

Abstract
Background
ZyCoV-D, a DNA-based vaccine, showed promising safety and immunogenicity in a phase 1/2 trial. We now report the interim efficacy results of phase 3 clinical trial with ZyCoV-D vaccine in India.
Methods
We conducted an interim analysis of a multicentre, double-blind, randomised, placebo-controlled phase 3 trial at 49 centres in India. Healthy participants aged at least 12 years were enrolled and randomly assigned (1:1) to receive either ZyCov-D vaccine (Cadila Healthcare; 2 mg per dose) or placebo. An interactive web response system was used for randomisation (blocks of four) of participants as well as to enrol those aged 60 years and older with or without comorbid conditions, and those aged 12–17 years. It was also used to identify 600 participants for immunogenicity (blocks of six). Participants, investigators, and outcome assessors were masked to treatment assignment. Three doses of vaccine or placebo were administered intradermally via a needle-free injection system 28 days apart. The primary outcome was the number of participants with first occurrence of symptomatic RT-PCR-positive COVID-19 28 days after the third dose, until the targeted number of cases (interim analysis n=79, full analysis n=158) have been achieved. The analysis was done in the per-protocol population, which consisted of all participants with negative baseline SARS-CoV-2 status who received three doses of vaccine or placebo. Assessment of safety and tolerability was based on the safety population, which consisted of all enrolled participants who were known to have received at least one dose of study vaccine or placebo. This trial is registered with Clinical Trial Registry India, CTRI/2021/01/030416, and is ongoing.
Findings
Between Jan 16, and June 23, 2021 (data cutoff), 33 194 individuals were screened, of whom 5241 did not meet screening criteria and 27 703 were enrolled and randomly assigned to receive ZyCoV-D (n=13 851) or placebo (n=13 852). Per-protocol, 81 cases were eligible and included in efficacy analysis (20 of 12 350 in the ZyCoV-D group and 61 of 12 320 in placebo group). The ZyCoV-D vaccine efficacy was found to be 66·6% (95% CI 47·6–80·7). The occurrence of solicited adverse events was similar between the treatment groups (623 [4·49%] in the ZyCoV-D group vs 620 [4·47%] in the placebo group). There were two deaths (one in each group) reported at the data cutoff, neither of which was considered related to the study treatments.
Interpretation
In this interim analysis, ZyCoV-D vaccine was found to be efficacious, safe, and immunogenic in a phase 3 trial.
Funding
National Biopharma Mission, Department of Biotechnology, Government of India and Cadila Healthcare, Ahmedabad, Gujarat India.
Introduction
SARS-CoV-2 is highly infectious, and has affected millions of people globally since the COVID-19 pandemic was declared in March, 2020.1 Patients infected with SARS-CoV-2, especially patients older than 60 years and people with pre-existing respiratory or cardiovascular conditions, are at greater risk of severe complications and death.2, 3
The COVID-19 pandemic has caused substantial excess mortality and plunged national economies into deep recessions. Although the spread of the virus can be mitigated through physical distancing, face coverings, and testing and tracing—and potentially with therapeutics—the risk of outbreaks and disruption to economic and social life will probably remain until effective vaccines are administered to a large global population to prevent hospitalisation and severe disease, and achieve herd immunity to halt transmission of the virus. Several COVID-19 vaccines have now been approved by various global agencies, with many more in development. Yet, having licensed vaccines is not enough to achieve global control of COVID-19. Vaccines need to be produced at scale, affordably priced, globally allocated, and widely deployed in local communities.4 Attention has now turned to expanding production capacity to promote the widespread rollout of successful vaccines, as well as efficiently distributing them to administration facilities.4 The development of vaccines that are temperature stable can help to improve the global distribution to countries with little or no capacity for cold storage.5 Cadila Healthcare has developed a DNA-based vaccine, called ZyCoV-D, to overcome the logistics and manufacturing challenges of other RNA-based vaccines, which they have found to be safe and effective in a preclinical animal model.6 Temperature stability is a key attribute of this vaccine candidate. ZyCoV-D was stored at 2–8°C and has stability data at room temperature for 3 months. In an open-vial study,7 ZyCoV-D was stable and sterile up to 28 days; such stability and sterility could reduce vaccine wastage by removing the need to wait for multiple people to be vaccinated from a single vial.
Research in context.
Evidence before this study
We searched PubMed on March 23, 2021, using the searchterms “COVID-19”, “SARS-CoV-2”, “vaccine”, and “clinical trial”. Scarce data have been published on DNA based vaccines for SARS-CoV-2 infection. In 2020, a DNA vaccine candidate (INO-4800) was developed, which demonstrated safety and tolerability in a phase 1 study. In a phase 1 trial, the vaccine was found to be immunogenic in 38 (100%) of 38 vaccinated participants by eliciting humoral or cellular immune responses, or both. In a phase 2 trial, involving 400 participants, the majority of adverse events were grade 1 and grade 2 in severity, which did not appear to increase in frequency with the second dose. INO-4800 generated balanced humoral and cellular immune responses measured at week 6 compared with the baseline levels at day 0 (pre-dose) or the placebo participants in all age groups tested. Cadila Healthcare, India, developed a candidate vaccine, ZyCoV-D, comprising a DNA plasmid vector carrying the gene encoding the spike protein (S) of the SARS-CoV-2 virus. A preliminary animal study demonstrated that the candidate DNA vaccine induces antibody response including neutralising antibodies against SARS-CoV-2 and provided Th-1 response as evidenced by elevated IFN-g levels. The DNA vaccine ZyCoV-D is among several vaccines being developed to prevent the spread of SARS-CoV-2 infection. ZyCoV-D, delivered intradermally using a needle-free injection device, showed good safety and immunogenicity in a phase 1/2 clinical trial conducted in India in 1048 participants. Vaccination with ZyCoV-D resulted in generation of both humoral and cellular response. The seroconversion rate for neutralising antibody titres were greater than 88% and the IFN-γ levels measured by enzyme-linked immunospot (ELISpot) showed almost 10–12-fold increase after vaccination.
