

Abstract
The improper maintenance of the bioactivated form of vitamin-D (1α,25-(OH)2D) may result in vitamin-D insufficiency and therefore compromise the absorption of dietary calcium. A significant regulator of vitamin-D metabolism is the inactivating function of the mitochondrial enzyme cytochrome P450 24A1 (CYP24A1). In humans, CYP24A1 carries out hydroxylation of carbon-23 (C23) or carbon-24 (C24) of the 1α,25(OH)2D side chain, eventually resulting in production of either an antagonist of the vitamin-D receptor (C23 pathway) or calcitroic acid (C24 pathway). Despite its importance to human health, the human isoform (hCYP24A1) remains largely uncharacterized due in part to the difficulty in producing the enzyme using recombinant means. In this study, we utilize a cleavable fusion with the cognate redox partner, human Adx (hAdx), to stabilize hCYP24A1 during production. The subsequent cleavage and isolation of active hCYP24A1 allowed for an investigation of substrate and analog binding, enzymatic activity, and redox partner recognition. We demonstrate involvement of a nonpolar contact involving Leu-80 of hAdx and a nonconserved proximal surface of hCYP24A1. Interestingly, shortening the length of this residue (L80V) results in enhanced binding between the CYP-Adx complex and 1α,25(OH)2D yet unexpectedly results in decreased catalysis. The same mutation has a negligible effect on rat CYP24A1 (a C24-hydroxylase), indicating the presence of a species-specific requirement that may correlate with differences in regioselectivity of the reaction. Taken together, this work presents an example of production of a challenging human CYP as well as providing details regarding hydrophobic modulation of a CYP-Adx complex that is critical to human vitamin-D metabolism.
Graphical Abstract
INTRODUCTION
The maintenance of proper levels of bioactivated vitamin-D hormone (1α,25(OH)2D) relies on the metabolic activities of multiple membrane-embedded cytochrome P450 (CYP) enzymes.1,2 Among these, the activity of the multifunctional mitochondrial enzyme cytochrome P450 24A1 (CYP24A1) represents the route of primary metabolic inactivation. As such, dysregulation of human CYP24A1 (hCYP24A1) results in disruption of homeostatic concentrations of vitamin-D and calcium, as represented by cases of idiopathic infantile hypercalcemia3–6 and chronic renal insufficiency.7
The enzyme hCYP24A1 decreases circulating concentrations of 1α,25(OH)2D in two ways (Figure 1). First, it hydroxylates the side-chain carbon-24 (C24) position of the inactive precursor 25(OH)D prior to its activation by the 1α-hydroxylase CYP27B1.1,2 The product of this reaction, 24R,25(OH)2D, is reported to promote bone fracture repair in CYP24A1-null mice.8 However, the primary hCYP24A1 pathway consists of a C24-targeted modification of activated 1α,25(OH)2D to produce 1α,24,25(OH)3D. Subsequent catalysis by hCYP24A1 eventually produces calcitroic acid, the inactive water-soluble form of vitamin-D. Notably, while CYP24A1 in most species appears to function in a regiospecific fashion (targeting either C24 or carbon-23 (C23)), the human isoform displays both C24 and C23 activity at an approximate 8:2 ratio.9 The distinction is important as the C23 pathway results in production of 1α,25(OH)2D-26,23-lactone, a metabolite with a demonstrated role in mediating the resorption of bones in patients with bone remodeling disorders.10,11
Figure 1.
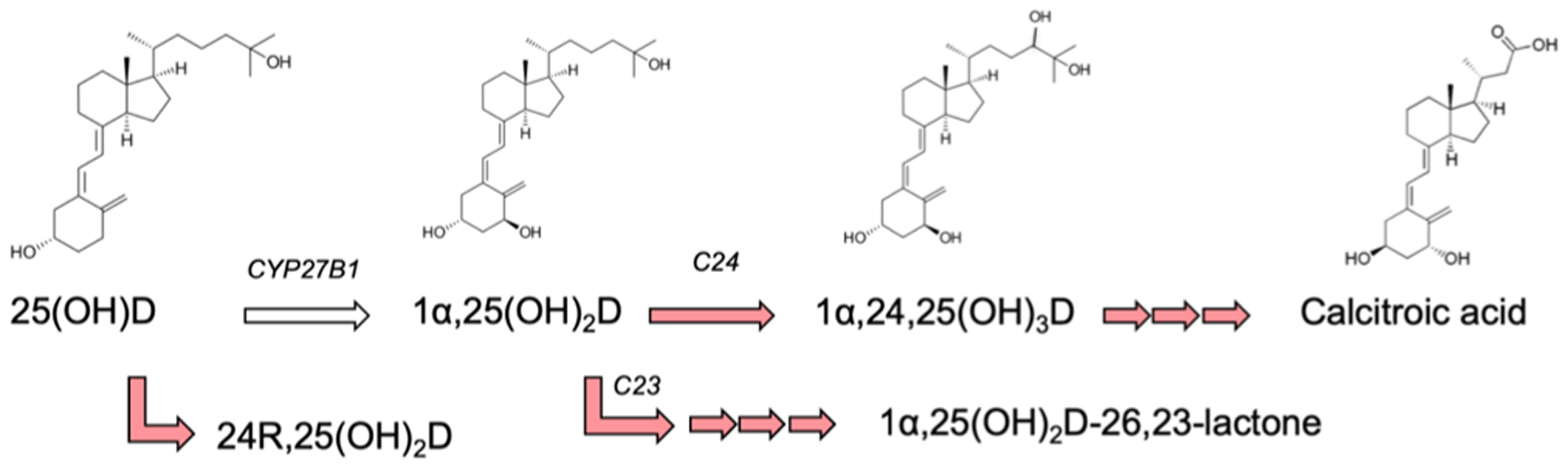
Overview of human vitamin-D metabolism. CYP24A1-mediated reactions are shown in red.
Despite its significance to human health, there exists a gap in our structural and biochemical understanding of hCYP24A1. To date, only structures of the rat isoform (rCYP24A1) have been reported.12 Moreover, detailed features of the essential CYP enzyme function, such as descriptions of substrate binding and same-species molecular recognition of the ferredoxin adrenodoxin (Adx), have also only been reported for rCYP24A1. Of note, rCYP24A1 is an imperfect model for human vitamin-D metabolism since it does not perform the C23 hydroxylation reaction, despite containing 84% sequence identity to hCYP24A1.
A likely reason for the scarcity of data on hCYP24A1 is the difficulty of recombinant expression and purification of the enzyme in Escherichia coli. Until the current study, our own efforts at chaperone-assisted expression and isolation of hCYP24A1 had not been successful. In this study, we report the use of a cleavable fusion construct consisting of an N-terminal copy of human Adx (hAdx) and a C-terminal copy of hCYP24A1. The intervening linker contains a tobacco etch virus (TEV) protease recognition site, thus allowing for the isolation of ligand-free and functional hCYP24A1. Using this platform, we have carried out a biochemical characterization of hCYP24A1 that includes spectral binding data for three vitamin-D substrates: 25(OH)D, 1α,25(OH)2D, and 1α,24,25-(OH)3D, along with the vitamin-D supplement 1α(OH)D. We also combine reconstituted functional assays, absorbance spectroscopy, and chemical cross-linking to characterize the required interaction with (nonfused) human Adx. These data support the presence of an Adx-induced conformational change in hCYP24A1 that affects ligand binding in a differential manner. Moreover, data from point mutations at the hCYP24A1-hAdx interface support the presence of a specific binding geometry between nonpolar contacts near the nonconserved outer ring of the proximal surface of hCYP24A1. This study provides mechanistic insight regarding a vital step in human vitamin-D metabolism. Moreover, utilization of a cleavable fusion construct illustrates an example of recombinant production of CYP enzymes that are otherwise difficult to produce.
MATERIALS AND METHODS
Protein Production.
A single construct encoding the full sequence of human Adx, the linker sequence containing the underlined TEV recognition sequence (HRHQPVTIGEPAENLYFQGENLYFQG), and the mature form of human CYP24A1 (UniProt ID Q07973) was synthesized (GenScript) and cloned into a pTrc expression vector (Invitrogen). The DNA insert was codon-optimized for expression in E. coli and was cloned into the vector using NcoI and HindIII restriction sites. The full hAdx_hCYP24A1 fusion construct also contained a six-residue histidine tag on the amino terminus. The plasmid was transformed into JM109 chemically competent E. coli (Promega) and selected for using 100 μg/mL carbenicillin. Cultures were grown from a single colony in LB containing 50 μg/mL carbenicillin and then used to inoculate 500 mL cultures of terrific broth media in 2.8 L Fernbach flasks. The cultures were grown at 37 °C with orbital shaking at 180 RPM until log phase growth and then induced with 1 mM isopropyl β-d-1-thiogalactopyranoside and 80 mg/L 5-aminolevulinic acid. Expression was carried out at 22 °C for 48 h with continued shaking at 180 RPM. Cells were harvested and washed with 50 mM potassium phosphate and 1 mM ethylenediaminetetraacetic acid (EDTA) and stored as paste at −80 °C until purification. After thawing, the cells were resuspended in 100 mL of lysis buffer consisting of 50 mM potassium phosphate (pH 7.4), 300 mM NaCl, 20% glycerol, 2 mM BME, and 300 μL of ProBlock protease inhibitor cocktail (GoldBio). Cell lysis was initiated by the addition of 0.5 mg/mL lysozyme and 2 μg/mL DNaseI while stirring at 4 °C for 30 min. Membrane extraction was carried out at 1.0% (w/v) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), with stirring at 4 °C for 1 h followed by sonication in 30 s bursts for a total time of 3 min and using a Branson SFX Sonifier set to 60% amplitude. The solubilized protein was then subjected to ultracentrifugation at 104,000g for 1 h. An extra 200 μL of a ProBlock protease inhibitor was added to the lysate before loading on the column.
