

Summary
Hepatic lipid accumulation is closely associated with nonalcoholic fatty liver disease (NAFLD). Adipose-triglyceride-lipase (ATGL) regulates triglyceride hydrolysis and maintains energy homeostasis in hepatocytes. Identifying key factors in the regulation of ATGL will help tackle hepatic lipid accumulation and related metabolic diseases. Herein, we demonstrate that syntaxin11 (STX11), a member of the SNARE family, generally expressed in immune cells, mediates lipid metabolism by binding to ATGL and inhibiting lipid droplet degradation and lipid autophagy in hepatocytes. Our data show that the C-terminal of STX11 and the patatin domain-containing segment of ATGL have direct physical interactions. Thus, STX11 overexpression prevents spatial translocation of ATGL onto LDs by recruitment of ATGL to the ER. Conversely, STX11 deficiency in hepatocytes promotes lipid hydrolysis, and the ATGL-SIRT1 signaling pathway enhances lipophagy. Overall, this study uncovered that the regulation of lipolysis and lipophagy is achieved by STX11 through the attenuation of ATGL action in hepatocytes.
Subject areas: Biological sciences, Physiology, Molecular biology, Endocrinology, Cell biology
Graphical abstract
Highlights
-
•
STX11 inhibits lipid droplet degradation via ATGL in hepatocytes
-
•
Interaction of ATGL and STX11 affects trafficking of ATGL
-
•
STX11 suppresses lipophagy in association with ATGL and SIRT1
Biological sciences; Physiology; Molecular biology; Endocrinology; Cell biology
Introduction
Lipids such as glycerol-lipids, sterol-lipids, glycerol-phospholipids, sphingolipids, and bile salts in the form of fatty acids are essential entrepots for energy within cells (Fahy et al., 2009). Excessive amounts of fatty acids resulting from oxidation are converted to triglycerides (TGs) in the endoplasmic reticulum (ER) and stored in the form of highly dynamic organelles termed lipid droplets (LDs) (Gross and Silver, 2014). The dynamic regulation of LDs is determined by their growth and consumption because of lipolysis or autophagy/lipophagy and de novo synthesis of TGs, respectively. In addition, the surfaces of LDs contain signaling proteins that control lipid trafficking and flux (Olzmann and Carvalho, 2019; Schulze et al., 2019; Yang et al., 2012). Lipids are found in somatic adipose and liver tissue while their metabolism is mainly regulated in the liver (Nguyen et al., 2008). Aberrant lipid metabolism in the liver can cause hepatic steatosis, where the accumulation of neutral lipids, usually triacylglycerol (TAG) occurs in LDs, leading to their accumulation in the liver. This LD accumulation is a major feature of alcoholic fatty liver disease and nonalcoholic fatty liver disease (NAFLD), the second most common form of liver disease (Kohjima et al., 2007; Perlemuter et al., 2007).
Currently, two mechanisms exist that are responsible for the breakdown of stored TAG in the form of LDs, lipolysis and autophagy (Mashek, 2020). The classical pathway of lipolysis is mediated by three enzymes: adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MGL). TAGs are catabolized by these three enzymes into a final end product comprising one glycerol and three fatty acid (FAs) residues (Jenkins et al., 2004; Villena et al., 2004; Zimmermann et al., 2004). Free FAs released by lipolysis are transported to the mitochondria as a substrate for β-oxidation or as signaling molecules for other cellular processes. These can be re-esterified into TAG and then stored until needed (Bolsoni-Lopes and Alonso-Vale, 2015; Ong et al., 2011). In this pathway, ATGL is considered as the rate-limiting enzyme (Cerk et al., 2018). In addition to the classical pathway of lipolysis, during starvation, a second route of autophagy-mediated hydrolysis of LDs can occur. Autophagy is a lysosomal-dependent catabolic process referred to as lipophagy when involving lipid metabolism (Ward et al., 2016). During lipophagy, the LDs are transported into lysosomes or the mitochondria for degradation and oxidation (Singh et al., 2009). Recent studies have revealed that ATGL can mediate lipophagy in hepatocytes through the SIRT1 signaling pathway (Sathyanarayan et al., 2017). Multiple proteins regulate the activity of this ATGL in the classical pathway; G0/G1 switch protein 2 (G0S2) and Hypoxia-inducible lipid droplet-associated protein (HILPDA) represent potent inhibitors of ATGL that can influence hepatic energy metabolism (Padmanabha Das et al., 2018; Sugaya and Satoh, 2017; Yang et al., 2010). However, lipid droplet-binding protein, comparative gene identification 58 (CGI-58), and pigment epithelium-derived factor (PEDF) can activate enzymes involved in lipolysis (Lass et al., 2006; Notari et al., 2006). These proteins regulate LD hydrolysis indirectly through interaction with ATGL. Although the hydrolase activity of ATGL is regulated by multiple proteins, the trafficking and spatial distribution of ATGL within the cytoplasm are poorly understood.
Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins are common cellular fusogenic factors that transfer intracellular cargo and promote membrane fusion (Hong and Lev, 2014). Syntaxin11 (STX11) is a member of the SNARE protein family, which is predominantly expressed in the immune system, and its mutation can lead to familial hemophagocytic lymphohistiocytosis type 4 (FHL4) (Dabrazhynetskaya et al., 2012; Kogl et al., 2013). Strikingly, STX11 deficiency has been reported to cause weight decrease in mice (Kogl et al., 2013). However, its role in lipid regulation, especially for hepatic lipid metabolism, has never been established. In this study, we investigated the function of STX11 in hepatic lipid metabolism in human hepatocyte THLE-2 cells and found that STX11 directly interacts with ATGL and can regulate hepatic lipid metabolism by influencing the spatial distribution of ATGL, thereby facilitating LD lipolysis and lipophagy.