Added value of this study
To our knowledge, this is the first time a DNA vaccine has been tested in a phase 3 trial in a large population in India, the first phase 3 trial globally to deliver a DNA vaccine using a needle-free delivery device, and the first time that a COVID-19 vaccine is being tested in the age group of 12–17 years in India. Overall, our study demonstrated that intradermal injection of ZyCoV-D vaccine is safe and feasible and might achieve successful prevention of COVID-19 diseases in a large population. Additionally, the DNA vaccine is based on a plasmid DNA platform, which allows rapid generation of new constructs; the ZyCoV-D vaccine can therefore pave the way for next-generation DNA vaccines capable of handling mutant strains.
Implications of all the available evidence
This study provides the first evidence that induction of immune responses against the spike protein using a plasmid construct carrying the spike-S-gene provides protection against COVID-19 in humans. We anticipate that the major implication of this study will be the introduction of DNA-based prophylactic therapy against highly infectious diseases such as SARS-CoV-2. The ZyCoV-D vaccine has the potential to make a significant contribution to containing the COVID-19 pandemic in India and globally.
We previously reported the results of an adaptive phase 1/2 study of ZyCoV-D in healthy participants.8 43 (90%) of 48 participants completed the phase 1 study up to day 84, and 911 (91%) of 1000 participants completed the phase 2 study up to day 224. No safety concerns were noted in the phase 1/2 study with ZyCov-D vaccine administered up to 2 mg via a needle-free injection system or via needle, and the vaccine was found to be safe and well tolerated. The phase 1/2 study result showed that ZyCoV-D also elicited a good immune response. Vaccination with ZyCoV-D resulted in a 10–12-fold rise IFN-γ spot-forming cells per million peripheral blood mononuclear cells (PBMCs), suggesting a strong cellular response. Considering the safety and efficacy profile in the phase 1/2 study, we conducted a phase 3 study to evaluate efficacy, safety, and immunogenicity of a 2 mg dose of ZyCoV-D vaccine administered via a needle-free injection system. We report interim efficacy, safety, and immunogenicity findings from this trial.
Methods
Study design and participants
We investigated the efficacy, safety, and immunogenicity of ZyCoV-D at 2 mg dose administered intradermally via a needle-free injection system, compared with placebo, in a multicentre, double-blind, randomised, controlled trial at 49 hospitals in India. Healthy male and female individuals aged 12 years or older were eligible to participate in the study. Individuals with febrile illness, confirmed SARS-CoV-2, a history of SARS/MERS infection, history of contact with SARS-CoV-2-infected patients within the previous 14 days, confirmed immunosuppressive or immunodeficiency disorder, on any immunosuppressive or immunostimulant therapy, or other clinically significant systemic disorder were excluded from the study.
The study was initiated after approval from an ethics committee at each study site and local regulatory authorities. Written informed consent from all the participants was obtained before initiation of any study-related procedures. Additionally, assent was taken from participants aged 12–17 years and parents or guardians were asked for consent. The study was done in accordance with all national and local regulations as well as Indian Good Clinical Practice. The study was monitored by an independent data safety monitoring board.
Randomisation and masking
Eligible participants were randomly assigned (1:1) to ZyCoV-D vaccine or placebo using a randomisation schedule generated using SAS software (version 9.4 or higher) with the help of an interactive web response systems (IWRS). The IWRS was used for randomisation (blocks of four) of participants as well as to enrol individuals aged 60 years and older with or without comorbid conditions, and those aged 12–17 years. It was also used to identify 600 participants for immunogenicity (blocks of six).
Investigators at sites received random allocation information through the IWRS. The randomisation sequence was generated by an independent statistician using SAS and fed into the IWRS. Participants were enrolled by investigators with the help of the IWRS.
The population also included a subgroup of participants aged 12–17 years, participants aged older than 60 years, and participants with comorbidities (ie, hypertension, diabetes, obesity, chronic respiratory diseases, chronic kidney disease, or chronic heart disease).
The study medication was supplied in identical packages. The study medications were similar in colour and appearance, thereby maintaining double-blind conditions. The masking of individual allocation was maintained for all investigators and participants.
Procedures
ZyCoV-D is comprised of a DNA plasmid vector pVAX1 carrying gene-expressing spikeS protein of SARS-CoV-2 and IgE signal peptide. The spike gene region was selected from submitted Wuhan Hu1 isolate (Genebank accession number MN908947.3).
All enrolled participants received three doses of ZyCoV-D vaccine (2 mg per dose) or placebo intradermally via a needle-free injection system 28 days apart (days 0, 28, and 56). Participants were kept under medical observation for 30 min after vaccine administration to assess adverse reactions.
After vaccine administration, a series of telephone follow-up visits were scheduled to detect suspected symptomatic COVID-19. Participants were then grouped in to one of three categories to identify asymptomatic and symptomatic individuals with SARS-CoV-2 infection as well as to assess the immunogenicity response.
Category 1 included participants who were symptomatic (pre-planned number of participants n=17 616). Telephone follow-up visits were done on days 70, 98, 112, 126, 140, 154, 168, 182, 196, 210, 224, 238, 252, 266, 280, 294, 308, 322, 336, and 350 (plus or minus 3 days) after the first vaccine dose to detect symptomatic COVID-19 cases following doses. If COVID-19 was suspected, nasopharyngeal and oropharyngeal swabs were collected for RT-PCR testing to confirm SARS-CoV-2 infection. The study used a case adjudication committee to review the source data of suspected cases, where RT-PCR results were equivocal, or the symptomology was suspected and not recorded correctly.
Category 2 included participants who were asymptomatic, and remained asymptomatic, or became symptomatic (pre-planned number of participants n=10 000). In addition to the procedures listed for category 1, in this group nasopharyngeal and oropharyngeal swabs to detect asymptomatic COVID-19 were collected every 4 weeks for up to 32 weeks after the third vaccine dose. During the telephone follow-up visits, if a suspected case of COVID-19 was identified, a nasopharyngeal and oropharyngeal sample was collected for RT-PCR testing to confirm SARS-CoV-2 infection. Telephone follow-up visits were scheduled for days 70, 98, 126, 154, 182, 210, 238, 252, 266, 280, 294, 308, 322, 336, and 350 (plus or minus 3 days) after the first vaccine dose.