A Ni-NTA affinity column (GE) was pre-equilibrated with buffer A (50 mM potassium phosphate, 300 mM NaCl, 20% glycerol, 0.5% CHAPS, 2 mM BME, pH 7.4). The fractionation lysate was loaded onto the column and washed with 12 column volumes of additional buffer A. Early optimizations of the protocol indicated that the hAdx_hCY-P24A1 fusion protein binds and retains imidazole, as indicated by a low-spin form of the heme that persisted following removal of unbound imidazole. Therefore, wash and elution buffers were modified to avoid imidazole. The bound protein was washed in two stages, beginning with 10 column volumes of wash buffer B (50 mM potassium phosphate, 300 mM NaCl, 0.5% CHAPS, 20% glycerol, 100 mM glycine, pH 6.5) followed by 10 column volumes of a 10% wash with elution buffer C (50 mM potassium phosphate, 300 mM NaCl, 0.5% CHAPS, 20% glycerol, 100 mM glycine, 50 mM histidine, pH 6.5). Protein was then eluted from the column using 100% buffer C. Peak fractions were concentrated and diluted 20-fold with TEV reaction buffer (50 mM potassium phosphate, 20% glycerol, 0.1% CHAPS, 0.5 mM DTT, pH 7.4). The cleavage was carried by the addition of 0.5 μg of TEV protease and without agitation at 4 °C for at least 24 h. The cleavage products were then resolved using a size exclusion column with a 120 mL bed volume and with a running buffer consisting of 500 mM potassium phosphate (pH 7.4), 300 mM NaCl, 20% glycerol, and 0.5% CHAPS. Purity of hCYP24A1 was assessed by an absorption ratio of A420/A280 greater than 0.8 and confirmed by SDS-PAGE. Production of unfused hAdx and AdR was carried out as described previously.13–15 Mutations for both free hAdx and the hAdx_hCYP24A1 fusion were generated by GenScript, and the mutant proteins were generated using the same protocols described for wild-type proteins. CYP24A1 from rats was produced as described in our earlier protocol.16 TEV protease was produced in-house using the available protocol.17
Spectral Ligand Binding Assays.
Vitamin-D ligands (25(OH)D, 1α,25(OH)2D, 1α,24,25(OH)3D, and 1α(OH)D) were purchased from ApexBio. Ligand titrations were carried out at ambient temperature on a Shimadzu UV-2700 spectrophotometer with minor modifications from the previous protocol.13 Briefly, the enzyme was diluted to 1 μM in 10 mM potassium phosphate buffer prior to binding. Each titration point was incubated for 8 min prior to recording a wavelength scan. Ligands were added from a stock dissolved into dimethyl sulfoxide (DMSO), with solvent concentrations not exceeding a 2% threshold. Titrations with 1α,25(OH)2D and 1α(OH)D) were carried out between 0.1 and 8 μM. For assays carried out in the presence of unfused hAdx, 10 μM hAdx was baselined in the cuvette prior to hCYP24A1 addition and ligand binding. Data were collected in triplicate for each sample and were analyzed by one-way analysis of variance (ANOVA) to determine statistical significance. For CO difference spectra, hCYP24A1 was first reduced with sodium dithionite, baselined on the spectrophotometer, and the absorption spectra were acquired between 400 and 500 nm following the introduction of CO gas. CO-bound spectra were recorded at regular intervals between 50 and 120 min. CO-difference spectra were also acquired upon the conclusion of ligand titrations to monitor the stability of hCYP24A1.
CYP24A1 Functional Assays.
Reconstituted hCYP24A1 assays were carried out as described previously with minor modifications.13 Briefly, assays contained 0.8 μM hCYP24A1 and 1.5 μM hAdx and AdR (from bovine). CYP concentrations were calculated using values from the reduced, CO-bound spectra. For reactions involving the uncleaved hAdx_hCYP24A1 protein, no additional hAdx was added. 1α,25(OH)2D was added at 5 μM, and the reactions were pre-incubated for 5 min at 37 °C followed by initiation by 1 mM NADPH. Reactions were terminated after 1.5, 2.5 or 5 min by the additions of two volumes of acetonitrile. Zero minute control reactions were treated with acetonitrile prior to addition of NADPH. Addition of 25(OH)D (2 μM) was added as an internal standard prior to samples being processed and resolved by high-performance liquid chromatography on an Agilent 1260 Infinity II liquid chromatography system with a Poroshell 120 EC-C18 column (4.6 × 25 mm) and using an isocratic mobile phase of 70% acetonitrile and 30% water. The substrate and the internal reference were detected by monitoring absorbance at 264 nm. All samples were run in triplicate. Substrate depletion was calculated as a percentage of the substrate remaining following a 2.5 or 5 min reaction time compared to 0 min reactions. Replicate values were tested for statistical significance with one-way ANOVA compared to the WT hCYP24A1/hAdx values.
EDC Cross-Linking.
Chemical cross-linking was carried out as described previously.13 The zero-length cross-linker EDC (ethyl-3-[3(dimethylamino)propyl]carbodiimide) was used to trap the interaction between amines and carboxylate groups of hCYP24A1 and hAdx, respectively. All cross-linking reactions were incubated with 2 mM EDC for 2 h at ambient temperature with 5 μM hCYP24A1 or rCYP24A1 and 60 μM hAdx. The reactions were quenched by addition of 2× Laemmli sample buffer and boiling for 5 min. An SDS-PAGE gel was run to detect cross-linked products.
RESULTS
Adx-Assisted Production of Human CYP24A1.
A comprehension of vitamin-D inactivation in humans would benefit from the production and analysis of recombinant purified hCYP24A1. However, the human enzyme is difficult to express in E. coli, possibly due to its more labile structure relative to the rat enzyme. We have previously produced human CYP24A1 for purely structural analyses by supplementing growth media with the high-affinity CYP inhibitor clotrimazole.16 However, the presence of a strong-binding inhibitor, as required to stabilize the protein through purification, also prevents a functional evaluation of the enzyme.
To stabilize the structure while retaining a ligand-free state, we expressed hCYP24A1 as a fusion construct with the natural electron transfer partner human Adx (Figure 2A). The N-terminal Adx domain is separated from hCYP24A1 by an 18-residue linker domain that consists of an amino acid sequence derived from the natural CYP fusion protein P450RhF18 and modified to contain a recognition sequence for the TEV protease. Thus, the design of the construct allows for Adx-stabilization of ligand-free hCYP24A1 during expression and metal affinity purification followed by the TEV cleavage and separation of the two domains by size exclusion chromatography. The overlaid absorbance spectra for the Adx-linked and separated hCY24A1 are shown in Figure 2B.
Figure 2.
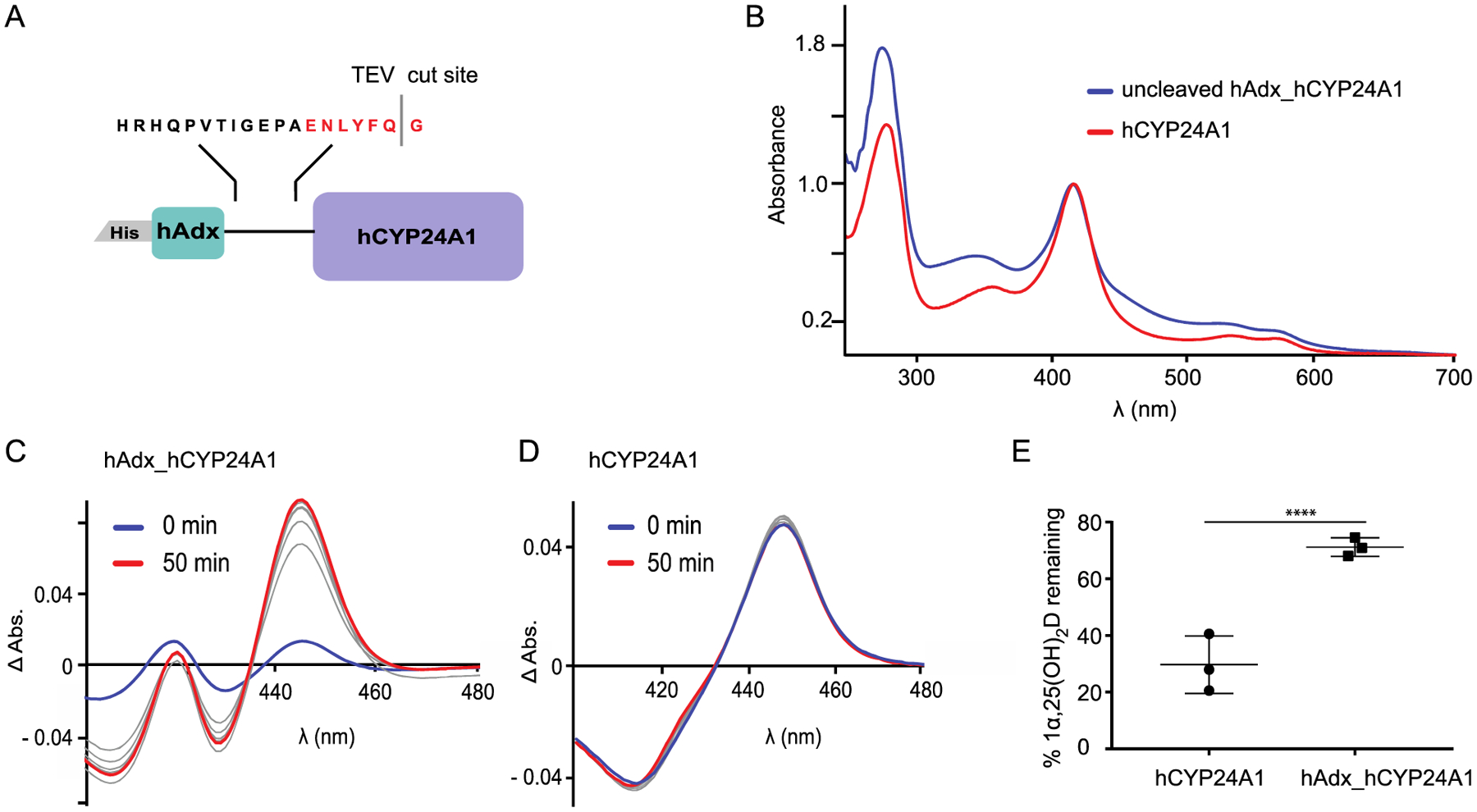
(A) Schematic of the fusion construct used to produce hCYP24A1. (B) UV–vis wavelength scan of hAdx_hCYP24A1 (blue) and separated hCYP24A1 (red). (C, D) CO difference spectra developed over 50 min for hAdx_hCYP24A1 and hCYP24A1, respectively. (E) 1α,25(OH)2D depleted over 5 min with hCYP24A1 and hAdx_hCYP24A1. Asterisk (*), one-way ANOVA analysis α = 0.05, n = 3, p < 0.05.
