

Abstract
The small GTPase ADP-ribosylation factor 6 (ARF6) mediates chemokine (C-C motif) ligand 18 (CCL18)-induced activation of breast cancer (BC) metastasis through its downstream effector AMAP1. However, the molecular mechanisms underlying CCL18 up-regulating ARF6 remain largely unclear. Here, microRNAs (miRNAs) that target ARF6 were predicted and selected in high metastatic BC cells treated with CCL18. Next, we assessed the role of exosomal miR-760 in vitro and in vivo. We further analyzed the expression of ARF6, AMAP1, and phosphorylated (p)-AMAP1 in tumor and adjacent normal tissues. We first observed that CCL18 increased the expression of ARF6 and p-AMAP1 and activated the Src/phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway. ARF6 knockdown significantly impaired CCL18-induced malignant cellular behaviors and the Src/PI3K/Akt signaling pathway. Next, ARF6 was confirmed as a target gene of miR-760 in exosomes derived from CCL18-stimulated high metastatic BC cells. Moreover, recipient MCF-7 cells could effectively uptake these miR-760-rich exosomes that significantly promoted proliferation, tumor growth in vivo, migration, invasion, and chemoresistance by activating ARF6-mediated Src/PI3K/Akt signaling and the epithelial-mesenchymal transition (EMT) pathway. Together, our results support that exosomal miR-760 secreted by CCL18-stimulated high metastatic BC cells promoted the malignant behaviors in low metastatic BC cells by up-regulating the ARF6-mediated Src/PI3K/Akt signaling pathway.
Key words: breast cancer, exosome, CCL18, miR-760, ARF6/Src/PI3K/Akt signaling pathway
Graphical abstract
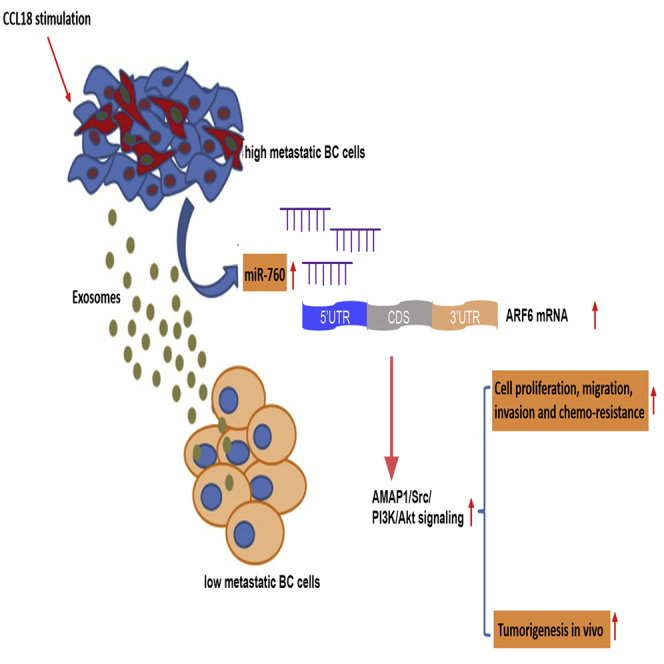
Exosomal miR-760 secreted by CCL18-stimulated high metastatic breast cancer cells promoted the malignant behaviors in low metastatic breast cancer cells by up-regulating the ARF6-mediated Src/PI3K/Akt signaling pathway.
Introduction
Breast cancer (BC) represents the most common malignant tumor and one of the leading causes of tumor-related mortality worldwide.1 The standard therapeutic methods have elongated the survival of BC patients. However, recurrence and acquired therapy resistance are common in BC, which is mainly ascribed to distal metastasis of highly invasive BC cells.2,3 Therefore, it is urgent to identify promising molecular targets associated with BC progression to improve BC prognosis.
The tumor microenvironment plays a vital role in cancer progression.4 Chemokine (C-C motif) ligand 18 (CCL18), produced mainly by tumor-associated macrophages (TAMs), has been reported to be a crucial regulator of tumor development and metastasis, such as in ovarian cancer,5 gastric cancer,6 and esophageal squamous cell carcinoma.7 In BC, CCL18 significantly promotes tumor progression and metastasis by inducing epithelial-mesenchymal transition (EMT) via the phosphatidylinositol 3-kinase (PI3K)/Akt/glycogen synthase kinase-3beta (GSK3β)/Snail signaling pathway.8 Besides, CCL18-mediated down-regulation of miR-98 and miR-27b promotes BC metastasis.9 Moreover, we have previously demonstrated that CCL18 induced AMAP1 phosphorylation and translocation to activate nuclear factor κB (NF-κB) and promote BC metastasis.10 AMAP1 is a phospholipid-dependent Arf GTPase-activating protein and could be phosphorylated by Src.11 AMAP1 is a downstream effector of the small GTPase ADP-ribosylation factor 6 (ARF6), which is a master regulator of membrane trafficking.12 As illustrated by Sabe et al.13 and Hashimoto et al.,14 ARF6 and AMAP1 are frequently overexpressed in BC and constitute a central pathway to induce BC invasion and metastasis. However, the upstream mechanisms of ARF6/AMAP1 under CCL18 stimulation in BC pathogenesis remain elusive.
MicroRNAs (miRNAs) are a class of endogenous, small non-coding RNAs 18–25 nt in length, which could stabilize target mRNA or activate translation via binding to the 5′UTR of target mRNAs.15, 16, 17 Exosomes are membrane vesicle-like structures sized 20–100 nm and particularly enriched in tumor microenvironments, which are key mediators of cell-cell communication via carrying proteins, mRNAs, and miRNAs in bodily fluids.18,19 It has been reported that exosomes deliver specific miRNAs to tumor cells to control their biological function. For example, exosome-mediated transfer of miR-193a-3p, miR-210-3p, and miR-5100 could promote invasion of lung cancer cells by activating STAT3 signaling-induced EMT.20 Exosomal miR-222-3p enhanced the proliferation, gemcitabine resistance, migration, invasion, and anti-anoikis of parental sensitive cancer cells by directly targeting the promoter of SOCS3.21 Moreover, cancer-associated fibroblasts and cancer cells can secrete exosomal miRNAs to affect each other. miRNA dysregulation in cancer-associated fibroblasts is considered to be associated with a secretory phenotype change, tumor invasion, tumor migration and metastasis, drug resistance, and poor prognosis.22 However, exosomal miRNAs in the BC microenvironment need further investigation.
In the present study, we first investigated the expression of ARF6/AMAP1 and Src/PI3K/Akt signaling pathways and the biological function of ARF6 under CCL18 stimulation. Via bioinformatics analysis, we predicted miRNAs that target ARF6 and explored whether certain CCL18-derived exosomal miRNAs play an important role in BC progression by regulating the ARF6/Src/PI3K/Akt signaling pathway. Our findings will provide new insights to the treatment of BC.
