

Abstract
The coronavirus disease 2019 (COVID-19) epidemic in Brazil was driven mainly by the spread of Gamma (P.1), a locally emerged variant of concern (VOC) that was first detected in early January 2021. This variant was estimated to be responsible for more than 96 per cent of cases reported between January and June 2021, being associated with increased transmissibility and disease severity, a reduction in neutralization antibodies and effectiveness of treatments or vaccines, and diagnostic detection failure. Here we show that, following several importations predominantly from the USA, the Delta variant rapidly replaced Gamma after July 2021. However, in contrast to what was seen in other countries, the rapid spread of Delta did not lead to a large increase in the number of cases and deaths reported in Brazil. We suggest that this was likely due to the relatively successful early vaccination campaign coupled with natural immunity acquired following prior infection with Gamma. Our data reinforce reports of the increased transmissibility of the Delta variant and, considering the increasing concern due to the recently identified Omicron variant, argues for the necessity to strengthen genomic monitoring on a national level to quickly detect the emergence and spread of other VOCs that might threaten global health.
Keywords: Genomic monitoring, variants replacment, Brazil, Gamma, Delta
Since late 2020, the evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been characterized by the appearance of mutations on the Spike protein leading to the emergence of so-called ‘variants of concern’ (VOCs), some of which have spread globally (Safari and Elahi 2021; Tao et al. 2021). In particular, some of the mutations in VOCs are suggested to impact viral transmissibility (Nonaka et al. 2021), resistance to neutralizing antibodies (Greaney et al. 2021), and virulence. The identification of such variants has recently challenged public health authorities with respect to tracking transmission and mitigating the impact in the ongoing pandemic. To date, the most important VOCs documented are Alpha, Beta, Gamma, Delta, and Omicron (described in late November 2021), first detected in the UK, South Africa, Brazil, India, and South Africa/Botswana, respectively (Faria et al. 2021; Meng et al. 2021; Tegally et al. 2021b; Viana et al. 2022).
In the context of continuous surveillance of SARS-CoV-2 clinical samples on behalf of the National Pandemic Alert Network based in the State of São Paulo (Brazil) since early January 2021, our team has been monitoring the proportion of circulating variants in Brazil. Data from this initiative show that after July 2021, the Delta variant has become highly prevalent. It has been suggested that the Delta variant might be more transmissible (Planas et al. 2021), having led to public health emergencies in other countries due to overwhelming increases in the number of cases, hospitalizations, and deaths when compared to previous circulating variants. Here, we describe how Delta became the predominant SARS-CoV-2 variant in Brazil, rapidly replacing the previously dominant Gamma, and how this displacement was not associated with an increase in reported case or death numbers.
The COVID-19 epidemic in Brazil can generally be characterized by two epidemic waves accounting for more than 22 million cases and 616,251 deaths until early December 2021 (WHO | World Health Organization 2022). The first epidemic wave was characterized by the circulation of multiple SARS-CoV-2 lineages (including B.1.1.28 and B.1.1.33), as a direct consequence of multiple independent introduction events between February 2020 and March 2020 (Fig. 1A) (Giovanetti et al. 2021). By the end of October 2020, even with the implementation of non-pharmaceutical interventions (NPIs), a second wave associated with a dramatic resurgence in case and death numbers took place. This wave was fuelled by the emergence and circulation of several variants under monitoring (VUMs), such as P.2 (i.e. Zeta), and some VOCs including Alpha and later Gamma (i.e. P.1), which became widespread by January 2021 and dominated the viral population for nearly 8 consecutive months (Fig. 1A). Despite the national vaccination roll-out beginning on 17 January 2021, the COVID-19 death toll in the country steadily rose in March 2021, reaching a peak in April 2021 (Fig. 1A). This was followed by a decrease in the number of daily cases and deaths by April (Fig. 1A and Supplementary Figs S1–S2), likely as a consequence of a gradual increase in population immunity. Zeta (P.2) largely dominated the first epidemic wave persisting up to March 2021 when it was replaced by Gamma (P.1) (Fig. 1A–C). This period was characterized by an upsurge in the number of total cases with a peak registered between February and June 2021. The Alpha variant was also detected from January 2021 onwards, but it remained at a very low frequency nationally (less than 2 per cent) (PAHO/WHO 2021).
Figure 1.