Category 3 included participants who were symptomatic and had an immunogenicity assessment (pre-planned number of participants n=600). In addition to the procedures outlined in category 1, blood samples were collected for serum analysis for immunological assessment. Telephone follow-up visits were scheduled for days 70, 98, 126, 154, 182, 210, 224, 238, 266, 280, 294, 308, 322, 336, and 350 (plus or minus 3 days) after the first dose. During these follow-up visits, if COVID-19 was suspected, nasopharyngeal and oropharyngeal samples were collected for RT-PCR testing to confirm SARS-CoV-2 infection. Of the 600 participants, 100 participants were enrolled in the group aged 12–17 years and older than 60 years. Enrolment was ongoing at the time of interim analysis. The data cutoff date for the interim analysis was June 23, 2021, and interim analysis was done on June 26, 2021.
A diary card was issued to all participants for self-recording of solicited adverse events. The local solicited adverse events listed in the diary card were pain, redness, swelling, and itching. The systemic solicited adverse events listed in the diary card were fever, headache, tiredness, nausea, vomiting, diarrhoea, joint pain, chills, and muscle pain.
Outcomes
The primary efficacy endpoint was the number of participants with first occurrence of symptomatic RT-PCR-positive COVID-19, 28 days after the third dose of ZyCoV-D vaccine, until the targeted number of cases (n=158) had been achieved. The interim analysis of the primary endpoint was conducted once 50% (79 cases) of the target number of cases was met. Participants infected with SARS-CoV-2 were categorised as asymptomatic (RT-PCR-positive without any signs and symptoms), mild (RT-PCR-positive with signs and symptoms of COVID-19), moderate (RT-PCR-positive and pneumonia with no signs of severe disease), or severe (RT-PCR positive with severe pneumonia). For primary outcome assessment, the symptomatic mild, moderate, and severe cases were combined. Nasopharyngeal and oropharyngeal swabs were collected from the participants with any one symptom of COVID-19 lasting for at least 48 h.
Secondary endpoints were first occurrences of asymptomatic COVID-19 cases, severe COVID-19 cases, moderate COVID-19 cases, mild COVID-19 cases, and virologically confirmed COVID-19 deaths 28 days after the third dose of vaccine until the targeted number of cases had been achieved. Immunogenicity assessment included seroconversion rate based on IgG against S1 antigen by ELISA, geometric mean titre (GMT), geometric mean fold rise (GMFR), and neutralising antibody titres as described previously.8 Seroconversion was defined as antibody-negative participants at baseline who became antibody positive after vaccination. The IgG concentration was analysed with a standard S1 ELISA using NIBSC standard with a concentration range of 45·23–1·4 EU. Additionally, a plaque reduction neutralisation test (PRNT50; ie, the concentration of serum to reduce plaques by 50% compared with the serum-free virus) was used for estimation of neutralising antibody titre in human serum samples against anti-SARS-CoV-2 on days 0, 56, and 84. In this assay, a reduction in the number of plaques formed by the virus correlates with the presence of neutralising antibody in the serum samples. The cellular response assessment included assessment of IFN-γ from PBMC samples. The methods for cellular assays has been previously described.8
Safety assessment included incidence and severity of solicited and unsolicited adverse events after each dose and incidence of serious adverse events throughout the study. Assessment of safety, tolerability, and baseline characteristics was based on the safety population, which consisted of all enrolled participants who were known to have received at least one dose of study vaccine or placebo.
Statistical analysis
Assuming vaccine efficacy of 60% after the third dose, approximately 158 confirmed COVID-19 cases would provide 90% power to conclude true vaccine efficacy of more than 30%, allowing early cessation for efficacy at interim analysis. A total of 11 286 evaluable participants per group (80% of 14 108 participants, accounting for 20% dropout or non-evaluable participants when randomly assigned [1:1] with placebo), means a total sample size of 28 216 was required. We assumed a 1% attack rate per year in the placebo group; thus, accrual of 158 COVID-19 cases within 6 months was required.
An interim analysis was done by independent statisticians after 50% of the target events (79 cases) were achieved. The interim analysis of a primary efficacy endpoint was based on the per-protocol population, which consisted of all participants with negative baseline SARS-CoV-2 status and who received three doses of vaccine or placebo. COVID-19 cases were adjudicated by a masked committee (comprising two independent members: one internal medicine physician and one pulmonologist).
Vaccine efficacy for the primary endpoint was analysed by calculating the infection rate ratio (IRR). The vaccine efficacy was compared using a standard statistical conditional exact test, based on the conditional binomial distribution of the number of infected cases in the vaccine group, given the total number of cases in both groups. Vaccine efficacy was defined as 100 × (1 – IRR), where IRR was calculated as the ratio of the first occurrence of symptomatic RT-PCR-positive COVID-19 cases, 28 days after the third dose of ZyCoV-D vaccination.
The secondary efficacy endpoints were evaluated using the same methods as the primary endpoint. Descriptive summary statistics for local reactions, systemic events, adverse events or serious adverse events, and laboratory parameters are presented.
The immunogenicity assessment including GMT, GMFR, and associated 95% CIs was presented for SARS-CoV-2 IgG on days 0, 56, and 84. Neutralisation titre (GMT, GMFR, and associated 95% CIs) was analysed in subgroups on days 0, 56, and 84. Cellular responses were analysed at days 0, 56, and 84.
This trial is registered with Clinical Trial Registry India, CTRI/2021/01/030416.
Role of the funding source
Cadila Healthcare designed and conducted the study and was involved in the study design, data collection, data analysis, data interpretation, and writing of the report.