Results
CCL18 increased the expression of ARF6 and phosphorylated (p)-AMAP1, as well as activated the Src/PI3K/Akt signaling pathway
Our previous work showed that TAM-secreted CCL18 induced the phosphorylation of AMAP1. With the evidence that AMAP1 is an effector of ARF6 in invasion by binding to GTP-ARF6 through its zinc-finger domain,23,24 we thus wonder whether TAM-derived cytokines could affect the expression of ARF6. Here, CCL18 and CCL20 (two members from the same chemokine family) were used to stimulate MCF-7 cells. As shown in Figure 1A, CCL18, but not CCL20, significantly elevated the expression of ARF6 mRNA. Although the expression of AMAP1 mRNA was not obviously changed on stimulation with CCL18 or CCL20 (Figure 1B, p > 0.05), the phosphorylation of AMAP1 was increased by CCL18 stimulation (Figure 1C). In addition, both western blot analysis (Figure 1C) and immunofluorescence (Figure 1D) demonstrated that ARF6 protein expression was remarkably up-regulated after CCL18 treatment compared with PBS or CCL20. Moreover, we investigated the effect of CCL18 on the Src/PI3K/Akt pathway. As illustrated in Figure 1E, much more phosphorylated Src, PI3K, and Akt proteins were exhibited by CCL18 stimulation compared with PBS and CCL20 treatment.
Figure 1.

CCL18 increased the expression of ARF6 and p-AMAP1, as well as activated the Src/PI3K/Akt pathway
MCF-7 cells were treated with PBS, CCL18, or CCL20. qRT-PCR analysis was performed to determine the mRNA expression of ARF6 (A) and AMAP1 (B) in MCF-7 cells. Data were expressed as mean ± SD. ∗∗p < 0.01, compared with PBS or CCL20. (C) The protein expression levels of ARF6, AMAP1, and p-AMAP1 were measured using western blot analysis in MCF-7 cells. (D) Immunofluorescence staining of ARF6 in MCF-7 cells in PBS, CCL20, and CCL18 groups. Scale bars: 20 μm. (E) Western blotting of MCF-7 cells treated with PBS, CCL18, or CCL20 for the expression of the phosphorylated and total proteins of Src, PI3K, and AKT.
ARF6 knockdown significantly impaired CCL18-induced malignant cellular behaviors
Because ARF6 was up-regulated in CCL18-stimulated MCF-7 cells, we next investigated the biological function of ARF6 in vitro by loss-of-function assays. CCK-8 assay (Figure 2A) and Edu staining (Figures 2B and 2D) first confirmed cell proliferation that significantly enhanced after CCL18 treatment was obviously attenuated after ARF6 knockdown. Colony formation assay further showed that CCL18-induced increased colonies were significantly reduced after ARF6 knockdown (Figures 2C and 2E). According to the above results, we selected si-ARF6-2 for the subsequent experiments. Next, we evaluated cell motility by wound healing assay and found that knockdown of ARF6 strongly decreased cell motility, which was induced by CCL18 (Figure 2F). Similarly, transwell assay indicated that CCL18-induced cell migration and invasion were significantly suppressed by ARF6 knockdown (Figure 2G).
Figure 2.

ARF6 knockdown significantly impaired CCL18-induced proliferation, migration, and invasion in MCF-7 cells
MCF-7 cells were transfected with si-ARF6-1, si-ARF6-2, si-ARF6-3, or si-NC, followed by CCL18 treatment. (A) Cell growth was assessed by CCK-8 assay. (B and D) Cell proliferation was analyzed by Edu staining. Scale bars: 100 μm. (C and E) Colony formation assay was conducted in MCF-7 cells in different groups. (F) Cell motility in MCF-7 cells by wound healing assay. Scale bars: 100 μm. (G) Cell migration and invasion ability in MCF-7 cells by transwell assay. Scale bars: 100 μm. Data were expressed as mean ± SD. ∗∗p < 0.01, compared with CCL18.
Furthermore, we assessed the effects of ARF6 knockdown on drug sensitivity in CCL18-stimulated MCF-7 cells. CCK-8 assay showed that CCL18-treated cells displayed higher cell viability than control cells, which was alleviated after ARF6 knockdown under the same concentration of doxorubicin (Figure 3A), paclitaxel (Figure 3B), or tamoxifen (Figure 3C). Next, we investigated the effect of ARF6 on cell apoptosis and found that knockdown of ARF6 remarkably ameliorated the low apoptosis status of CCL18-stimulated MCF-7 cells after treatment with 10 μg/mL doxorubicin (Figure 3D), 100 nM paclitaxel (Figure 3E), or 1 μM tamoxifen (Figure 3F). These findings indicated that knockdown of ARF6 improved drug sensitivity of CCL18-stimulated MCF-7 cells.
Figure 3.

ARF6 knockdown could improve drug sensitivity of CCL18-stimulated MCF-7 cells
CCK-8 assay was carried out to analyze cell viability of MCF-7 cells in control, CCL18, CCL18 + si-NC, or CCL18 + si-ARF6-2 groups after treatment with increasing concentrations of (A) doxorubicin (0, 5, 10, and 20 μg/mL), (B) paclitaxel (0, 50, 100, and 200 nM), and (C) tamoxifen (0, 0.5, 1.0, and 5 μM), respectively. Cell apoptotic rate was determined using flow cytometry in MCF-7 cells in control, CCL18, CCL18 + si-NC, or CCL18 + si-ARF6-2 groups after treatment with 10 μg/mL doxorubicin (D), 100 nM paclitaxel (E), or 1 μM tamoxifen (F). Data were expressed as mean ± SD. ∗∗p < 0.01, compared with CCL18.
ARF6 knockdown significantly impaired the CCL18-induced Src/PI3K/Akt signaling pathway
qRT-PCR (Figure 4A) and western blot (Figure 4C) confirmed that si-ARF6-2 efficiently knocked down ARF6 expression in CCL18-stimulated MCF-7 cells. Although AMAP1 mRNA did not significantly change (Figure 4B, p > 0.05), its phosphorylation was obviously down-regulated after ARF6 knockdown (Figure 4C). In addition, the significantly down-regulated E-cadherin and up-regulated vimentin, p-Src, p-PI3K, and p-AKT in CCL18-stimulated MCF-7 cells were all notably reversed after ARF6 knockdown (Figures 4C and 4D). Collectively, these data indicated that the CCL18-activated Src/PI3K/Akt signaling pathway was attenuated after ARF6 knockdown.
Figure 4.

ARF6 knockdown significantly impaired the CCL18-induced Src/PI3K/Akt signaling pathway
CCL18-stimulated MCF-7 cells were transfected with si-NC or si-ARF6-2. qRT-PCR was performed to measure the mRNA expression of ARF6 (A) and AMAP1 (B). Data were expressed as mean ± SD. ∗∗p < 0.01, compared with CCL18. (C) Western blot analysis was conducted to analyze the protein expression of ARF6, AMAP1, p-AMAP1, E-cadherin, and vimentin. (D) Western blot analysis was conducted to analyze the phosphorylated and total proteins of Src, PI3K, and AKT. (E) CCL18-treated MCF-7 cells were treated with Src activator MCB-613, PI3K activator 740 Y-P, or solvent control (SC), followed by si-ARF6-2 transfection. Western blot analysis was conducted to analyze the phosphorylated and total proteins of Src, PI3K, and AKT. (F and G) Representative images of migratory and invasive cells (scale bars: 100 μm) (F) and quantification of migratory and invasive cells in different groups were shown (G). Data were expressed as mean ± SD. ∗∗p < 0.01, compared with CCL18 + si-ARF6-2.