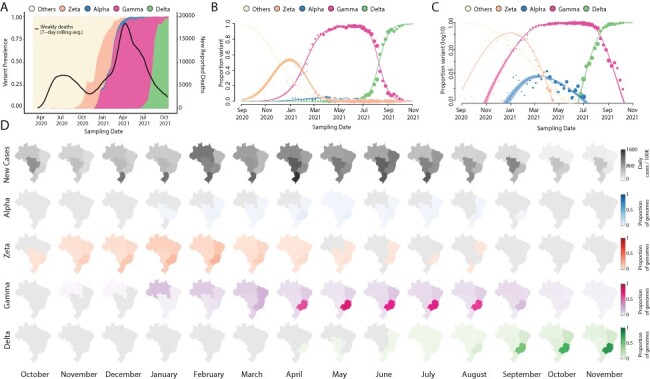
Replacement of the Gamma by the Delta variant in Brazil. (A) Dynamics of the SARS-CoV-2 epidemic in Brazil showing the number of daily COVID-19 deaths and the progression in the proportion of circulating variants in the country over time, with the rapid replacement of the Gamma by the Delta variant. (B–C) The modelled proportion of SARS-CoV-2 variants over time in Brazil in a linear and in a logarithmic scale (respectively), showing that Gamma became the dominant variant in Brazil by the beginning of 2021 and was rapidly outcompeted by Delta from June 2021. Model fits are based on a multinomial logistic regression. The size of the dots corresponds to the weekly sample size of SARS-CoV-2 genomes (independently of variants, such that each week will have the same point size across all variant curves). (D) Prevalence maps following the progression in the monthly average of a daily number of cases and proportions of each variant (Alpha, Zeta, Gamma, and Delta) per region (North, Northeast, Midwest, Southeast, and South) in Brazil from October 2020 to October 2021. The scale 0–1 used represents the time-relative prevalence of each variant calculated independently of all the others (i.e. variant prevalence divided by its maximum across time). It shows the spatio-temporal evolution of each variant’s spread.
In April 2021, through the intensified sampling of likely imported cases associated with returning travellers, the Delta variant was detected. The Delta variant first spread in the southeast of Brazil—the most populous region that contains the largest urban centres and airports with the greatest amount of national and international travel that continued to operate throughout 2021 (Rodriguez-Morales et al. 2020). Between July and August 2021, Delta rapidly displaced Gamma, becoming the dominant variant circulating nationally during August 2021 (Fig. 1A and Supplementary Fig. S1). Notably, however, this variant displacement was not associated with a concurrent increase in reported COVID-19 cases or deaths (Fig. 1A and Supplementary Fig. S2A). We estimate that Delta had a growth advantage of 0.064 (95 per cent confidence interval [CI] 0.058–0.071) per day compared to Gamma (Fig. 1B–C). Assuming that the variants have the same generation time of 5.2 days (Ganyani et al. 2020), this corresponds to a growth advantage of 33 per cent (95 per cent CI 30–37 per cent) per generation of viral transmission, which is in good agreement with earlier findings based on sequence data from multiple countries (Campbell et al. 2021).
We hypothesize that a relatively successful early vaccination campaign in all Brazilian macroregions, which reached more than 50 per cent of vaccination coverage with both first and second shots (Supplementary Fig. S3B), coupled with a reasonable percentage of the population with natural immunity acquired by prior infection with Gamma, contributed to a continuous decline in reported COVID-19 deaths and cases during the emergence of the Delta variant (Fig. 1A and Supplementary Fig S2A). In fact, after Delta became dominant in August 2021, reported COVID-19 deaths reached a minimum not seen since October 2020. In contrast, after Gamma became dominant in February 2021, reported COVID-19 deaths and cases increased to a historical maximum, which would have been attributed to a relatively naïve population to infection with any SARS-CoV-2 variant at the time. This epidemiological scenario of rapid switching of new variants without an increase in the number of deaths or cases seems not to be uncommon and has also been observed elsewhere (Merhi et al. 2021; Naveca et al. 2021; Olsen et al. 2021; Tegally et al. 2021a; Marques et al. 2022).
We further estimated phylogenetic trees to explore the relationship of the sequenced Brazilian genome to those of other isolates across the world. For this purpose, we retrieved 11,147 Delta genomes from Brazil, from which 6,626 were generated by our National Pandemic Alert Network and a globally representative set of other Delta genomes (n = 13,261). Our time-stamped phylogeny revealed that the Brazilian Delta isolates are scattered throughout the phylogeny suggesting multiple independent introductions (Fig. 2A and Supplementary Fig. S4).
Figure 2.
