

Abstract
Assessment of sphingosine-1-phosphate receptor 1 (S1PR1) expression could be a unique tool to determine the neuroinflammatory status for central nervous system (CNS) disorders. Our preclinical results indicate that PET imaging with [11C]CS1P1 radiotracer can quantitatively measure S1PR1 expression changes in different animal models of inflammatory diseases. Here we developed a multiple step F-18 labeling strategy to synthesize the radiotracer [18F]FS1P1, sharing the same structure with [11C]CS1P1. We explored a wide range of reaction conditions for the nucleophilic radiofluorination starting with the key ortho-nitrobenzaldehyde precursor 10. The tertiary amine additive TMEDA proved crucial to achieve high radiochemical yield of ortho-[18F]fluorobenzaldehyde [18F]12 starting with a small amount of precursor. Based on [18F]12, a further four-step modification was applied in one-pot to generate the target radiotracer [18F]FS1P1 with 30∼50% radiochemical yield, >95% chemical and radiochemical purity, and a high molar activity (37∼166.5 GBq/μmol, decay corrected to end of synthesis, EOS). Subsequently, tissue distribution of [18F]FS1P1 showed a high brain uptake (ID%/g) of 0.48 ± 0.06 at 5 min, and bone uptake of 0.27 ± 0.03, 0.11 ± 0.02 at 5, and 120 min respectively, suggesting no in vivo defluorination. MicroPET studies showed [18F]FS1P1 has high macaque brain uptake with a standard uptake value (SUV) of ~2.3 at 120 min. Radiometabolite analysis in nonhuman primate plasma samples indicated that [18F]FS1P1 has good metabolic stability, and no major radiometabolite confounded PET measurements of S1PR1 in nonhuman primate brain. Overall, [18F]FS1P1 is a promising F-18 S1PR1 radiotracer worthy of further clinical investigation for human use.
Graphical Abstract
A promising Sphingosine-1-Phosphate Receptor 1 PET radiotracer [18F]FS1P1 was synthesized using a facile multiple step F-18 radiochemistry procedure.
Introduction
Sphingosine-1-phosphate (S1P) is a natural high-affinity ligand that binds to the five members of the S1P receptor family (S1PR1, 2, 3, 4, and 5). S1PR1 is the most abundant of the five members of S1P receptors and is expressed in a broad range of tissues including the central nervous system (CNS).1 It plays a key role in many physiological and cellular processes. For example, it is involved in the activation of the immune response by regulating differentiation, egress, and migration of immune cells.2 S1PR1 is widely accepted as a therapeutic target for treating inflammatory diseases,3 such as multiple sclerosis (MS),4, 5 colitis,6 inflammatory bowel diseases,7 and atherosclerotic disease.8 To date, the mechanism of S1PR1 modulation in CNS remains not fully understood. An S1PR1 specific PET radiotracer may provide a unique non-invasive tool to advance our understating of S1PR1 function in CNS and other diseases (Figure 1).
Figure 1.

Chemical structures of selected modulators of S1PR1.
To identify a clinical suitable S1PR1 specific radiotracer, we previously reported the synthesis and evaluation of a carbon-11 labeled S1PR1 radiotracer [11C]CS1P1 in three animal models of diseases including MS,9 carotid injury,10 and vascular injury.11 With FDA approval of an exploratory Investigational New Drug (eIND) of [11C]CS1P1 for human use, we recently completed dosimetry and safety studies of [11C]CS1P1 in 10 healthy volunteers (5 female and 5 male), suggesting [11C]CS1P1 is safe for human use to investigate neuroinflammation.12 The proof of mechanism study in MS patients is currently underway in our PET imaging center. Although C-11 labeled radiotracer confers many advantages such as low radiation exposure for patients and the short half-life of the isotope (20.38 minutes) permits multiple studies on the same subject in the same day but also constrains production and distribution for multicenter clinical trials using PET. On the other hand, F-18 labeling is most widely used in clinical PET imaging studies of cardiology, oncology, and neurology. Compared to C-11 radiotracers, the relatively long half-life of F-18 isotope (109.7 minutes) allows for the multiple step synthesis and longer scan sessions and F-18 radiotracers that can increase target-to-reference ratios.13 In addition, F-18 radiotracers facilitate radiotracer distribution for multi-center clinical trials. Therefore, identification of a clinically suitable F-18 S1PR1 radiotracer is imperative. Although a few F-18 radiotracers have been reported for S1PR1,14, 15 none have been transferred for clinical investigation due to either high non-specific binding, fast metabolism in vivo, or other concerns.16, 17 Inspired by promising preclinical animal study results, and human dosimetry, tissue distribution, and safety studies of [11C]CS1P1 from whole body PET scans in healthy volunteer subjects, we herein report a multiple step approach to incorporate F-18 through the aromatic ring of the CS1P1 structure that yields an F-18 labeled radiotracer, 3-((2-[18F]fluoro-4-(5-(20-methyl-2-(trifluoromethyl)-[1,10-biphenyl]-4-yl)-1,2,4-oxadiazol-3-yl)-benzyl)methyl)-amino)-propanoic acid, named as [18F]FS1P1. We further performed tissue distribution of [18F]FS1P1 using Sprague-Dawley rats and confirmed no in vivo defluorination. We also performed PET brain studies of [18F]FS1P1 to compare with [11C]CS1P1 in nonhuman primate, and showed that [18F]FS1P1 and [11C]CS1P1 have almost identical pharmacokinetics in nonhuman primate brain. Radiometabolite analysis of nonhuman primate plasma samples indicated negligible formation of radiometabolites in vivo post-injection of [18F]FS1P1. Thus, we concluded that [18F]FS1P1 is a promising S1PR1 F-18 radiotracer to investigate neuroinflammation worthy of further clinical investigation for human use.
Results
Chemistry
To radiosynthesize [18F]FS1P1, we started with direct nucleophilic radiofluorination of the nitroarenes including uncycled precursor 5 or the cycled precursor 6, and then removed the t-butyl protection group using trifluoroacetic acid (TFA). Therefore, corresponding uncycled nitro precursor 5, and the cycled precursor 6 were prepared as depicted in Scheme 1. Compound 2 was prepared from 4-bromo-3-(trifluoromethyl)benzoic acid 1, which underwent Suzuki cross-coupling with ortho-tolylboronic acid and subsequent base hydrolysis.18 Nucleophilic substitution between 4-(bromomethyl)-3-nitrobenzonitrile 319 and tert-butyl 3-(methylamino)propanoate, followed by treatment with hydroxylamine hydrochloride in the presence of sodium bicarbonate yielded the intermediate amidoxime 4. Compound 2 was coupled with 4 to generate the uncycled intermediate 5 at room temperature. Then potassium hydroxide was used to promote the intramolecular cyclization of 5 to provide the nitro oxadiazole compound 6 in 82% yield.20
Scheme 1.

Reagents and conditions: (a) o-tolylboronic acid, Pd(PPh3)4, K2CO3, toluene/H2O, 110 °C, 75%; (b) NaOH, THF/H2O, RT, 95%; (c) tert-butyl 3-(methylamino)propanoate, NEt3, MeOH, RT; (d) NH2OH·HCl, NaHCO3, MeOH, reflux, 55% for 2 steps; (e) EDCI, HOBt, DMF, RT, 56%; (f) KOH (5 M), DMSO, RT, 82%.
However, starting with either precursor 5 or 6 to synthesize the key 18F-intermediate through the nitro/[18F]F- replacement reaction was not successful although a variety of conditions were tested. The decomposing of 5 or 6 is faster than radiofluorination at our test reaction conditions. We then developed a multiple step 18F-labeling procedure to produce [18F]FS1P1, via two sequential reductive amination reactions (Scheme 2).