A comparison of carbon monoxide (CO)-bound difference spectra from E. coli cell lysate following induction of either the fusion construct or just hCYP24A1 on its own indicates a significant increase in the expression of the CYP enzyme, as represented by the CO-bound peak at 450 nm (Figure S1). The ability of CYPs to form a complex with CO is typically consistent with functionality in the enzyme.19 Interestingly, despite a detectible CO-bound signal in the purified fusion protein (Figure 2C), our reconstituted functional assays measure only minimal depletion of 1α,25(OH)2D (Figure 2E). In contrast, following removal of the Adx domain, the catalytic activity of the enzyme is restored. This also correlates with a stronger CO-bound spectrum for hCYP24A1 (all P450) following its separation from fused Adx (a mixture of P420 and P450) (Figure 2D). These data indicate that, despite conferring stability on the CYP, the presence of linked Adx is detrimental to catalysis by hCYP24A1, possibly by disrupting efficient reduction of Adx by AdR. However, since the objective of obtaining ligand-free hCYP24A1 had been achieved, we did not further optimize the length or chemistry of the linker.
Vitamin-D Ligands Binding to hCYP24A1.
The prior unavailability of isolated hCYP24A1 means that all quantification of binding data between CYP24A1 and primary or secondary vitamin-D substrates or analogs is derived from the rat isoform of the enzyme. Here, we carried out spectral binding assays, first using cleaved and separated hCYP24A1 and then in the presence of a 10-fold excess of unfused hAdx that had been separately expressed and purified.
As expected, titration with the primary substrate 1α,25-(OH)2D produces a type-I transition of the Soret band, with difference spectra indicating an increase at 387 nm and a calculated dissociation constant of 1.1 μM (Figure 3A and Table 1). Other binding data included the clinically relevant vitamin-D supplement 1α(OH)D. Supplementation with 1α(OH)D is more effective in treating bone density disorders than the natural vitamin-D hormone.20,21 Its effectiveness may be attributed to its metabolic stability as it is a poor substrate for rat CYP24A113 but is instead metabolized into the active hormone by microsomal CYP2R1.22,23 Using the same ligand concentrations, titration with 1α(OH)D also produces a type-I response, albeit with a more noticeable increase in the high-spin transition (Figure 3B).
Figure 3.
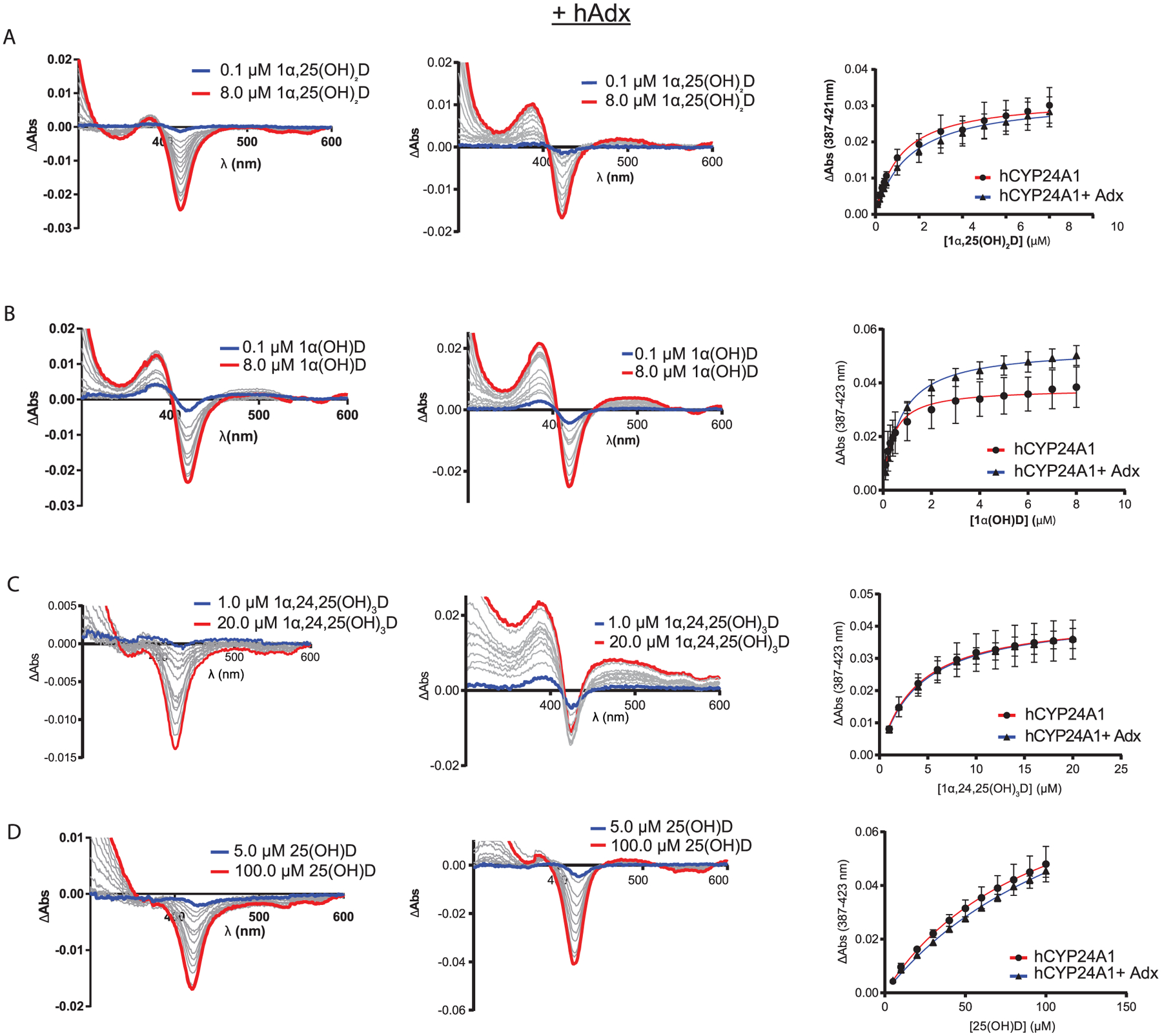
(A–D) Ligands binding to hCYP24A1 with and without hAdx present. Difference spectra of ligands binding to hCYP24A1 (1 μM) (left-hand panels). Difference spectra of ligands binding to hCYP24A1 (1 μM) in the presence of hAdx (10 μM) (middle panels). The blue line is the initial titration point, and the red line is the final addition. Binding curves measuring ΔAbs between ~390 and ~420 nm are shown in the right-hand panels. Titrations were run in triplicate with error bars indicating one standard deviation.
Table 1.
Effect of 10-Fold Addition of hAdx on Ligand Binding to hCYP24A1a
| +hAdx | ||||
|---|---|---|---|---|
| KD (μM) | ΔAmax (AU) | KD (μM) | ΔAmax (AU) | |
| 1α,25(OH)2D | 1.1 ± 0.2 | 0.03 ± 0.004 | 1.5 ± 0.2 | 0.03 ± 0.004 |
| 1α,24,25(OH)3D | N/A | N/A | 4.0 ± 0.04 | 0.04 ± 0.003 |
| 1α(OH)D | 0.4 ± 0.1 | 0.04 ± 0.008 | 0.7 ± 0.05 | 0.055 ± 0.004 |
One-way ANOVA, α = 0.05, n = 3, p < 0.05, error represents SD.
However, addition of the secondary substrate (and C24-hydroxylation product) 1α,24,25(OH)3D results in only a minimal type-I response (Figure 3C), and in the case of the pre-activated hormone, 25(OH)D, no measureable high-spin form is detected (Figure 3D). This prevents an accurate measurement of dissociation constants for these two substrates.
Interestingly, for most ligands, the presence of hAdx promotes a noticeable increase in the percentage of the high-spin transition upon binding (Figure 3, middle column) while not having a significant effect on ligand affinity (Table 1). One possible explanation for these differences is that hAdx has a stabilizing effect on hCYP24A1, therefore leading to more pronounced spectral transitions. This would be supported by our ability to produce the enzyme recombinantly as a fusion. Additional stability could in theory counter the effects of accumulating the DMSO solvent during the titration. To account for this possibility, we also recorded the CO difference spectra at the end of each titration (data not shown) and observed that, in the case of 1α,25(OH)2D and 1α(OH)D, no appreciable amount of P420 accumulates either with or without hAdx, as predicted by destabilization of hCYP24A1. This indicates that the increase in high spin is due primarily to the presence of an hAdx-induced conformational change in hCYP24A1. We have previously reported on the presence of long-range communication between the active site and the proximal Adx-binding surface of the corresponding interaction in rCYP24A1.13,14
An interesting feature of hCYP24A1 is that, in contrast to other mitochondrial CYPs,24,25 titration with unfused hAdx alone does not introduce a detectible high-spin character of the heme (Figure S2). This is also true for the spectra of the uncleaved hCYP24A1-hAdx protein. We interpret this finding to be consistent with conformational changes reported previously for rCYP24A1, in which the addition of the ligand and the redox partner in concert are required to promote a cumulative high-spin response.
Although for some ligands, we are only able to accurately quantify dissociation constants while in the presence of hAdx, we also note that, in general, the rank ordering of ligand affinity data for hCYP24A1 is consistent with that reported previously for the rat enzyme (Table 1, right column); the analog 1α(OH)D binds strongest followed by the primary substrate 1α,25(OH)2D and then the secondary substrate 1α,24,25-(OH)3D.