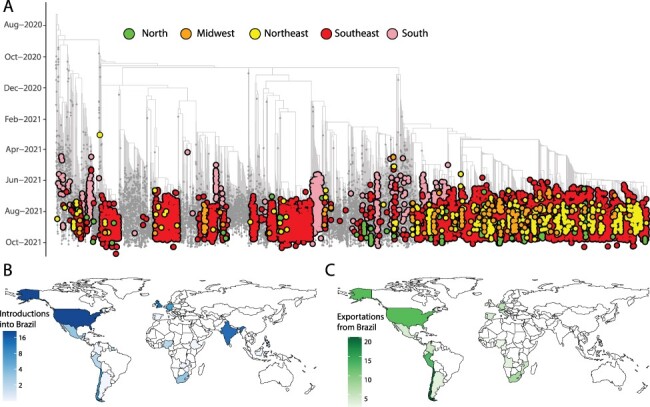
Phylogenetic analysis of the Delta variant in Brazil. (A) Time-resolved maximum clade credibility phylogeny of 11,147 SARS-CoV-2 Delta sequences from Brazil, from which 6,626 were generated by our National Network for Pandemic Alert of SARS-CoV-2 along with global Delta sequences (grey). Only Brazilian genomes have been highlighted in the figure. Colours indicate different Brazilian regions (North in green; Midwest in orange; Northeast in yellow; Southeast in red; and South in light pink). (B) Inferred locations of importations of the Delta variant into Brazil. (C) Inferred locations of exportation of the Delta variant from Brazil.
Our analysis revealed at least 124 independent introductions of the Delta variant into Brazil between October 2020 and October 2021 (Fig. 2B), with the majority (30 per cent) originating from North America, followed by India (17 per cent), European countries (mostly UK 13 per cent), and other South American countries.
Following introductions into Brazil and in line with recent findings (Arantes et al. 2021), the Delta variant appears to have further spread within Brazilian regions (Fig. 2A), highlighting the complex local transmission dynamics maintained by domestic travel. Importantly, the Delta variant appears to have been introduced into each region of Brazil through multiple ports of entry, making it challenging to accurately reconstruct precise transmission pathways across the country. Our analysis further shows that the Delta variant subsequently spread from Brazil into other countries in South America and also to North America, Europe, and Africa (Fig. 2C) (Candido et al. 2020).
The first months of the Brazilian epidemic were fuelled by the circulation of multiple lineages, as a direct consequence of multiple viral independent introductions from overseas. As the epidemic progressed, the observed large-scale community transmission led to the local emergence of VOCs and VUMs (Giovanetti et al. 2021). These variants dominated the end of the first and second epidemic waves in the country, resulting in an exponential increase in the number of daily cases and deaths, making Brazil one of the countries hardest hit by the SARS-CoV-2 pandemic worldwide.
In this study, we analysed the displacement of Gamma by the Delta variant in Brazil, showing how this replacement was not associated with an increase in reported case and death numbers. Given that during the observed displacement NPIs remained relatively relaxed locally, it is plausible that a significant proportion of the Brazilian population had developed immunity (through either natural infection or vaccination) before the emergence of Delta, which would have helped to prevent a rise in reported cases and deaths. We estimated that in Brazil, Delta had a transmission advantage of 33 per cent (95 per cent CI 30–37 per cent) compared to Gamma. The spread of future variants will be possible if they exhibit increased transmissibility or immunity evasiveness, both of which seem likely in the case of the Omicron variant (Viana et al. 2022). However, as shown here, displacement does not necessarily equate to new (large) epidemic waves of reported cases or deaths, since the outcome of the spread of a new variant will depend on a complex interplay between local interventions, infrastructure, and the immunity landscape of the population due to the circulation of previous variants. Considering that past replacement events have taken place so rapidly in Brazil, future epidemiological assessments of new VOC must be conducted rapidly and regularly. The capacity to detect and respond to new variants requires continued support and funding for molecular surveillance and sequencing capacity more generally. Moreover, enhanced sampling efforts in countries like Brazil are needed to ensure better geographical representativeness of available SARS-CoV-2 sequences and the rapid detection of emerging variants when their frequencies are still low. These factors will be key to detect, understand, and respond to the likely upcoming Omicron wave that has already been detected in more than 87 countries, including Brazil, and which will possibly alter the landscape of variants currently circulating in the country.
1. Materials and methods
1.1. Ethics statement
This research was approved by the Ethics Review Committee of the Pan American Health Organization (PAHOERC.0344.01) and by the Federal University of Minas Gerais (CEP/CAAE: 32912820.6.1001.5149). The availability of these samples for research purposes during outbreaks of national concern is allowed under the terms of the 510/2016 Resolution of the National Ethical Committee for Research—Brazilian Ministry of Health (BrMoH; Comissão Nacional de Ética em Pesquisa, Ministério da Saúde) that authorize, without the necessity of an informed consent, the use of clinical samples collected in the Brazilian Central Public Health Laboratories to accelerate knowledge building and contribute to surveillance and outbreak response. The samples processed in this study were obtained anonymously from material exceeding the routine diagnosis in Brazilian public health laboratories that belong to the public network within BrMoH.