To confirm whether ARF6 directly regulated the Src/PI3K/Akt pathway, CCL18-treated MCF-7 cells were transfected by si-ARF6-2 and then treated with Src activator MCB-613, PI3K activator 740 Y-P, or solvent control (SC). As shown in Figure 4E, MCB-613 increased the expression of p-Src, p-PI3K, and p-AKT in si-ARF6-2-transfected cells. Meanwhile, MCB-613 and 740 Y-P obviously reversed the suppressive effect of ARF6 knockdown on CCL18-treated MCF-7 cells. Transwell assay showed either MCB-613 or 740 Y-P significantly increased migratory or invasive cells after ARF6 knockdown (Figures 4F and 4G). The above results indicated that activation of the Src/PI3K/Akt signaling pathway reversed the effects of ARF6 knockdown on cell migration and invasion.
Identification of miRNAs that targeted ARF6 in exosomes derived from CCL18-stimulated high metastatic BC cells
To investigate the upstream regulator of ARF6 and the Src/PI3K/Akt pathway in CCL18-induced cells, we used bioinformatics analysis to search potential miRNAs that target ARF6, and we identified three miRNAs, miR-760, miR-1246, and miR-1290. Considering exosomes are the main transporters of miRNAs, we then isolated exosomes from CCL18-stimulated high metastatic BC cell lines (MBA-MB-231, MBA-MB-453, and MBA-MB-468). The exosomes isolated from cell culture supernatants have a diameter of approximately 100 nm by electron microscopy. Interestingly, CCL18-stimulated cells generated more exosomes than control cells (Figure 5A). NanoSight particle tracking analysis showed sharp peaks at 50–100 nm in derived exosomes, which indicated extracted nanovesicles were exosomes (Figure 5B). Next, we detected the levels of miR-760, miR-1246, and miR-1290 in high metastatic BC-derived exosomes. As shown in Figure 5C, CCL18 stimulation significantly increased the levels of miR-760, miR-1246, and miR-1290 in exosomes. Notably, the increasing level of miR-760 reached the highest in MDA-MB-468 cells. We thus selected miR-760 and MDA-MB-468 cells for further analysis. Western blot analysis further confirmed that exosome markers, including CD63, ALIX, and TSG101, were highly expressed in derived exosomes (Figure 5D). The presence of exosomal markers and absence of Calnexin, which would be detected only in total cellular lysates and not in exosomes, verified the purity of our exosome preparations.
Figure 5.

Identification of miRNAs that targeted ARF6 in exosomes derived from CCL18-stimulated high metastatic BC cells
(A) Representative electron microscopic images of exosomes in three high metastatic BC cell-derived exosomes. Scale bars: 100 nm. (B) Particle size distribution of exosomes was measured by NanoSight particle tracking analysis. (C) qRT-PCR analysis of miR-760, miR-1246, and miR-1290 levels in three high metastatic BC cell-derived exosomes. (D) Western blot analysis showing the presence of exosome markers CD63, ALIX, and TSG101 and absence of endoplasmic reticulum-specific protein Calnexin in high metastatic MDA-MB-468-derived exosomes.
CCL18-induced exosomal miR-760 promoted proliferation, migration, invasion, and chemoresistance by activating the ARF6-mediated Src/PI3K/Akt signaling pathway
As mentioned above, miR-760 was remarkably up-regulated in CCL18-stimulated exosomes derived from MDA-MB-468 cells (Exo-CCL18). We thus speculated that CCL18 regulated the development of BC through exosomal miR-760. To validate this hypothesis, we co-cultured PKH67-labeled exosomes (Exo-CCL18) with MCF-7 cells. We used flow cytometry to measure fluorescence at different time points and found the uptake of PKH67 + exosomes reached 95% at 6 h (Figure 6A). At the same time, MCF-7 cells were transfected with miR-760 mimics or inhibitor. Subsequently, Edu staining (Figure 6B), colony formation (Figure 6C), transwell assay (Figures 6D and 6E), and apoptosis assay (Figure 7A) were employed to examine cell proliferation, migration, invasion, and apoptosis. The results indicated that MCF-7 cells co-cultured with Exo-CCL18 or miR-760 mimics presented accelerated proliferation and increased number of migratory/invasive cells but did not change the apoptotic rate compared with the mock group. On the contrary, miR-760 inhibitor transfection significantly suppressed cell proliferation, migration, and invasion and promoted cell apoptosis. In addition, we analyzed the effects of miR-760 on cell drug sensitivity. Following co-culture with Exo-CCL18 or miR-760 mimics, cell apoptosis was significantly decreased in response to 10 μg/mL doxorubicin (Figure 7B), 100 nM paclitaxel (Figure 7C), or 1 μM tamoxifen (Figure 7D). However, miR-760 knockdown enhanced cell drug sensitivity in response to the above three drugs.
Figure 6.

CCL18-induced exosomal miR-760 promoted proliferation, migration, and invasion in MCF-7 cells
(A) Flow cytometry was applied to analyze the fluorescence intensity in MCF-7 cells after co-culture with PKH67-labeled exosomes derived from CCL18-simulated MDA-MB-468 cells at 10 min, 1 h, 6 h, and 24 h, respectively. (B–E) Edu staining (scale bars: 100 μm) (B), colony formation (C), and transwell assay (D and E) (scale bars: 100 μm) were performed to analyze cell proliferation, migration, and invasion in MCF-7 cells co-cultured with Exo-CCL18 or miR-760 mimics/inhibitor. ∗∗p < 0.01, compared with mock.
Figure 7.

CCL18-induced exosomal miR-760 promoted chemoresistance in MCF-7 cells
(A) Flow cytometry was performed to analyze cell apoptosis in MCF-7 cells co-cultured with Exo-CCL18 or miR-760 mimics/inhibitor transfection. The drug sensitivity was analyzed in MCF-7 cells co-cultured with Exo-CCL18 or miR-760 mimics/inhibitor transfection in response to doxorubicin (B), paclitaxel (C), or tamoxifen (D). ∗∗p < 0.01, compared with mock.