Results
STX11 mediates lipid metabolism in the liver
The liver is the primary site of energy metabolism and to initiate functional studies, we treated C57BL/6J mice with HFD for 90 days to determine the effect of HFD on liver STX11 protein levels. H&E staining and IHC staining illustrated that the LDs and expression of STX11 in the liver of mice fed with HFD were increased when compared to those mice on the control diet (Figures 1A and 1B). Immunoblotting also illustrated that perilipin2, a lipid droplet-specific protein marker, and STX11 were both increased in the HFD group (Figure 1C). Therefore, we performed endogenous expression of STX11 in different cell types. Immunoblotting demonstrated an abundant expression of STX11 in human hepatocyte THLE-2 cells and HeLa cells and, to a lesser degree, in HEK293T cells (Figures 1D and 1E). High expression levels of STX11 were also confirmed in THLE-2 and HeLa cells treated with oleic acid (OA 400 μM) for 24 h (Figures S1A and S1B). This suggests that TG promotes the expression of STX11 in vivo and in vitro; therefore, STX11-Myc overexpression vector was used to transiently express STX11 in 24 h OA (400 μM)-treated cells. When we looked at immunofluorescence in THLE-2 cells, we found that LD content in the STX11 overexpression group was significantly higher than that seen in the control group (Figures 1F and 1G), confirming that STX11 participates in lipid metabolism in hepatocytes. These same results were also observed in the HeLa cells (Figure 1G). In addition, STX11 was knocked down in THLE-2 and HeLa cells (Figures 1H and 1I), the content of LD significantly reduced when compared to the control group (Figure 1J), but this phenotype caused by the knockdown of STX11 could be rescued by the co-expression of STX11-Myc, which is consistent with the above results. These results demonstrated that STX11 could influence lipid metabolism in hepatocytes.
Figure 1.

Involvement of STX11 in lipid metabolism
(A) H&E staining and IHC staining of liver sections of C57BL/6J mice fed with a control diet or HFD for 90 days. IHC staining with the anti-STX11 antibody. LDs (black arrows).
(B) Quantitative analysis of STX11 staining and LDs.
(C) Endogenous STX11 protein in the liver from mice treated with a control diet or HFD for 90 days was measured by immunoblotting. Perilipin2, a lipid droplet-specific protein marker. β-tubulin was used as the loading control.
(D) Cellular distribution of STX11 protein assessed by immunoblotting analysis. 1, HepG2; 2, THLE-2; 3, HeLa; 4, HK2; 5, HEK293T. β-tubulin was used as the loading control.
(E) Relative expression of STX11 in different cells.
(F and G) Overexpression of STX11 affects LD accumulation in cells. STX11-Myc proteins immunofluorescence staining with anti-Myc (red) antibodies was performed to reveal LD changes in THLE-2 and HeLa cells overexpressing STX11-Myc. Cells were incubated under normal growth conditions with 400 μM OA complexed to albumin for 24 h. Lipid droplets were stained with BODIPY 493/503 fluorescent dye. Quantification of LDs under the fluorescence microscope (G) (n = 6 cells for Ctrl; n = 6 cells for STX11 overexpression).
(H and I) Knock down of STX11 or simultaneous overexpression of STX11-Myc in THLE-2 and HeLa cells treated as described in (F and G). STX11 proteins measured by immunoblotting using STX11 antibodies (I). β-actin was used as the loading control.
(J) Relative intensity of LDs under the fluorescence microscope. Data are shown as mean ± SEM (n = 3). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, t test.
STX11 affects lipid metabolism through specific interaction with ATGL
ATGL is extensively expressed in various cell lines (Figures S1C and S1D). ATGL is the rate-limiting enzyme of lipid catabolism and is extremely important for the stability of the metabolic homeostasis of fats and the liver. We investigated the relationship between STX11 and ATGL to explore the function of STX11 in lipid metabolism. Our immunoprecipitation analysis revealed that STX11 specifically interacted with ATGL by physical attachment (Figure 2A) because syntaxin18 (STX18), which is a member of the same family as STX11, does not interact with ATGL (Figures 2A, 2B, and S2C). STX11-Myc and ATGL-HA were co-expressed in HeLa or THLE-2 cells and were labeled with different immunofluorescent dyes (Figures 2C–2E, and S2A). The results revealed that STX11 and ATGL had indeed co-localized. Furthermore, a similar result was seen using endogenous immunofluorescence analysis (Figures S3A and S3B). In the NAFLD mouse model and the THLE-2 cells, utilization of endogenous immunoprecipitation analysis also provided evidence of the physical interaction between STX11 and ATGL in vivo and in vitro (Figures 2F and S3C). To confirm a modulatory role for STX11 during lipid metabolism was via ATGL, we transiently co-expressed STX11-Myc and ATGL-HA vectors in HeLa cells and then starved the cells for 4 h after OA (400 μM) treatment for 24 h. The immunofluorescence data indicated that the LD content was significantly higher than in cells expressing ATGL alone when STX11 and ATGL were co-expressed (Figures 2G and 2H). This result suggests that STX11 could inhibit LD hydrolysis, and this was mediated by its effect on ATGL.
Figure 2.

STX11 affects lipid metabolism through interaction with ATGL
(A) STX11 specifically interacts with ATGL. HeLa cells were co-transfected with STX11-Myc or STX18-Myc and ATGL-HA vectors. STX11-Myc and STX18-Myc proteins were immunoprecipitated using anti-Myc antibodies. STX11 and ATGL proteins in both the immunoprecipitates and lysates were detected by immunoblotting with Myc and HA antibodies.
(B) No co-localization of STX18 and ATGL. HeLa cells were co-transfected with ATGL-HA and STX18-Myc. ATGL-HA protein immunofluorescence staining were performed by using anti-HA (red) and STX18-Myc proteins were stained with anti-Myc (green) antibodies.
(C) Co-localization of STX11 and ATGL in cells. HeLa cells transfected with ATGL-HA and STX11-Myc were incubated under normal growth conditions. Immunofluorescence staining with anti-HA and anti-Myc antibodies was performed to reveal co-localization between ATGL-HA (red) and STX11-Myc (green).
(D and E) Co-localization analysis of STX11 and ATGL using Plot Profile and Co-localization Finder from ImageJ software. Curve coincidence illustrates that the two proteins had co-localized in theory (D) and Pearson’s R = 0.926 (E) (R from 0 to 1.0, 0 indicates no significant correlation and 1.0 indicates complete negative correlation).
(F) STX11 interacts with ATGL in vivo. STX11 proteins were immunoprecipitated using STX11 antibodies in NAFLD mouse liver extracts. STX11 and ATGL proteins were detected by immunoblotting using STX11 and ATGL antibodies.
(G and H) Presence of STX11 inhibits lipolysis mediated by ATGL. STX11-Myc and ATGL-HA co-expression in HeLa cells after treatment with OA (400 μM) for 24 h, and starvation for 4 h. 1: Cell co-expressing STX11 and ATGL; 2: Cell expressing ATGL alone. STX11-Myc and ATGL-HA proteins were subjected to immunostaining with anti-Myc (violet) and anti-HA (red) antibodies. Lipid droplets were stained with BODIPY 493/503 fluorescent dye. Quantification of the number of LDs under the fluorescence microscope (H) (n = 6 cells for Ctrl; STX11 and ATGL co-expression and ATGL overexpression). Data are shown as mean ± SEM (n = 3). ∗∗p < 0.01, t test.