1.2. Epidemiological data
We analyzed daily COVID-19 cases and deaths in Brazil up to 2 November 2021 from the Official National repository available at covid.saude.gov.br. For convenience, the geographical locations were aggregated by Brazilian macroregions: North, Northeast, Southeast, South, and Midwest. The Official National repository releases daily updates on the number of confirmed new cases, deaths, and recoveries, with a breakdown by states and regions.
1.3. Sample collection and molecular diagnostic assays
As part of the National Pandemic Alert Network, since early January 2021, our team has been monitoring the proportion of circulating variants in Brazil. For this purpose, convenience clinical samples from public laboratories in Brazil were received and randomly selected for sequencing every week. Depending on the partner institution, library preparation and sequencing were done either on the Illumina and/or Oxford Nanopore Platform. Viral RNA was extracted from nasopharyngeal swabs using an automated protocol and tested for SARS-CoV-2 by multiplex real-time polymerase chain reaction (PCR) assays: (1) the Allplex 2019-nCoV Assay (Seegene) targeting the envelope (E), the RNA-dependent RNA polymerase (RdRp), and the nucleocapsid (N) genes; (2) the Charité: SARS-CoV2 (E/RP) assay (Bio-Manguinhos/Fiocruz) targeting the E gene, and (3) the GeneFinder COVID-19 Plus RealAmp Kit (Osang Healthcare, South Korea) supplied by the BrMoH, Butantan Institute, and the Pan American Health Organization.
1.4. cDNA synthesis and whole-genome sequencing
Samples were selected for sequencing based on the Ct value (≤30) and availability of epidemiological metadata, such as date of sample collection, sex, age, and municipality of residence. The preparation of SARS-CoV-2 genomic libraries was performed using two different strategies: (1) the Illumina COVIDSeq test following the manufacturer’s instructions (Giovanetti et al. 2021) and (2) the Oxford Nanopore sequencing using the ARTIC Network primer scheme (Artic Network 2019). The normalized libraries were loaded for the Illumina sequencing onto a 300-cycle MiSeq Reagent Kit v2 and run on the Illumina MiSeq instrument (Illumina, San Diego, CA, USA) and for the Nanopore strategy into an R9.4 flow cell (Oxford Nanopore Technologies) as previously described (Xavier et al. 2020). All experiments were performed in a biosafety level-2 cabinet. In each sequencing run, we used negative controls to prevent and check for possible contamination with less than 2 per cent mean coverage.
1.5. Genome assembly
Sequences generated on the Illumina and Nanopore platforms were assembled using Genome Detective 1.132/3 (Vilsker et al. 2019).
2. Estimating relative transmission advantage
We analysed 11,147 SARS-CoV-2 Brazilian sequences, from which n = 6,626 were generated by our National Pandemic Alert Network, that have been uploaded to Global Initiative on Sharing All Influenza Data (GISAID) from 26 April 2021 to 23 October 2021. We used a multinomial logistic regression model to estimate the growth advantage of Delta compared to Gamma in Brazil (Campbell et al. 2021; Davies et al. 2021). We added splines to account for time-varying growth rates in the model fit and estimated the overall growth advantage without splines. We fitted the model using the multinom function of the nnet package (Venables and Ripley 2021) in R.
2.1. Phylogenetic analysis
We analysed 11,147 Delta variants from Brazil, publicly available on GISAID up to 4 November 2021, of which n = 6,626 were generated by our National Pandemic Alert Network. These were put in their phylogenetic context through comparison with a globally representative (n = 13,261) data set of other SARS-CoV-2 Delta variants from around the world, sampled from 23 October 2020 to 23 October 2021. This was achieved by the inclusion of sequences in the NextStrain Delta build (https://nextstrain.org/groups/neherlab/ncov/21A.Delta) at the time of writing and a random selection of 9,000 Delta sequences from the GISAID database. We feel that this approach would capture enough of the global diversity observed in the Delta data set to contextualize the Brazilian sequences. During our downsampling strategy, we took into account that the different sequencing efforts employed by different Brazilian states resulted in different numbers of available genomes for each state/region. To maximize the temporal signal with minimal geographic bias, Brazilian genomes were selected such that the sequences included were distributed as evenly as possible in each epi-week for which samples were available. Within each week, sequences were sampled proportionally to the cumulative number of cases in that location, aiming at building a more representative data set (Supplementary Fig. S5).