Results
Between Jan 16 and June 23, 2021, 33 194 individuals were screened for inclusion; 5241 did not meet screening criteria. 27 703 participants were enrolled and randomly assigned (1:1) to receive ZyCoV-D (n=13 851) or placebo (n=13 852; figure 1 ). 18 592 (67·11%) participants were male and 9111 (32·89%) were female (table 1 ). The overall mean age was 36·5 years (SD 13·79); the mean age in the ZyCoV-D group was 36·4 years (13·83) and 36·6 years (13·76) in the placebo group. 935 (3·38%) participants were aged 12–17 years, 24 702 (89·17%) were aged 18–59 years, and 2066 (7·46%) were aged 60 years or older. 709 (5·12%) participants enrolled in the ZyCoV-D group and 740 (5·34%) participants enrolled in the placebo group had comorbid conditions, including stable chronic heart disease, stable chronic lung disease, controlled diabetes, stable liver disease, and severe obesity. All demographic characteristics were comparable between the treatment groups (table 1).
Figure 1.

Trial profile
Table 1.
Summary of demographics and baseline characteristics (safety population)
| ZyCoV-D (n=13 851) | Placebo (n=13 852) | Overall (N=27 703) | |
|---|---|---|---|
| Age, years | |||
| Mean (SD) | 36·4 (13·83) | 36·6 (13·76) | 36·5 (13·79) |
| Median (range) | 35·0 (12·00–88·00) | 35·0 (12·00–87·00) | 35·0 (12·00–88·00) |
| Age group, years | |||
| 12 to 17 | 448 (3·23) | 487 (3·52) | 935 (3·38) |
| 18 to <60 | 12 364 (89·26) | 12 338 (89·07) | 24 702 (89·17) |
| ≥60 | 1039 (7·50) | 1027 (7·41) | 2066 (7·46) |
| Gender | |||
| Female | 4506 (32·53%) | 4605 (33·24%) | 9111 (32·89%) |
| Male | 9345 (67·47%) | 9247 (66·76%) | 18 592 (67·11%) |
| Participant test for detection of antibody performed | |||
| No | NA | 2 (0·01%) | 2 (0·01%) |
| Yes | 12 043 (86·95%) | 12 020 (86·77%) | 24 063 (86·86%) |
| Missing | 1808 (13·05%) | 1803 (13·21%) | 3638 (13·13%) |
| SARS-CoV-2 antibody | |||
| Absent | 10 136 (73·18%) | 10 145 (73·24%) | 20 281 (73·21%) |
| Present | 1862 (13·44%) | 1846 (13·33%) | 3708 (13·38%) |
| Missing | 45 (0·32%) | 29 (0·21%) | 74 (0·27%) |
| Participant at risk (comorbidities) | |||
| No | 13 142 (94·88%) | 13 112 (94·66) | 26 254 (94·77) |
| Yes | 709 (5·12%) | 740 (5·34) | 1449 (5·23) |
| Stable chronic heart disease | 167 (1·21%) | 155 (1·12%) | 322 (1·16%) |
| Stable chronic lung disease | 13 (0·09%) | 7 (0·05%) | 20 (0·07%) |
| Controlled diabetic | 275 (1·99%) | 289 (2·09%) | 564 (2·04%) |
| Stable liver disease | 2 (0·01%) | 3 (0·02%) | 5 (0·02%) |
| Severe obesity | 18 (0·13%) | 14 (0·10%) | 32 (0·12%) |
| Other stable comorbidity | 295 (2·13%) | 293 (2·12%) | 588 (2·12%) |
Data are mean (SD), median (range), or n (%). NA=not applicable.
12 350 participants in the ZyCoV-D group and 12 320 participants in the placebo group, who completed 28 days after the third dose, were included in the per-protocol population. 81 participants with COVID-19 were eligible for interim primary efficacy endpoint analysis per protocol. 20 cases occurred in the ZyCoV-D group and 61 in the placebo group. ZyCoV-D vaccine efficacy was 66·6% (95% CI 47·6–80·7).
During the study, one severe COVID-19 case occurred after the second dose. The event was considered a fatal adverse event. As per the ethics committee recommendation, the participant's treatment code was unblinded, and the participant was found to have received placebo. Given no other severe COVID-19-related adverse events occurred in the vaccine group, ZyCoV-D was found to be 100% efficacious to prevent severe cases of COVID-19 after two doses. All three of the reported moderate COVID-19 cases were identified to have occurred in the placebo group. ZyCoV-D was therefore found to be 100% efficacious in moderate cases. 58 of 78 mild COVID-19 cases were identified in the placebo group and 20 cases in the ZyCoV-D group. ZyCoV-D therefore had an efficacy of 64·9% (95% CI 44·9–79·8) in mild cases.
The proportion of participants who achieved seroconversion at day 84 was higher in the ZyCoV-D group (n=126 [93·33%]) when compared with the placebo group (n=68 [52·31%]). The antibody concentration defined by GMT based on IgG at day 84 was higher in the ZyCoV-D group (952·67 EU, 95% CI 707·94–1282·00) than the placebo group (154·82 EU, 91·25–262·70; figure 2 ; table 2 ). The increase in antibody titre, as defined by GMFR, at day 84 was higher in the ZyCoV-D group (136·10, 95% CI 101·13–183·14]) than the placebo group (22·12, 13·04–37·53; figure 3 table 3 ). The immunogenicity response at day 84 in the group aged 12–17 years was higher than the overall participant population (IgG seroconversion 100% vs 93·33%, respectively; GMT 2083 EU vs 952·67 EU, respectively; and GMFR 297·65 vs 136·10, respectively).
Figure 2.