The C-terminal of STX11 directly interacts with the PT domain of ATGL
To further identify the specific region of the interaction between ATGL and STX11, we constructed three mutants, ATGL-Myc, ATGL-PT-Myc, and ATGL-ΔPT-Myc, based on the structural features of ATGL (Figure 3A). All ATGL mutants were independently co-expressed with STX11 in HEK293T cells, and their interactions were observed by immunoprecipitation. The results indicated that STX11 interacts with the full-length version of the ATGL and PT domain, but no interaction with ΔPT was observed (Figure 3B). This result illustrates that STX11 interacts with the PT domain of ATGL. To further determine the segment of STX11 which interacts with the PT domain, we used the structural characteristics of STX11 and found that the C-terminal sequence of STX11 is rich in cysteine, which is of great significance for the function of STX11. Next, we constructed two mutants, STX11-HA and STX11ΔC-HA (Figure 3C). These mutants were coprecipitated with the PT domain in HEK293T cells. The results showed that STX11 does indeed interact with the PT domain; however, STX11ΔC showed no interaction with the PT domain (Figures 3D and S3D). These findings demonstrated that the cysteine-rich region at the C-terminal of STX11 interacts with the PT domain of ATGL. In addition, ATGL-PT and STX11 were co-expressed in HeLa cells and a co-localization of the PT domain and STX11 in these cells was observed by immunofluorescence (Figures 3E and 3F). From the results of this study, we have concluded that the interaction between STX11 and ATGL occurs at the C-terminal region of STX11 and the PT domain of ATGL.
Figure 3.

STX11 directly interacts with ATGL
(A) Schematic representation of the structure of murine ATGL, its mutants, and the PT domain in ATGL is indicated as the gray region.
(B) STX11 interacts with the PT domain of ATGL. HEK293T cells were co-transfected with STX11-HA together with different ATGL constructs (ATGL-Myc + STX11-HA, ATGL-PT-Myc + STX11-HA, ATGL-ΔPT-Myc + STX11-HA). STX11-HA proteins were immunoprecipitated with anti-HA antibodies. STX11 and ATGL proteins in immunoprecipitated lysates were detected by immunoblotting with HA and Myc antibodies.
(C) Schematic representation of the structure of murine STX11, its mutants, and the Habc domain and the SNARE motif in STX11 are indicated as the gray region.
(D) C-terminal of STX11 interacts with the PT domain of ATGL. HEK293T cells were co-transfected with STX11ΔC-HA and ATGL-PT-Myc. Other groups and immunoprecipitation and immunoblotting analyses were performed as described in (B).
(E) STX11 co-localizes with PT domain of ATGL. Immunofluorescence staining of STX11-HA and ATGL-PT-Myc in HeLa cells was performed using anti-HA (green) and anti-Myc (red) antibodies. Intensity traces from the original images were plotted using ImageJ software. Curve coincidence illustrates that the two proteins have co-localized in theory.
(F) HeLa cells were co-transfected with STX11ΔC-HA and ATGL-Myc. STX11ΔC-HA proteins immunofluorescence staining was performed by using anti-HA (green) and ATGL-Myc proteins were stained with anti-Myc (red) antibodies.
(G and H) GST pulldown analysis between recombinant STX11-GST, ATGL-MBP-His, and ATGL-MBP-PT-His.
To investigate the nature of this interaction between STX11 and ATGL, we purified the fusion proteins of ATGL-MBP, ATGL-PT-MBP, and STX11-GST using an E. coli expression system, and we analyzed the interaction by GST pull-down experiments. The results revealed a direct interaction between STX11 and ATGL as shown in Figures 3G and 3H.
STX11 regulates ATGL-mediated hydrolysis
THLE-2 cells were treated with OA (400 μM) for 24 h to determine the effect of STX11 on ATGL hydrolase activity. STX11-Myc and ATGL-HA constructs were expressed in THLE-2 cells (Figure 4A). After starvation for 2 h, the cellular TG levels and the content of glycerol in the medium were determined. We found that the TG content in the STX11-overexpressing cells was significantly increased when compared to the controls, whereas the glycerol content in the culture medium was significantly decreased. In addition, when compared to the group overexpressing ATGL alone, the TG content in the group overexpressing both STX11 and ATGL was significantly increased, whereas the glycerol content was dramatically decreased (Figures 4B and 4C), demonstrating that STX11 affected the activity of ATGL hydrolase in the cells. To further confirm our above conclusion, we knocked down STX11 in THLE-2 cells and observed the effect on lipid hydrolysis (Figure 4D). The data from this experiment suggested that in cells having STX11 knocked down but with overexpression of ATGL, the content of TG in the cells was significantly decreased, but the glycerol in the medium was significantly increased when compared to the group overexpressing ATGL alone (Figures 4E and 4F). This result indicates that knockdown of STX11 can promote the hydrolysis of TG, and the results from STX11 knockdown and overexpression studies were consistent. Significantly, glycerol release of lipolysis in the liver was enhanced in hepatic STX11 knockdown mice (Figures S2F and S2G). Thus, we have shown that STX11 mediates lipid hydrolysis via ATGL.
Figure 4.

Knockdown and overexpression of STX11 affects ATGL mediated lipid hydrolysis
(A) THLE-2 cell clones for the positive or negative expression of STX11-Myc were treated with ATGL-HA or control plasmid. Protein expression was determined by immunoblotting using anti-HA and anti-Myc antibodies.
(B and C) Overexpression of STX11 inhibits TG hydrolysis and glycerol release. Cells were incubated in 400 μM of OA for 24 h, after which they were starved for 2 h, intracellular TG content and Glycerol content of the medium were determined as described in STAR Methods.
(D) THLE-2 cell clones with both positive or negative expression of ATGL-HA were treated with STX11 siRNA (siSTX11) or control mismatched siRNA. Protein expression was determined by immunoblotting using anti-STX11 and anti-HA antibodies.
(E and F) Knockdown of STX11 promotes hydrolysis of TG and glycerol release. Cellular TG and glycerol assays were performed as described in (B and C). β-actin was used as the loading control for the immunoblotting and the data were normalized to the total protein in the cellular extracts. Data are shown as mean ± SEM (n = 3). ∗p < 0.05, ∗∗p < 0.01, t test.
