protection against hippocampal volume loss in the sFKN treated group compared with the control GFP (Fig. 5A). Further, stereological counts of the granular cells of the dentate gyrus show that sFKN has a protective effect against neuron loss compared with the GFP control group (Fig. 5B).
Fig. 3.
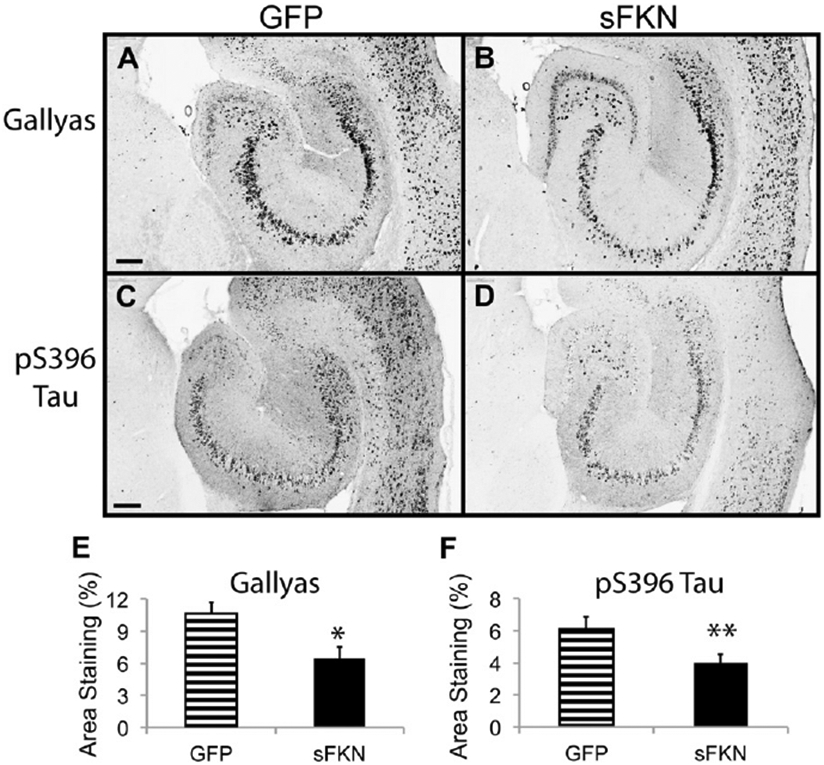
Tau pathology is reduced after sFKN transduction. Gallyas staining of rAAV-GFP (A) and rAAV-sFKN (B) -injected mice hippocampi. Staining with anti-phosphotau 396 antibody in rAAV-GFP (C) and rAAV-sFKN (D) -injected mice. Percentage area positive staining for Gallyas (E) and anti-phopsphotau 396 (F). ** P < 0.01; * P < 0.05. Scale = 200 μm. Abbreviations: FKN, fractalkine; GFP, green fluorescent protein; rAAV, recombinant adeno-associated virus serotype; sFKN, secreted FKN.
Fig. 4.
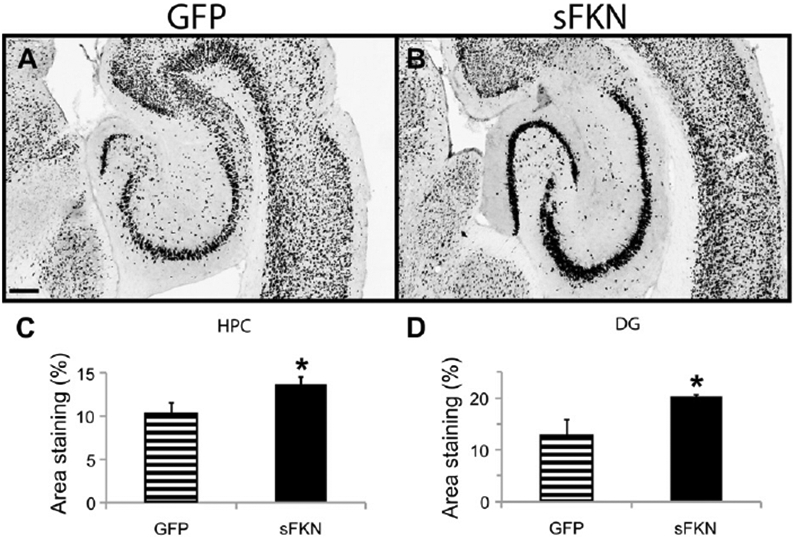
Anti-NeuN immunohistochemistry is increased after rAAV-sFKN treatment. Staining with anti-NeuN antibody in rAAV-GFP (A) and rAAV-sFKN (B) -injected mice. Percentage area positive staining for anti-NeuN in the CA1–CA3 of HPC (C) or DG subfield (D). * P < 0.05. Scale = 200 μm. Abbreviations: DG, dentate gyrus; FKN, fractalkine; GFP, green fluorescent protein; HPC, hippocampus; rAAV, recombinant adeno-associated virus serotype; sFKN, secreted FKN.