The ARF6 5′UTR contains putative binding sites for miR-760 (Figure 8A). Schematic diagrams of ARF6 5′UTR-LUC and P1-P5 LUC plasmids were shown in Figure 8B. Mechanically, luciferase reporter assay confirmed that ARF6 was a target gene of miR-760 (Figure 8C). The levels of ARF6 mRNA (Figure 8D) and protein (Figure 8F) were significantly increased after co-culture with Exo-CCL18 or miR-760 mimics, but notably decreased after miR-760 inhibitor transfection, indicating ARF6 was positively regulated by miR-760. We also found that not AMAP1 mRNA (Figure 8E, p > 0.05), but the phosphorylation of AMAP1, was up-regulated after co-culture with Exo-CCL18 or miR-760 mimics and down-regulated after miR-760 inhibitor transfection (Figure 8F). In accord with altered cell migration and invasion, E-cadherin was reduced, while vimentin was elevated after co-culture with Exo-CCL18 or miR-760 mimics, which was reversed by miR-760 inhibitor transfection (Figure 8G). What is more, we found the Src/PI3K/Akt signaling pathway activated after co-culture with Exo-CCL18 or miR-760 mimics and suppressed after miR-760 inhibitor transfection (Figure 8H). The aforementioned results suggested that miR-760, carried by Exo-CCL18, can promote proliferation, migration, invasion, and chemoresistance by targeting ARF6 and activating the Src/PI3K/Akt signaling pathway.
Figure 8.

CCL18-induced exosomal miR-760 activated the ARF6-mediated Src/PI3K/Akt signaling pathway
(A) Putative miR-760 binding sites within the ARF6 gene 5′UTR. (B) Schematic diagram of ARF61 5′ UTR-LUC and P1-P5 LUC plasmid. (C) The association between miR-760 and ARF6 was validated using luciferase reporter assay. ∗∗p < 0.01 compared with WT + miR-mimics. qRT-PCR was applied to determine the mRNA expression of ARF6 (D) and AMAP1 (E) in MCF-7 cells co-cultured with Exo-CCL18 or miR-760 mimics/inhibitor transfection. ∗p < 0.05, ∗∗p < 0.01, compared with mock. Western blot analysis was employed to detect the protein expression of (F) ARF6, AMAP1, and p-AMAP1, (G) EMT markers (E-cadherin and vimentin), as well as (H) the phosphorylated and total proteins of Src, PI3K, and AKT.
CCL18-induced exosomal miR-760 promoted in vivo tumorigenesis by up-regulating the ARF6-mediated Src/PI3K/Akt signaling pathway
In addition, we assessed the role of miR-760 in tumor growth in vivo by constructing a xenograft mouse model. As shown in Figure 9A, the tumor size was larger in Exo-CCL18 and miR-760 mimics groups. Also, knockdown of ARF6 obviously reduced the tumor size. Moreover, the time-dependent analysis showed that miR-760 overexpression elevated, while ARF6 knockdown markedly suppressed, the tumor volume (Figure 9B). Moreover, the expression levels of ARF6, p-AMAP1, Vimentin, p-Src, p-PI3K, and p-AKT were up-regulated, whereas E-cadherin was down-regulated in Exo-CCL18 and miR-760 mimics groups. In addition, knockdown of ARF6 suppressed the expression of ARF6, p-AMAP1, Vimentin, p-Src, p-PI3K, and p-AKT but promoted E-cadherin expression (Figure 9C). The above data suggested that CCL18-induced exosomal miR-760 promoted in vivo tumorigenesis by promoting the ARF6-mediated Src/PI3K/Akt signaling pathway.
Figure 9.

CCL18-induced exosomal miR-760 promoted in vivo tumorigenesis by up-regulating the ARF6-mediated Src/PI3K/Akt signaling pathway
(A) Representative images of xenograft tumors excised from nude mice 28 days after subcutaneous inoculation of MCF-7 cells from five groups (Mock, Exo, miR-760 mimics, CCL18, and CCL18 + shARF6, n = 5 in each group). (B) Tumor volumes were measured at 0, 3, 7, 14, and 28 days, respectively. ∗∗p < 0.01, compared with mock; ##p < 0.01, compared with CCL18. (C) Representative immunohistochemistry images of ARF6, p-AMAP1, E-cadherin, Vimentin, p-Src, p-PI3K, and p-AKT were shown. Scale bars: 50 μm. ∗p < 0.05, ∗∗p < 0.01, compared with mock; ##p < 0.01, compared with CCL18.
Analysis of miR-760, ARF6, and AMAP1 expression in BC patient tissues
We next examined miR-760 level in BC patient tumor and adjacent normal tissues. Fluorescence in situ hybridization (FISH) demonstrated increased miR-760 level in tumor, which mainly expressed in the cytoplasm (Figure 10A). Moreover, we found exosomal miR-760 level was significantly elevated in tumor patient peripheral blood specimens (Figure 10B). In addition, immunohistochemistry (IHC) showed that the expression of ARF6 and p-AMAP1, but not AMAP1, was elevated in tumor tissues (Figure 10C). Similarly, immunofluorescence assay also showed enhanced ARF6 and p-AMAP1 expression in tumor tissues (Figure 10D).
Figure 10.

Analysis of miR-760, ARF6, and AMAP1 expression in BC patient tissues
(A) FISH was used to locate and reflect the expression of miR-760 in BC tumor and adjacent normal tissues. The red fluorescence represented miR-760, and blue fluorescence represented the nucleus. Scale bars: 50 μm. (B) The relative expression of exosomal miR-760 was determined in peripheral blood specimens obtained from 16 tumor patients and healthy controls. (C) IHC staining of ARF6, AMAP1, and p-AMAP1 in BC tumor and adjacent normal tissues. (D) Immunofluorescence of ARF6, AMAP1, and p-AMAP1 expression in BC tumor and adjacent normal tissues. Scale bars: 50 μm.
Discussion
TAMs, a crucial component in the tumor microenvironment, can secrete a range of growth factors, cytokines, and chemokines to initiate and promote tumor progression.25 Among these secretions, CCL18 is an important cytokine derived from M2-type TAMs that were involved in diverse malignant processes, including BC lymph node metastasis and worse prognosis.26 Here, we showed that CCL18 significantly enhanced the proliferation, migration, invasion, and EMT and decreased apoptosis and multidrug sensitivity of BC cells. Similarly, CCL18 promotes osteosarcoma proliferation and metastasis via the EP300/UCA1/Wnt/β-catenin pathway.27 In gallbladder cancer, CCL18 could activate PI3K/AKT signaling and lead to cell migration, invasion, and EMT.28 Moreover, Src phosphorylation is initiated by CCL18, causing BC metastasis via PITPNM3.29 Based on these facts, we speculated that CCL18 might promote BC malignant behaviors through activating Src/PI3K/AKT signaling, as reflected by up-regulated p-Src, p-PI3K, and p-AKT.
Interestingly, we found the expression levels of ARF6 and p-AMAP1 were significantly increased in CCL18-stimulated MCF-7 cells and BC patient tumor tissues. Moreover, knockdown of ARF6 remarkably suppressed CCL18-induced malignant cellular behaviors and the Src/PI3K/Akt signaling pathway. We previously showed that AMAP1 not only strongly predicted poor BC prognosis but also mediated CCL18-induced activation of NF-κB to promote cell migration, invasion, and EMT.10 Our present data further indicated that ARF6 knockdown suppressed the phosphorylation of AMAP1. In fact, the regulatory relation between ARF6 and AMAP1 has been reported in recent studies: frequently overexpressed ARF6 and its downstream effector AMAP1 could promote tumor metastasis and drug resistance.13,30 Moreover, ARF6 regulates CCL18-elicited BC cell migration via the acetyltransferase PCAF-mediated acetylation.31 The aforementioned evidence suggested that CCL18 activated cell proliferation, migration, invasion, EMT, and drug resistance through up-regulating ARF6/p-AMAP1 expression in BC cells.