STX11 regulates lipid hydrolysis by affecting the spatial distribution of ATGL
In hepatocytes, ATGL is homogeneously distributed in the cytoplasm and can be activated under conditions of starvation or isoproterenol treatment. When activation occurs, ATGL moves from the cytoplasmic matrix to the surface of LDs, causing their hydrolysis to promote FA oxidation. When STX11 was expressed alone, its distribution became patchy within the cytoplasm. Furthermore, when ATGL was expressed alone, it exhibited a diffuse distribution in the cytoplasm. However, when STX11 was co-localized with ATGL, the ATGL fraction co-localizing with STX11 was also patchy (Figures 2A and 2F), indicating that STX11 affected the cytoplasmic distribution of ATGL. Therefore, we speculated that the regulation of LD hydrolysis by STX11 was caused by affecting the distribution of ATGL in the cytoplasm. To verify this idea, the co-localization of STX11 and ATGL was visualized in living cells using STX11-EGFP and ATGL-mcherry tagged vectors (Figure S2B). We found that the co-localization of STX11 with ATGL increased gradually over time (Figures 5A and 5B and Video S1), whereas the distribution of ATGL in the cytoplasm remained unchanged in the presence of ATGL alone (Video S2). This confirmed that the distribution of ATGL in the cytoplasm was affected by STX11, providing evidence for our conjecture.
Figure 5.

STX11 affects the spatial localization of ATGL
(A and B) Live-cell confocal imaging of HeLa cells transiently expressing STX11-EGFP and ATGL-mcherry cultured under basal growth conditions or treated with isoproterenol (1 μM) (see corresponding Video S1). STX11 and ATGL co-localization analysis using ImageJ software (B) (n = 3 cells for live-cell confocal imaging).
(C and D) Representative images and quantification of ATGL co-localization with LDs. HeLa cells were transfected with ATGL-HA and incubated with OA (400 μM) 24 h, followed by treatment with 1 μM isoproterenol for 30 min or starvation for 4 h. ATGL were co-stained with anti-HA (red) and Lipid droplets were co-stained with BODIPY 493/503. Histogram in (D) showing the percentage of ATGL co-localization with LDs in C (n = 18 cells for isoproterenol; n = 21 cells for starvation).
(E and F) Representative images and quantification of ATGL co-localization with LDs demonstrate that STX11 prevents ATGL movement to the surface of LDs. STX11-Myc and ATGL-HA co-expressed in HeLa cells and treated as in (C). STX11-Myc protein immunofluorescence staining was performed using anti-Myc (violet) antibodies and ATGL-HA proteins were stained with anti-HA (red) antibodies. Lipid droplets were stained with BODIPY 493/503 fluorescent dye. Histogram in (F) showing the percentage of ATGL co-localization with LDs in E (n = 18 cells for isoproterenol; n = 22 cells for starvation). Data are shown as mean ± SEM (n = 3). ∗∗∗p < 0.001, t test.
HeLa cells transfected with STX11-EGFP and ATGL-mcherry and then treated with isoproterenol (1 μM). Images were taken at 0.5 min intervals for 2 h using a Leica confocal microscope. Bar, 10 μm.
HeLa cells transfected with ATGL-mcherry and then treated with isoproterenol (1 μM). Images were taken at 0.5 min intervals for 2 h using a Leica confocal microscope. Bar, 10 μm.
Moreover, when ATGL and STX11-expressing HeLa cells were treated with OA (400 μM) for 24 h, the distribution of ATGL in the cytoplasm under starvation and isoproterenol conditions was observed. We found that ATGL aggregation from the cytoplasm to the surface of LDs promoted lipolysis after starvation and isoproterenol treatment in the presence of ATGL alone (Figures 5C and 5D), but this observation had not occurred when STX11 was co-expressed with ATGL (Figures 5E, 5F, and S2E). This illustrates that the regulation of lipolysis by STX11 was indeed caused by altering the trafficking of ATGL from the cytoplasm to LDs.
C-terminal palmitoylation site of STX11 mediates co-localization with ATGL at the ER
Studies have reported that STX11 in immune cells is mainly localized to the cell membrane and endosomes and is involved in transporting intracellular cytotoxic granules (Dabrazhynetskaya et al., 2012; Valdez et al., 1999). The distribution of STX11 in cells was mainly concentrated to the periphery of the nucleus (Figures 1F and 2F), similar to the localization of STX18 in the ER (Figure 2E), demonstrating that STX11 might also localize to the ER. To determine the subcellular localization of STX11, fluorescently tagged STX11 protein was expressed in THLE-2 cells and labeled with ER-Tracker, a marker of the ER. Our results revealed a significant co-localization of STX11 with the ER, indicating that this localization at the ER in THLE-2 cells (Figure 6A) is consistent with our hypothesis. In contrast, an apparent co-localization of ATGL to the ER could not be observed (Figures 6B and 6C). However, the co-expression of STX11 and ATGL and then labeling the ER revealed a co-localization of STX11 and ATGL at this organelle (Figure 6D). The same results were obtained in HeLa cell lines (Figures S4A–S4C). The C-terminal cysteine of STX11 is palmitoylated, which is vital for the physiological function of STX11. When we mutated five cysteines in the C-terminal of STX11 to alanine (Figures 6E and S4D), the membrane localization of STX11 was abolished, as was the physical interaction and co-localization with ATGL (Figures 6F and 6G). This result suggests that the regulation of lipolysis by STX11 was achieved through its dynamic binding and release of ATGL by palmitoylation at the ER in hepatocytes. The co-localization of STX11 with the Golgi body, however, was not detected (Figure S4E). In addition, we found that STX11 interacts with DGAT2 (Figures 6H, 6I, and S2D), an important protein involved in the regulation of LD synthesis at the ER; this result confirmed that STX11 localizes to the ER and also that STX11 not only regulates LD hydrolysis but may also be involved in LD synthesis.
Figure 6.

Co-localization of STX11 with ATGL at the ER
(A) STX11 localizes to the ER. THLE-2 cells were transfected with STX11-mcherry, and the ER was stained with ER-Tracker Blue-White DPX followed by confocal microscopic analysis.
(B) No significant co-localization of ATGL with the ER. THLE-2 cells were transfected with ATGL-mcherry and then the ER were stained with ER-Tracker Blue-White DPX followed by confocal microscopic analysis.
(C) Analysis of STX11 and ATGL co-localization with ER is shown as Rr.
(D) Analysis using confocal microscopy demonstrates that co-localization of STX11 with ATGL at the ER. STX11-EGFP and ATGL-mcherry were co-transfected into THLE-2 cells and ER were stained with ER-Tracker Blue-White DPX.
(E) Schematic of the amino acid mutation in the C-terminal region of STX11.
(F) No co-localization of STX11-C5A and ATGL. HEK293T cells were co-transfected with ATGL-HA together with STX11-Myc or STX11-C5A-Myc and were immunoprecipitated with anti-Myc antibodies. STX11 and ATGL proteins in immunoprecipitated lysates were detected by immunoblotting with Myc and HA antibodies.