Proximal Surface Mutations of hCYP24A1.
The production of active hCYP24A1 also provided an opportunity to evaluate features of molecular recognition of Adx without the presence of the linker. First, we attempted a charge-neutralizing substitution of the highly conserved residue Arg-466, which is located on the putative Adx recognition site and has been implicated previously as an important CYP27B1-Adx contact that, when mutated, correlates with the onset of vitamin-D-dependent rickets-type I.26 However, attempts to use the fusion construct to produce an R466Q mutant were not entirely successful. Upon separation from cleaved Adx, the R466Q mutant produced a mix of 420 and 450 nm from CO difference spectra but failed to bind 1α,25(OH)2D (data not shown). Predictably, the R466Q mutant is also not active (Figure 4A). This outcome may implicate this cationic side chain in binding to Adx and is therefore an indicator of the degree to which the complex with Adx stabilizes hCYP24A1 during its production.
Figure 4.
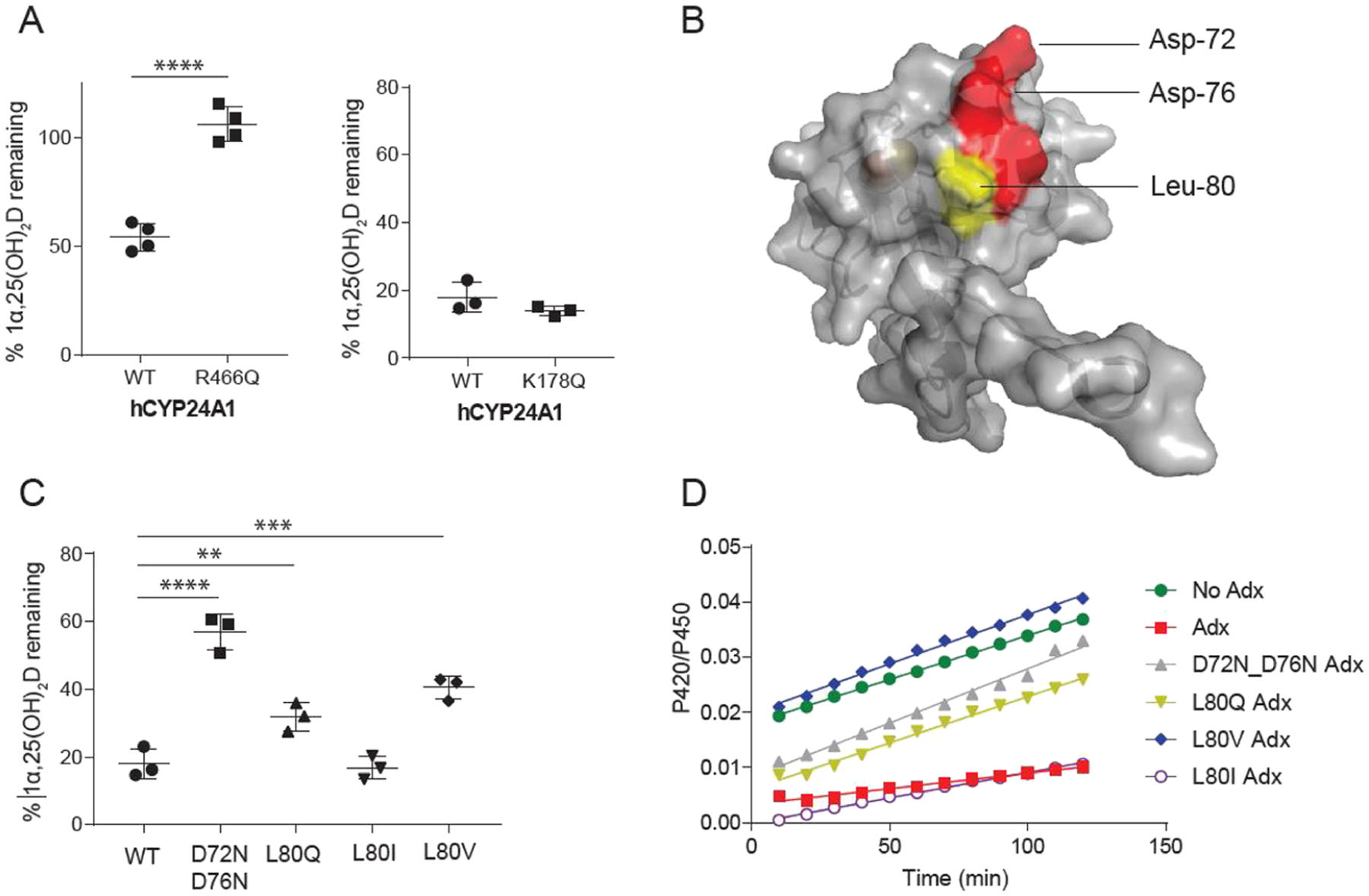
Effects of surface mutations of hCYP24A1 and hAdx. (A) 1α,25(OH)2D depleted in the presence of hCYP24A1 R466Q and K178Q. Reactions were run for 5 and 2.5 min each compared with a control of WT hCYP24A1. (B) Structure of hAdx (PDB 3P1M) with orientations of Asp-72 and Asp-76 indicated in red and Leu-80 in yellow. (C) 1α,25(OH)2D depleted in the presence of hAdx mutants. (D) Time-dependent change in the CO-bound spectra in the presence of each hAdx mutant. Asterisk (*), one-way ANOVA analysis α = 0.05, n = 3, p < 0.05.
Next, we incorporated a charge-neutralizing substitution on the proximal residue Lys-178 of the hCYP24A1 D-helix. In a recent study, Lys-177 of rat CYP24A1 was identified by LC–MS as forming a cross-link with Glu-109 of Adx.14 Since the corresponding Lys-177 residue is not conserved in humans, we instead mutated the neighboring residue, Lys-178, which is completely conserved across species. The fusion construct with a K178Q mutation expressed and purified with comparable yields to the parent construct and the catalytic domain remained stable following separation from Adx. Additionally, substrate binding and overall catalytic activity were not affected by the mutation (Figure 4A), suggesting that, in hCYP24A1, it does not participate in Adx recognition.
Interfacial Hydrophobic Contacts in the hCYP24A1 and hAdx Complex.
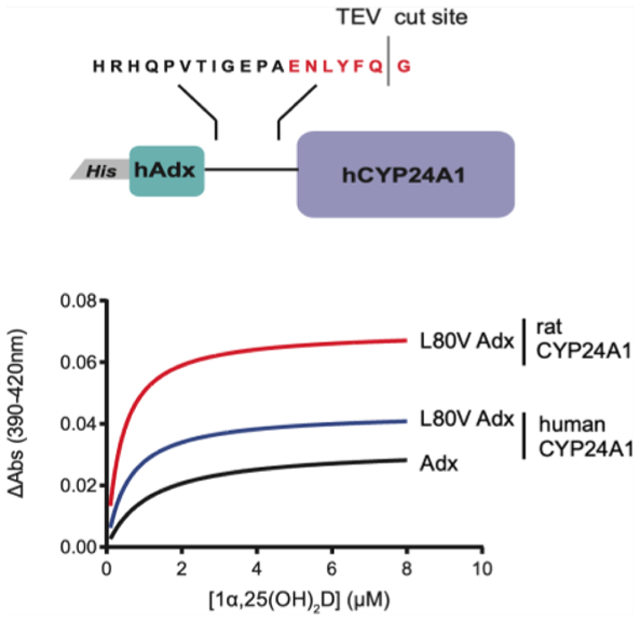
The interaction between Adx and mitochondrial CYPs is driven primarily by a complementarity of charge between a conserved anionic surface of Adx and conserved cationic side chains on the proximal surface of the CYP27–29 (Figure 4B). However, a hydrophobic contact involving Leu-80 of Adx has been reported previously for the corticosteroid metabolizing enzymes CYP11B1 and CYP11B2.30 Recently, a docked model of the rat CYP24A1 complex with Adx also implicated a nonpolar interaction between Leu-80 of Adx and Val-170 of rCYP24A1.14 Since Val-170 is replaced by a glycine residue in hCYP24A1, we hypothesized that the modulation of the Leu-80 hydrophobic contact would differentially impact hCYP24A1 activity. We substituted the Leu-80 residue of human Adx with a polar side chain (L80Q), with a different nonpolar side chain of similar size (L80I) and with a shortened nonpolar side chain (L80V). For a comparison with the effects of disruption of the electrostatic contacts, we also included the Adx double mutant D72N_D76N, which neutralizes the charges on two of the solvent-facing residues on the conserved recognition site. As expected, the aspartate double mutant effectively disrupts depletion of 1α,25(OH)2D, with approximately 60% of the substrate remaining (Figure 4C). However, despite the absence of a valine in position 170 of hCYP24A1, the L80Q substitution also had a negative effect on catalysis, suggesting that a nonpolar contact remains and plays a role in productive binding of Adx. Interestingly, the L80V substitution also significantly disrupts substrate depletion, with overall activity similar to that of L80Q. A comparison between L80V and L80I, which supports activity similar to wild-type Adx, suggests that merely shortening this nonpolar side chain by a single carbon bond length is sufficient to disrupt proper formation of the CYP-Adx complex.
Next, we used the ability of Adx to stabilize the CO-bound form of hCYP24A1 to further analyze the Leu-80 mutants. The presence of 10-fold Adx slows the transition of the 450 nm CO-bound peak into a 420 nm peak, thus indicating Adx-stabilization of the hCYP24A1-CO complex (Figure 4D). Here, we observed that L80V fails to contribute toward stabilization of the CO-bound complex, similar to the aspartate double mutant and even to a lesser extent than L80Q. These data further reinforce a strict distance requirement at the Leu-80 side chain in the hCYP24A1-Adx complex.