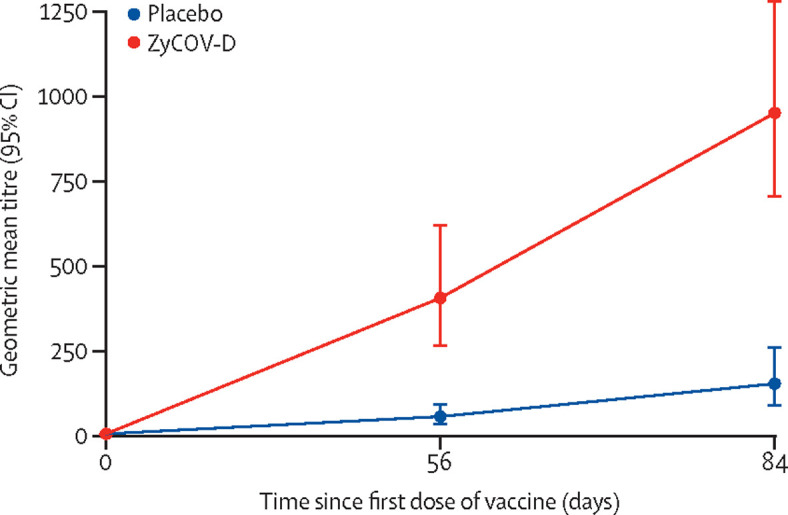
IgG comparison of geometric mean titre of ZyCoV-D and placebo at days 0, 56, and 84
Table 2.
Analysis of antibody titre (immunogenicity population)
| ZyCoV-D (n=135) | Placebo (n=130) | |
|---|---|---|
| Day 0 | ||
| GMT (95% CI) | 7 (7·00–7·00) | 7 (7·00–7·00) |
| Day 56 | ||
| GMT (95% CI) | 407·58 (266·73–622·83) | 57·97 (36·10–93·07) |
| GMFR (95% CI) | 58·23 (38·10–88·98) | 8·28 (5·16–13·30) |
| Day 84 | ||
| GMT (95% CI) | 952·67 (707·94–1282·00) | 154·82 (91·25–262·70) |
| GMFR (95% CI) | 136·10 (101·13–183·14) | 22·12 (13·04–37·53) |
GMT=geometric mean titre. GMFR=geometric mean fold rise.
Figure 3.
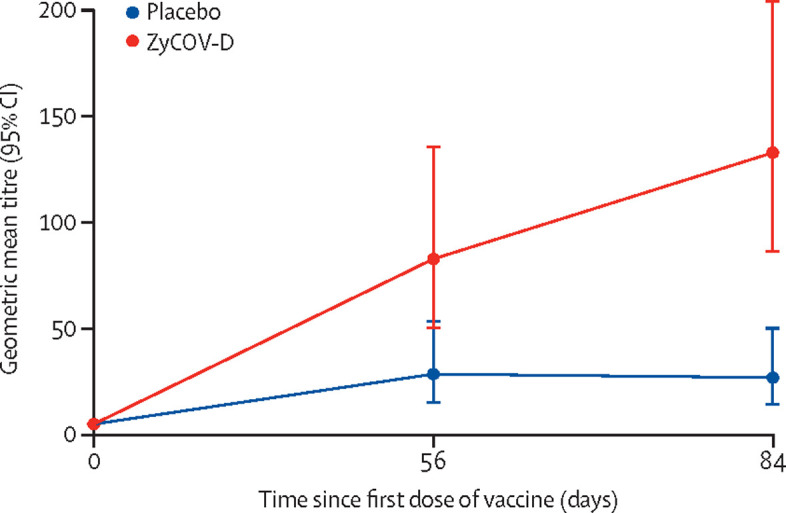
NAB(PRNT50) comparison of geometric mean titre of ZyCoV-D and placebo at days 0, 56, and 84
Table 3.
Summary and comparison of seroconversion of neutralising antibodies (immunogenicity population)
| ZyCoV-D (n=50) | Placebo (n=47) | p value | |
|---|---|---|---|
| Day 84 | |||
| No | 6 (12·00%) | 27 (57·45%) | .. |
| Yes | 44 (88·00%) | 20 (42·55%) | <0·0001 |
| Day 56 | |||
| No | 12 (24·00%) | 25 (53·19%) | .. |
| Yes | 38 (76·00%) | 22 (46·81)% | 0·0031 |
Data are n (%). GMT=geometric mean titre. GMFR=geometric mean fold rise. Day-wise comparison of proportion of participants, with seroconversion rate based on IgG antibodies as compared with baseline, between ZyCov-D and placebo is evaluated using χ2 test. Significant p value (<0·05) indicates that there is a significant difference between ZyCov-D and placebo based on proportion of participants with seroconversion rate.
The proportion of participants who achieved seroconversion of neutralising antibodies at day 84 was significantly (p<0·0001) higher in the ZyCoV-D group (44 [88·00%]) than the placebo group (20 [42·55%]; table 3). The neutralising antibody concentration defined by GMT at day 84 was higher in the ZyCoV-D group (133·39 PRNT50, 95% CI 86·88–204·81) than the placebo group (30·40 PRNT50, 16·35–56·53; table 4 ). The increase in neutralising antibody titre as defined by GMFR at day 84 was higher in the ZyCoV-D group (26·68, 95% CI 17·38–40·96) than the placebo group (5·74, 3·14–10·48).
Table 4.
Analysis of neutralising antibodies (PRNT50; immunogenicity population)
| ZyCoV-D (n=50) | Placebo (n=47) | |
|---|---|---|
| Day 0 | ||
| GMT (95% CI) | 5·00 (5·00–5·00) | 5·00 (5·00–5·00) |
| Day 56 | ||
| GMT (95% CI) | 83·10 (50·70–136·20) | 28·64 (15·32–53·53) |
| GMFR (95% CI) | 16·62 (10·14–27·24) | 5·73 (3·06–10·71) |
| Day 84 | ||
| GMT (95% CI) | 133·39 (86·88–204·81) | 26·99 (14·48–50·32) |
| GMFR (95% CI) | 26·68 (17·38–40·96) | 5·40 (2·90–10·06) |
GMT=geometric mean titre. GMFR=geometric mean fold rise.