Fig. 5.
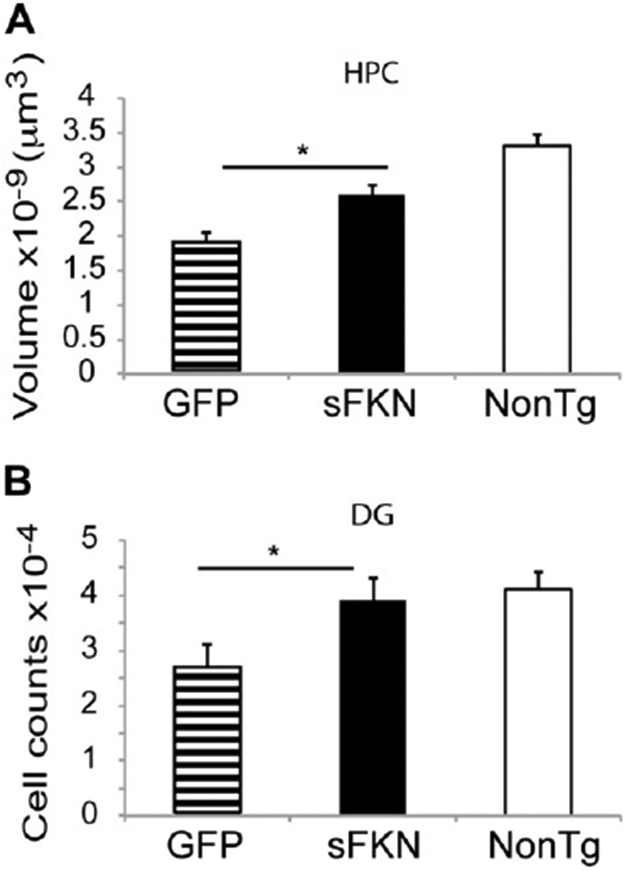
Stereological analysis demonstrates a reduction in neuron loss in the AAV-sFKN treated group compared with the AAV-GFP treated group. HPC volume (μm3) (A) and granular cell counts of the DG (B). * P < 0.02. Abbreviations: AAV, adeno-associated virus serotype; DG, dentate gyrus; FKN, fractalkine; GFP, green fluorescent protein; HPC, hippocampus; NonTg, nontransgenic; sFKN, secreted FKN.
Western blot analysis of phospho-tau species showed reductions in the sFKN-treated compared with the control group (Fig. 6A and B). Interestingly, we did not observe reductions in total tau (H150) but we observed significant reductions in pS199/S202, pS262, and pS396. We also observed reductions in the active form of GSK3 αβ kinase. There were also significant reductions in tau species that are recognized by the paired helical filament antibodies AT8 (pSer202/Thr205), AT180 (pThr231), and AT270 (pThr181) (Fig. 6A and B). Insoluble tau was extracted with formic acid. In the formic acid tau fraction we observed reductions in pS396 and pS262 but not pS199/202 (Fig. 6C). Total tau (H150) showed a trend for reduction as observed with Gallyas staining, but this was not statistically significant.
Fig. 6.