Scheme 2.

Indirect appraoch plan for [18F]FS1P1 by sequential reductive amination reactions.
The critical ortho-nitrobenzaldehyde precursors 10 and 11 were made as shown in Scheme 3.21–24 Firstly, amidoxime 7 was prepared from 4-(bromomethyl)-3-nitrobenzonitrile 3 by nucleophilic substitution with potassium acetate, followed by treating with hydroxylamine hydrochloride in the presence of sodium bicarbonate. Using a similar protocol of making compounds 5 and 6, the uncycled nitro benzyl alcohol 8, and its cycled compound 9 were produced at room temperature. The oxidation of 8 and 9 using Dess-Martin reagent afforded the substituted ortho-nitrobenzaldehyde precursors 10 and 11, and they were used as precursors for preparing [18F]FS1P1.
Scheme 3.

Reagents and conditions: (a) KOAc, DMF, RT; (b) NH2OH·HCl, NaHCO3, MeOH, reflux, 79% for 2 steps; (c) 2, EDCI, HOBt, DCM, RT, 60%; (d) KOH (5 M), DMSO, RT, 90%; (e) Dess-Martin reagent, DCM, 0 °C-RT, 85% for 10, 88% for 11.
Radiosynthesis
To radiosynthesis [18F]FS1P1, we initially focused on optimizing conditions to improve the yield of making substituted ortho-[18F]fluorobenzaldehyde [18F]12 through the NO2/[18F]/F- replacement of the precursors 10 or 11. As shown in Table 1, when directly using substituted ortho-nitrobenzaldehyde 11 (4 mg) reacting with [18F]KF in DMSO at 150 °C, no product of [18F]12 was detected by radioactive TLC (entry 1). Further analysis indicated that both the uncycled precursor 10 and cycled precursor 11 were easily decomposed under high temperature that reduced the radiochemical yield for [18F]12. Therefore, we increased the amount of precursor to increase radiochemical yield (entries 2–5). Using 18 mg of the substituted ortho-nitrobenzaldehyde 10 or 11 led to a radiochemical yield of [18F]12 that was ~50% based on radioactive TLC monitoring (entry 4–5). Nevertheless, the purification of the radioactive product [18F]12 through high-performance liquid chromatography (HPLC) became challenging because the excess amount of precursor 10 or 11 generated a substantial mass of side products. The tertiary amines such as 1, 4-diaza[2.2.2]bicyclooctane (DABCO) have been reported to enhance the nucleophilic radiofluorination efficiency.25–27 Therefore, we added DABCO to the radiolabeling reaction vial (entry 6). Starting with 4 mg of the uncycled precursor 10 at the presence of DABCO, gave 45% radiochemistry yield of [18F]12 (entry 6 versus entry 3). Inspired by the remarkable efficiency of DABCO addition, different tertiary amine additives including 1, 8-diazabicyclo[5.4.0]undec-7-ene (DBU), N,N,N’,N’-tetramethyl-ethylenediamine (TMEDA), N,N-diisopropylethylamine (DIPEA) were further tested (entries 7–9). The experiment data suggested that TMEDA was the optimal additive to prepare [18F]12 with 65 ± 10% radiochemical yield (entry 8). Further condition testing by lowering reaction temperature or decreasing precursor loading did not improve the reaction output (entry 10–12). In the end, the optimized reaction condition for [18F]12 was determined (entry 13), starting with ~4.0 mg of precursor 10, utilizing TMEDA (30 μL) as an additive, combined with H2O (1 μL) and heating 5 min at 150 °C in DMSO (300 μL), the synthesis of [18F]12 was accomplished in 70% radioactive TLC yield. Furthermore, organic solvent ether extraction was employed to replace the HPLC separation for purification of the intermediate [18F]12, which reduced the total time for the whole radiosynthesis procedure. Together, utilizing a small amount of precursor 10 combined with TMEDA as the additive led to a significant improvement of this radiolabeling procedure and resolved the challenge of [18F]12 purification by HPLC.
Table 1.
Optimization of 18F-radiolabeling conditions from ortho-nitro benzaldehyde 10 or 11. a
|
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | 10 or 11 (mg) | Additive (μL) | T (°C) | [18F]12 RCY (%)b |
|
| ||||
| 1 | 11 (4.5) | none | 150 | 0 |
| 2 | 11 (9.2) | none | 150 | <5 |
| 3 | 10 (9.5) | none | 150 | <5 |
| 4 | 11 (18.2) | none | 150 | 50 ± 5 |
| 5 | 10 (18.5) | none | 150 | 55 ± 8 |
| 6 | 10 (4.5) | DABCO (10) | 150 | 45 ± 13 |
| 7 | 10 (4.5) | DBU (10) | 150 | 30 ± 6 |
| 8 | 10 (4.5) | TMEDA (10) | 150 | 65 ± 10 |
| 9 | 10 (4.5) | DIPEA (10) | 150 | 43 ± 12 |
| 10 | 10 (4.5) | TMEDA (10) | 120 | 20 ± 5 |
| 11 | 10 (2.3) | TMEDA (10) | 150 | 60 ± 13 |
| 12 | 10 (1.2) | TMEDA (10) | 150 | 12 ± 3 |
| 13c | 10 (4.5) | TMEDA (30) | 150 | 70 ± 16 |
[18F]KF/K222, K2CO3, DMSO (300 μL), 5 min.
RCY was determined based on radio-TLC (n = 3).
1 μL H2O added.
DABCO: 1, 4-diaza[2.2.2]bicyclooctane; DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene; TMEDA: N, N, N′, N′-tetramethylethylenediamine; DIPEA: N,N-diisopropylethylamine.
After a reliable procedure of synthesizing and purifying the key intermediate [18F]12 was established, we further explored a multiple step F-18 labeling strategy for making [18F]FS1P1 as shown in Scheme 4. [18F]12 firstly went through reductive amination by reacting with β-alanine, and then methylation by treating with formalin in one pot (See ESI, Figure S12), the radiotracer [18F]FS1P1 was obtained with 10% radiochemical yield from [18F]12 (decay corrected to end of synthesis). However, substituted ortho-[18F]fluorobenzyl alcohol was the major radioactive product when purifying [18F]FS1P1. The lower solubility of β-alanine in ethanol may lower the conversion rate from [18F]12 to the imine; instead the aldehyde intermediate [18F]12 was mainly reduced to the corresponding ortho-[18F]fluorobenzyl alcohol as a side product. To address this issue, the β-alanine methyl ester was used to replace β-alanine, and we found no side products of ortho-[18F]fluorobenzyl alcohol. Subsequently, using aqueous sodium hydroxide solution to hydrolyze the methyl ester yielded the intended radioactive product [18F]FS1P1. After purification using a reverse-phase HPLC system, the final product [18F]FS1P1 was obtained with 30∼50% radiochemical yield, >95% chemical and radiochemical purity, and a high molar activity (37∼166.5 GBq/μmol, decay corrected to end of synthesis, EOS). It took approximately 120 min from drying the [18F]KF to formulate the radioactive dose of [18F]FS1P1 for in vitro and in vivo animal study. The lipophilicity of [18F]FS1P1 was measured using 1-octanol/PBS buffer method and the LogD7.4 value was 2.62 ± 0.31 (n = 3), suggesting a good blood-brain barrier (BBB) permeability.
Scheme 4.