To better understand the physiological role of CCL18 in BC, we further explored the upstream miRNAs that target ARF6. As a result, three miRNAs (miR-760, miR-1246, and miR-1290) were predicted. A recent study has confirmed exosomes as mediators of intercellular communication that can carry and deliver miRNAs to recipient cells.32 As demonstrated by Li et al.,33 exosome-mediated transfer of miR-1246 induced a tumor-promoting phenotype, including increased cell proliferation, migration, and drug resistance by directly targeting CCNG2 in BC. Exosomal miR-223 and miR-501-3p facilitates the development of epithelial ovarian cancer34 and pancreatic ductal adenocarcinoma,35 respectively. In addition, miR-76036,37 and miR-129038,39 have been reported to associate with BC poor clinical outcomes and malignant behaviors. The oncogenic role of miR-760 has also been reported in ovarian cancer to facilitate cell proliferation by targeting PHLPP2.40 Here, we found the levels of miR-760, miR-1246, and miR-1290 were significantly increased in CCL18-stimulated exosomes from high metastatic BC cells. More importantly, further analysis showed that high metastatic BC cells deliver miR-760 through exosomes to low metastatic BC cells, thereby promoting BC malignant behaviors through activating the ARF6-mediated Src/PI3K/Akt signaling pathway.
In summary, our findings indicated that CCL18 up-regulated miR-760 levels in exosomes derived from high metastatic BC cells, and uptake of these miR-760-rich exosomes in low metastatic BC cells could effectively promote malignant progression, including chemoresistance phenotype. Mechanically, we identified a new exosomal miR-760/ARF6/Src/PI3K/Akt signaling pathway promoting BC proliferation, migration, invasion, and chemoresistance.
Materials and methods
MCF-7 cell treatment and transfection
BC cell line MCF-7 was grown in MEM medium (SH30024.01B; Hyclone) supplemented with 10% FBS (10099-141; Gibco) in a humidified atmosphere with 5% CO2 at 37°C. Three different small interfering RNAs (siRNAs) targeting ARF6 (see Table S1), miR-760 mimics, inhibitor, and corresponding controls were synthesized by RiboBio (Guangzhou, China). For chemokine treatment, MCF-7 cells were exposed to 20 ng/mL CCL18 or CCL20 (both from R&D, Shanghai, China) for 1 h. For ARF6 function, MCF-7 cells were transfected with si-ARF6-1, si-ARF6-2, si-ARF6-3, or si-NC, followed by CCL18 treatment. For Src/PI3K/Akt signaling, CCL18-treated MCF-7 cells were treated with Src activator MCB-613 (8 μΜ), PI3K activator 740 Y-P (10 μΜ), or SC for 2 h, followed by si-ARF6-2 transfection. For miR-760 function, MCF-7 cells were transfected with miR-760 mimics or inhibitor. All transfection protocols were conducted according to the instructions provided by Lipofectamine 3000 (Invitrogen, USA) for 48 h.
Isolation and identification of exosomes derived from high metastatic BC cells
Prior to CCL18 treatment, three high metastatic BC cell lines (MBA-MB-231, MBA-MB-453, and MBA-MB-468) were cultured in serum-free medium for 12 h. Conditioned media were harvested 2 h after CCL18 treatment, and the supernatant was collected by centrifuging at 3,000 × g for 30 min. Then, the supernatant was incubated with ExoQuick exosome precipitation solution overnight at 4°C and gently aspirated by centrifuging the ExoQuick mixture at 1,500 × g for 30 min. After further centrifugation at 1,500 × g for 5 min, exosomes were isolated from the remaining ExoQuick solution and re-suspended in PBS. The morphology of exosomes derived from CCL18-untreated and CCL18-treated high metastatic BC cells was observed by transmission electron microscopy. The exosomes surface markers (CD63, ALIX, and TSG101) and endoplasmic reticulum-specific protein Calnexin were detected by western blot analysis. The corresponding size distribution of exosomes was measured using the NanoSight NS300 instrument (Malvern Instruments, UK).
Uptake of PKH67-labeled exosomes
MDA-MB-468-derived exosomes (2 μg) were collected and labeled with PKH67 Fluorescent Cell Linker kits (Sigma-Aldrich, St. Louis, MO, USA) as per the manufacturer’s protocol. MCF-7 cells were incubated with PKH67-labeled vesicles for 10 min, 1 h, 6 h, or 24 h, respectively. Next, cells were washed with PBS and fixed in 2% paraformaldehyde. The percentage of cells positive for PKH67 + exosomes was analyzed by flow cytometry (BD Biosciences, San Diego, CA).
Quantitative real-time PCR analysis
Total RNA was isolated using TRIzol reagent (Invitrogen) or a miRNeasy Mini Kit I (QIAGEN, Hilden, Germany), and reverse transcription was performed by PrimeScript RT kit (Applied Biosystems) according to the manufacturer’s instructions. qRT-PCR was undertaken using SYBR Premix Ex Taq II (TaKaRa, Shiga, Japan) on Applied Biosystems 7500 Fast Real-Time PCR System. The sequences of the primers used in this study were designed and synthesized by Life Technologies (Table S2). The relative gene expression levels were calculated with the 2−ΔΔCT formula with GAPDH or U6 as the internal reference.
Western blot analysis
Total protein was extracted using radioimmunoprecipitation assay (RIPA) buffer (Sigma, USA), and the corresponding protein concentration was determined with Pierce BCA Protein assay kit. Protein samples were subjected to 12% SDS-PAGE and transferred onto a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with 5% skim milk powder for 2 h at room temperature. After being rinsed with PBS twice, the membrane was incubated with specific primary antibodies overnight at 4°C, followed by incubation with appropriate horseradish peroxidase (HRP)-linked secondary antibody for 2 h at room temperature. Finally, the protein bands were visualized using enhanced chemiluminescence (ECL; Millipore, Bedford, MA, USA), and relative grayscale quantification was conducted by ImageJ software.
Drug resistance experiment
Doxorubicin, paclitaxel, and tamoxifen were purchased from Sigma Chemical (St. Louis, MO, USA). MCF-7 cells in different groups were plated in 96-well plates at an initial density of 3,000 cells per well and treated with increasing concentrations of doxorubicin (0, 5, 10, and 20 μg/mL), paclitaxel (0, 50, 100, and 200 nM), and tamoxifen (0, 0.5, 1.0, and 5 μM), respectively. Afterward, CCK-8 assay and apoptosis assay were performed to assess the drug resistance of MCF-7 cells under different conditions.
Colony formation assay
MCF-7 cells at an initial density of 1,000 cells per well were seeded in six-well plates after specific transfection or treatment and cultured in complete medium for 2 weeks. Cell colonies were fixed with 4% paraformaldehyde for 30 min and stained with 0.5% crystal violet for 30 min at room temperature. The number of colonies (≥50 cells/colony) was counted and averaged in three independent experiments.