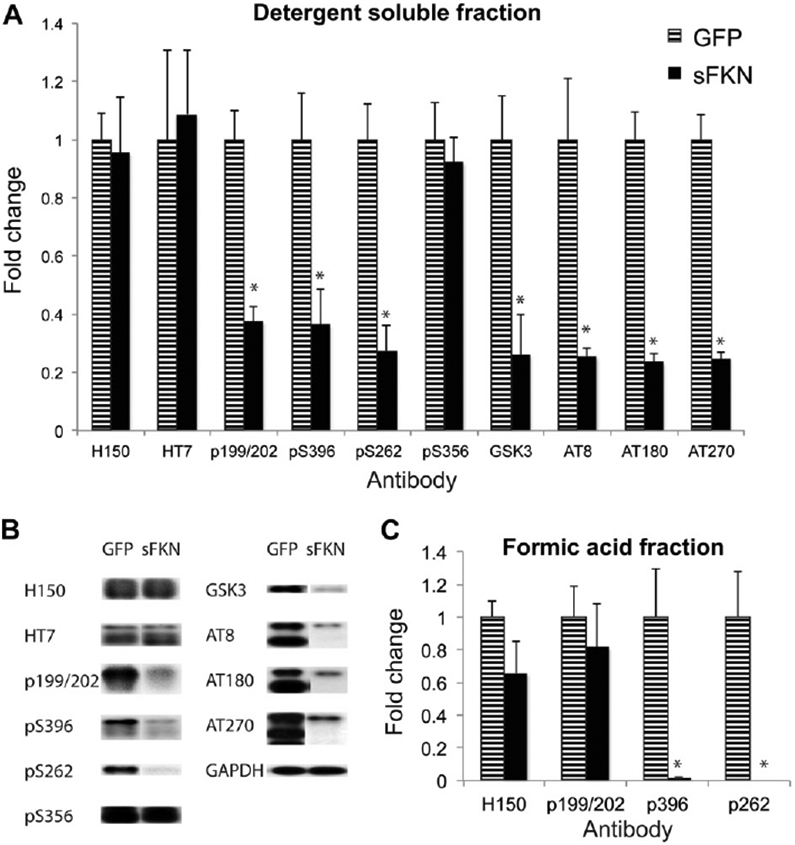
Western blot analysis shows reduced phospho-tau levels. (A) Intensity of staining is represented as fold change over the control GFP treated group after standardizing to GAPDH staining. Total tau (H150 and HT7 antibody), anti-pS199/S202, pS262, pS396, and pS356 tau; anti-GSK3 α;*216/β279, and anti-PHF tau antibodies AT8, AT180, and AT270 are shown. (B) Representative Western blot images of panel A. (C) Fold change in intensity for the formic acid tau fraction. * P < 0.01. Abbreviations: FKN, fractalkine; GAPDH, glyc-eraldehyde-3-phosphate dehydrogenase; GFP, green fluorescent protein; PHF, pair helical filament; sFKN, secreted FKN.
We have previously shown that sFKN reduces microglial activation (Morganti et al., 2012), therefore, in this study we examined the level of CD45 positive staining in our 2 animal groups, a microglial marker we have found previously elevated in Tg4510 mice (Lee et al., 2010a). The sFKN group had a reduction of approximately 70% compared with the control GFP-treated mice (Fig. 7A, B, and D). The morphology of stained cells in the GFP- treated group included many spherical cells, possibly perivascular macrophages, and more ramified cells (Fig. 7C).
Fig. 7.