Synthesis of radiotracers [18F]FS1P1. Reagents and conditions: (a) [18F]KF, Kryptofix 222, TMEDA, DMSO, H2O, 150 °C, 5 min; (b) β-alanine, AcOH, EtOH, 100 °C, 5 min; (c) NaCNBH3, RT, 2 min; (d) formalin, 100 °C, 5 min, then NaCNBH3, RT, 2 min; (e) β-alanine methyl ester, AcOH, EtOH, 100 °C, 5 min; (f) NaOH (5 M), 100 °C, 5 min, then AcOH for neutralization.
Tissue distribution study of [18F]FS1P1 in rats
To evaluate the kinetics and the tissue distribution of [18F]FS1P1 in rodents, Sprague Dawley (SD) male rats (6∼7 weeks old; 200∼300 g) were used and euthanized at 5, 30, 60, and 120 min post-injection. As shown in Figure 2, the initial tracer uptake was high in most tissues at 5 min. The liver had the highest uptake (%ID/g) at 3.04 ± 0.18, whereas the heart, lung, spleen, kidney, and small intestine had moderate to high uptake at 1.39 ± 0.15, 1.44 ± 0.13, 1.14 ± 0.13, 1.76 ± 0.15, and 1.00 ± 0.21 respectively. The radioactivity was rapidly washed out from heart, lung, pancreas, spleen, kidney, and liver, at 30 min post injection with ID%/g values decreasing to 0.55 ± 0.02, 0.84 ± 0.05, 0.66 ± 0.04, 0.54 ± 0.03, 1.13 ± 0.66, and 2.51 ± 0.10 respectively as shown in Figure 2a. Importantly, the bone uptake was relatively low with a %ID/g value of 0.27 ± 0.03 at 5 min, and no significant change was observed from 5 min to 120 min (0.11 ± 0.02), indicating no defluorination of [18F]FS1P1 occurred in vivo. Similar to [11C]CS1P1, a high rat brain uptake of [18F]FS1P1 was observed as 0.48 ± 0.06, 0.48 ± 0.02, 0.60 ± 0.04, and 0.65 ± 0.03 at 5, 30, 60, and 120 min respectively, indicating the brain retention of [18F]FS1P1 were relatively stable. The uptake in the brain regions of interest including the brain stem, cerebellum, cortex, striatum, thalamus, and hippocampus were 0.55 ± 0.06, 0.55 ± 0.07, 0.47 ± 0.08, 0.42 ± 0.05, 0.56 ± 0.12, and 0.42 ± 0.05 at 5 min respectively; and a slight increase was observed from 5 min to 120 min as shown in Figure 2b. The tissue distribution data suggested that [18F]FS1P1 has a high rat brain uptake and no defluorination happened in vivo.
Figure 2.

Tissue distribution of [18F]FS1P1 in normal SD rats. (a) The uptake (ID%/gram) of [18F]FS1P1 was high in the tissues including heart, lung, pancreases, spleen, kidney, liver, brain, small intestine, and large intestine; (b) The uptake (ID%/gram) of [18F]FS1P1 in brain regionals of interest was relative high ranging from 0.4 to 0.8. Data represents mean ± SEM.
MicroPET studies of the cynomolgus macaque brain
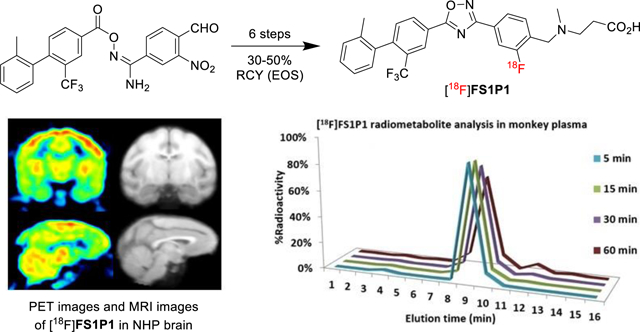
To further confirm if [18F]FS1P1 is suitable for PET imaging study of S1PR1 in the brain, microPET studies in the brain of a male cynomolgus macaque was performed. The microPET brain imaging scans were carried out in the same animal to precisely compare the pharmacokinetics of [18F]FS1P1 and [11C]CS1P1, which shared the same chemical structure, but were labeled with different isotope. As shown in Figure 3a, the time tissue activity curves of the brain uptake (standard uptake value, SUV) of [18F]FS1P1 and [11C]CS1P1 were almost identical. Both [18F]FS1P1 and [11C]CS1P1 penetrated the BBB very well and the brain uptake reached a maximum SUV value of ~2.3 from 20 to 120 min post-injection, indicating [18F]FS1P1 and [11C]CS1P1 have identical pharmacokinetics in the macaque brain.
Figure 3.

The uptake of [18F]FS1P1 in the macaque brain. (a) The total brain uptakes of [18F]FS1P1 and [11C]CS1P1 were very similar; (b) The uptake of [18F]FS1P1 in different regions of the macaque brain; (c) Representative PET images and co-registered MRI images of [18F]FS1P1 in macaque brain; (d) The total brain uptakes (SUV) of [18F]FS1P1 using different doses were also identical.
Among different brain regions, [18F]FS1P1 showed a high uptake in thalamus, putamen, caudate, and prefrontal cortex, whereas cerebellum and basal frontal cortex had only moderately high uptake (Figure 3b and 3c). In addition, the initial studies of [18F]FS1P1 in the brains of different macaques indicated a good reproducibility, the high and low injection doses of ~8.13 MBq (2.17 mCi) and ~25.2 MBq (6.79 mCi) injection provided similarly good quality visualization of brain and almost identical time tissue activity curves as shown in Figure 3d. Further PET imaging modeling and binding potential studies will determine the optimal dose of [18F]FS1P1 for particular applications.
HPLC Radiometabolite analysis of macaque plasma samples
HPLC radiometabolism analysis of macaque plasma samples collected at different time points post-injection of [18F]FS1P1 permitted analysis of the stability of [18F]FS1P1 and the radiometabolites in vivo that can be detected. As shown in Figure 4, the parent radioactive compound [18F]FS1P1 was the only major radioactive peak with a retention time of ~9 min on the HPLC (Figure 4a), the percentage of the parental radiotracer [18F]FS1P1 was 96%, 96%, 94%, and 88% of total radioactivity at 5, 15, 30, and 60 min post-injection, respectively (Figure 4a and 4b). Nonobvious radiometabolite peak was detected in the 5 and 15 min plasma samples. For 30 and 60 min plasma samples, only a negligible lipophilic radioactive peak with a retention time of ~12 min, was observed with 2% and 6% of the total radioactivity respectively (Figure 4a and 4b). No hydrophobic radioactive peak was detected for all plasma samples collected from 5, 15, 30, and 60 min post-injection of [18F]FS1P1, suggesting [18F]FS1P1 has favorable in vivo stability, and no major radiometabolite will confound the PET with [18F]FS1P1 measurement of the S1PR1 in the brain.
Figure 4.

Radiometabolite analysis of macaque plasma samples post injection of [18F]FS1P1. No major metabolite was detected in macaque plasma samples collected at 5, 15, 30, and 60 min post injection.
Our biodistribution studies indicate that [18F]FS1P1 had good brain uptake without in vivo defluorination. Our PET brain studies in macaques indicated that [18F]FS1P1 had a good brain uptake and similar pharmacokinetics with [11C]CS1P1 in macaque. Our initial radiometabolite analysis of [18F]FS1P1 in macaque indicated [18F]FS1P1 has a good in vivo stability with no major radiometabolite emerged in vivo within 60 min post-injection. [18F]FS1P1 could be a promising S1PR1 radiotracer for investigating neuroinflammation as well as other inflammatory diseases.