CCK-8 assay
For cell growth curve assay, MCF-7 cells at a density of 3,000 cells per well were seeded in 96-well plates and cultured in complete medium overnight. At 24, 48, and 72 h, cells in each well were incubated for 2 h with CCK-8 solution (Dojindo, Japan) at 37 °C. Finally, the optical density (OD) value at 450 nm was measured using a microplate reader. For a drug-resistant experiment, MCF-7 cells in different groups were treated with doxorubicin, paclitaxel, and tamoxifen at an increasing concentration. After 48 h, cell viability was analyzed.
Apoptosis assessment
Cell apoptosis was determined using the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis kit (KeyGen Biotechnology, Nanjing, China) according to the manufacturer’s instructions. In brief, MCF-7 cells in different groups were seeded onto six-well plates with 40,000 cells per well and cultured overnight. The next day, cells were trypsinized, washed with ice-cold PBS, and fixed with 75% ethanol. The fixed cells were incubated with 5 μL Annexin V-FITC and 5 μL PI for 48 h. All samples were harvested, and the apoptotic rate was assessed by flow cytometry (BD Biosciences, USA). This experiment was performed three times independently.
5-Ethynyl-2′deoxyuridine staining
MCF-7 cells in different groups were incubated with medium containing Edu solution (RiboBio, Guangzhou, China) for 2 h and rinsed with PBS. Afterward, cells were fixed with 4% paraformaldehyde for 30 min and rinsed with PBS containing 0.5% Triton X-100. The cells were finally incubated with Hoechst 33342 staining solution for 30 min at room temperature in darkness. The stained cells were observed under a fluorescence microscope, and cells with a red nucleus were regarded as positive cells.
Wound healing assay
Approximately 2 × 106 MCF-7 cells were seeded into each well of six-well plates and cultured for 24 h. Then a straight scratch was carefully created with a 200-μL sterilized pipette tip. The wound area was examined at 0 and 48 h, respectively, under a light microscope (Leica, Germany) and analyzed by ImageJ software.
Transwell assay
Transwell assay was performed with MCF-7 cells using a 24-well transwell chamber (Corning, USA) pre-coated with Matrigel for invasion or without Matrigel (BD Bioscience, USA) for migration assay. In brief, MCF-7 cells re-suspended in serum-free medium were seeded onto the upper chamber, while the medium containing 10% FBS was added into the lower chamber. After 24-h incubation, the cells that adhered to the lower chamber were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet for 20 min. The number of migratory or invasive cells was quantified in five randomly selected fields under a light microscope.
Luciferase reporter assay
Based on the predicted binding sequences of miR-760 in the 5′UTR of ARF6 mRNA, ARF6 wild-type (WT) and five different mutant luciferase reporter vectors (MUT-P1-P5; see Table S3) targeting the miR-760 binding site were constructed using PmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega, Madison, WI, USA). Subsequently, these vectors were transfected into MCF-7 cells together with miR-760 mimics or miR-NC by Lipofectamine 3000 (Invitrogen, USA). After 48 h, luciferase activities were determined using the dual-luciferase reporter system (Promega, USA) with Renilla luciferase activity as a standardized control.
Patient samples
Primary tumor tissue and adjacent normal tissue samples were obtained from BC patients after surgical resection at the Sixth Affiliated Hospital of Sun Yat-sen University. For histological examinations, all tissues were fixed in 10% buffered formalin and embedded in paraffin for sectioning. In addition, peripheral blood specimens were obtained from tumor patients and healthy controls, which were stored in 1-mL aliquots at −80°C and were thawed immediately prior to exosome isolation. All patients signed written informed consents, and this study was performed in accordance with the Declaration of Helsinki principles under the approval provided by the Ethics Committee of the Sixth Affiliated Hospital of Sun Yat-sen University.
Fluorescence in situ hybridization
The paraffin-embedded block was cut into 4-μm pathology slides, and FISH for miR-760 was performed on these frozen slices. In brief, the slices were dewaxed in xylene, rehydrated in citrate buffer, and digested with proteinase K digestion. Next, slices were hydrated with a graded series of alcohol and air dried. Afterward, the slides were incubated with hybridization buffer supplemented with denatured 5′ CY3-labeled locked nucleic acid (LNA) probes directed against miR-760 (GenePharma, China) at 37°C overnight. After being washed twice with saline sodium citrate buffer, the slices were counterstained with DAPI for 20 min and visualized through a fluorescence microscope (Olympus, Tokyo, Japan).
IHC
IHC was performed according to standard procedures as previously described.41 In brief, the paraffin-embedded tissues were cut into 5 μm thick and dried out for 2 h at 65°C. Samples were deparaffinized in xylene and hydrated through a graded series of alcohol. After antigen retrieval, the sections were blocked with 3% hydrogen peroxide for 15 min and incubated with the primary antibodies against ARF6, AMAP1, and p-AMAP1 overnight at 4°C, followed by incubation with universal secondary antibody for 15 min at room temperature. Then, the immunoreactivity was visualized using 3, 3-diaminobenzidine tetrahydrochloride (DAB), and staining intensities were evaluated by two independent double-blinded investigators.
Immunofluorescence
MCF-7 cells were inoculated on sterilized coverslips and fixed with 4% formaldehyde for 15 min. Then, cells were permeabilized with 0.2% Triton X-100, blocked with 3% BSA, and incubated with ARF6 primary antibody (1:500) overnight at 4°C. For BC tissues, the sections (5 μm thick) were blocked with 10% goat serum for 1 h and incubated overnight with rabbit anti-ARF6, AMAP1, and p-AMAP1 at 4°C. Following incubation with the corresponding secondary antibody at 4°C, the sections were counterstained with DAPI (Sigma-Aldrich). Stained images were acquired under a fluorescence microscope (Olympus, Tokyo, Japan) and analyzed with ImageJ software.
Tumor xenograft model
Five-week-old female BALB/c nude mice weighing 25 ± 4 g were purchased from Shanghai Laboratory Animals Center of Chinese Academy of Sciences and fed under the standard environment with free access to food or water. Subsequently, five groups of MCF-7 cells were prepared: mock cells (5 μg Exo-NC), exosome-treated cells (5 μg Exo-CCL18), miR-760 mimics-transfected cells, CCL18-treated cells (20 ng/mL), and CCL18 + shARF6-transfected cells. Next, 2 × 107 MCF-7 cells from different groups were suspended in 1 mL physiological saline solution and subcutaneously inoculated into the lower right flank of each mouse (n = 5 in each group) to establish tumors. After that, Exo-NC (5 μg), Exo-CCL18 (5 μg), miR-760 mimics, or CCL18 (20 ng/mL) was injected every other day until the mice were sacrificed. At 0, 3, 7, 14, and 28 days, tumor volumes in each group were determined by digital caliper and calculated by the formula: length × width2/2. After 28 days, mice were sacrificed, and excised tumor tissues were collected for IHC analysis of ARF6, p-AMAP1, E-cadherin, Vimentin, p-Src, p-PI3K, and p-AKT expression following the previously described method.42 All the in vivo procedures were approved by the Institutional Committee for Animal Research and in accordance with the Care and Use of Laboratory Animals of the Sixth Affiliated Hospital of Sun Yat-sen University.