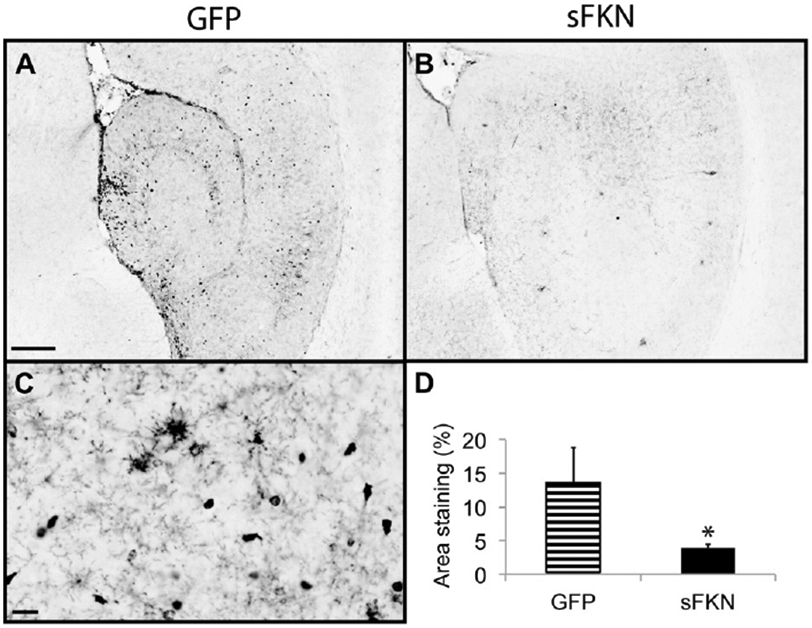
Anti-CD45 immunohistochemistry is reduced after rAAV-sFKN treatment. Anti-CD45 staining of rAAV-GFP (A) and rAAV-sFKN (B) -injected mice hippocampi (scale = 200 μm). Morphology of CD45-stained cells in an rAAV-GFP injected mouse (scale = 50 μm) (C). Percentage area positive staining for anti-CD45 (D). * P < 0.02. Abbreviations: AAV, adeno-associated virus serotype; FKN, fractalkine; GFP, green fluorescent protein; rAAV, recombinant AAV; sFKN, secreted FKN.
Cognitive performance was assessed with the radial arm water maze (Fig. 8). The addition of sFKN to the hippocampus did not significantly rescue the behavioral deficit observed in the Tg4510 mice compared with nontransgenic mice.
Fig. 8.
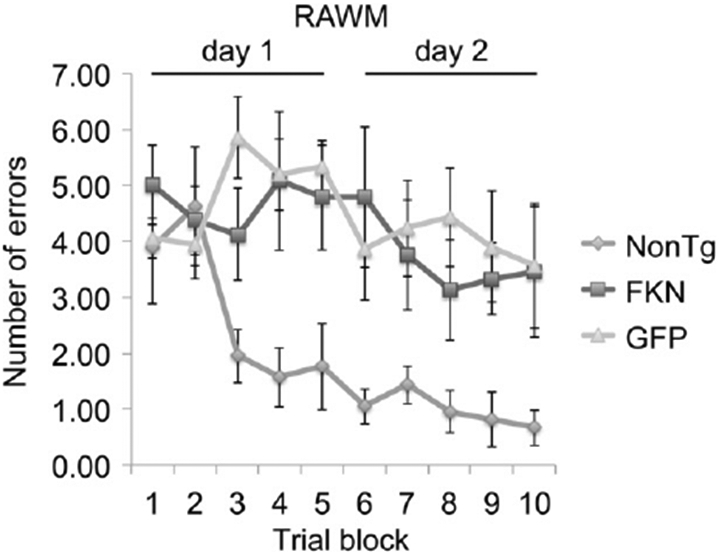
RAWM test demonstrates no phenotype rescue with AAV-sFKN treatment in the hippocampus compared with nonTg and AAV-GFP treated Tg4510 mice. Number of errors is shown as an average of every 3 trials as 1 block. Abbreviations: AAV, adeno-associated virus serotype; FKN, fractalkine; GFP, green fluorescent protein; nonTG, nontransgenic; RAWM, radial arm water maze; sFKN, secreted FKN.
Because loss of FKN signaling in the CX3CR1−/− null mice led to reduced amyloid deposition, we were concerned that increased signaling might cause the opposite effect. Our initial experiment examined effects of overexpression of sFKN in 13-month-old APP/PS1 mice for 2 months. However, no change was detected in either total Ab levels or Congo red stained compact plaques (data not shown). However, we were concerned that the substantial Aβ burden present at the beginning of the experiment, or the short duration of sFKN exposure might have masked significant effects that could be in mice with less starting amyloid or longer exposures. Therefore, we administered sFKN to 6-month-old APP/PS1 mice with less amyloid burden, and allowed them to survive for 4 months. As in the first experiment, we failed to observe significant changes in either Aβ (Fig. 9A, B, and E) or Congophilic compact plaques (Fig. 9C, D, and F).
Fig. 9.