Discussion
S1PR1 plays a crucial role in various physiological and pathophysiological processes. While most previous efforts aimed at the development of S1PR1 specific ligands for improving their therapeutic effect, our efforts focused on the identification of a S1PR1 specific radioligand for quantitative measurement of S1PR1 expression in response to inflammation. We previously reported a few of S1PR1 specific radioligands and our preclinical studies for rodent disease models including MS,9 carotid injury,10 vascular injury,11 and infection disease.28, 29 Importantly, with the FDA approval of [11C]CS1P1 for human use, we completed whole body dosimetry studies and tissue distribution studies in 10 human subjects, suggesting [11C]CS1P1 is safe for investigating S1PR1 expression for human CNS disorders and other diseases.28–30 Nevertheless, an F-18 labeled S1PR1 specific radiotracer may offer many advantages for clinical use and facilitate multiple center clinical trial studies of neuroinflammation in CNS and peripheral tissues. Because the structure of the CS1P1 molecule contains a fluorine atom in one of the three aromatic rings, we explored F-18 radiochemistry to develop an F-18 labeled [18F]FS1P1. If preclinical animal studies of [18F]FS1P1 demonstrated similar in vivo binding specificity with no defluorination, then [18F]FS1P1 could be a promising S1PR radiotracer that could be translated to human clinical investigations using the toxicology data that has already been generated for [11C]CS1P1. This plus the additional human experience with [11C]CS1P1 from human studies, should facilitate implementation of human studies with [18F]FS1P1.
We developed a multiple step, reliable strategy to synthesize [18F]FS1P1 with good yield and high quality for in vivo study. Using the tertiary amine, TMEDA as an additive for the NO2/[18F]F- replacement reaction at optimal conditions, yielded the key intermediate ortho-[18F]fuorobenzaldehyde [18F]12 with 70% radioactive TLC yield starting with a reasonable amount of precursor 10 (4 mg). After that, [18F]12 went through continuous twice reductive amination reactions, followed by hydrolysis and neutralization, [18F]FS1P1 was achieved with good radiochemical yield (30∼50%), >95% radiochemical and chemical purities and high molar activity (37∼166.5 GBq/μmol, EOS). This suggested [18F]FS1P1 synthesized by the multiple step strategy is suitable for in vivo evaluation in animals. If [18F]FS1P1 is selected for translational clinical investigation for human use, the scale-up radiosynthesis with current condition will be tested and optimized under for automation production under current good manufacturing practices (cGMP) fashion. Even current multiple step procedure is successful and effective, further exploration of a more efficient radiosynthesis with fewer steps allows to deliver large amount of [18F]FS1P1 from one batch production is necessary. A more efficient radiosynthesis procedure expedite multiple clinical trials of [18F]FS1P1 for clinical use.
Our tissue distribution study in Sprague-Dawley rats showed that [18F]FS1P1 has good brain uptake comparable to [11C]CS1P1 and similar uptake in other tissues.10, 11 For the brain regions of interests, [18F]FS1P1 also showed relatively high uptake and good retention, indicating good BBB permeability of [18F]FS1P1. This is consistent with its experimental measure of LogD7.4 value (2.62 ± 0.31), considered within the optimal range of 1 to 3 for most CNS drugs.31, 32 Importantly, a very low bone uptake was observed and has no increasing trend from 5 to 120 min post-injection, indicating [18F]FS1P1 has no defluorination in vivo, which is a critical concern for most F-18 radiotracers. Our microPET study indicates that [18F]FS1P1 and [11C]CS1P1 have similar pharmacokinetics in the nonhuman primate brain, as well as a similar distribution in the brain regions of interest as shown in Figure 4b.28 The brain uptake of [18F]FS1P1 in different macaques also showed good brain time-activity curve reproducibility. PET studies with different injection doses of 8.13 MBq (2.17 mCi) versus 25.2 MBq (6.79 mCi) led to similar visualization of [18F]FS1P1 in the brain with high resolution. The brain time-activity curves of tracer uptake (SUV) were almost identical as showed in Figure 4d, suggesting the lower dose of [18F]FS1P1 could be used in future studies. Our metabolite analysis of nonhuman primate plasma samples showed no major radiometabolite were formed within 60 min post-injection of [18F]FS1P1, eliminating the concern that potential radioactive metabolites may confound PET quantification of brain uptake. Since different species may have different metabolism, human plasma radiometabolite analysis is needed to confirm no new formed radiometabolite will impact precise PET with [18F]FS1P1 measurement in human beings.
Together, our preliminary studies suggest that [18F]FS1P1 has almost identical in vivo pharmacological properties as [11C]CS1P1. The reliable multiple-step procedure of producing [18F]FS1P1 with good F-18 radiochemistry yield allows sufficient doses of [18F]FS1P1 for multiple PET studies although further optimization of the procedure under current Good Manufacturing Practices (cGMP) fashion is necessary for automation production of [18F]FS1P1 for human use. Our data suggest that [18F]FS1P1 is a promising F-18 radiotracer for imaging S1PR1 in vivo for inflammatory diseases.
Conclusion
In this study, the radiosynthesis of [18F]FS1P1 was accomplished from the substituted ortho-nitro benzenaldehyde precursor 10 via a multiple step procedure with high radiochemical yield and good quality. [18F]FS1P1 has a high possibility to be a promising F-18 radiotracer for imaging of S1PR1 expression in response to neuroinflammation and other inflammatory diseases in vivo. Further translational clinical investigation of [18F]FS1P1 will confirm its suitability for human use.
Experimental Section
General
All reagents and chemicals were purchased commercially and used as received, unless otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) using silica gel 60 F254 (EMD Chemicals Inc, Billerica, MA). Flash column chromatography was conducted using 230–400 mesh silica gel (SiliCycle Inc, Quebec, Canada). Melting points were determined on a MEL-TEMP 3.0 apparatus without correction. All deuterated solvents were purchased from Cambridge Isotope Laboratories. 1H and 13C NMR spectra were recorded on a 400 MHz Varian instrument. Chemical shifts were reported in parts per million (ppm) and were calibrated using a residual undeuterated solvent as an internal reference (CDCl3: δ 7.26 ppm; CD3OD: δ 3.31 ppm; Acetone-d6: δ 2.05 ppm; DMSO-d6: δ 2.50 ppm). Coupling constants (J) are reported in Hertz (Hz). Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), h (hextet), m (multiplet) and br (broad). High-resolution positive ion mass was acquired by a Bruker MaXis 4G Q-TOF mass spectrometer with an electrospray ionization source. The Accell Plus QMA Cartridge (Inc Catalog # WAT023525), sodium sulfate Sep-Pak cartridges (Inc Catalog # WAT054265), and C18 Sep-Pak Plus cartridges (Inc Catalog # WAT020515) were purchased from Waters Corporation, Milford, MA.
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Washington University and approved by the Institutional Animal Care and Use Committee (IACUC) of Washington University in St. Louis, Missouri, USA. The animal studies were conducted in the Preclinical Imaging Facility at the Washington University School of Medicine.
Chemistry
2’-Methyl-2-(trifluoromethyl)-[1,1’-biphenyl]-4-carboxylic acid (2)
Synthesis of compound 2 followed the published procedure.18
Tert-butyl (Z)-3-((4-(N’-hydroxycarbamimidoyl)-2-nitrobenzyl)-(methyl)amino)propanoate (4)
To a round-bottom flask equipped with a stir bar was added 4-(bromomethyl)-3-nitrobenzonitrile 3 (1.20 g, 5.0 mmol), tert-butyl 3-(methylamino)propanoate (0.95 g, 6.0 mmol), and methanol (20 mL). After cooling to 0 °C, triethylamine (1.01 g, 10.0 mmol) was added to the mixture dropwise. The mixture was warmed to RT and stirred overnight. Then, the mixture was concentrated in vacuum, and the crude residue was used directly without further purification.