(G) HeLa cells were co-transfected with STX11-C5A-Myc and ATGL-HA. STX11-C5A-Myc protein immunofluorescence staining was performed using anti-Myc (green) and ATGL-HA proteins were stained with anti-HA (red) antibodies.
(H) Co-localization of STX11 and DGAT2 in cells (white arrows). HeLa cells were co-transfected with STX11-Myc and DGAT2-HA. Immunofluorescence staining with anti-HA and anti-Myc antibodies was performed to reveal co-localization between DGAT2-HA (green) and STX11-Myc (red).
(I) STX11 and DGAT2 have a direct physical interaction. STX11-Myc proteins were immunoprecipitated with anti-Myc antibodies. STX11 and DGAT2 proteins in immunoprecipitates and lysates were detected by immunoblotting using Myc and HA antibodies. Data are shown as mean ± SEM (n = 3).
STX11 is involved in the regulation of autophagy/lipophagy
The final step of the autophagy process is the autophagosome fusion with the lysosome, mediated by the STX17 and SNAP29 protein complexes of the SNARE protein family (Tian et al., 2021). To determine whether STX11 affects autophagy, we expressed LC3-EGFP and the STX11-mcherry vectors into THLE-2 cells and observed the distribution of STX11 and LC3 in living cells. STX11 and LC3 exhibited a lower co-localization under normal conditions, but the co-localization of STX11 and LC3 increased after the autophagy pathway was activated during starvation (Figures 7A and 7B). Quite clearly, STX11 was found to be involved in the regulation of autophagy in hepatocytes. The same results were obtained in HeLa cells (Figure S5A). In addition, we knocked down STX11 in THLE-2 cells and treated them with OA (400 μM) for 24 h and a starvation regimen for 2 h, and then we assessed the distribution of LDs and autophagosomes by TEM (Figures 7C and 7D). The knockdown of STX11 caused a dramatic loss of LDs when compared to the controls, whereas the number of autophagosomes was notably higher than the control group (Figures 7E and 7F). These findings showed that the siRNA-mediated reduction of STX11 promotes increased autophagy/lipophagy in hepatocytes. Identical results to these were seen when STX11 was knocked down in HeLa cells as the immunofluorescence detection showed a significantly higher level of endogenous LC3 than in controls (Figures S5B–S5D).
Figure 7.

The regulation of lipophagy by STX11 is mediated through ATGL and SIRT1
(A and B) STX11 co-localizes with LC3 under starvation conditions. THLE-2 cells were co-transfected with STX11-mcherry and LC3-EGFP. Cells were treated with starvation (2 h) or no starvation followed by confocal microscopic analysis.
Histogram in (B) showing the percentage of STX11 co-localization with LC3 in A (n = 6 cells for control; n = 6 cells for starvation).
(C and D) Representative EM images showing LDs (yellow arrows) and autophagosomes (APh) (red arrows) in siSTX11 THLE-2 cells and control THLE-2 cells. Cells were treated with OA (400 μM) 24 h and starved 2 h. Protein expression was analyzed by immunoblotting with anti-STX11 antibodies.
(E and F) Quantification of LD and autophagosome (APh) numbers from EM images.
(G) Overexpression of STX11 inhibits autophagy. Immunoblotting of THLE-2 cells transfected with STX11-Myc. Cells were treated with OA (400 μM) following serum-free starvation for 2 h. Protein form cell lysates were detected by immunoblotting with anti-STX11, anti-LAMP1, anti-ATGL, and anti-LC3B antibodies.
(H) Knockdown of STX11 promotes autophagy. THLE-2 cells were treated with STX11 siRNA. Cell treatments and immunoblotting were the same as described in (G).
(I) Presence of STX11 inhibits SIRT1 pathway. Immunoblotting of THLE-2 cells transfected with STX11-Myc. Cell treatments were the same as described in (G). Proteins were detected by immunoblotting with anti-STX11 and anti-SIRT1 antibodies.
(J) Deficiency of STX11 activates SIRT1 pathway. THLE-2 cells were treated with STX11 siRNA. Cell treatments and immunoblotting were the same as described in (G).
(K) STX11 knockdown activates lipophagy mediated by PKA pathway. THLE-2 cells were treated with STX11 siRNA, and cell treatments were as shown in (G) Immunoblots were assessed with anti-STX11, anti-p-PKA, anti-PKA, anti-SIRT1, anti-ATGL, and anti-LC3B antibodies. β-actin was used as the loading control for the immunoblots and the data are shown as mean ± SEM (n = 3). NS: not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, t test.
To further test the effect of STX11 on autophagic flux, we used the tandem GFP-RFP-LC3 sensor for cell labeling to measure the autophagic flux. Confocal microscopy analysis showed the decreased red punctate, which is indicative of reduced lysosomal activity and attenuated autophagic flux as observed with the STX11-overexpressed cells compared to control cells (Figures S6A and S6B). Moreover, we overexpressed STX11 in THLE-2 cells treated for 12 h with or without the lysosome inhibitor chloroquine (CQ 100 μM). Although the major lipase in autophagy decreased, it increased in STX11 knockdown cells. These results show that STX11 blunted autophagic flux in human hepatocytes (Figures S6C and S6D).
STX11 and ATGL regulate lipophagy via SIRT1
SIRT1 is an essential protein involved in the regulation of autophagy by the cAMP/PKA pathway (Song et al., 2015). Importantly, ATGL also regulates autophagy/lipophagy by regulating SIRT1 (Sathyanarayan et al., 2017). Our experiments concluded that STX11 is involved in ATGL-regulated lipolysis and can mediate autophagy/lipophagy. Therefore, we hypothesized that STX11 might be involved in the lipophagy pathway mediated by ATGL and SIRT1. To verify our conjecture, we overexpressed and knocked down STX11 in THLE-2 cells, and we starved the cells for 2 h after OA (400 μM) treatment for 24 h to determine if there any changes in proteins related to the autophagy pathway. When STX11 was overexpressed, LC3II/LC3I and LAMP1 were decreased under normal and starvation conditions (Figure 7G); however, opposite results were observed for STX11 knockdowns (Figure 7H). These results show that STX11 overexpression inhibits the frequency of lipophagy, and the knockdown activates lipophagy. Moreover, SIRT1 was decreased with overexpression of STX11 (Figure 7I) and was increased after STX11 knockdown (Figure 7J), but STX11 does not alter SIRT1 subcellular localization (Figure S7A). This demonstrated that STX11 is involved in lipophagy mediated by SIRT1. Furthermore, STX11 is involved in lipolysis and lipophagy and is regulated by the PKA pathway because p-PKA, SIRT1, FOXO1, LC3II/LC3I, and ATGL were all increased by STX11 knockdown (Figures 7K, S7B, and S7C), indicating activation of lipolysis and lipophagy. These results reinforce our view that STX11 mediates lipolysis by affecting the distribution of ATGL in the cytoplasm and the regulation of lipophagy by SIRT1 involves the PKA pathway.