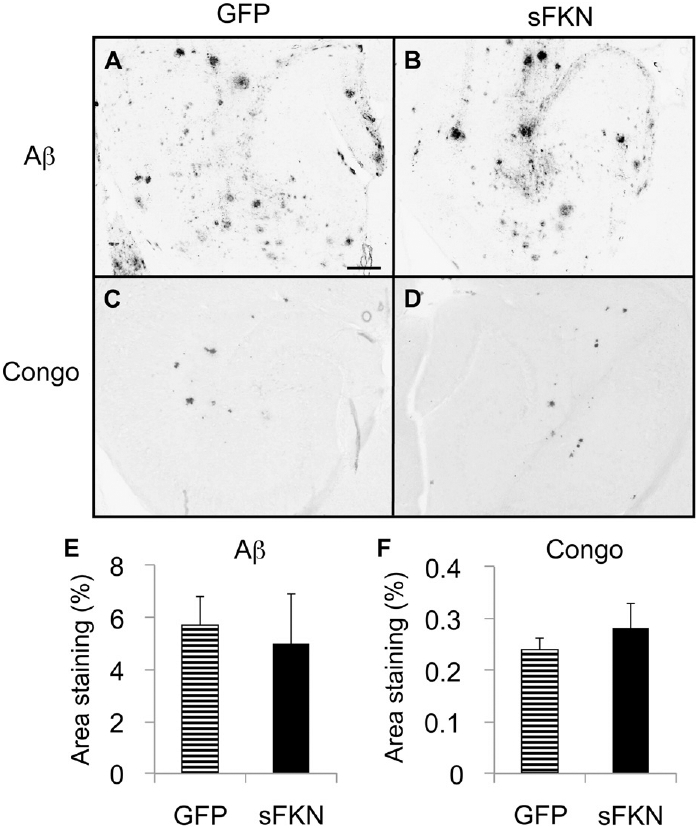
Amyloid levels in APP/PS1 mice treated with GFP or sFKN virus are unaltered. Anti-Aβ staining of rAAV-GFP (A) and rAAV-sFKN (B) -injected mice hippocampi. Congo red staining of rAAV-GFP (C) and rAAV-sFKN (D) -injected mice hippocampi. Percentage area positive staining for anti-Aβ (E) and Congo red staining (F). Scale = 200 μm. Abbreviations: Aβ, amyloid beta; AAV, adeno-associated virus serotype; FKN, fractalkine; GFP, green fluorescent protein; rAAV, recombinant AAV; sFKN, secreted FKN.
4. Discussion
It has long been recognized that microglial activation is a key neuropathologic feature of AD (Akiyama et al., 2000). There is considerable and highly consistent literature that individuals using high doses of nonsteroidal anti-inflammatory drugs (NSAIDs) have reduced risk of AD (McGeer et al., 2006). This observation has led a series of failed attempts to treat AD with immunosuppressive agents such as steroids (Aisen et al., 2000) or NSAIDs (Aisen et al., 2003; Thal et al., 2005). NSAIDs even failed in prevention studies, and might have actually increased conversion to AD when using random assignment (Group et al., 2007). The mismatch between epidemiology and controlled trials is not well explained.
Recent data from mouse models of select aspects of AD pathology, the amyloid- depositing and the tau-depositing mouse models, have indicated quite different responses of these 2 pathologies to the same treatments. Studies examining central LPS injections, genetic overexpression of IL-1, or elimination of FKN signaling all find that increasing microglial activation benefits amyloid pathology and the same manipulation exacerbates tau pathology (Ghosh et al., 2010; Herber et al., 2007; Lee et al., 2010a, 2010b; Shaftel et al., 2007). Thus, 1 possible issue with the attempts to control inflammation thus far is that the effects might not be uniformly beneficial on the different types of pathology found in AD. Our tools might be too blunt to cajole the microglial activation state into one that provides optimal benefits with few deleterious consequences (Morgan et al., 2005; Wyss-Coray, 2006). Thus, further examination of effector molecules, such as FKN, that modulate microglial activation might yield novel and important insights into AD pathology.