Statistical analysis
Statistical analyses were performed using the SPSS 21.0 software (IBM, Armonk, NY, USA). Quantitative data were expressed as mean ± standard deviation (SD) of three independent experiments. One-way analysis of variance (ANOVA), followed by Tukey’s post hoc test, was applied to evaluate differences among multiple groups, and statistical significance was accepted when p < 0.05.
Acknowledgments
This work was supported by the Science and Technology Planning Project of Guangzhou (805275295029) and the Natural Science Foundation of Guangdong Province (2020A1515010425).
Author contributions
H.L.: study design/planning and funds collection; X.H., S.L., and F.Q.: data collection/entry and preparation of manuscript; Z.L. and X.F.: data analysis/statistics and interpretation; Q.L. and X.Z.: figure preparation; C.W.: literature analysis/search; all authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.03.004.
Supplemental information
References
- 1.DeSantis C.E., Ma J., Goding Sauer A., Newman L.A., Jemal A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J. Clin. 2017;67:439–448. doi: 10.3322/caac.21412. [DOI] [PubMed] [Google Scholar]
- 2.Cavazzoni A., Bonelli M.A., Fumarola C., La Monica S., Airoud K., Bertoni R., Alfieri R.R., Galetti M., Tramonti S., Galvani E., et al. Overcoming acquired resistance to letrozole by targeting the PI3K/AKT/mTOR pathway in breast cancer cell clones. Cancer Lett. 2012;323:77–87. doi: 10.1016/j.canlet.2012.03.034. [DOI] [PubMed] [Google Scholar]
- 3.Karlsson E., Veenstra C., Emin S., Dutta C., Perez-Tenorio G., Nordenskjold B., Fornander T., Stal O. Loss of protein tyrosine phosphatase, non-receptor type 2 is associated with activation of AKT and tamoxifen resistance in breast cancer. Breast Cancer Res. Treat. 2015;153:31–40. doi: 10.1007/s10549-015-3516-y. [DOI] [PubMed] [Google Scholar]
- 4.Cheng C.J., Bahal R., Babar I.A., Pincus Z., Barrera F., Liu C., Svoronos A., Braddock D.T., Glazer P.M., Engelman D.M., et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518:107–110. doi: 10.1038/nature13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zohny S.F., Fayed S.T. Clinical utility of circulating matrix metalloproteinase-7 (MMP-7), CC chemokine ligand 18 (CCL18) and CC chemokine ligand 11 (CCL11) as markers for diagnosis of epithelial ovarian cancer. Med. Oncol. 2010;27:1246–1253. doi: 10.1007/s12032-009-9366-x. [DOI] [PubMed] [Google Scholar]
- 6.Leung S.Y., Yuen S.T., Chu K.-M., Mathy J.A., Li R., Chan A.S.Y., Law S., Wong J., Chen X., So S. Expression profiling identifies chemokine (C-C motif) ligand 18 as an independent prognostic indicator in gastric cancer. Gastroenterology. 2004;127(2):457–469. doi: 10.1053/j.gastro.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 7.Wang W., Wu D., He X., Hu X., Hu C., Shen Z., Lin J., Pan Z., He Z., Lin H., et al. CCL18-induced HOTAIR upregulation promotes malignant progression in esophageal squamous cell carcinoma through the miR-130a-5p-ZEB1 axis. Cancer Lett. 2019;460:18–28. doi: 10.1016/j.canlet.2019.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Zhao C., Zheng S., Yan Z., Deng Z., Wang R., Zhang B. CCL18 promotes the invasion and metastasis of breast cancer through Annexin A2. Oncol. Rep. 2020;43:571–580. doi: 10.3892/or.2019.7426. [DOI] [PubMed] [Google Scholar]
- 9.Lin X., Chen L., Yao Y., Zhao R., Cui X., Chen J., Hou K., Zhang M., Su F., Chen J., et al. CCL18-mediated down-regulation of miR98 and miR27b promotes breast cancer metastasis. Oncotarget. 2015;6:20485–20499. doi: 10.18632/oncotarget.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Zhang D., Yu J., Liu H., Chen Z., Zhong H., Wan Y. CCL18-dependent translocation of AMAP1 is critical for epithelial to mesenchymal transition in breast cancer: AMAP1 mediates CCL18-induced epithelial to mesenchymal transition. J. Cell Physiol. 2017;233:3207–3217. doi: 10.1002/jcp.26164. [DOI] [PubMed] [Google Scholar]
- 11.Brown M.T., Andrade J., Radhakrishna H., Donaldson J.G., Cooper J.A., Randazzo P.A. ASAP1, a phospholipid-dependent arf GTPase-activating protein that associates with and is phosphorylated by Src. Mol. Cell. Biol. 1998;18:7038–7051. doi: 10.1128/mcb.18.12.7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Otsuka Y., Oikawa T., Yoshino H., Hashimoto S., Handa H., Yamamoto H., Hashimoto A., Sabe H. Frequent overexpression of AMAP1, an Arf6 effector in cell invasion, is characteristic of the MMTV-PyMT rather than the MMTV-Neu human breast cancer model. Cell Commun. signal. 2018;16:1. doi: 10.1186/s12964-017-0212-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabe H., Hashimoto S., Morishige M., Ogawa E., Hashimoto A., Nam J.M., Miura K., Yano H., Onodera Y. The EGFR-GEP100-Arf6-AMAP1 signaling pathway specific to breast cancer invasion and metastasis. Traffic. 2009;10:982–993. doi: 10.1111/j.1600-0854.2009.00917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashimoto A., Hashimoto S., Ando R., Noda K., Ogawa E., Kotani H., Hirose M., Menju T., Morishige M., Manabe T., et al. GEP100-Arf6-AMAP1-cortactin pathway frequently used in cancer invasion is activated by VEGFR2 to promote angiogenesis. PloS One. 2011;6:e23359. doi: 10.1371/journal.pone.0023359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ørom U.A., Nielsen F.C., Lund A.H. MicroRNA-10a binds the 5'UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell. 2008;30:460–471. doi: 10.1016/j.molcel.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Shimakami T., Yamane D., Jangra R.K., Kempf B.J., Spaniel C., Barton D.J., Lemon S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. U S A. 2012;109:941–946. doi: 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu M., Roth A., Yu M., Morris R., Bersani F., Rivera M.N., Lu J., Shioda T., Vasudevan S., Ramaswamy S., et al. The IGF2 intronic miR-483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes Dev. 2013;27:2543–2548. doi: 10.1101/gad.224170.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pluchino S., Smith J.A. Explicating exosomes: reclassifying the rising stars of intercellular communication. Cell. 2019;177:225–227. doi: 10.1016/j.cell.2019.03.020. [DOI] [PubMed] [Google Scholar]