Discussion
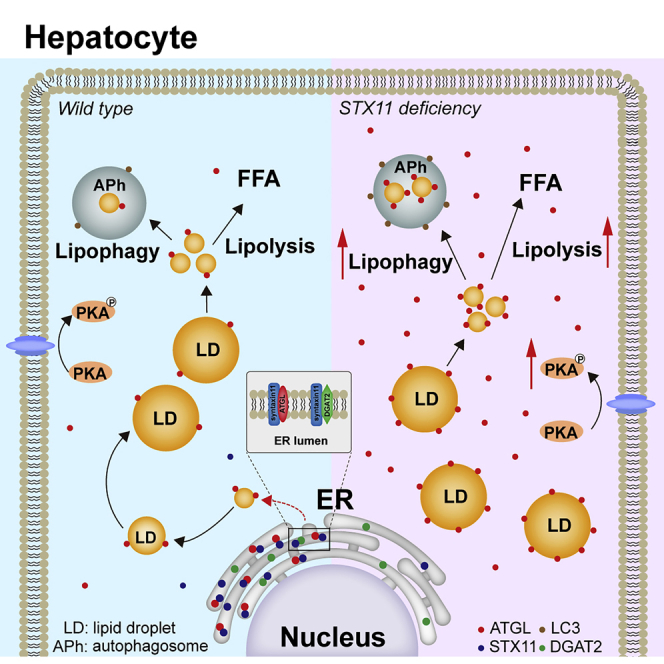
ATGL is a key regulatory enzyme involved in lipolysis and plays a critical role in maintaining the balance between lipid storage and mobilization in the liver. The mechanism of enzymatic activity of ATGL in regulating lipid metabolism has been extensively studied (Schreiber et al., 2019). However, its cellular distribution and transport mechanism are mostly unknown. The present study provides compelling evidence for a role for STX11 as an important negative regulator of ATGL-mediated lipolysis and lipophagy by affecting its distribution in the cytoplasm. It is well-known that ATGL is distributed uniformly in the cytoplasm under basal conditions, where it regulates the dynamics of LD breakdown and maintains homeostatic balance. Nevertheless, upon stimulation (e. g., Isoproterenol application or starvation), perilipin1 is phosphorylated by PKA, causing the activation of ATGL to promote lipolysis and provide cellular energy (Granneman et al., 2009). Our present data illustrates that under normal conditions, ATGL specifically binds to STX11 and translocates to the ER (Figure 6D), which affects the recruitment of ATGL to LDs and reduces the extent of lipolysis and lipophagy in hepatocytes. In the absence of STX11, p-PKA increases, leading to ATGL translocation from the ER to LDs, thereby accelerating lipolysis and lipophagy (Figure S8).
In the current study, we have demonstrated by various methods that the C-terminal region of STX11 directly interacts with the N-terminal patatin (PT) domain of ATGL (Figure 3). STX11 is an atypical member of the T-SNARE family and is composed of an N-terminal peptide (N peptide), a three-helical bundle (Habc domain), a single helical SNARE motif, and a C-terminal peptide (C peptide). STX11 has a cysteine-rich domain at the C-terminal, which is different from that seen with other SNARE family members (Tang et al., 1998). Previous studies have shown that the C-terminal cysteine of STX11 is palmitoylated, which results in the regulation of protein function in natural killer cells (Hellewell et al., 2014). Here, our results also show that the interaction between STX11 and ATGL is regulated by palmitoylation (Figures 6F and 6G), which is a posttranslational modification, and that this precise regulatory mechanism needs to be further investigated.
Our findings revealed that STX11 localizes with ATGL at the ER (Figure 6) and that STX18 is an ER-localized soluble NSF attachment protein receptor of the T-SNARE family, best known as a member of a SNARE complex regulating Golgi-ER retrograde transport consisting of STX18, BNIP1, Use1, and Sec22b (Harriff et al., 2016; Hatsuzawa et al., 2000). We used STX18 as a control to analyze the interaction between STX11 and ATGL. Based on our IP and IF data, only ATGL was detected in the STX11 immunoprecipitates, suggesting a specific interaction between STX11 and ATGL at the ER (Figures 2D and 2E). The intracellular vesicle transport protein, STX11 has also been reported to mediate late endosome to lysosome fusion in macrophages (Offenhauser et al., 2011). Moreover, the interaction between ATGL and LC3 can combine to promote the occurrence of lipophagy in the liver (Martinez-Lopez et al., 2016; Schwerbel et al., 2020). Our immunofluorescence staining demonstrated that STX11 and LC3 were co-localized during starvation (Figure 7A). Therefore, from our results, we conclude that STX11 plays a vital role in the process of autophagy/lipophagy.
Multiple signaling pathways exist which are involved in the regulation of lipid metabolism; however, among these, the AMPK/cAMP/PKA signaling pathway can activate the lipid metabolism pathway mediated by ATGL (Kim et al., 2016; Yang and Mottillo, 2020; Zimmermann et al., 2009). Similarly, AMPK/cAMP/PKA signaling can also regulate lipophagy mediated by ATGL and SIRT1 (Sathyanarayan et al., 2017; Zechner et al., 2017). Our western blot results had revealed that when STX11 was knocked down in hepatocytes, the level of p-PKA was increased (Figure 7K), illustrating that the lipid metabolism pathway regulated by p-PKA was enhanced. Therefore, we conclude that STX11 regulates lipolysis and lipophagy in the liver, which is mediated by the PKA pathway. Under stimulatory conditions, we speculate that the STX11 binding to ATGL is inhibited, then ATGL is released from the ER and participates in lipase-mediated lipolysis and SIRT1 ultimately results in lipophagy.