The full set of sequences were aligned with NextAlign (Aksamentov et al. 2021) to obtain a good codon quality alignment against the Wuhan-Hu-1 universal reference sequence. The subsequent alignment was then used to infer a Maximum Likelihood tree topology in IQTREE2 (Nguyen et al. 2015) (-m GTR, -b 100). Transfer bootstrap support for splits in the topology was inferred using Booster (Lemoine et al. 2018). The resulting consensus Maximum Likelihood (ML) tree topology was assessed for molecular clock signal in TempEst (Rambaut et al. 2016). Potential outlier sequences or sequences lacking required metadata (e.g. date and location of sampling) were pruned off the topology with the ape package (Popescu et al. 2012) in R prior to dating. The branches in the ML tree topology were then converted into units of calendar time in TreeTime (Sagulenko, Puller, and Neher 2018) using a constant rate of 0.0008 substitutions/site/year (Wilkinson et al. 2021), with a clock standard deviation of 0.0004 substitutions/site/year. Following the dating of the phylogeny, we annotated the tips and internal nodes using the ‘mugration’ package extension of TreeTime and then counted the state changes from one country to another and their inferred time points. Using the resulting annotated tree topology, we were able to count the number of transitions (i.e. virus importations and exportations) of SARS-CoV-2 Delta isolates entering Brazil over time. Importantly, this analysis was not dependent on a monophyletic clustering. To provide a measure of confidence in the time and source of viral transitions, we performed the discrete ancestral state reconstruction on ten bootstrap replicate trees using the same approach already described by Giovanetti et al. (2021), Tegally et al. (2021b), and Wilkinson et al. (2021).
Supplementary Material
Acknowledgements
The authors acknowledge the National Network for Pandemic Alert of SARS-CoV-2 and the contribution of all employees of General Coordination of Public Health Laboratories and professionals of Public Health Laboratories of Brazil for their contribution towards the sequencing effort and for their commitment and work during the fight of the COVID-19 pandemic. We also thank all the authors who have kindly deposited and shared genome data on GISAID.
Contributor Information
Vagner Fonseca, Laboratorio de Genética Celular e Molecular, Instituto de Ciências Biologicas, Universidade Federal de Minas Gerais, Av. Pres. Antônio Carlos, 6627 - Pampulha, Belo Horizonte, Minas Gerais 31270-901, Brazil; Organização Pan-Americana da Saúde/Organização Mundial da Saúde, Lote 19 - Avenida das Nações, SEN - Asa Norte, Brasília, Distrito Federal 70312-970, Brazil; KwaZulu-Natal Research Innovation and Sequencing Platform (KRISP), School of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, 238 Mazisi Kunene Rd, Glenwood, Durban 4041, South Africa; Centre for Epidemic Response and Innovation (CERI), School of Data Science and Computational Thinking, Stellenbosch University, Banhoek Road & Joubert Street, Stellenbosch 7600, South Africa.
Eduan Wilkinson, KwaZulu-Natal Research Innovation and Sequencing Platform (KRISP), School of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, 238 Mazisi Kunene Rd, Glenwood, Durban 4041, South Africa; Centre for Epidemic Response and Innovation (CERI), School of Data Science and Computational Thinking, Stellenbosch University, Banhoek Road & Joubert Street, Stellenbosch 7600, South Africa.
Houriiyah Tegally, KwaZulu-Natal Research Innovation and Sequencing Platform (KRISP), School of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, 238 Mazisi Kunene Rd, Glenwood, Durban 4041, South Africa; Centre for Epidemic Response and Innovation (CERI), School of Data Science and Computational Thinking, Stellenbosch University, Banhoek Road & Joubert Street, Stellenbosch 7600, South Africa.
Emmanuel James San, KwaZulu-Natal Research Innovation and Sequencing Platform (KRISP), School of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, 238 Mazisi Kunene Rd, Glenwood, Durban 4041, South Africa; Centre for Epidemic Response and Innovation (CERI), School of Data Science and Computational Thinking, Stellenbosch University, Banhoek Road & Joubert Street, Stellenbosch 7600, South Africa.
Christian L Althaus, Institute of Social and Preventive Medicine, University of Bern, Hochschulstrasse 6, Bern 3012, Switzerland.
Joilson Xavier, Laboratorio de Genética Celular e Molecular, Instituto de Ciências Biologicas, Universidade Federal de Minas Gerais, Av. Pres. Antônio Carlos, 6627 - Pampulha, Belo Horizonte, Minas Gerais 31270-901, Brazil; Laboratório Central de Saúde Pública do Estado de Minas Gerais, Fundação Ezequiel Dias, Rua Conde Pereira Carneiro, 80 Gameleira, Belo Horizonte, Minas Gerais 30510-010, Brazil.
Svetoslav Nanev Slavov, Ribeirão Preto Medical School, Blood Center of Ribeirão Preto, University of São Paulo, R. Quintino Bocaiuva, nº 470 - Centro, Ribeirão Preto, SP 14015-160, Brazil.
Vincent Louis Viala, Butantan Institute, Avenida Doutor Vital Brasil, 1500 - Butantã, Sao Paulo - SP, Brazil.