To a round-bottom flask equipped with a stir bar and the crude residue was added hydroxylamine hydrochloride (0.70 g, 10 mmol), NaHCO3 (1.68 g, 20 mmol), and methanol (20 mL). The reaction was refluxed and stirred in a pre-heated 70 °C oil-bath for 8 h. The reaction mixture was cooled to room temperature, and the precipitate was filtered off and washed with methanol. The filtrate was concentrated under reduced pressure, and the crude residue was purified by flash chromatography, eluted with ethyl acetate/methanol (3/2, v/v) to afford the product 4 (1.05 g). Yield: 55%, yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 9.73 (s, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.39 (d, J = 11.5 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 5.83 (s, 2H), 3.47 (s, 2H), 2.55 (t, J = 6.5 Hz, 2H), 2.33 (t, J = 6.6 Hz, 2H), 2.08 (s, 3H), 1.34 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 171.62, 162.03, 150.04, 134.61, 131.37, 126.10, 121.28, 112.18, 79.89, 53.93, 52.77, 41.89, 33.92, 28.12.
Tert-butyl (Z)-3-(methyl(4-(N’-((2’-methyl-2-(trifluoromethyl)-[1,1’-biphenyl]-4- carbonyl) oxy)carb-amimidoyl)-2-nitro-benzyl)-amino)propanoate (5)
To a round-bottom flask equipped with a stir bar was added acid 2 (280 mg, 1.0 mmol), HOBt (135 mg, 1.0 mmol), EDCI (287 mg, 1.5 mmol), and DMF (10 mL). The reaction mixture was stirred for 0.5 h followed by adding amidoxime 4 (422 mg, 1.2 mmol). The reaction mixture was stirred overnight at room temperature and monitored by TLC. After finish, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine, and dried over anhydrous MgSO4. After filtration and concentration, the residue was purified on a silica gel column to afford 5. Yield: 56%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.37 (s, 1H), 8.19 (d, J = 7.8 Hz, 1H), 8.11 (s, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.30 (d, J = 7.9 Hz, 1H), 7.22 (d, J = 7.3 Hz, 1H), 7.20 – 7.09 (m, 2H), 7.02 (d, J = 7.4 Hz, 1H), 5.69 (s, 2H), 3.72 (s, 2H), 2.61 (t, J = 6.8 Hz, 2H), 2.31 (q, J = 7.1 Hz, 2H), 2.08 (s, 3H), 1.94 (d, J = 4.0 Hz, 3H), 1.35 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 203.84, 171.83, 162.83, 155.57, 149.26, 145.75 (d, JC-F = 2.0 Hz), 137.68 (d, JC-F = 3.0 Hz), 135.45, 132.29 (d, JC-F = 4.0 Hz), 132.16, 131.31, 130.85, 130.75, 129.72, 129.33 (d, JC-F = 30.3 Hz), 128.85, 128.60, 128.36, 127.24 (d, JC-F = 6.0 Hz), 124.96, 124.72, 123.36 (q, JC-F = 275.7 Hz), 122.84 (d, JC-F = 2.0 Hz), 80.51, 58.17, 53.15, 42.07, 42.04, 33.86, 28.02, 19.97; HRMS (ESI) calcd for C31H34F3N4O6 [M + H]+ 615.2430, found 615.2425.
Tert-butyl 3-(methyl(4-(5-(2’-methyl-2-(trifluoro methyl)-[1,1’-biphenyl]-4-yl)-1,2,4- oxadiazol-3-yl)-2-nitro-benzyl)amino)-propanoate (6)
To a round-bottom flask equipped with a stir bar was added 5 (307 mg, 0.5 mmol) and DMSO (5 mL). A solution of potassium hydroxide (0.6 mL, 5 M) was added dropwise. The reaction mixture was stirred for 0.5 h at room temperature and monitored by TLC. The reaction was diluted with ethyl acetate and water, the ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtering and concentrated in vacuum, the crude residue was purified on a silica gel column to afford 6. Yield: 82%, yellow oil. 1H NMR (400 MHz, Acetone-d6) δ 8.45 (s, 1H), 8.41 (s, 1H), 8.36 (d, J = 7.8 Hz, 1H), 8.26 – 8.19 (m, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.49 (d, J = 7.9 Hz, 1H), 7.21 (q, J = 7.4 Hz, 2H), 7.13 (t, J = 7.1 Hz, 1H), 7.04 (d, J = 7.4 Hz, 1H), 3.75 (s, 2H), 2.58 (t, J = 7.1 Hz, 2H), 2.26 (t, J = 7.1 Hz, 2H), 2.08 (s, 3H), 1.93 (s, 3H), 1.29 (s, 9H); 13C NMR (101 MHz, Acetone-d6) δ 174.93, 171.04, 167.33, 150.10, 145.50, 137.92, 137.60, 135.40, 133.42 (d, JC-F = 5.0 Hz), 132.17, 131.16, 130.66, 129.78, 129.40 (d, JC-F = 31.3 Hz), 128.84, 128.53, 126.63, 125.64 (q, JC-F = 5.0 Hz), 125.09, 123.50 (q, JC-F = 275.7 Hz), 123.44, 122.88, 79.48, 58.01, 53.07, 41.59, 41.54, 33.46, 27.40, 19.30, 19.27; HRMS (ESI) calcd for C31H32F3N4O5 [M + H]+ 597.2325, found 597.2321.
(Z)-N’-hydroxy-4-(hydroxymethyl)-3-nitrobenzimid amide (7)
To a round-bottom flask equipped with a stir bar was added 4-(bromomethyl)-3-nitrobenzonitrile 3 (1.20 g, 5.0 mmol), potassium acetate (0.74 g, 7.5 mmol), and DMF (20 mL). The mixture was stirred overnight at RT. Then, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine, and dried over anhydrous MgSO4. After filtration and concentration, the residue was used directly without further purification.
To a round-bottom flask equipped with a stir bar and the crude residue was added hydroxylamine hydrochloride (0.70 g, 10 mmol), NaHCO3 (1.68 g, 20 mmol), and methanol (20 mL). The reaction was refluxed and stirred in a pre-heated 70 °C oil-bath for 8 h. The reaction mixture was cooled to room temperature, and the precipitate was filtered off and washed with methanol. The filtrate was concentrated under reduced pressure, and the crude residue was purified by flash chromatography, eluted with ethyl acetate/methanol (3/1, v/v) to afford the product 7 (0.83 g), Yield: 79%, yellow solid, MP: 101–103 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.87 (s, 1H), 8.29 (s, 1H), 8.01 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 6.02 (s, 2H), 5.56 (t, J = 5.2 Hz, 1H), 4.79 (d, J = 4.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 149.41, 147.05, 139.10, 133.37, 130.49, 128.69, 121.38, 60.26.