Last, to further evaluate the interaction between hCYP24A1 and hAdx with the L80V mutation, we carried out additional functional assays representing a steady-state condition and using subsaturating and supersaturating concentrations of hAdx relative to hCYP24A1. Initial time-course experiments using equimolar concentrations of hAdx and hCYP24A1 indicated that a linear decrease in 1α,25(OH)2D occurs between 60 and 90 s (data not shown). Therefore, all assays were quenched at 90 s and the remaining substrate was quantified as described previously in this study. As expected, depletion of 1α,25(OH)2D increases linearly with increasing ratios of wild-type hAdx (Figure 5). In contrast, increasing the relative amounts of L80V does not significantly promote hCYP24A1 activity at hAdx concentrations below 1:1. However, a 2-fold addition of the mutant does promote activity similar to that of the wild type. This outcome is consistent with a decreased affinity of the protein–protein complex due to the L80V mutation, which appears to be overcome at higher concentrations of hAdx. These data further reinforce a strict distance requirement at the Leu-80 side chain in the hCYP24A1:hAdx complex.
Figure 5.
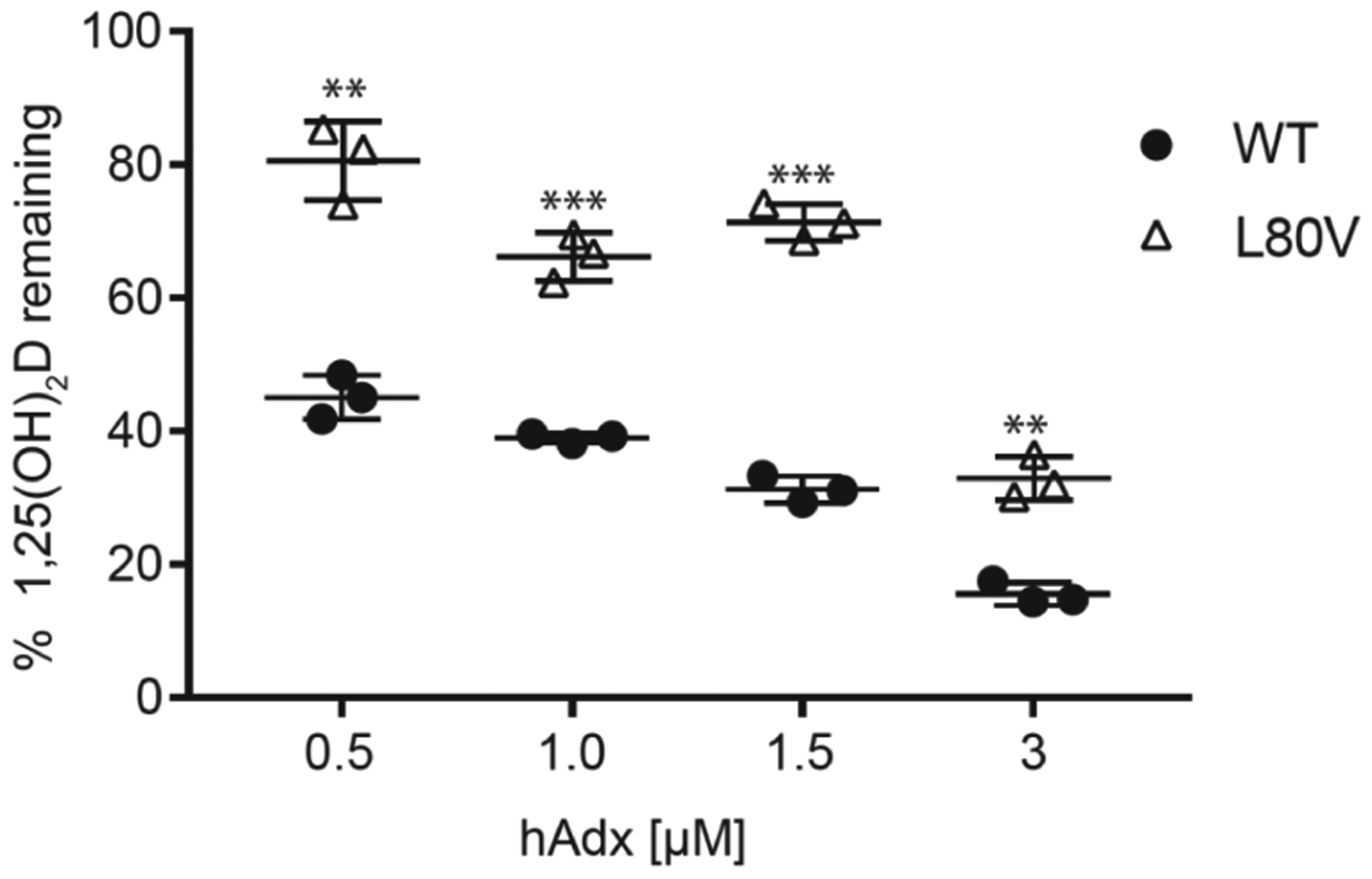
Substrate depletion with varying concentrations WT and L80V hAdx with hCYP24A1 (1.5 μM). Assays were run in triplicate and quenched at 90 s. Significance indicated using one-way ANOVA, α = 0.05, n = 3, p < 0.05.
Disruption of the hCYP24A1-Adx Interface Results in Nonfunctional Redox Complexes.
Modulation of the stability of the CO-hCYP24A1 complex (Figure 4D) implies the existence of an Adx-induced change in conformation on the distal (ligand-binding) side of the heme. This is consistent with enhanced binding of 1α(OH)D in the presence of Adx (Figure 3B). It follows that substrate binding may also be affected by modulation of the hydrophobic and electrostatic contacts at the CYP-Adx interface. Here, we carried out spectral binding assays between hCYP24A1 and 1α,25(OH)2D in the presence of a 10-fold excess of hAdx mutants. As shown previously (Figure 3A), addition of wild-type hAdx does not promote further binding of 1α,25(OH)2D. However, the presence of D72N_D76N hAdx actually results in a modest decrease in overall binding as measured by low-spin to high-spin transitions (Figure 6 and Table 2) but moderately higher affinity (0.7 μM +/− 0.1) than without hAdx. Similarly, L80Q also results in less overall binding. Interestingly, the presence of L80I and L80V induces an opposite response. Here, we observed a marked increase in overall binding, reflecting a 40% relative increase in the high-spin form at saturating concentrations of 1α,25(OH)2D (Table 2).
Figure 6.
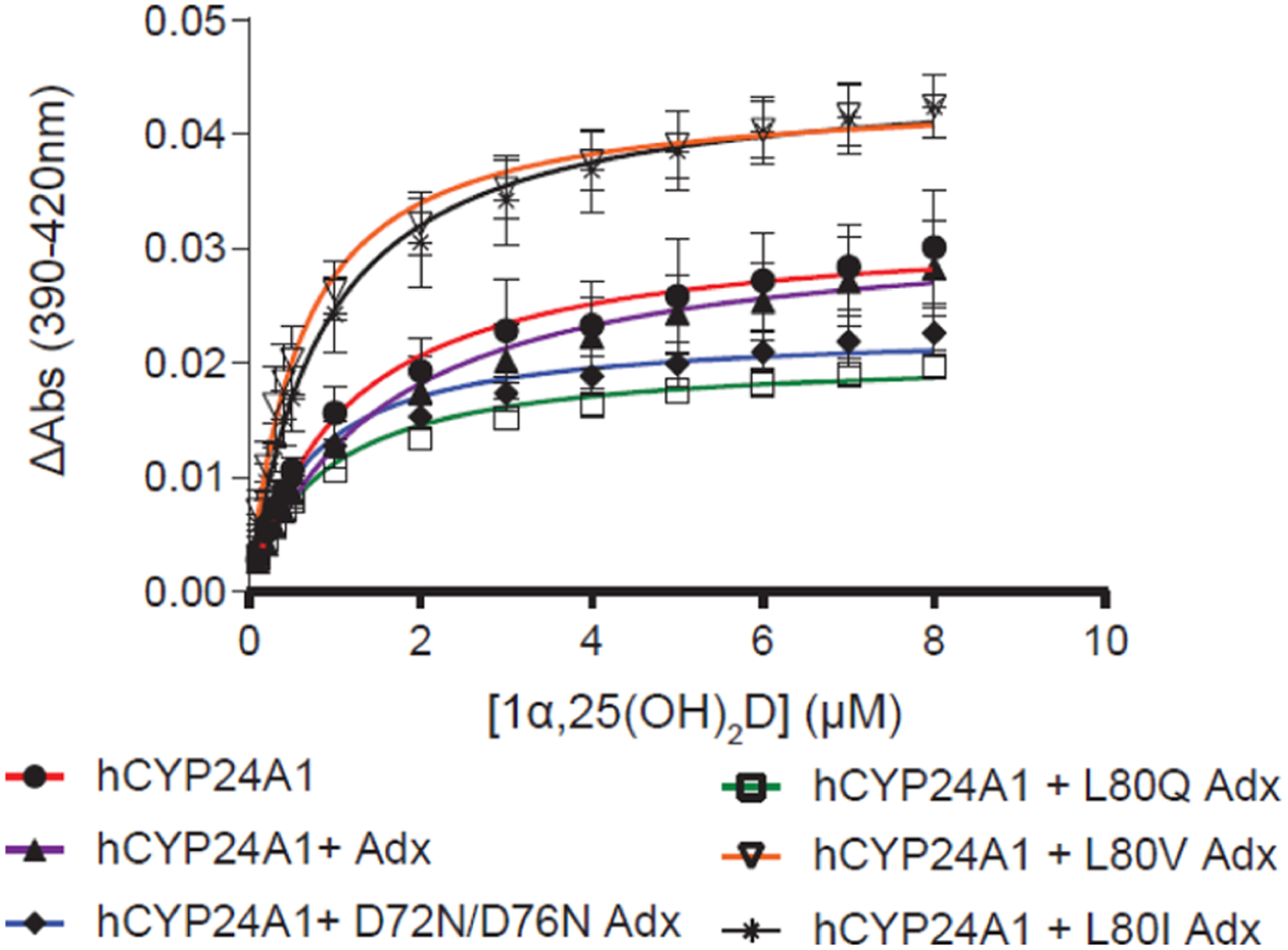
Effect of Adx surface mutations on 1α,25(OH)2D binding to hCYP24A1. Binding curves are shown for titrations in the presence of the 10-fold excess of hAdx mutants. n = 3, error bars represent SD.