Alex Ranieri Jerônimo Lima, Butantan Institute, Avenida Doutor Vital Brasil, 1500 - Butantã, Sao Paulo - SP, Brazil.
Gabriela Ribeiro, Butantan Institute, Avenida Doutor Vital Brasil, 1500 - Butantã, Sao Paulo - SP, Brazil.
Jayme A Souza-Neto, School of Agricultural Sciences, São Paulo State University (UNESP), R. Quintino Bocaiuva, nº 470, Botucatu 05508-900, Brazil.
Heidge Fukumasu, Department of Veterinary Medicine, School of Animal Science and Food Engineering, University of Sao Paulo, Pirassununga, R. Quintino Bocaiuva, nº 470, Sao Paulo, Brazil.
Luiz Lehmann Coutinho, Centro de Genômica Funcional da ESALQ, University of São Paulo, R. Quintino Bocaiuva, nº 470, Piracicaba, SP, Brazil.
Rivaldo Venancio da Cunha, Bio-Manguinhos, Fundação Oswaldo Cruz, Rio de Janeiro, Av. Brasil, 4365, Rio de Janeiro 21040-360, Brazil.
Carla Freitas, Coordenacão Geral dos Laboratórios de Saúde Publica/Secretaria de Vigilância em Saúde, Ministério da Saúde (CGLAB/SVS-MS), Esplanada dos Ministérios - Bloco G - Edifício Sede - CEP, Brasília, Distrito Federal 70058-900, Brazil.
Carlos F Campelo de A e Melo, Organização Pan-Americana da Saúde/Organização Mundial da Saúde, Lote 19 - Avenida das Nações, SEN - Asa Norte, Brasília, Distrito Federal 70312-970, Brazil.
Wildo Navegantes de Araújo, Organização Pan-Americana da Saúde/Organização Mundial da Saúde, Lote 19 - Avenida das Nações, SEN - Asa Norte, Brasília, Distrito Federal 70312-970, Brazil.
Rodrigo Fabiano Do Carmo Said, Organização Pan-Americana da Saúde/Organização Mundial da Saúde, Lote 19 - Avenida das Nações, SEN - Asa Norte, Brasília, Distrito Federal 70312-970, Brazil.
Maria Almiron, Organização Pan-Americana da Saúde/Organização Mundial da Saúde, Lote 19 - Avenida das Nações, SEN - Asa Norte, Brasília, Distrito Federal 70312-970, Brazil.
Tulio de Oliveira, KwaZulu-Natal Research Innovation and Sequencing Platform (KRISP), School of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, 238 Mazisi Kunene Rd, Glenwood, Durban 4041, South Africa; Centre for Epidemic Response and Innovation (CERI), School of Data Science and Computational Thinking, Stellenbosch University, Banhoek Road & Joubert Street, Stellenbosch 7600, South Africa; Centre for the AIDS Programme of Research in South Africa (CAPRISA), Doris Duke Medical Research Institute, Nelson R Mandela School of Medicine, University of KwaZulu-Natal Private Bag X7, Congella 4013, South Africa; Department of Global Health, University of Washington, Schmitz Hall 1410 NE Campus Parkway Box 355852, Seattle, WA 98195-5852, USA.
Sandra Coccuzzo Sampaio, Butantan Institute, Avenida Doutor Vital Brasil, 1500 - Butantã, Sao Paulo - SP, Brazil.
Maria Carolina Elias, Department of Zoology, Peter Medawar Building, University of Oxford, 1a Mansfield Rd, Oxford OX1 3SZ, UK.
Dimas Tadeu Covas, Ribeirão Preto Medical School, Blood Center of Ribeirão Preto, University of São Paulo, R. Quintino Bocaiuva, nº 470 - Centro, Ribeirão Preto, SP 14015-160, Brazil; Butantan Institute, Avenida Doutor Vital Brasil, 1500 - Butantã, Sao Paulo - SP, Brazil.
Edward C Holmes, Sydney Institute for Infectious Diseases, School of Life and Environmental Sciences and School of Medical Sciences, University of Sydney, Sydney, NSW, Australia.
José Lourenço, Department of Zoology, Peter Medawar Building, University of Oxford, 1a Mansfield Rd, Oxford OX1 3SZ, UK; Biosystems and Integrative Sciences Institute (BioISI), Universidade de Lisboa, Campo Grande, Lisbon 1749-016, Portugal.
Simone Kashima, Ribeirão Preto Medical School, Blood Center of Ribeirão Preto, University of São Paulo, R. Quintino Bocaiuva, nº 470 - Centro, Ribeirão Preto, SP 14015-160, Brazil.