In this experiment, we examined the effect of increasing FKN signaling over baseline on tau and amyloid pathology. Therefore, we examined the effects of sFKN in 2 animal models, APP/PS1 (a model of amyloid deposition) and rTg4510 (a model of tau deposition with the frontal temporal dementia tau mutation P301L). We used a viral vector that expresses the soluble ectodomain of the CX3CL1 protein (Morganti et al., 2012). This domain was reported to have bioactivity previously, and should spread further from the injection site because of diffusion, thus allowing a greater affected volume in vivo. We estimate by enzyme-linked immunosorbent assay, that in the hippocampus, we achieved a 2-fold increase over endogenous FKN expression. This did not seem to cause any overt physiological issues in these mice, as indicated by maintained weight.
Examination of the tau pathology in Tg4510 mice expressing supraphysiological sFKN demonstrates a decrease in tau pathology, consistent with the increased tau pathology found when eliminating FKN signaling in the CX3CR1 null mice. We observed a decrease in phoshotau-Ser396 immunostaining, a mid- to late-stage marker of tau en route to formation of neurofibrillary tangles. We further observed a similar decrease in Gallyas silver staining, thought to represent mature tangles in the brains of AD patients. By Western blot analysis we also observed reductions of pS199/S202, pS396, and pS262 tau, but not pS356 tau. Antibodies AT8, AT180, and AT270, that recognize paired helical filaments, all showed reduced staining. Consistent with the reduction in tau tangles stained with Gallyas. Reductions in phospho-tau were also observed in the insoluble fraction. More importantly, this reduction in tau pathology was associated with an increase in the staining of NeuN positive neurons in the hippocampus. Typically in Tg4510 mice of this age there is a thinning of the neuronal layers in the pyramidal and granule cell layers of hippocampus. These mice appeared to retain staining for most of these cells, implying either absence of cell loss or at least retention of phenotype. This is further supported by a stereological count that demonstrates a preservation of granular cells of the dentate gyrus when compared with nontransgenic and GFP-injected controls. We also observed a decrease in the reduction of hippocampal volume compared with the GFP control, suggesting that tau-associated neurodegeneration appears abated
In the radial arm water maze cognitive analysis, there was no rescue of the Tg4510 phenotype with AAV-sFKN treatment. This might be because of the aggressive nature of the Tg4510 model or the limited transduction of the viral expression of fractalkine to the dentate region of the hippocampus. The latter is consistent with the rescue of granular cells in the dentate but not a complete rescue of total hippocampal volume. Further, rescue might require injection into the cortical brain regions where this model also has significant pathology and neuron loss. Injection of the sFKN at an earlier time point in the animals’ life, or suppression of the tau expression with doxycycline before expression of the sFKN might also be required to achieve a significant behavioral rescue in this animal model. Other animals that have a more progressive pathology with age might also offer a better model to examine the benefits of fractalkine expression.
As stated earlier in this report, the loss of FKN signaling in the CX3CR1−/− × APP mice caused a decrease in the deposition of amyloid associated with increased microglial activation. This would suggest that increasing FKN signaling with sFKN might accelerate amyloid pathology, thus reducing enthusiasm for FKN signaling as a therapeutic target. Interestingly, we observed no changes in amyloid deposition with increases of sFKN expression in either a 2- or 4-month study. One explanation is that the endogenous levels of FKN found in APP/PS1 mice are sufficient to maximally suppress any microglia phenotype that would lead to accelerated amyloid deposition; additional FKN signaling has no additive effect. However, the phenotype that is exacerbating tau pathology is not maximally suppressed by endogenous FKN, and that additional FKN signaling can still have benefits. This would be consistent with the increase in CX3CR1 in Tg4510 mice that was not observed in APP/PS1 mice. More receptors in Tg4510 mice would suggest that the receptors are less likely to be saturated by endogenous FKN thus allowing for recombinant FKN ligation and microglial suppression. A second alternative is that only loss of membrane-associated fractalkine signaling leads to clearance of amyloid deposits. Because we are only increasing soluble fractalkine activity, any potential negative effects on amyloid pathology might be avoided.