We assessed cellular response in the ZyCoV-D vaccine group and in the placebo group at baseline (day 0), at day 56, and at day 84. Cellular response was assessed using an ex vivo IFN-γ ELISpot assay to enumerate antigen-specific T cells. ZyCoV-D, administered intradermally via a needle-free injection system at 2 mg dose showed a peak in IFN-γ response at day 56 after two vaccine doses, with a 13-times rise in the median value of spot forming cells per million PBMCs compared with baseline. On day 84, the IFN-γ cellular response was maintained at a 9·6-times rise in median spot forming cells per million PBMCs compared with baseline in the vaccinated group. In the placebo group, no significant fold change was observed (figure 4 ).
Figure 4.

Cellular response (IFN-γ) to ZyCoV-D and placebo at days 0, 56, and 84
PBMC=peripheral blood mononuclear cells.
At the data cutoff, 13 849 participants had received a first dose of ZyCoV-D vaccine and 13 852 had received a first dose of placebo; 13 153 had received a second dose of ZyCoV-D and 13 129 had received a second dose of placebo; and 12 773 had received a third dose of ZyCoV-D and 12 733 had received a third dose of placebo.
924 participants experienced 1243 solicited adverse events (appendix p 3). The occurrence of solicited adverse events was similar between the treatment groups (623 [4·49%] in the ZyCoV-D group vs 620 [4·47%] in the placebo group). In the ZyCoV-D group; 191 participants reported 313 events after the first dose, 148 participants reported 170 events after the second dose, and 120 participants reported 140 events after the third dose. In the placebo group, 190 participants reported 314 events after the first dose, 156 participants reported 182 events after the second dose, and 118 participants reported 124 events after the third dose. The majority of adverse events were mild to moderate in intensity in both the treatment groups. The majority of solicited adverse events were either possibly related (test group; dose 1: 118 [37·70%], dose 2: 56 [32·94%], dose 3: 35 [17·86%], placebo group; dose 1: 118 [37·58%], dose 2: 75 [41·21%], dose 3: 29 [23·39%]), probably related (test group; dose 1: 110 [35·14%], dose 2: 37 [21·76%], dose 3: 37 [26·43%], placebo group; dose 1: 109 [34·71%], dose 2: 38 [20·88%], dose 3: 28 [22·58%]), or definitely related (test group; dose 1: 78 [24·92%], dose 2: 52 [30·59%], dose 3: 50 [35·71%], placebo group; dose 1: 78 [24·84%], dose 2: 43 [23·63%], dose 3: 35 [28·23%]) in both groups after each dose. No action was taken for the majority of adverse events in both groups and the adverse events resolved. The study intervention was withdrawn for one participant in the ZyCoV-D group and two participants in the placebo group after the adverse events. The adverse events that led to withdrawal of intervention per the investigator's discretion were myalgia, pyrexia, and fatigue.
508 participants experienced 917 unsolicited adverse events during the study. The occurrence of unsolicited adverse events was also similar between the treatment groups (454 [3·27%] in the ZyCoV-D group vs 463 [3·34%] in the placebo group). In the ZyCoV-D group, 18 participants reported 43 events after the first dose, 26 participants reported 74 events after the second dose, and 198 participants reported 337 events after the third dose. In the placebo group, 20 participants reported 46 events after the first dose, 26 participants reported 64 events after the second dose, and 220 participants reported 353 events after the third dose.
Overall, there was no difference between the treatment groups in solicited or unsolicited adverse events. The most frequently reported solicited local adverse events in both the treatment groups (ZyCoV-D and placebo, respectively) were pain at injection site (92 [0·66%] and 82 [0·62%] after dose 1, 45 [0·34%] and 46 [0·35%] after dose 2, and 35 [0·27%] and 33 [0·26%] after dose 3), redness (43 [0·31%] and 39 [0·28%] after dose 1, 25 [0·19%] and 12 [0·09%] after dose 2, and 22 [0·17%] and 11 [0·09%] after dose 3), swelling (38 [0·27%] and 39 [0·28%] after dose 1, ten [0·08%] and eight [0·06%] after dose 2, and 12 [0·09%] and seven [0·05%] after dose 3), and itching (11 [0·08%] and 20 [0·14%] after dose 1, seven [0·05%] and nine [0·07%] after dose 2, and three [0·02%] and seven [0·05%] after dose 3). Most of the adverse events were mild or moderate in intensity. These events were similar between ZyCoV-D and placebo groups. The most commonly reported solicited systemic adverse events in both treatment groups (ZyCoV-D and placebo, respectively) were headache (34 [0·25%] and 30 [0·22%] after dose 1, 26 [0·20%] and 31 [0·24%] after dose 2, and 21 [0·16%] and 22 [0·17%] after dose 3), fever (28 [0·20%] and 19 [0·14%] after dose 1, 19 [0·14%] and 27 [0·21%] after dose 2, and 16 [0·13%] and 13 [0·10%] after dose 3), muscle pain (27 [0·19%] and 39 [0·28%] after dose 1, 15 [0·11%] and 24 [0·18%] after dose 2, and 14 [0·11%] and 12 [0·09%] after dose 3), and fatigue (26 [0·19%] both groups after dose 1, 18 [0·14%] and 21 [0·16%] after dose 2, and 11 [0·09%] and 17 [0·13%] after dose 3).
Most of the adverse events were mild or moderate in intensity. No different was observed with respect to successive dosing within each group or between the treatment groups.
Some of the commonly reported unsolicited adverse events were arthralgia, back pain, muscle spasms, myalgia, musculoskeletal pain, neck pain, vertigo, diarrhoea, gastritis, gastro-oesophageal reflux disease, nausea, vomiting, asthenia, chills, eye irritation, abdominal distension, abdominal pain, fatigue, pain, pyrexia, nasopharyngitis, pain in extremity, ageusia, anosmia, cerebral infarction, dizziness, headache, cough, dyspnoea, nasal dryness, oropharyngeal pain, rhinorrhoea, sneezing.
The safety profile in terms of solicited and unsolicited adverse events in adolescent participants (12–17 years), participants older than 60 years, and participants with comorbidities was similar to the general study population.