(Z)-4-(hydroxymethyl)-N’-((2’-methyl-2-(trifluoromethyl)-[1,1’-biphenyl]-4-carbonyl)oxy)-3-nitro benzimidamide (8)
To a round-bottom flask equipped with a stir bar was added acid 2 (280 mg, 1.0 mmol), HOBt (135 mg, 1.0 mmol), EDCI (287 mg, 1.5 mmol), and dichloromethane (10 mL). The reaction mixture was stirred for 0.5 h followed by adding amidoxime 7 (253 mg 1.2 mmol). The reaction mixture was stirred for overnight at room temperature and monitored by TLC. After finish, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with 1 M HCl, saturated brine, and dried over anhydrous MgSO4. After filtration and concentration, the residue was purified on a silica gel column to afford 8. Yield: 60%, yellow solid, MP: 151–154 °C. 1H NMR (400 MHz, CD3OD) δ 8.54 (d, J = 7.9 Hz, 2H), 8.47 (d, J = 6.7 Hz, 1H), 8.16 (d, J = 7.5 Hz, 1H), 7.99 (d, J = 7.4 Hz, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.30 (s, 2H), 7.23 (s, 1H), 7.12 (s, 1H), 5.01 (s, 2H), 2.04 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 163.60, 156.89, 147.02, 145.62, 140.50, 137.88, 135.25, 132.32, 131.63, 131.27, 129.39, 128.89 (d, JC-F = 31.3 Hz), 128.69, 128.58, 128.38, 128.07, 126.90 (q, JC-F = 5.0 Hz), 124.67, 123.56 (d, JC-F = 274.7 Hz), 123.06, 60.37, 18.74.
(4-(5-(2’-Methyl-2-(trifluoromethyl)-[1,1’-biphenyl]-4-yl)-1,2,4-oxadiazol-3-yl)-2-nitro-phenyl)methanol (9)
To a round-bottom flask equipped with a stir bar was added 8 (473 mg, 1.0 mmol) and DMSO (10 mL). A solution of potassium hydroxide (1.2 mL, 5 M) was added dropwisely. The reaction mixture was stirred for 0.5 h at room temperature and monitored by TLC. The reaction was diluted with ethyl acetate and water, the ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtering and concentrated in vacuum, the crude residue was purified on a silica gel column to afford 9. Yield: 90%, yellow solid, MP: 143–146 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.70 (d, J = 1.3 Hz, 1H), 8.56 (s, 1H), 8.51 – 8.35 (m, 2H), 8.15 (d, J = 8.1 Hz, 1H), 7.61 (d, J = 7.9 Hz, 1H), 7.39 – 7.29 (m, 2H), 7.26 (t, J = 7.2 Hz, 1H), 7.18 (d, J = 7.5 Hz, 1H), 5.06 (s, 2H), 4.85 (s, 1H), 2.06 (d, J = 4.8 Hz, 3H); 13C NMR (101 MHz, Acetone-d6) δ 174.88, 167.31, 147.27, 145.48, 141.98, 137.59, 135.42, 133.40 (d, JC-F = 5.0 Hz), 131.67, 131.11, 129.77, 129.38 (d, JC-F = 30.3 Hz), 129.34, 128.85, 128.53, 126.16, 125.59 (q, JC-F = 5.0 Hz), 125.08, 123.50 (q, JC-F = 275.7 Hz), 123.38, 122.96 (d, JC-F = 3.0 Hz), 60.68, 19.27 (d, JC-F = 4.0 Hz).
(Z)-4-formyl-N’-((2’-methyl-2-(trifluoromethyl)-[1,1’-biphenyl]-4-carbonyl)oxy)-3-nitrobenz imidamide (10)
To a round-bottom flask equipped with a stir bar was added 8 (237 mg, 0.5 mmol) and dichloromethane (5 mL). The reaction mixture was stirred at 0 °C for 0.5 h followed by adding Dess-Martin reagent (254 mg, 0.6 mmol). The reaction mixture was stirred at room temperature and monitored by TLC. After finish, the reaction was diluted with dichloromethane and water, the ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtering and concentrated in vacuum, the crude residue was purified on a silica gel column to afford 10. Yield: 85%, yellow solid, MP: 160–163 °C. 1H NMR (400 MHz, Acetone-d6) δ 10.43 (s, 1H), 8.63 (s, 1H), 8.55 (s, 1H), 8.50 (d, J = 7.9 Hz, 1H), 8.42 (d, J = 8.0 Hz, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.52 (d, J = 7.9 Hz, 1H), 7.35 (d, J = 6.9 Hz, 2H), 7.27 (t, J = 7.0 Hz, 1H), 7.17 (d, J = 7.5 Hz, 1H), 7.06 (s, 2H), 2.06 (s, 3H); 13C NMR (101 MHz, Acetone-d6) δ 189.12, 163.05, 155.93, 150.46, 146.21, 142.09, 138.85, 137.89, 136.27, 133.65, 133.48, 133.37, 132.89, 130.82, 130.65, 130.60, 130.16, 129.75, 129.46 (d, JC-F = 30.3 Hz), 129.26, 128.04 (d, JC-F = 6.0 Hz), 125.92, 124.65 (d, JC-F = 274.7 Hz), 123.66, 20.15; HRMS (ESI) calcd for C23H17F3N3O5 [M + H]+ 472.1120, found 472.1115.
4-(5-(2’-Methyl-2-(trifluoromethyl)-[1,1’-biphenyl]-4-yl)-1,2,4-oxadiazol-3-yl)-2-nitrobenz aldehyde (11)
To a round-bottom flask equipped with a stir bar was added 9 (228 mg, 0.5 mmol) and dichloromethane (5 mL). The reaction mixture was stirred at 0 °C for 0.5 h followed by adding Dess-Martin reagent (254 mg, 0.6 mmol). The reaction mixture was stirred at room temperature and monitored by TLC. After finish, the reaction was diluted with dichloromethane and water, the ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtering and concentrated in vacuum, the crude residue was purified on a silica gel column to afford 11. Yield: 88%, yellow solid, MP: 152–155 °C. 1H NMR (400 MHz, Acetone-d6) δ 10.46 (s, 1H), 8.85 (d, J = 1.0 Hz, 1H), 8.74 – 8.62 (m, 2H), 8.57 (d, J = 7.9 Hz, 1H), 8.19 (d, J = 7.8 Hz, 1H), 7.68 (d, J = 7.9 Hz, 1H), 7.37 (d, J = 7.6 Hz, 2H), 7.30 (t, J = 7.1 Hz, 1H), 7.21 (d, J = 7.4 Hz, 1H), 2.09 (s, 3H); 13C NMR (101 MHz, Acetone-d6) δ 189.04, 176.32, 167.87, 151.01, 146.61, 138.49, 136.31, 134.43, 134.08, 133.10, 132.46, 132.18, 131.64, 130.71, 130.36 (d, JC-F = 30.3 Hz), 129.75, 129.47, 126.65 (q, JC-F = 5.0 Hz), 126.02, 124.41 (q, JC-F = 274.7 Hz), 124.23, 123.86, 20.14; HRMS (ESI) calcd for C23H15F3N3O4 [M + H]+ 454.1015, found 454.1009.
2-Fluoro-4-(5-(2’-methyl-2-(trifluoromethyl)-[1,1’-biphenyl]-4-yl)-1,2,4-oxadiazol-3-yl)benzaldehyde (12)
Synthesis of compound 12 followed the procedure in Electronic Supplementary Material (Scheme S1).
Yield: 53%, white solid, MP: 121–123 °C. 1H NMR (400 MHz, Acetone-d6) δ 10.38 (s, 1H), 8.61 (s, 1H), 8.52 (d, J = 8.0 Hz, 1H), 8.16 (d, J = 8.0 Hz, 1H), 8.11 – 7.97 (m, 2H), 7.66 (d, J = 7.9 Hz, 1H), 7.41 – 7.31 (m, 2H), 7.27 (t, J = 7.1 Hz, 1H), 7.19 (d, J = 7.5 Hz, 1H), 2.06 (s, 3H); 13C NMR (101 MHz, Acetone-d6) δ 186.22 (d, JC-F = 6.0 Hz), 175.13, 167.43, 164.13 (d, JC-F = 258.6 Hz), 145.54, 137.58, 135.41, 133.74 (d, JC-F = 10.1 Hz), 133.48, 131.20, 129.88 (d, JC-F = 2.0 Hz), 129.79, 129.38 (d, JC-F = 30.3 Hz), 128.83, 128.56, 126.15 (d, JC-F = 9.0 Hz), 125.66 (q, JC-F = 6.0 Hz), 125.11, 124.87, 123.58 (d, JC-F = 4.0 Hz), 123.50 (q, JC-F = 275.7 Hz), 115.34 (d, JC-F = 24.2 Hz), 19.23; HRMS (ESI) calcd for C23H15F4N2O2 [M + H]+ 427.1070, found 427.1067.