Thus, this study demonstrates that increased FKN signaling could be a potential target for therapeutic intervention for tau-mediated neurodegeneration. Moreover, this can be achieved without any detrimental consequences to amyloid deposition. We propose that immune modulation with compounds like FKN to slow development of tau pathology in combination with other therapies for amyloid removal might be an effective way to slow progression of AD. Further, treatments increasing FKN signaling might have similar beneficial effects in other tauopathies such as frontal temporal dementia, and potentially in other diseases with intraneuronal protein deposition such as synucleinopathies.
Acknowledgements
The authors thank Dr Peter Mouton for his expert assistance with stereological counts. This work was funded in part by NIH/NIA R01 AG025509, NIH NS76308, and NIH AG15470.
Footnotes
Disclosure statement
The authors have no conflicts of interest.
Ethics Statement
All animal testing procedures were approved by the Institutional Animal Care and Use Committee of the University of South Florida and followed the NIH guidelines for the care and use of laboratory animals (Approval ID no. A4100-01).
References
- Aisen PS, Davis KL, Berg JD, Schafer K, Campbell K, Thomas RG, Weiner MF, Farlow MR, Sano M, Grundman M, Thal LJ, 2000. A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology 54, 588–593. [DOI] [PubMed] [Google Scholar]
- Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ, 2003. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289, 2819–2826. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T, 2000. Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D, 2006. Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat. Protoc 1,1671–1679. [DOI] [PubMed] [Google Scholar]
- Arendash GW, King DL, Gordon MN, Morgan D, Hatcher JM, Hope CE, Diamond DM, 2001. Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res. 891, 42–53. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT, 2010. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, Reier PJ, Mandel RJ, Muzyczka N, 2004. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol. Ther 10, 302–317. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee J-C, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM, 2006. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci 9, 917–924. [DOI] [PubMed] [Google Scholar]
- Carroll JC, Iba M, Bangasser DA, Valentino RJ, James MJ, Brunden KR, Lee VMY, Trojanowski JQ, 2011. Chronic stress exacerbates tau pathology, neurodegeneration, and cognitive performance through a corticotropin-releasing factor receptor-dependent mechanism in a transgenic mouse model of tauopathy. J. Neurosci 31, 14436–14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty N, Lee D, Dickey C, Ceballos-Diaz C,Jansen-West K, Golde TE, Gordon MN, Morgan D, Nash K, 2010. Convection-enhanced delivery and systemic mannitol increase gene product distribution of AAV vectors 5, 8, and 9 and increase gene product in the adult mouse brain. J. Neurosci. Methods 194,144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ, 2000. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage.J. Neurosci 20, RC87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey C, Kraft C, Jinwal U, Koren J, Johnson A, Anderson L, Lebson L, Lee D, Dickson D, de Silva R, Binder LI, Morgan D, Lewis J, 2009. Aging analysis reveals slowed tau turnover and enhanced stress response in a mouse model of tauopathy. Am. J. Pathol 174, 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield JS, 2003. The inflammatory macrophage: a story of Jekyll and Hyde. Clin. Sci 104, 27–38. [DOI] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CKE, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J, 2010. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci 13, 411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, Raines EW, 2001. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J. Biol. Chem 276, 37993–38001. [DOI] [PubMed] [Google Scholar]
- Ghosh SW, Trojanczyk JA, Olschoka JA, O’Banion K, 2010. Sustained over-expression of Interleukin-1b ameliorates plaque pathology but exacerbates tangle pathology in the triple transgenic mouse model of Alzheimer’s disease. Neuroscience Abstracts, San Diego, Program number 50.8/J13. [Google Scholar]
- Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KL, Connor K, Melachrino JO, O’Callaghan JP, Morgan D, 2002. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp. Neurol 173,183–195. [DOI] [PubMed] [Google Scholar]