Previous studies have demonstrated that when LDs are synthesized at the ER, ATGL is directly transported from the ER to LDs through the Golgi-ER transport protein complex GBF1-Arf1-COPI (Wilfling et al., 2014). However, the regulation of other proteins involved is yet to be characterized. Besides, the DGAT2 is an important fat synthase located in the ER and is responsible for the mediation of LD formation (Bhatt-Wessel et al., 2018; Wilfling et al., 2013). We have identified that the content of STX11 protein is increased in the liver of mice fed with HFD and THLE-2 cells treated with OA (Figures 1C–1E, and S1E), indicating that STX11 may be involved in LD generation. In addition, we have found that STX11 interacts with DGAT2 (Figures 6H and 6I), which confirms our previous assumptions; however, the exact mechanism needs to be further explored in-depth. These findings also revealed that STX11 might be involved in targeting ATGL from the ER to lipid droplets in the process of LD formation.
In summary, STX11 acts as a vesicular transport protein and is principally involved in the mediation of the body’s immune response. We found that STX11 also affects lipid metabolism in the liver and achieves this by regulating the spatial distribution of ATGL. It appears that the immune response is closely related to the lipid metabolism of organisms. Importantly, we found that STX11 mediates lipid metabolism in hepatocytes and this may have an important role in the pathogenesis of NAFLD in hepatocytes; therefore, this interesting phenomenon needs further investigation in animal models.
Limitations of the study
The current study reveals that STX11 is a negative regulator of lipolysis in human hepatocytes. The results demonstrated that STX11 directly binds to ATGL and inhibits its recruitment to LD, which in turn inhibits lipolysis and lipophagy. Although this study identified the involvement of STX11 in lipid metabolism in the human hepatocyte, its in vivo, relevant role remains to be proven. Currently, our results show that STX11 expression increases during hepatocytes LD accumulation and interacts with the lipogenic factor DGAT2. Further studies are required to determine the role of STX11 in adipogenesis. Lipid metabolism and immune regulation are intimately linked, and STX11 is also an essential regulatory protein in the immune responses. The role of STX11 regarding the interrelationship between lipid metabolism and immune regulations further needs to be explored in future studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ATGL | Proteintech | Cat# 10957-1-AP; RRID: AB_11182818 |
| Rabbit polyclonal anti-STX11 | Proteintech | Cat# 13301-1-AP, RRID: AB_2198214 |
| Rabbit polyclonal anti-LAMP1 | Proteintech | Cat# 21997-1-AP, RRID: AB_2878966 |
| Rabbit polyclonal anti-LC3B specific | Proteintech | Cat# 18725-1-AP, RRID: AB_2137745 |
| Rabbit polyclonal anti-perilipin2 | Proteintech | Cat# 15294-1-AP |
| Mouse polyclonal anti-SIRT1 | Proteintech | Cat# 60303-1-Ig, RRID: AB_2881417 |
| Mouse monoclonal anti-Beta-Actin | Proteintech | Cat# Biotin-60008, RRID: AB_2883062 |
| Mouse monoclonal anti-Beta-Tubulin | Proteintech | Cat# CL488-66240, RRID: AB_2883292 |
| Rabbit polyclonal anti-PKAC | Cell Signaling Technology | Cat# 4782, RRID: AB_2170170 |
| Rabbit polyclonal anti-phospho-PKAC(THr197) | Cell Signaling Technology | Cat# 4781, RRID: AB_2300165 |
| Rabbit polyclonal anti-HA | Proteintech | Cat# 51064-2-AP, RRID: AB_11042321 |
| Rabbit polyclonal anti-Myc | Proteintech | Cat# 10059-1-AP, RRID: AB_2282407 |
| Mouse monoclonal anti-HA | Proteintech | Cat# 66006-2-Ig, RRID: AB_2881490 |
| Mouse monoclonal anti-Myc | Proteintech | Cat# 60003-2-Ig, RRID: AB_2734122 |
| Donkey anti-Mouse IgG(H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat# A-21202, RRID: AB_141607 |
| Donkey anti-Rabbit IgG(H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | Cat# A-21207, RRID: AB_141637 |
| Donkey anti-Mouse IgG(H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | Cat# A-21203, RRID: AB_141633 |
| Donkey anti-Rabbit IgG(H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat# A-31573, RRID: AB_2536183 |
| Donkey anti-Mouse IgG(H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat# A-31571, RRID: AB_162542 |
| Chemicals, peptides, and recombinant proteins | ||
| Oleic acid (OA) | Sigma-Aldrich | Cat# O1383 |
| Lipo 8000 Transfection Reagent | Beyotime | Cat# C0533 |
| BODIPY 493/503 | Invitrogen | Cat# D3922 |
| Fetal bovine serum | BI | Cat# 04-001-1A |
| DMEM | BI | Cat# 01-052-1A |
| Opti-MEM | GIBCO | Cat# 11058-021 |
| DMEM (No-Glucose, No Phenol Red) | GIBCO | Cat# A1443001 |
| Phosphate Buffered Saline (PBS) | BI | Cat# 02-024-1A |
| ER-Tracker Blue-White DPX | Invitrogen | Cat# E12353 |
| Golgi-Tracker Red | Beyotime | Cat# C1043 |
| DAPI | Beyotime | Cat# C1002 |
| TRIzol reagent | Invitrogen | Cat# 11668019 |
| 1640 media | BI | Cat# 01-100-1A |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# P8340 |
| chloroquine (CQ) | Sigma-Aldrich | Cat# 105M4035V |
| Isoproterenol (ISO) | Sigma-Aldrich | Cat# I5879 |
| Critical commercial assays | ||
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| EndoFree Plasmid Maxi Kit | QIAGEN | Cat# 12362 |
| Triglyceride Assay Kit | Sigma | Cat# MAK266 |
| Glycerol Assay Kit | Abcam | Cat# ab133130 |
| Experimental models: Cell lines | ||
| THLE-2 | ATCC | ATCC CRL-2706 |
| HeLa | ATCC | ATCC CCL-2 |
| HEK293T | ATCC | ATCC CRL-321 |
| Oligonucleotides | ||
| Control siRNA: UUC UCC GAA CGU GUC ACG UTT | This paper | N/A |
| siSTX11: GCA UUA AGC GCG ACA CCA ATT | This paper | N/A |
| Recombinant DNA | ||
| pCDNA3.1-STX11-HA | This Paper | Expression construct generated in the lab |
| pCDNA3.1-STX11-Myc | This Paper | Expression construct generated in the lab |
| pCDNA3.1-ATGL-HA | This Paper | Expression construct generated in the lab |
| pCDNA3.1-ATGL-Myc | This Paper | Expression construct generated in the lab |
| pCDNA3.1-DGAT2-HA | This Paper | Expression construct generated in the lab |
| pEGFP-N2-STX11 | This Paper | Expression construct generated in the lab |
| pEGFP-N2-ATGL | This Paper | Expression construct generated in the lab |
| pCDNA3.1-STX11ΔC-HA | This Paper | Expression construct generated in the lab |
| pCDNA3.1-ATGL-PT-Myc | This Paper | Expression construct generated in the lab |
| pCDNA3.1-ATGL-ΔPT-Myc | This Paper | Expression construct generated in the lab |
| pCDNA3.1-STX11-mcherry | This Paper | Expression construct generated in the lab |
| pGEX5X-3-STX11-GST | This Paper | Expression construct generated in the lab |
| pET30a-ATGL-MBP | This Paper | Expression construct generated in the lab |
| pCDNA3.1-ATGL-mcherry | This Paper | Expression construct generated in the lab |
| pET30a-ATGL-PT-MBP | This Paper | Expression construct generated in the lab |
| Software and algorithms | ||
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| GraphPad Prism 8 | GraphPad | www.graphpad.com |
| Adobe Illustrator 2020 | Adobe | http://aotucad2.xmjfg.com/pg/230.html |
Resource availability
Lead contact
Further information and requests for resources and materials should be directed to and will be fulfilled by the Lead Contact, Xingyuan Yang (xingyuan@ahu.edu.cn).