Luiz Carlos Junior de Alcantara, Laboratorio de Flavivirus, Fundação Oswaldo Cruz, Rio de Janeiro, Instituto Oswaldo Cruz /IOC /FIOCRUZ - Av. Brasil, 4365, Rio de Janeiro, Rio de Janeiro 21040-360, Brazil; Laboratorio de Genética Celular e Molecular, Instituto de Ciências Biologicas, Universidade Federal de Minas Gerais, Av. Pres. Antônio Carlos, 6627 - Pampulha, Belo Horizonte, Minas Gerais 31270-901, Brazil; Department of Science and Technology for Humans and the Environment, University of Campus Bio-Medico di Roma, Via Álvaro del Portillo, 21, Rome 00128, Italy.
Data availability
All SARS-CoV-2 whole-genome sequences produced by the National Network for Pandemic Alert of SARS-CoV-2 have been deposited in the GISAID sequence database and are publicly available subject to the terms and conditions of the GISAID database. The GISAID accession numbers of sequences used in the phylogenetic analysis are provided in Supplementary Table S1.
Code availability
All input files along with all resulting output files and scripts used in the present study will be made available upon request and publicly shared on GitHub at final publication.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
This work was supported in part through National Institutes of Health USA grant U01 AI151698 for the United World Arbovirus Research Network, the CRP-ICGEB RESEARCH GRANT 2020 Project CRP/BRA20-03, Contract CRP/20/03, the Oswaldo Cruz Foundation VPGDI-027-FIO-20-2-2-30, the Brazilian Ministry of Health (SCON2021-00180), the CNPq (426559/2018-5), and the Faperj E-26/202.930/2016. M.G. and L.C.J.A. are supported by Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro [E-26/202.248/2018(238504); E26/202.665/2019(247400)]. C.L.A. received funding from the European Union’s Horizon 2020 research and innovation programme—project EpiPose (No. 101003688). Research reported in this publication/article was also supported by the South African Department of Science and Innovation and the South African Medical Research Council under the BRICS JAF #2020/049. E.C.H. is funded by an Australian Research Council Laureate Fellowship (FL170100022). The content and findings reported/illustrated herein are the sole deduction, view, and responsibility of the researcher(s) and do not reflect the official position and sentiments of the funders.
Conflict of interest:
The authors declare no competing interests.
Author contributions
Molecular screening and produced SARS-CoV-2 genomic data: M.G., J.X., S.N.S., V.L.V., A.R.J.L., G.R., J.A.S.-N., H.F., L.L.C., S.C.S., M.C.E., and S.K.; collected samples and curated metadata: S.C.S., M.C.E., V.F., D.T.C., S.K., L.C.J.A., C.F., C.F.C.A.M., W.N.A., R.F.C.S., and M.A.; analyzed the data: M.G., V.F., E.W., H.T., E.J.S., and C.L.A.; helped with study design and data interpretation: M.G., V.F., E.W., H.T., E.J.S., C.L.A., E.C.H., J.L., and L.C.J.A.; wrote the initial manuscript, which was reviewed by all authors: M.G., C.L.A., E.C.H., J.L., and L.C.J.A.
References
- Aksamentov I. et al. (2021) ‘Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes’, Journal of Open Source Software, 6: 37–73. [Google Scholar]
- Arantes L. et al. (2021) ‘Emergence and Spread of the SARS-CoV-2 Variant of Concern Delta across Different Brazilian Regions’, MedRxiv.doi: 10.1101/2021.11.25.21266251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artic Network (2019), <https://artic.network/ncov-2019> accessed 20 Oct 2021.
- Campbell C. et al. (2021) ‘Increased Transmissibility and Global Spread of SARS-CoV-2 Variants of Concern as at June 2021’, Euro Surveillance, 24: 21–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candido D. et al. (2020) ‘Routes for COVID-19 Importation in Brazil’, Journal of Travel Medicine, 27: 21–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAHO/WHO | Pan American Health Organization (2021), COVID-19 Situation Reports. <https://www.paho.org/en/covid-19-situation-reports> accessed 19 Oct 2021.