Radiochemistry
Procedure for the radiosynthesis of [18F]FS1P1 mediated by [18F]12 (Scheme 4)
The [18F]fluoride in a 0.2~2.5 mL bolus of [18O]H2O and was trapped on a pre-conditioned QMA cartridge (WAT023525, Waters) to remove [18O]H2O and other aqueous impurities. [18F]Fluoride was eluted into the reaction vessel using aqueous potassium carbonate solution (3.0 mg/mL).
[18F]KF (~7.4 GBq) aqueous solution was added to a vial containing Kryptofix 222 (K222) (6∼7 mg), and dried by azeotropic evaporation with acetonitrile (3 × 1 mL) under N2 flow at 100 °C. The vial was cooled to room temperature, and a solution of the precursor 10 (4∼5 mg) in DMSO (300 μL) and TMEDA (30 mg), H2O (1 μL) were added and heated at 150 °C for 5 min, and then cooled to room temperature. The reaction mixture was diluted with 3 mL saturated sodium chloride solution and extracted with ether (3 × 2 mL). The ether solution was collected and then passed through two stacked plus long sodium sulfate Sep-Pak cartridges (WAT054265, Waters) to remove the residual water. After removing ether, using N2 flow at room temperature, a solution of β-alanine methyl ester (5.0 mg) and acetic acid (5 μL) in anhydrous ethanol (300 μL) was added into the reaction vial and heated at 100 °C for 5 min. Upon cooling to room temperature, sodium cyanoborohydride (3 mg) was added to the reaction mixture. The vial was capped, shaken, and allowed to stand at room temperature for 2 min. Formalin (50 μL) was added to the reaction mixture. The vial was capped, shaken occasionally, and stand at room temperature for 2 min. Sodium cyanoborohydride (3 mg) was added to the reaction mixture. The vial was capped, shaken, and heated at 100 °C for 5 min. Upon cooling to room temperature, sodium hydroxide (100 μL, 5 M) was added to the reaction mixture. The vial was capped, shaken, and then heated at 100 °C for 5 min. After cooling by water bath, the reaction mixture was quenched by acetic acid (50 μL) and HPLC mobile phase (3.0 mL, 51% acetonitrile in 0.1 M ammonium formate buffer, pH = 4.5). The solution was loaded onto a reverse semi-preparative HPLC system for purification. The HPLC system contains a 5 mL injection loop, a Phenomenex Luna column (250 × 9.6 mm, 5 μm), a UV detector at 254 nm wavelength, and a radioactivity detector. Using acetonitrile/0.1 M ammonium formate buffer (51/49, v/v, pH 4.5) as the eluent with a flow rate of 4 mL/min, the retention time of the radioactive product was collected from 25 to 28 min. The radioactive product fraction collection was diluted using sterile water (∼50 mL) and then passed through a C18 Sep-Pak Plus short cartridge (WAT020515, Waters). The trapped product was eluted using 10% ethanol in 0.9% saline. After sterile filtration into a glass vial, [18F]FS1P1 was ready for quality control (QC) analysis and animal studies. QC HPLC was conducted following the conditions: Phenomenex SB-C18 column (250 × 4.6 mm, 5 μm), mobile phase 75% acetonitrile in ammonium formate buffer (0.1 M, pH 4.5) as mobile phase, flow rate at 1.5 mL/min, UV wavelength at 254 nm, and tR at 3.8 min. The decay corrected radiochemical yield of making [18F]FS1P1 from [18F]/fluoride was 30∼50% (decay corrected to the end of synthesis), with >95% chemical and radiochemical purity, and molar activity ranged from 37∼166.5 GBq/μmol (1000∼4500 Ci/mmol, decay corrected to the end of synthesis). The synthesis of [18F]FS1P1 took about 120 min including the [18F]fluorine drying step.
The other procedures for condition optimization of radiolabelling were similar with the process above.
Log D7.4 Measurement
Partition coefficient was measured by mixing the [18F]FS1P1 sample with 3 mL each of 1-octanol and buffer that is 0.1 M phosphate and pH equals 7.4 in a test tube. The mixture in the test tube was vortexed for 20 sec followed by centrifugation for 1 min at room temperature. Then 2 mL of the organic layer was transferred to a second test tube, and 1 mL of 1-octanol and 3 mL of PBS buffer were added. The resulting mixture was vortexed for 20 sec, followed by centrifugation for 1 min at room temperature. Then 1 mL of the organic and aqueous layer were taken separately for measurement. The radioactivity content values (count per minute) of two samples (1 mL each) from the 1-octanol and buffer layers were counted using a gamma counter. The partition coefficient LogD7.4 was determined by calculating as the decimal logarithm the ratio of cpm/mL between 1-octanol and PBS buffer. The measurements were repeated three times. The value of the partition coefficient is 2.62 ± 0.31.
Ex vivo biodistribution study in Sprague Dawley rats
A dose of [18F]FS1P1 (~3.7 MBq/100 μL) was injected via the tail vein into SD rats (male; 6∼7 weeks old; 200∼300 g). Rats were euthanized under anesthesia at 5, 30, 60, and 120 min post-injection (n = 4 per group). Tissues of interest including blood, heart, lung, muscle, fat, pancreas, spleen, kidney, liver, brain, bone, thymus, small intestine, and large intestine were collected, weighed, and counted on an automated Beckman Gamma counter (Beckman, Brea, CA). To evaluate the uptake of [18F]FS1P1 within the brain, brain dissection was performed and different brain regions including brain stem, cerebellum, cortex, striatum, thalamus, and hippocampus were collected and evaluated. The uptake of each organ was calculated and expressed as a percentage of the injection dose per gram of wet tissue (%ID per gram).
MicroPET brain study in cynomolgus macaque
Male macaques (∼10 kg) were used for PET imaging data acquisition with a microPET Focus 220 scanner (Siemens Inc., Knoxville, TN). Animals were anesthetized using ketamine and glycopyrrolate and maintained with inhalation of isoflurane. Core temperature was kept at 37 °C with a heated water blanket. The head was secured in a customized head holder. Subsequently, a 2 h dynamic PET scan was performed after administration of radiotracers (8.13 MBq (2.17 mCi) or ~25.2 MBq (6.79 mCi)) via the venous catheter. PET scans data were collected from 0∼120 min with the following time frames: 3 × 1 min, 4 × 2 min, 3 × 3 min, and 20 × 5 min. Emission data were corrected for dead time, scatter, and attenuation and then reconstructed. For quantitative analyses, dynamic PET images were co-registered to a standardized monkey MRI template using PMOD software 4.02 (PMOD Technologies, Zürich, Switzerland).28 Predefined brain regions of interest from the template were applied to the co-registered PET image to obtain regional time-activity curves. The measurement of the brain uptake of radiotracer was standardized to body weight and the dose of radioactivity injected to yield a standardized uptake value (SUV).