- Group AR, Lyketsos CG, Breitner JC, Green RC, Martin BK, Meinert C, Piantadosi S, Sabbagh M, 2007. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 68, 1800–1808. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L, 1998. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U. S. A 95, 10896–10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herber DL, Mercer M, Roth DL, Symmonds K, Maloney J, Wilson N, Freeman MJ, Morgan D, Gordon MN, 2007. Microglial activation is required for Abeta clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J. Neuroimmune Pharmacol 2, 222–231. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K, 1998. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med 4, 97–100. [DOI] [PubMed] [Google Scholar]
- Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen K-J, Rose-John S, Ludwig A, 2003. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 102, 1186–1195. [DOI] [PubMed] [Google Scholar]
- Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O, 1997. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 91, 521–530. [DOI] [PubMed] [Google Scholar]
- Kim KW, Vallon-Eberhard A, Zigmond E, Farache J, Shezen E, Shakhar G, Ludwig A, Lira SA, Jung S, 2011. In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood 118, e156–e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Tatom JB, Henderson KM, Henning PP, 2008. AAV8, 9, Rh10, Rh43 vector gene transfer in the rat brain: effects of serotype, promoter and purification method. Mol. Ther 16, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DC, Rizer J, Selenica M-LB, Reid P, Kraft C, Johnson A, Blair L, Gordon MN, Dickey CA, Morgan D, 2010a. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J. Neuroinflammation 7, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT, 2010b. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol 177, 2549–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Weber C, 2007. Transmembrane chemokines: versatile ‘special agents’ in vascular inflammation. Thromb. Haemost 97, 694–703. [PubMed] [Google Scholar]
- Lyons A, Lynch AM, Downer EJ, Hanley R, O’Sullivan JB, Smith A, Lynch MA, 2009. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J. Neurochem 110,1547–1556. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M, 2004. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Rogers J, McGeer EG, 2006. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J. Alzheimers Dis 9 (3 suppl), 271–276. [DOI] [PubMed] [Google Scholar]
- Morales I, Farias G, Maccioni RB, 2010. Neuroimmunomodulation in the pathogenesis of Alzheimer’s disease. Neuroimmunomodulation 17, 202–204. [DOI] [PubMed] [Google Scholar]
- Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM, 2005. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J. Neuropathol. Exp. Neurol 64, 743–753. [DOI] [PubMed] [Google Scholar]
- Morganti J, Nash K, Grimmig B, Ranjit S, Small B, Bickford P, Gemma C, 2012. The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson’s disease. J. Neurosci 32, 14592–14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC, 2011. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J. Neuroinflammation 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH, 2005. Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O’Banion MK, 2007. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Invest 117, 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan S, Hong-Min T, Yi F, Jun-Peng G, Yue F, Yan-Hong T, Yun-Ke Y, Wen- Wei L, Xiang-Yu W, Jun M, Guo-Hua W, Ya-Ling H, Hua-Wei L, Ding- Fang C, 2011. New evidences for fractalkine/CX3CL1 involved in substantia nigral microglial activation and behavioral changes in a rat model of Parkinson’s disease. Neurobiol. Aging 32, 443–458. [DOI] [PubMed] [Google Scholar]
- Streit WJ, 2004. Microglia and Alzheimer’s disease pathogenesis. J. Neurosci. Res 77, 1–8. [DOI] [PubMed] [Google Scholar]
- Streit WJ, 2006. Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci. 29, 506–510. [DOI] [PubMed] [Google Scholar]
- Thal LJ, Ferris SH, Kirby L, Block GA, Lines CR, Yuen E, Assaid C, Nessly ML, Norman BA, Baranak CC, Reines SA, 2005. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuro-psychopharmacology 30, 1204–1215. [DOI] [PubMed] [Google Scholar]
- van Rossum D, Hanisch UK, 2004. Microglia. Metab. Brain Dis 19, 393–411. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, 2006. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Med 12, 1005–1015. [DOI] [PubMed] [Google Scholar]
- Yoneda O, Imai T, Nishimura M, Miyaji M, Mimori T, Okazaki T, Domae N, Fujimoto H, Minami Y, Kono T, Bloom ET, Umehara H, 2003. Membrane-bound form of fractalkine induces IFN-gamma production by NK cells. Eur. J. Immunol 33, 53–58. [DOI] [PubMed] [Google Scholar]
- Zolotukhin S, Potter M, Zolotukhin I, Sakai Y, Loiler S, Fraites TJ, Chiodo VA, Phillipsberg T, Muzyczka N, Hauswirth WW, Flotte TR, Byrne BJ, Snyder RO, 2002. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods 28,158–167. [DOI] [PubMed] [Google Scholar]
- Zujovic V, Benavides J, Vige X, Carter C, Taupin V, 2000. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia 29, 305–315. [PubMed] [Google Scholar]