Table 2.
Characterization of 1α,25(OH)2D Binding to CYP24A1 in the Presence of Adx Mutantsa
| KD (μM) | ΔAmax (AU) | |
|---|---|---|
| hCYP24A1 | ||
| no Adx | 1.1 ± 0.2 | 0.03 ± 0.005 |
| WT | 1.5 ± 0.2 | 0.03 ± 0.004 |
| D72N_D76N | 0.7 ± 0.1 | 0.02 ± 0.002 |
| L80Q | 0.8 ± 0.1 | 0.02 ± 0.001* |
| L80V | 0.6 ± 0.05 | 0.04 ± 0.003* |
| L80I | 0.8 ± 0.08 | 0.04 ± 0.003* |
| rCYP24A1 | ||
| no Adx | 0.3 ± 0.01 | 0.05 ± 0.004 |
| WT | 0.3 ± 0.03 | 0.07 ± 0.003** |
| L80V | 0.4 ± 0.03 | 0.07 ± 0.004** |
One-way ANOVA analysis is indicated by one asterisk (P ≤ 0.05) or two (P ≤ 0.01).
In theory, complete disruption of the hCYP24A1-hAdx complex should result in neither an increase nor a decrease in the interaction between the enzyme and 1α,25(OH)2D since this would be equivalent to a substrate titration with hCYP24A1 in the absence of Adx. However, modification of the interface does appear to modulate substrate binding. This indicates that the single and double mutations that were introduced are not sufficient to prevent formation of a complex that reports less activity than the native complex and is likely due to an unfavorable change in hCYP24A1 conformation that can result in less binding (D72_D76N and L80Q) or, unexpectedly, more binding (L80I/V). To address whether these variant complexes can be detected, we followed up with chemical cross-linking of the purified proteins using the zero-length cross-linker EDC. EDC forms covalent cross-linkages between both carboxylates and primary amines, making it an ideal cross-linking agent for the investigation of electrostatic CYP-Adx complexes. Here, we resolved the cross-linked product on SDS-PAGE for the wild type as well as all mutant forms of Adx (Figure S3). The ability of even the D72N_D76N double mutant to form a cross-linked product implies the presence of a larger binding surface that likely also involves distance-dependent hydrophobic contacts at Leu-80.
Leu-80 of Adx Forms an Isoform-Dependent Non-polar Contact with CYP24A1.
There is evidence that isoform-specific complexes between CYP24A1 and Adx are not equivalent. Specifically, isoforms that display distinct regioselectivity for the vitamin-D side chain also rely on unique contacts with Adx.16 We have previously noted that a nonconserved site is located near the proximal surface of CYP24A1, in which Gly-170 in hCYP24A1 is replaced by a valine residue in rCYP24A1. Through docking studies that rely on the crystal structure rCYP24A1, there appears to be an interaction between this nonpolar site and Leu-80 of Adx.14 Therefore, here, we evaluated the impact of the L80V hAdx mutation on the rat enzyme to determine whether this contact is variable by species.
First, we observed a differential effect of WT hAdx on substrate binding in rCYP24A1 vs hCYP24A1. When hAdx is present with rCYP24A1, there is a 30% increase in ΔAmax of binding from 0.05 to 0.07 (Figure 7A and Table 2). This outcome is in contrast to that obtained with hCYP24A1 (Table 1). Another difference was with respect to the role played by Leu-80. With rCYP24A1, the L80V hAdx mutation behaved similarly to WT hAdx, both in terms of ΔAmax of binding and in activity assays, in which the mutation has only a modest effect on the rCYP24A1 function (Figure 7B). Last, we used EDC cross-linking to confirm that the complex formation between rCYP24A1 and WT and L80V hAdx is also unaffected (Figure S3). Taken together, these data indicate that the complex with rCYP24A1 is less sensitive to changes in Leu-80 when compared to the human isoform complex and reinforces the differences in redox partner recognition between C24 and C23/C24 vitamin-D hydroxylases.
Figure 7.
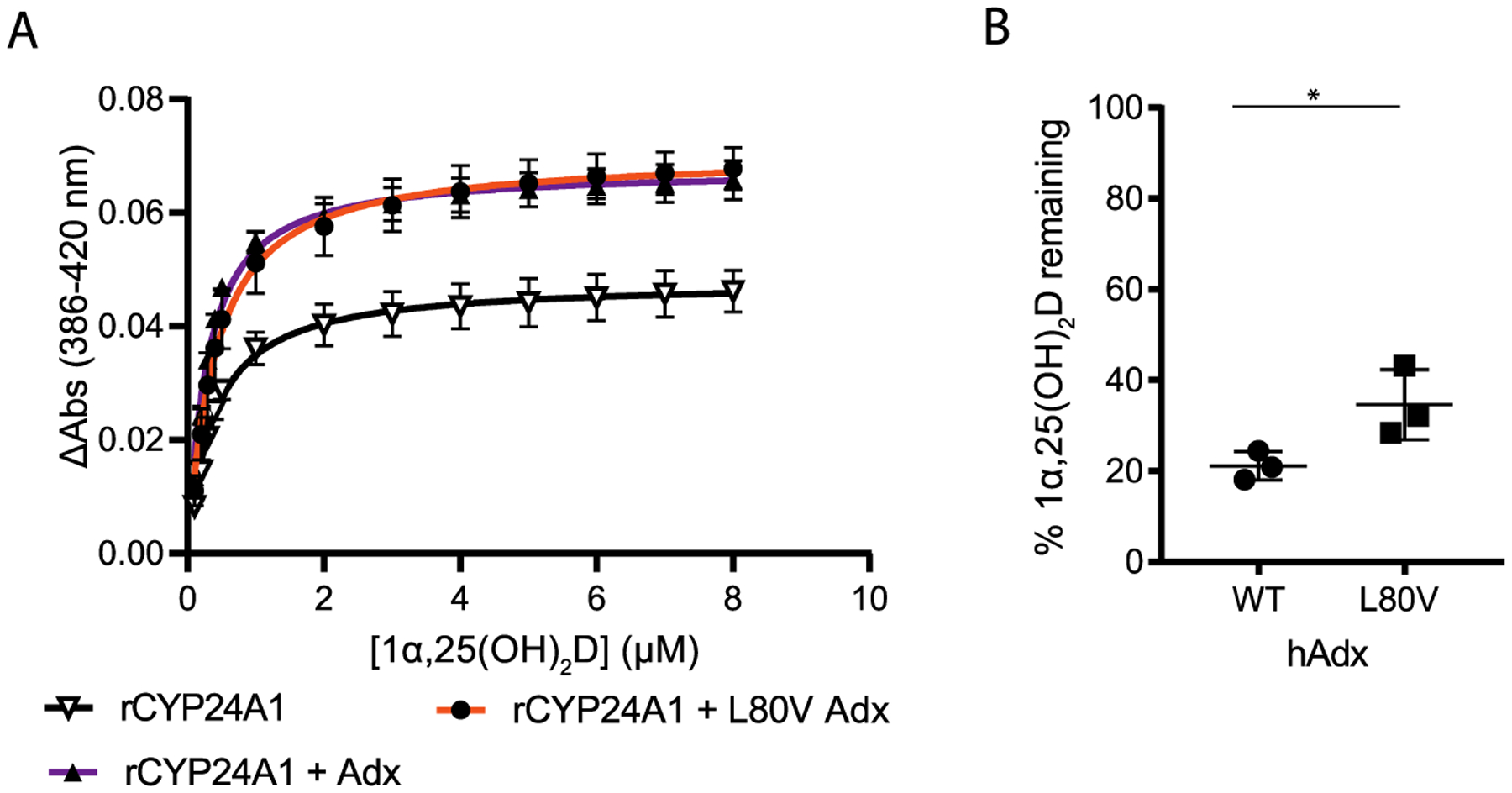
Effect of hAdx (L80V) on rat CYP24A1. (A) Binding of 1α,25(OH)2D in the presence of WT and L80V hAdx, increased overall binding. (B) 1α,25(OH)2D depletion with rCYP24A1 combined with WT and L80V hAdx. Asterisk (*), one-way ANOVA analysis α = 0.05, n = 3, p < 0.05.
DISCUSSION
The inability to produce the purified human isoform of CYP24A1 has long been a significant obstacle in furthering the understanding of molecular regulation of human vitamin-D metabolism. In this work, we relied on production of hCYP24A1 as a cleavable recombinant fusion with hAdx to first produce and then separate isolated and functional hCYP24A1. CYP fusions with redox partner proteins occur naturally,18,31 and mammalian CYP-Adx fusions have been used previously for the purpose of capturing the crystallographic interface with the redox partner complex.32,33 However, to our knowledge, this use represents the first application of a fusion construct that is cleavable and specifically designed to promote stability in a mammalian CYP prior to its separation from Adx, thus overcoming the difficulty of production of the enzyme in E. coli. Moreover, this strategy obviates the requirement for an Adx-affinity column during purification, as has been the case for previous preparations of CYP24A1.16,34
Using this platform, the affinity values for primary and secondary hCYP24A1 ligands were determined. In general, the rank ordering of ligand affinity mirrors that reported using rCYP24A1,35 albeit with dissociation constants with hCYP24A1 in the low micromolar regime compared to the tighter binding reported in rCYP24A1. As the primary substrate, 1α,25(OH)2D binds much tighter than the secondary ligands followed by 1α,24,25(OH)3D and 25(OH)D (Table 1). We should note here that, after adjusting for sample dilution, we were not able to saturate the interaction with 25(OH)D and there was an absence of type-I response (conversion to high spin) upon its titration; therefore, it was not possible to estimate accurate binding data for this substrate, other than the observation that it likely binds with a weaker affinity than the other substrates tested.