- 19.Thery C., Zitvogel L., Amigorena S. Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X., Sai B., Wang F., Wang L., Wang Y., Zheng L., Li G., Tang J., Xiang J. Hypoxic BMSC-derived exosomal miRNAs promote metastasis of lung cancer cells via STAT3-induced EMT. Mol. Cancer. 2019;18:40. doi: 10.1186/s12943-019-0959-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei F., Ma C., Zhou T., Dong X., Luo Q., Geng L., Ding L., Zhang Y., Zhang L., Li N., et al. Exosomes derived from gemcitabine-resistant cells transfer malignant phenotypic traits via delivery of miRNA-222-3p. Mol. Cancer. 2017;16:132. doi: 10.1186/s12943-017-0694-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang F., Ning Z., Ma L., Liu W., Shao C., Shu Y., Shen H. Exosomal miRNAs and miRNA dysregulation in cancer-associated fibroblasts. Mol. Cancer. 2017;16:148. doi: 10.1186/s12943-017-0718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Onodera Y., Hashimoto S., Hashimoto A., Morishige M., Mazaki Y., Yamada A., Ogawa E., Adachi M., Sakurai T., Manabe T., et al. Expression of AMAP1, an ArfGAP, provides novel targets to inhibit breast cancer invasive activities. EMBO J. 2005;24:963–973. doi: 10.1038/sj.emboj.7600588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashimoto S., Hashimoto A., Yamada A., Onodera Y., Sabe H. Assays and properties of the ArfGAPs, AMAP1 and AMAP2, in Arf6 function. Methods Enzymol. 2005;404:216–231. doi: 10.1016/S0076-6879(05)04021-8. [DOI] [PubMed] [Google Scholar]
- 25.Pollard J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 26.Chen J., Yao Y., Gong C., Yu F., Su S., Chen J., Liu B., Deng H., Wang F., Lin L., et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell. 2011;19:541–555. doi: 10.1016/j.ccr.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su Y., Zhou Y., Sun Y.J., Wang Y.L., Yin J.Y., Huang Y.J., Zhang J.J., He A.N., Han K., Zhang H.Z., et al. Macrophage-derived CCL18 promotes osteosarcoma proliferation and migration by upregulating the expression of UCA1. J. Mol. Med. 2019;97:49–61. doi: 10.1007/s00109-018-1711-0. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Z., Peng Y., Wu X., Meng S., Yu W., Zhao J., Zhang H., Wang J., Li W. CCL18 secreted from M2 macrophages promotes migration and invasion via the PI3K/Akt pathway in gallbladder cancer. Cell Oncol. 2019;42:81–92. doi: 10.1007/s13402-018-0410-8. [DOI] [PubMed] [Google Scholar]
- 29.Li H.Y., Cui X.Y., Wu W., Yu F.Y., Yao H.R., Liu Q., Song E.W., Chen J.Q. Pyk2 and Src mediate signaling to CCL18-induced breast cancer metastasis. J. Cell. Biochem. 2014;115:596–603. doi: 10.1002/jcb.24697. [DOI] [PubMed] [Google Scholar]
- 30.Hashimoto A., Oikawa T., Hashimoto S., Sugino H., Yoshikawa A., Otsuka Y., Handa H., Onodera Y., Nam J.M., Oneyama C., et al. P53- and mevalonate pathway-driven malignancies require Arf6 for metastasis and drug resistance. J. Cell Biol. 2016;213:81–95. doi: 10.1083/jcb.201510002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song X., Liu W., Yuan X., Jiang J., Wang W., Mullen M., Zhao X., Zhang Y., Liu F., Du S., et al. Acetylation of ACAP4 regulates CCL18-elicited breast cancer cell migration and invasion. J. Mol. Cell Biol. 2018;10:559–572. doi: 10.1093/jmcb/mjy058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J.J., Lötvall J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 33.Li X.J., Ren Z.J., Tang J.H., Yu Q. Exosomal MicroRNA MiR-1246 promotes cell proliferation, invasion and drug resistance by targeting CCNG2 in breast cancer. Cell Physiol. Biochem. 2017;44:1741–1748. doi: 10.1159/000485780. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y., Zhang X., Lin J., Chen Y., Qiao Y., Guo S., Yang Y., Zhu G., Pan Q., Wang J., et al. CCT3 acts upstream of YAP and TFCP2 as a potential target and tumour biomarker in liver cancer. Cell Death Dis. 2019;10:644. doi: 10.1038/s41419-019-1894-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin Z., Ma T., Huang B., Lin L., Zhou Y., Yan J., Zou Y., Chen S. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-beta signaling pathway. J. Exp. Clin. Cancer Res. 2019;38:310. doi: 10.1186/s13046-019-1313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu S.H., Wang C.H., Huang Z.J., Liu F., Xu C.W., Li X.L., Chen G.Q. miR-760 mediates chemoresistance through inhibition of epithelial mesenchymal transition in breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2016;20:5002–5008. [PubMed] [Google Scholar]
- 37.Han M.L., Wang F., Gu Y.T., Pei X.H., Ge X., Guo G.C., Li L., Duan X., Zhu M.Z., Wang Y.M. MicroR-760 suppresses cancer stem cell subpopulation and breast cancer cell proliferation and metastasis: by down-regulating NANOG. Biomed. Pharmacother. 2016;80:304–310. doi: 10.1016/j.biopha.2016.03.024. [DOI] [PubMed] [Google Scholar]
- 38.Endo Y., Toyama T., Takahashi S., Yoshimoto N., Iwasa M., Asano T., Fujii Y., Yamashita H. miR-1290 and its potential targets are associated with characteristics of estrogen receptor alpha-positive breast cancer. Endocr. relat. Cancer. 2013;20:91–102. doi: 10.1530/ERC-12-0207. [DOI] [PubMed] [Google Scholar]
- 39.Endo Y., Yamashita H., Takahashi S., Sato S., Yoshimoto N., Asano T., Hato Y., Dong Y., Fujii Y., Toyama T. Immunohistochemical determination of the miR-1290 target arylamine N-acetyltransferase 1 (NAT1) as a prognostic biomarker in breast cancer. BMC Cancer. 2014;14:990. doi: 10.1186/1471-2407-14-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao Y., Deng Y., Liu J., Ye Z., You Z., Yao S., He S. MiR-760 overexpression promotes proliferation in ovarian cancer by downregulation of PHLPP2 expression. Gynecol. Oncol. 2016;143:655–663. doi: 10.1016/j.ygyno.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J., Yang Y., Zhang Z., He Y., Liu Z., Yu Y., Wu S., Cai B., Feng Y. Gankyrin plays an essential role in estrogen-driven and GPR30-mediated endometrial carcinoma cell proliferation via the PTEN/PI3K/AKT signaling pathway. Cancer Lett. 2013;339:279–287. doi: 10.1016/j.canlet.2012.10.037. [DOI] [PubMed] [Google Scholar]
- 42.Wang J., Yu S., Cui L., Wang W., Li J., Wang K., Lao X. Role of SMC1A overexpression as a predictor of poor prognosis in late stage colorectal cancer. BMC Cancer. 2015;15:90. doi: 10.1186/s12885-015-1085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.