As of data cutoff, 15 serious adverse events were reported, including two fatal serious adverse events. These serious adverse events included cerebrovascular stroke (one event), cardiorespiratory arrest with septicaemia and alcoholic liver disease (one event), COVID-19 positive (seven events), cerebral infarct (one event), COVID-19 pneumonia (three events), hypoxaemia with COVID-19 (one event), and gram-negative enteritis with early shock (one event). All serious adverse events were mild to moderate in intensity except for the event of cerebrovascular stroke, COVID-19 pneumonia, and cardiorespiratory arrest, which were severe in intensity. All serious adverse events resolved, except for one event of COVID-19 pneumonia which remained unchanged at data cutoff as well as two deaths. None of the serious adverse events was considered causally related to vaccine or placebo.
The death (cardiorespiratory arrest with septicaemia and alcoholic liver disease) occurred after first dose and death event COVID-19 pneumonia occurred after second dose of study intervention. Both the events were considered unrelated to study intervention. For the death due to cardiorespiratory arrest, the treatment code was not unblinded, whereas for the death due to COVID-19 pneumonia, the treatment code was unblinded and the participant was found to be in the placebo group.
Discussion
We report interim efficacy, safety, and immunogenicity findings from the phase 3 clinical study conducted in India for the DNA vaccine ZyCoV-D to prevent COVID-19. A three-dose regimen of ZyCoV-D, administered intradermally via a needle-free injection system, was found to be 66·6% effective against COVID-19. To our knowledge, this is the first time a DNA vaccine has been tested in a large population in India
The study was ongoing during the peak of the second wave of COVID-19 in India, which was mainly due to the B.1.617.2 (delta) variant. In late April, 2021, there was a rapid increase in the proportion of people infected with the delta variant, which has since become the dominant strain (>99% of all sequenced genomes) in India.9 The number of delta variant cases detected in sample sequencing in April, May, and June, 2021, were 5356, 4947, and 3184, respectively. Cumulatively, India reported 19 766 (38%) delta sequences from Oct 15, 2020, to Sept 12, 2021.10 Therefore, we conclude that the ZyCoV-D vaccine is also effective against the delta variant. Considering that no severe or moderate COVID-19 cases were reported in the ZyCoV-D group, and based on the interim analysis results, the vaccine was found to be 100% effective against severe and moderate COVID-19 cases and 64·9% effective against mild COVID-19 cases. Therefore, it is possible that the severe and moderate cases that might result in fatalities and put enormous pressure on health-care systems could be prevented to a great extent with full vaccination using the ZyCoV-D vaccine.
The immunogenicity response of ZyCoV-D seen in phase 1/2 was maintained in the phase 3 study as well. The ZyCoV-D vaccine elicited a significantly high immunogenicity response at day 84 as evident from the seroconversion rate (93·33%) based on IgG against S1 antigen (by ELISA) and GMT (952·67 EU, 95% CI 707·9–1282·0) and GMFR (136·09, 95% CI 101·11–183·1) and neutralising antibody titre (GMT: 133·39 PRNT50, 95% CI 86·88–204·81; GMFR: 26·68, 95% CI 17·38–40·96). The vaccine also induced a significant cellular response as evident from higher IFN-γ levels in the ZyCoV-D group compared with the placebo group.
The favourable safety profile of ZyCoV-D observed in the phase 1/2 trial was further confirmed in this phase 3 interim analysis. Our results showed that the tolerability profile of ZyCoV-D in people aged 12–17 years, people older than 60 years, and in people with comorbidities was similar to that observed in the general study population. The three-dose vaccine was generally well tolerated, without any serious toxicity. The majority of adverse events reported were mild to moderate in intensity and resolved. The most commonly reported local solicited adverse events were pain at injection site, redness, swelling, and itching. The most commonly reported solicited systemic adverse events were headache, fever, muscle pain, and fatigue. These local and systemic adverse events after ZyCoV-D administration were comparable with the placebo group, indicating that there was no increased risk of adverse events with the vaccine. The type and severity of adverse events reported were similar to those seen in the phase 2 study of another DNA-based vaccine (INO-4800; Inovio Pharmaceuticals).5
The use of a needle-free injection system for vaccine administration should result in a reduction of side-effects typically associated with needle use (eg, injection site pain). ZyCoV-D is stored at 2–8°C, but is stable at 25°C for at least 3 months, retaining all the specifications set by the US Food and Drug Administration and other international guidelines.11 The thermostability of the vaccine will aid transportation and storage of the vaccine and reduce any cold chain breakdown challenges, thereby preventing vaccine wastage. The plasmid DNA platform provides ease of manufacturing with minimal biosafety requirements. Being a plasmid DNA vaccine, ZyCoV-D does not share the problems often associated with vector-based immunity, such as poor immune response to target antigens following vaccination due to pre-existing antibody titres to vectors resulting from natural infections from vectors such as adenoviruses, measles, and influenza viruses. Other problems associated with vector-based immunity include a requirement for very high doses resulting in several side-effects, and a requirement for long periods between booster doses to minimise the effect of vector immunity interference. The plasmid DNA platform also allows for the rapid generation of new constructs to deal with mutations in the virus. The three-dose regimen that might be perceived as cumbersome is actually equivalent to a third booster dose, which is being considered by many countries in existing vaccine platforms.
The study has several limitations: it was conducted only in a predominantly male population in India; the sample size was not calculated on the basis of subgroup analysis; the efficacy analysis was not performed on the basis of age group; the efficacy analysis was not done after the first and second dose for mild, moderate, and severe cases; and the common laboratory investigations such as haematology, renal function tests, and liver function tests were not evaluated. The sample size was small and the duration of the study was short at the time of the interim analysis when the efficacy of ZyCoV-D in prevention of severe COVID-19 was assessed as 100%.
To our knowledge, no previous phase 3 studies have been published on the efficacy of a DNA plasmid vaccine on SARS-CoV-2. Our study therefore provides the first evidence that induction of immune responses against the spike protein using a plasmid construct carrying spike-S-gene provides protection against COVID-19 in humans. From the interim analysis of this phrase 3 trial, ZyCoV-D was found to be efficacious, safe, and immunogenic, and it could significantly contribute to the efforts to contain the COVID-19 outbreak in India and globally.