- Davies N. G. et al. (2021) ‘Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 In England’, Science, 37: 2–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria N. et al. (2021) ‘Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil’, Science, 372: 815–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganyani T. et al. (2020) ‘Estimating the Generation Interval for Coronavirus Disease (COVID-19) Based on Symptom Onset Data, March 2020’, Euro Surveillance, 25: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanetti M. et al. (2021) ‘Genomic Epidemiology Reveals How Restriction Measures Shaped the SARS-CoV-2 Epidemic in Brazil’, MedRxiv.doi: 10.1101/2021.10.07.21264644. [DOI] [Google Scholar]
- Greaney A. J. et al. (2021) ‘Comprehensive Mapping of Mutations in the SARS-CoV-2 Receptor-binding Domain that Affect Recognition by Polyclonal Human Plasma Antibodies’, Cell Host & Microbe, 29: 463–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine F. et al. (2018) ‘Renewing Felsenstein’s Phylogenetic Bootstrap in the Era of Big Data’, Nature, 556: 452–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques A. D. et al. (2022) ‘SARS-CoV-2 Variants Associated with Vaccine Breakthrough in the Delaware Valley through Summer 2021’, mBio, 22: 8–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng B. et al. (2021) ‘Recurrent Emergence of SARS-CoV-2 Spike Deletion H69/V70 and Its Role in the Alpha Variant B.1.1.7’, Cell Reports, 35: 10.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merhi L. et al. (2021) ‘Replacement of the Alpha Variant of SARS-CoV-2 by the Delta Variant in Lebanon between April and June 2021’, MedRxiv.doi: 10.1101/2021.08.10.21261847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveca F. et al. (2021) ‘The SARS-CoV-2 Variant Delta Displaced the Variants Gamma and Gamma Plus in Amazonas, Brazil - SARS-CoV-2 Coronavirus / nCoV-2019 Genomic Epidemiology’, Virological. <https://virological.org/t/the-sars-cov-2-variant-delta-displaced-the-variants-gamma-and-gamma-plus-in-amazonas-brazil/765> accessed 24 Dec 2021. [Google Scholar]
- Nguyen L.-T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka C. K. et al. (2021) ‘Genomic Evidence of SARS-CoV-2 Reinfection Involving E484K Spike Mutation, Brazil’, Emerging Infectious Diseases Journal, 2: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen R. J. et al. (2021) ‘Trajectory of Growth of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Variants in Houston, Texas, January through May 2021, Based on 12,476 Genome Sequences’, The American Journal of Pathology, 191: 1754–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas D. et al. (2021) ‘Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization’, Nature, 596: 276–80. [DOI] [PubMed] [Google Scholar]
- Popescu A. et al. (2012) ‘Ape 3.0: New Tools for Distance-based Phylogenetics and Evolutionary Analysis in R’, Bioinformatics, 1: 15–36. [DOI] [PubMed] [Google Scholar]
- Rambaut A. et al. (2016) ‘Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen)’, VirusEvolution, 2: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Morales A. J. et al. (2020) ‘COVID-19 in Latin America: The Implications of the First Confirmed Case in Brazil’, Travel Medicine and Infectious Disease, 35: 16–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safari I., and Elahi E. (2021) ‘Evolution of the SARS-CoV-2 Genome and Emergence of Variants of Concern’, Archives of Virology, 10: 10–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagulenko P., Puller V., and Neher R. A. (2018) ‘TreeTime: Maximum-likelihood Phylodynamic Analysis’, VirusEvolution, 4: 1–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao K. et al. (2021) ‘The Biological and Clinical Significance of Emerging SARS-CoV-2 Variants’, Nature Reviews Genetics, 22: 757–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H. et al. (2021a) ‘Rapid Replacement of the Beta Variant by the Delta Variant in South Africa’, MedRxiv.doi: 10.1101/2021.09.23.21264018. [DOI] [Google Scholar]
- —— et al. (2021b) ‘Detection of a SARS-CoV-2 Variant of Concern in South Africa’, Nature, 592: 438–43. [DOI] [PubMed] [Google Scholar]
- Venables W. N., and Ripley B. D. (2021) Modern Applied Statistics with S, 4th edn. <https://www.stats.ox.ac.uk/pub/MASS4/> accessed 20 Oct 2021. [Google Scholar]
- Viana M. et al. (2022) ‘Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa’, Nature, 2: 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilsker M. et al. (2019) ‘Genome Detective: An Automated System for Virus Identification from High-Throughput Sequencing Data’, Bioinformatics, 35: 871–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO | World Health Organization (2022), <https://www.who.int/> accessed 20 Oct 2021.
- Wilkinson E. et al. (2021) ‘A Year of Genomic Surveillance Reveals How the SARS-CoV-2 Pandemic Unfolded in Africa’, Science, 1: 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier J. et al. (2020) ‘The Ongoing COVID-19 Epidemic in Minas Gerais, Brazil: Insights from Epidemiological Data and SARS-CoV-2 Whole Genome Sequencing’, Emerging Microbes & Infections, 9: 1824–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All SARS-CoV-2 whole-genome sequences produced by the National Network for Pandemic Alert of SARS-CoV-2 have been deposited in the GISAID sequence database and are publicly available subject to the terms and conditions of the GISAID database. The GISAID accession numbers of sequences used in the phylogenetic analysis are provided in Supplementary Table S1.