A key difference in human and rat vitamin-D metabolism is the presence of a minor C23 hydroxylation pathway that is performed by hCYP24A1 but is absent in the regioselective preference of rCYP24A1.36 Earlier characterization of unpurified hCYP24A1 indicated that at least one active site residue, Ala-326 of the I-helix, participates in determining the regioselectivity of hydroxylation of the 1α,25(OH)2D side chain.37 However, the human and rat isoforms share an 84% sequence identity in which the active site environment, including Ala-326, is largely conserved and with many of the nonconserved residues lining the solvent-exposed regions of the structure. This suggests that nonconserved surface contacts, including those located near the proximal (Adx-binding) surface, exert some influence on the degree of the C23 pathway that is present in the human enzyme. This notion is supported by NMR studies, indicating that species isoforms of CYP24A1 rely on distinct secondary binding sites for proper molecular recognition16 and that, in the case of the rat enzyme matched with rat Adx, the redox partner and the substrate together exert a concerted change in the CYP24A1 structure.14
In light of these data, here, we sought to understand the hCYP24A1 function in the context of a ternary complex consisting of hCYP24A1, hAdx, and the vitamin-D ligand. The ability of Adx to bring about changes in ligand positioning within the active site has been demonstrated previously in the case of 22-hydroxycholesterol binding to CYP11A1.38 A key finding from this study is that the human ternary complex with Adx and the substrate is more distinct than the one formed with the rat enzyme. Specifically, ΔAmax of binding substrate to hCYP24A1 is not affected by the presence of hAdx but is significantly increased when hAdx is added into rCYP24A1 (Figure 7A). Since the active site environment is largely conserved, this points toward differences in redox partner binding. The least conserved position on the proximal (redox-binding) side of the enzyme is the Val-170 that is present in rats but is substituted with a glycine in humans (Figure 8). Based on protein docking from a previous study, this valine is predicted to form a conditional conformation-dependent contact with Leu-80 of Adx.14 Leu-80 has also been implicated in binding to the corticosteroid metabolizing enzymes CYP11B1 and CYP11B2.30 Therefore, we then tested a variety of substitutions of the Leu-80 side chain to probe different chemistry at this site and its role in the hCPY24A1 function.
Figure 8.
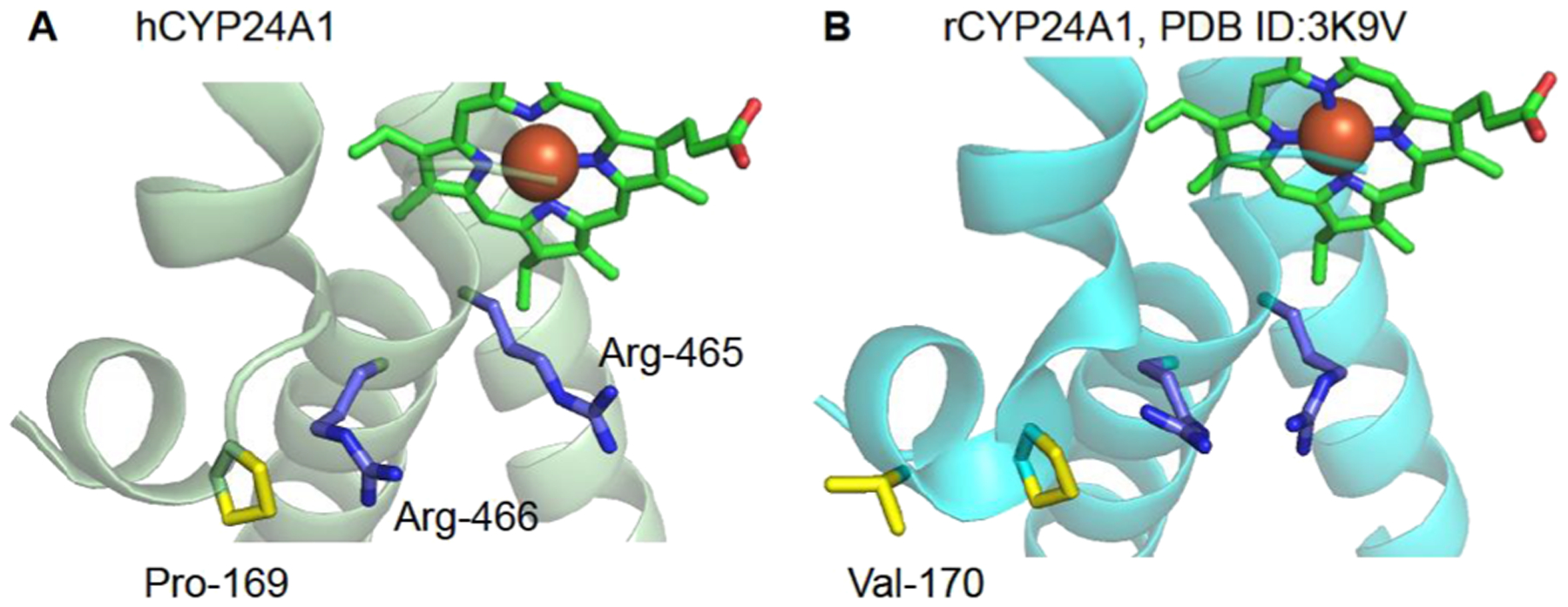
Proximal surface comparison of CYP24A1. The human enzyme (A) was modeled using RoseTTaFold.39 In hCYP24A1 and rCYP24A1 (B) PDB ID 3K9V,12 the distribution of nonpolar contacts (yellow) is shown in relation to the conserved basic surface residues Arg-465 and Arg-466 (in blue).
Here, we observed that the L80V mutation in the human system causes a decrease in substrate depletion to a similar extent compared to removing the nonpolar contact altogether (Figure 4C) yet unexpectedly induces more binding (increased ΔAmax) to the substrate (Figure 6). A similarly conservative mutation (L80I) also enhances substrate binding yet does not affect catalysis. These findings point toward a requirement for a specific nonpolar contact with hCYP24A1. Since hCYP24A1 lacks the Val-170 residue, we also tested the L80V mutation in rCYP24A1. Here, we found that L80V behaves similarly to WT hAdx in that it also enhances binding (Figure 7) but only marginally affects depletion of 1α,25(OH)2D. We interpret these species-variable results to indicate that a conditional contact occurs between Leu-80 of hAdx and Val-170 of rCYP24A1 that is then absent in hCYP24A1. Instead, the nonpolar contact in hCYP24A1 likely only occurs between Leu-80 and the aliphatic side chain of Pro-169. This contact would in theory be more sensitive to changes in distance and binding geometry since an alternative contact with Val-170 is not available. Importantly, Leu-80, along with the rest of the anionic alpha helix-3 of Adx, is highly conserved in all species.
Last, the data presented in this study provides additional evidence of Adx-induced conformational changes in CYP24A1, only this time demonstrated in a human CYP-Adx complex, and in such a way that reflects meaningful changes in ligand binding due to the modulation of the redox interface. Promotion of a closed conformation of the enzyme in response to Adx has been suggested previously.14 Notably, in early attempts to purify the hAdx_hCYP24A1 fusion using metal affinity chromatography, imidazole became trapped in the active site, leading to a persistent low-spin form of the enzyme that could not be removed by dialysis. This would also be consistent with the closing of the structure due to the presence of hAdx. Interestingly, mutations of hAdx that were designed to disrupt the interaction (L80Q and D72_D76N) were found to induce a decrease in substrate binding. In theory, the complete disruption of the complex should have no effect on the interaction between hCYP24A1 and 1α,25(OH)2D. Instead, these outcomes indicate that some version of the redox complex still forms (as evidenced by EDC cross-linking; Figure S3) but that the resulting conformational change in hCYP24A1 is not productive due to improper positioning of the substrate.
CONCLUSIONS
This study provides a functional characterization for hCYP24A1. Stabilization of hCYP24A1 was achieved using a cleavable fusion with hAdx, which then allowed for the investigation of hCYP24A1 as a ternary complex with hAdx and vitamin-D ligands. This platform was used to highlight species-variable differences in the ability of Adx to induce changes in substrate binding. Specifically, hCYP24A1 displays a strict requirement at the Leu-80 side chain, which, together with the anionic residues of alpha helix-3, forms the CYP recognition site of Adx. These differences indicate that nonconserved surfaces on the proximal surface of hCYP24A1 lead to specific recognition of its redox partner that results in functionally important conformational changes in hCYP24A1.
Supplementary Material
ACKNOWLEDGMENTS
Dr. Amit Kumar, University at Buffalo, is acknowledged for the contribution of the rat CYP24A1 enzyme. Dr. Emily Scott, University of Michigan, is acknowledged for the contribution of the wild-type human Adx plasmid.
Funding
Funding for this work was provided by R35GM133375.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.1c00770.
CO-binding assay with hCYP24A1 lysate, absorbance changes of hCYP24A1 titrated with hAdx, and EDC cross-linking with CYP24A1 and hAdx mutants (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biochem.1c00770
The authors declare no competing financial interest.
Contributor Information
Natalie Jay, Department of Biochemistry, University at Buffalo, Buffalo, New York 14203, United States.