Data sharing
As the study is ongoing, de-identified data are in the process of being deposited on the data repository for Cadila Healthcare, and the corresponding author can be contacted for data access.
Declaration of interests
JS, AD, KK, and TMCR are employees of Cadila Healthcare. All other authors declare no competing interests.
Acknowledgments
Acknowledgments
Phase 3 clinical development of ZyCoV-D was supported by a grant-in-aid from Mission COVID Suraksha under National Biopharma Mission, Department of Biotechnology, Government of India, to Cadila Healthcare (grant number BT/CS0037/CS/01/20). The authors acknowledge Deven Parmar and Ravindra Mittal for conceptualisation of the study; Kapil Maithal for conceptualising, designing, and developing the vaccine and guiding data analysis; Harish Chandra for the development of analytical procedures for testing of the vaccine and data analysis for ELISA neutralisation; Jayesh Bhatt for project management activities; Anjali Narkhede for quality assurance and regulatory support; Kuldipsinh Zala for manuscript writing support; Jatin Patel for medical writing support, Purav Trivedi for clinical operation management; Deepak Sahu for intellectual property management; Dipesh Pabrekar for inventory management; Vishal Nakrani for quality check support; Sita Verma for project management assistance; Tech Observer for statistics and data management support; and Octalsoft for interactive web response system support. The authors would like to thank PharmaJet, Golden, CO, USA for providing the PharmaJet Tropis needle-free injection system for vaccine delivery. The authors acknowledge all of the trial participants.
Acknowledgments
ZyCoV-D phase 3 Study Investigator Group
AK, SB, VR, SD, KG, HP, PS, IG, RR, RN, PK, Kalpesh Talati, Manish Hathila, Hari Shankar Gupta, Sharad Agarkhedkar, Sanjay Lalwani, Deepak Langade, Samir Gami, Rajendra Nerli, A G Srinivas Murthy, Vipul Khandelwal, Sandeep Jain, Dinesh Agarwal, Swapnav Borthakur, Tanmoy Mandal, Pankaj Bhardwaj, Parul Bhatt, Satyanarayan N Sharma, Animesh Choudhary, Shiva Narang, Vijay Kumar Shukla, Saurabh Agarwal, Sandeep Kumar Gupta, R Kulandaivel, Ravindra Mehta, A Vankateshwar Rao, K M Shivkumar, Raman Sharma, Dharmendra Gupta, Arepalli Sreedevi, N Srinivas Rao, V Rama Krishna CH, Veer Bahadur Singh, Manish Jain, Prashant Khandgave, Bhaskar Jedhe Deshmukh.
Contributors
JS and KK were involved in conceptualisation and designing of clinical study. AD was involved in designing and developing vaccine candidate, and doing data analysis for ELISPOT and Luminex assay. TMCR was involved in immunogenicity analysis. Each author contributed important intellectual content during manuscript drafting or revision and accepts accountability for the overall work by ensuring that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved. All authors approved the final version of the manuscript for submission.
Supplementary Material
References
- 1.WHO WHO Director-General's opening remarks at the media briefing on COVID-19. March 11, 2020. https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020
- 2.Grein J, Ohmagari N, Shin D, et al. Compassionate use of remdesivir for patients with severe COVID-19. N Engl J Med. 2020;382:2327–2336. doi: 10.1056/NEJMoa2007016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiss P, Murdoch DR. Clinical course and mortality risk of severe COVID-19. Lancet. 2020;395:1014–1015. doi: 10.1016/S0140-6736(20)30633-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wouters OJ, Shadlen KC, Salcher-Konrad M, et al. Challenges in ensuring global access to COVID-19 vaccines: production, affordability, allocation, and deployment. Lancet. 2021;397:1023–1034. doi: 10.1016/S0140-6736(21)00306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mammen MP, Jr, Tebas P, Agnes J, et al. Safety and immunogenicity of INO-4800 DNA vaccine against SARS-CoV-2: a preliminary report of a randomized, blinded, placebo-controlled, phase 2 clinical trial in adults at high risk of viral exposure. medRxiv. 2021 doi: 10.1101/2021.05.07.21256652. published online May 7. (preprint). [DOI] [Google Scholar]
- 6.Dey A, Chozhavel Rajnathan TM, Chandra H, et al. Immunogenic potential of DNA vaccine candidate, ZyCoV-D against SARS-CoV-2 in animal models. Vaccine. 2021;39:4108–4116. doi: 10.1016/j.vaccine.2021.05.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Revising global indicative wastage rates: a WHO initiative for better planning and forecasting of vaccine supply needs. 2019. https://www.who.int/immunization/programmes_systems/supply_chain/resources/Revising_Wastage_Concept_Note.pdf
- 8.Momin T, Kansagra K, Patel H, et al. Safety and immunogenicity of a DNA SARS-CoV-2 vaccine (ZyCoV-D): results of an open-label, non-randomized phase I part of phase I/II clinical study by intradermal route in healthy subjects in India. EClinicalMedicine. 2021;38 doi: 10.1016/j.eclinm.2021.101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.GISAID Tracking of variants—April 26, 2021. https://www.gisaid.org/hcov19-variants/
- 10.Salvatore M, Bhattacharyya R, Purkayastha S, et al. Resurgence of SARS-CoV-2 in India: potential role of the B.1.617.2 (Delta) variant and delayed interventions. medRxiv. 2021 doi: 10.1101/2021.06.23.21259405. published online June 30. (preprint). [DOI] [Google Scholar]
- 11.US Food and Drug Administration Considerations for plasmid DNA vaccine for infectious disease indication. Guidance for industry. 2007. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-plasmid-dna-vaccines-infectious-disease-indications
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
As the study is ongoing, de-identified data are in the process of being deposited on the data repository for Cadila Healthcare, and the corresponding author can be contacted for data access.