Radiometabolite analysis of nonhuman primate plasma samples
HPLC radiometabolite analysis of the macaque plasma samples collected at different time points post-injection of [18F]FS1P1 was performed as previously reported.16 A male macaque (∼10 kg) was intravenously injected with ~0.35 GBq of [18F]FS1P1. Arterial blood samples (~1.5 mL) were collected using heparinized syringes at 5, 15, 30, and 60 min post-injection. Plasma (400 μL) was then collected and mixed with 1.2 mL ice-cold acetonitrile to deproteinize. After centrifuge, 200 μL of the supernatant was loaded onto an analytical HPLC system with an SB C-18 analytical HPLC column (Agilent Technologies, Santa Clara, CA) and eluted with acetonitrile/0.1 M ammonium formate buffer pH 4.5, (75/25, v/v) with a flow rate of 1.13 mL/min. The eluted fractions were collected at one-minute intervals for a total of 16 minutes, the radioactivity of each fraction collection was counted in an automated gamma counter (Beckman, Brea, CA), and results were corrected by background radioactive counts and physical decay. The chromatography was regenerated represented by the percentage that was calculated by radioactivity count of every minute collection divided by the total injection amount on the HPLC system and multiplied by 100.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health including the National Institute of Neurological Disorders and Stroke [NS103988, NS75527, NS0103957], and the National Institute of Biomedical Imaging and Bioengineering [EB025815].
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Lee M-J, Brocklyn JRV, Thangada S, Liu CH, Hand AR, Menzeleev R, Spiegel S and Hla T, Science, 1998, 279, 1552–1555. [DOI] [PubMed] [Google Scholar]
- 2.Chi H, Trends Pharmacol. Sci, 2011, 32, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aoki M, Aoki H, Ramanathan R, Hait NC and Takabe K, Mediators Inflamm, 2016, 2016, 8606878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, Foster CA, Zollinger M and Lynch KR, J. Biol. Chem, 2002, 277, 21453–21457. [DOI] [PubMed] [Google Scholar]
- 5.Rivera J, Proia RL and Olivera A, Nat. Rev. Immunol, 2008, 8, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montrose DC, Scherl EJ, Bosworth BP, Zhou XK, Jung B, Dannenberg AJ and Hla T, J. Lipid Res, 2013, 54, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karuppuchamy T, Behrens E. h., González-Cabrera P, Sarkisyan G, Gima L, Boyer JD, Bamias G, Jedlicka P, Veny M, Clark D, Peach R, Scott F, Rosen H and Rivera-Nieves J, Mucosal Immunol, 2017, 10, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nofer J-R, Bot M, Brodde M, Taylor PJ, Salm P, Brinkmann V, Berkel T. v., Assmann G and Biessen EAL, Circulation, 2007, 115, 501–508. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Jin H, Yue X, Luo Z, Liu C, Rosenberg AJ and Tu Z, Mol. Imaging Biol, 2016, 18, 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu H, Jin H, Yue X, Han J, Baum P, Abendschein DR and Tu Z, Mol. Imaging, 2017, 16, 1536012116689770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin H, Yang H, Liu H, Zhang Y, Zhang X, Rosenberg AJ, Liu Y, Lapi SE and Tu Z, J. Nucl. Cardiol, 2017, 24, 558–570. [DOI] [PubMed] [Google Scholar]
- 12.Mansor S, Laforest R, Luo Z, Gaehle G, Nickels M, Benzinger T and Tu Z, J. Nuc. Med, 2021, 62, 1591–1591. [Google Scholar]
- 13.Jacobson O, Kiesewetter DO and Chen X, Bioconjug. Chem, 2015, 26, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prasad VP, Wagner S, Keul P, Hermann S, Levkau B, Schäfers M and Haufe G, Bioorg. Med. Chem, 2014, 22, 5168–5181. [DOI] [PubMed] [Google Scholar]
- 15.Shaikh RS, Schilson SS, Wagner S, Hermann S, Keul P, Levkau B, Schäfers M and Haufe G, J. Med. Chem, 2015, 58, 3471–3484. [DOI] [PubMed] [Google Scholar]
- 16.Liu H, Luo Z, Gu J, Jiang H, Joshi S, Shoghi KI, Zhou Y, Gropler RJ, Benzinger TLS and Tu Z, Mol. Imaging Biol, 2020, 22, 1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo Z, Han J, Liu H, Rosenberg AJ, Chen DL, Gropler RJ, Perlmutter JS and Tu Z, Org. Biomol. Chem, 2018, 16, 9171–9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quattropani A, Sauer WHB, Crosignani S, Dorbais J, Gerber P, Gonzalez J, Marin D, Muzerelle M, Beltran F, Nichols A, Georgi K, Schneider M, Vitte P-A, Eligert V, Novo-Perez L, Hantson J, Nock S, Carboni S, de Souza ALS, Arrighi J-F, Boschert U and Bombrun A, ChemMedChem, 2015, 10, 688–714. [DOI] [PubMed] [Google Scholar]
- 19.Jin CH, Krishnaiah M, Sreenu D, Subrahmanyam VB, Rao KS, Lee HJ, Park S-J, Park H-J, Lee K, Sheen YY and Kim D-K, J. Med. Chem, 2014, 57, 4213–4238. [DOI] [PubMed] [Google Scholar]
- 20.Baykov S, Sharonova T, Osipyan A, Rozhkov S, Shetnev A and Smirnov A, Tetrahedron Lett, 2016, 57, 2898–2900. [Google Scholar]
- 21.van der Born D, Pees A, Poot AJ, Orru RVA, Windhorst AD and Vugts DJ, Chem. Soc. Rev, 2017, 46, 4709–4773. [DOI] [PubMed] [Google Scholar]
- 22.Lemaire C, Libert L, Plenevaux A, Aerts J, Franci X and Luxen A, J. Fluorine Chem, 2012, 138, 48–55. [Google Scholar]
- 23.Shen B, Löffler D, Reischl G, Machulla H-J and Zeller K-P, J. Fluorine Chem, 2009, 130, 216–224. [Google Scholar]
- 24.Shen B, Löffler D, Zeller K-P, Übele M, Reischl G and Machulla H-J, J. Fluorine Chem, 2007, 128, 1461–1468. [Google Scholar]
- 25.Gómez AB, Cortés González MA, Lübcke M, Johansson MJ, Halldin C, Szabó KJ and Schou M, Chem. Comm, 2016, 52, 13963–13966. [DOI] [PubMed] [Google Scholar]
- 26.Naumiec GR, Cai L, Lu S and Pike VW, Eur. J. Org. Chem, 2017, 2017, 6593–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee SJ, Morales-Colón MT, Brooks AF, Wright JS, Makaravage KJ, Scott PJH and Sanford MS, J. Org. Chem, 2021, 86, 14121–14130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang H, Joshi S, Liu H, Mansor S, Qiu L, Zhao H, Whitehead T, Gropler RJ, Wu GF, Cross AH, Benzinger TLS, Shoghi KI, Perlmutter JS and Tu Z, ACS Chem. Neurosci, 2021, 12, 3733–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang H, Gu J, Zhao H, Joshi S, Perlmutter JS, Gropler RJ, Klein RS, Benzinger TLS and Tu Z, Mol. Imaging, 2021, 2021, 9982020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu H, Laforest R, Gu J, Luo Z, Jones LA, Gropler RJ, Benzinger TLS and Tu Z, Mol. Imaging Biol, 2019, 22, 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rankovic Z, J. Med. Chem, 2017, 60, 5943–5954. [DOI] [PubMed] [Google Scholar]
- 32.Rankovic Z, J. Med. Chem, 2015, 58, 2584–2608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.