Sean R. Duffy, Department of Biochemistry, University at Buffalo, Buffalo, New York 14203, United States
D. Fernando Estrada, Department of Biochemistry, University at Buffalo, Buffalo, New York 14203, United States.
REFERENCES
- (1).Tieu EW; Tang EKY; Tuckey RC Kinetic analysis of human CYP24A1 metabolism of vitamin D via the C24-oxidation pathway. FEBS J. 2014, 281, 3280–3296. [DOI] [PubMed] [Google Scholar]
- (2).Prosser DE; Jones G Enzymes involved in the activation and inactivation of vitamin D. Trends Biochem. Sci 2004, 29, 664–673. [DOI] [PubMed] [Google Scholar]
- (3).Pronicka E; Ciara E; Halat P; Janiec A; Wójcik M; Rowińska E; Rokicki D; Płudowski P; Wojciechowska E; Wierzbicka A; Książyk JB; Jacoszek A; Konrad M; Schlingmann KP; Litwin M Biallelic mutations in CYP24A1 or SLC34A1 as a cause of infantile idiopathic hypercalcemia (IIH) with vitamin D hypersensitivity: molecular study of 11 historical IIH cases. J. Appl. Genet 2017, 58, 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).DeLuca HF; Schnoes HK Metabolism and mechanism of action of vitamin D. Annu. Rev. Biochem 1976, 45, 631–666. [DOI] [PubMed] [Google Scholar]
- (5).Meusburger E; Mündlein A; Zitt E; Obermayer-Pietsch B; Kotzot D; Lhotta K Erratum: Medullary nephrocalcinosis in an adult patient with idiopathic infantile hypercalcaemia and a novel CYP24A1 mutation. Cli.n Kidney J 2013, 6, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ross AC; Taylor CL; Yaktine AL; Del Valle HB Overview of vitamin D. Dietary Reference Intakes for Calcium and Vitamin D; National Academies Press; 2011. [PubMed] [Google Scholar]
- (7).Mawer EB; Taylor CM; Backhouse J; Lumb GA; Stanbury SW Failure of formation of 1,25-dihydroxycholecalciferol in chronic renal insufficiency. Lancet 1973, 301, 626–628. [DOI] [PubMed] [Google Scholar]
- (8).Martineau C; Naja RP; Husseini A; Hamade B; Kaufmann M; Akhouayri O; Arabian A; Jones G; St-Arnaud R Optimal bone fracture repair requires 24R,25-dihydroxyvitamin D3 and its effector molecule FAM57B2. J. Clin. invest 2018, 128, 3546–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kusudo T; Sakaki T; Abe D; Fujishima T; Kittaka A; Takayama H; Hatakeyama S; Ohta M; Inouye K Metabolism of A-ring diastereomers of 1alpha,25-dihydroxyvitamin D3 by CYP24A1. Biochem. Biophys. Res. Commun 2004, 321, 774–782. [DOI] [PubMed] [Google Scholar]
- (10).Ishizuka S; Kurihara N; Reddy SV; Cornish J; Cundy T; Roodman GD (23S)-25-Dehydro-1{alpha}-hydroxyvitamin D3–26,23-lactone, a vitamin D receptor antagonist that inhibits osteoclast formation and bone resorption in bone marrow cultures from patients with Paget’s disease. Endocrinology 2005, 146, 2023–2030. [DOI] [PubMed] [Google Scholar]
- (11).Ishizuka S; Kurihara N; Miura D; Takenouchi K; Cornish J; Cundy T; Reddy SV; Roodman GD Vitamin D antagonist, TEI-9647, inhibits osteoclast formation induced by 1alpha,25-dihydroxyvitamin D3 from pagetic bone marrow cells. J. Steroid Biochem. Mol. Biol 2004, 89–90, 331–334. [DOI] [PubMed] [Google Scholar]
- (12).Annalora AJ; Goodin DB; Hong WX; Zhang Q; Johnson EF; Stout CD Crystal structure of CYP24A1, a mitochondrial cytochrome P450 involved in vitamin D metabolism. J. Mol. Biol 2010, 396, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kumar A; Estrada DF Specificity of the Redox Complex between Cytochrome P450 24A1 and adrenodoxin relies on carbon-25 hydroxylation of vitamin-D Substrate. Drug Metab. Dispos 2019, 47, 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kumar A; Wilderman PR; Tu C; Shen S; Qu J; Estrada DF Evidence of allosteric coupling between substrate binding and Adx recognition in the vitamin-D carbon-24 hydroxylase CYP24A1. Biochemistry 2020, 59, 1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Brixius-Anderko S; Scott EE Structure of human cortisol-producing cytochrome P450 11B1 bound to the breast cancer drug fadrozole provides insights for drug design. J. Biol. Chem 2019, 294, 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Estrada DF The cytochrome P450 24A1 interaction with adrenodoxin relies on multiple recognition sites that vary among species. J. Biol. Chem 2018, 293, 4167–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tropea JE; Cherry S; Waugh DS Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol Biol. 2009, 498, 297–307. [DOI] [PubMed] [Google Scholar]
- (18).Nodate M; Kubota M; Misawa N Functional expression system for cytochrome P450 genes using the reductase domain of self-sufficient P450RhF from Rhodococcus sp. NCIMB 9784. Appl. Microbiol. Biotechnol 2006, 71, 455–462. [DOI] [PubMed] [Google Scholar]
- (19).Guengerich FP; Martin MV; Sohl CD; Cheng Q Measurement of cytochrome P450 and NADPH-cytochrome P450 reductase. Nat. Protoc 2009, 4, 1245–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Shiraishi A; Higashi S; Ohkawa H; Kubodera N; Hirasawa T; Ezawa I; Ikeda K; Ogata E The advantage of alfacalcidol over vitamin D in the treatment of osteoporosis. Calcif. Tissue Int 1999, 65, 311–316. [DOI] [PubMed] [Google Scholar]
- (21).Shiraishi A; Takeda S; Masaki T; Higuchi Y; Uchiyama Y; Kubodera N; Sato K; Ikeda K; Nakamura T; Matsumoto T; Ogata E Alfacalcidol inhibits bone resorption and stimulates formation in an ovariectomized rat model of osteoporosis: distinct actions from estrogen. J. Bone Miner. Res 2000, 15, 770–779. [DOI] [PubMed] [Google Scholar]
- (22).Shinkyo R; Sakaki T; Kamakura M; Ohta M; Inouye K Metabolism of vitamin D by human microsomal CYP2R1. Biochem. Biophys. Res. Commun 2004, 324, 451–457. [DOI] [PubMed] [Google Scholar]
- (23).Cheng JB; Motola DL; Mangelsdorf DJ; Russell DW De-orphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxilase. J. Biol. Chem 2003, 278, 38084–38093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hanukoglu I; Spitsberg V; Bumpus JA; Dus KM; Jefcoate CR Adrenal mitochondrial cytochrome P-450scc. Cholesterol and adrenodoxin interactions at equilibrium and during turnover. J. Biol. Chem 1981, 256, 4321–4328. [DOI] [PubMed] [Google Scholar]
- (25).Hanukoglu I; Privalle CT; Jefcoate CR Mechanisms of ionic activation of adrenal mitochondrial cytochromes P-450scc and P-45011 beta. J. Biol. Chem 1981, 256, 4329–4335. [DOI] [PubMed] [Google Scholar]
- (26).Zalewski A; Ma NS; Legeza B; Renthal N; Flück CE; Pandey AV Vitamin D-Dependent Rickets Type 1 Caused by Mutations in CYP27B1 Affecting Protein Interactions With Adrenodoxin. J. Clin. Endocrinol. Metab 2016, 101, 3409–3418. [DOI] [PubMed] [Google Scholar]
- (27).Coghlan VM; Vickery LE Site-specific mutations in human ferredoxin that affect binding to ferredoxin reductase and cytochrome P450scc. J. Biol. Chem 1991, 266, 18606–18612. [PubMed] [Google Scholar]
- (28).Coghlan VM; Vickery LE Electrostatic interactions stabilizing ferredoxin electron transfer complexes. Disruption by ″conservative″ mutations. J. Biol. Chem 1992, 267, 8932–8935. [PubMed] [Google Scholar]
- (29).Adamovich TB; Pikuleva IA; Chashchin VL; Usanov SA Selective chemical modification of cytochrome P-450SCC lysine residues. Identification of lysines involved in the interaction with adrenodoxin. Biochim. Biophys. Acta 1989, 996, 247–253. [DOI] [PubMed] [Google Scholar]
- (30).Peng HM; Auchus RJ Molecular Recognition in Mitochondrial Cytochromes P450 That Catalyze the Terminal Steps of Corticosteroid Biosynthesis. Biochemistry 2017, 56, 2282–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).McLean KJ; Girvan HM; Munro AW Cytochrome P450/redox partner fusion enzymes: biotechnological and toxico-logical prospects. Expert Opin. Drug Metab. Toxicol 2007, 3, 847–863. [DOI] [PubMed] [Google Scholar]
- (32).Strushkevich N; MacKenzie F; Cherkesova T; Grabovec I; Usanov S; Park HW Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc. Natl. Acad. Sci. U.S. A 2011, 108, 10139–10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Brixius-Anderko S; Scott EE Structural and functional insights into aldosterone synthase interaction with its redox partner protein adrenodoxin. J. Biol. Chem 2021, 296, 100794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hartfield KA; Stout CD; Annalora AJ The novel purification and biochemical characterization of a reversible CYP24A1:adrenodoxin complex. J. Steroid Biochem. Mol. Biol 2013, 136, 47–53. [DOI] [PubMed] [Google Scholar]
- (35).Annalora A; Bobrovnikova-Marjon E; Serda R; Lansing L; Chiu ML; Pastuszyn A; Iyer S; Marcus CB; Omdahl JL Rat cytochrome P450C24 (CYP24A1) and the role of F249 in substrate binding and catalytic activity. Arch. Biochem. Biophys 2004, 425, 133–146. [DOI] [PubMed] [Google Scholar]
- (36).Sakaki T; Sawada N; Komai K; Shiozawa S; Yamada S; Yamamoto K; Ohyama Y; Inouye K Dual metabolic pathway of 25-hydroxyvitamin D3 catalyzed by human CYP24. Eur. J. Biochem 2000, 267, 6158–6165. [DOI] [PubMed] [Google Scholar]
- (37).Prosser DE; Kaufmann M; O’Leary B; Byford V; Jones G Single A326G mutation converts human CYP24A1 from 25-OH-D3–24-hydroxylase into −23-hydroxylase, generating 1alpha,25-(OH)-2D3–26,23-lactone. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 12673–12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mast N; Annalora AJ; Lodowski DT; Palczewski K; Stout CD; Pikuleva IA Structural basis for three-step sequential catalysis by the cholesterol side chain cleavage enzyme CYP11A1. J. Biol. Chem. 2011, 286, 5607–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, Millán C, Park H, Adams C, Glassman CR, DeGiovanni A, Pereira JH, Rodrigues AV, van Dijk AA, Ebrecht AC, Opperman DJ, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy MK, Dalwadi U, Yip CK, Burke JE, Garcia KC, Grishin NV, Adams PD, Read RJ, Baker D Accurate prediction of protein structures and interactions using a three-track neural network, Science; New York, N.Y.., 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.