Materials availability
Materials and protocols used in this study are available from the authors upon request.
Experimental model and subject details
Animal experiments
C57BL/6 female mice (Cyagen, China) were housed under standard temperature and humidity in the specific pathogen-free facilities of Anhui University. Six-week-old mice were fed a high fat diet (HFD) (60% fat, 20% protein, 20% carbohydrate) or normal chow diet for 12 weeks (n = 3 mice/group). All animal protocols were approved by the Institutional Animal Care and Use Committee of the Experimental Animal Center, Anhui University.
Cell culture
The human hepatocyte cell line THLE-2 was obtained from the American Type Culture Collection (ATCC, CRL-2706) and was cultured in Roswell Park Memorial Institute medium 1640 (RPMI-1640) (011001A, BI, Israel). HeLa and HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (010521A, BI, Israel). The RPMI-1640 medium and DMEM medium were supplemented with 1 mM glutamine, penicillin (100 U/mL), streptomycin (100 U/mL), and 10% fetal bovine serum (FBS) (040011A, BI, Israel). The cells were incubated at 37°C under a humidifying atmosphere with 5% CO2 and 95% air.
Method details
Animal tissue extraction
100 mg of mouse liver tissues collected from 18-week-old C57BL6 mice (HFD mice or normal chow diet mice) were placed in a tissue grinder vessel containing 1 ml lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 1% Triton X-100, 0.1% SDS, and protease inhibitor cocktail) and ground well on ice. Completely homogenized tissues were incubated on ice for 30 min and centrifuged at 12000 x g at 4°C for 30 min. The clarified lysates were detected by immunoprecipitation or mixed with 5 × SDS sample buffer, and the solubilized proteins were detected using immunoblotting.
Plasmid expression and siRNA mediated knockdown
The STX11, ATGL, LC3B, STX18, and DGAT2 genes were amplified from a mouse (C57BL6) liver cDNA library. The genes were cloned into either pcDNA3.1-Myc/HA, pcDNA3.1-mcherry, or pEGFP-N2 vectors, and transfection was performed using Lipo 8000 reagent (C0533, Beyotime, China). The overexpressing plasmids (pcDNA3.1-Myc/HA, pcDNA3.1-mcherry or pEGFP-N2 vector) and Lipo 8000 reagent were combined carefully in Opti-MEM Medium (11058021, GIBCO, USA) to develop plasmid-lipid complexes. After this, the complexes were added to the cells and incubated for 24 h; the cells were ready for experimentation. The sequences of the STX11 siRNA oligonucleotides (General Biol, China) were as follows: sense 5′-CACUCAAAUUGAAGUAUCATT-3′, and antisense 5′-UGAUACUUCAAUUUGAGUGTT-3′ and were targeted against human STX11. For the STX11 knockdown experiments in HeLa and THLE-2 cells, 100 pmol of mixed oligonucleotides was transfected into cells and plated into 35 mm dishes using Lipo 8000. After three days of incubation, cells were used for the designated assays.
Western blotting
THLE-2 and HeLa cell lysates were prepared using ice-cold RIPA lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 2 mM EDTA, 10% glycerol, and protease inhibitor cocktail) and incubated on the ice for 30 min and their protein concentrations were quantified using a BCA protein assay kit (23225, Thermo Fisher Scientific, USA). The cell lysate proteins (40 μg) were then resolved using 10–15% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Nitrocellulose (NC) (PN66485, PALL, USA) membranes at 100 V for 3 h under ice-cold condition. The NC membranes were blocked with 5% semi-skimmed milk in phosphate-buffered saline (PBS) containing 0.1% Tween 20 (TBST) for 1 h. The membranes were then exposed to primary antibodies overnight and then incubated with horseradish peroxidase conjugated secondary antibody for 1 h. The resulting protein bands were visualized on a luminescence detecting image analyzer (Tanon 5200, China), and the signal intensities of the target protein bands were analyzed by ImageJ software (NIH, Bethesda, USA), and β-actin or localization β-tubulin were used to determine the integral optical density (IOD) ratio.
Triglyceride and glycerol measurement
The rate of lipolysis was determined as the rate of glycerol release in the medium and the concentration of the TGs remaining in the cells. Briefly, cells were seeded into 35 mm plates and incubated with 400 μM oleic acid (OA) for 24 h followed by a starvation protocol for 2 or 4 h in serum and glucose-free medium. Next, the cells were collected and lysed ultrasonically in PBS buffer and the TGs were measured using a TG assay kit along with the glycerol of in the medium using a glycerol assay Kit according to the manufacturer’s instructions. Total cellular protein content was also quantified using the BCA Protein Assay kit. Intracellular TG concentration and medium’s glycerol were normalized to total cellular protein concentration and expressed as μmol/μg protein.
Immunoprecipitation (IP)
HEK293T cells were transfected with the overexpression-plasmid for 24 h and then scraped and lysed in RIPA lysis buffer. The lysates were placed on ice for 30 min, following centrifugation at 12,000 g for 20 min. The supernatants were collected into new tubes and incubated overnight with primary antibodies at 4°C. The Protein A/G-Sepharose beads (P2055, Beyotime, China) were added to the overnight incubation tube which was rotated for 2 h at room temperature (RT). Next, the beads were washed four times with cold PBS buffer and boiled in 1 × SDS loading buffer at 100°C for 5 min. For the in vitro pull-down, 1.5 μg GST tagged protein, or 1.5 μg STX11-GST tagged protein was incubated with ATGL-MBP-His protein in protein binding buffer (20 mM Tris-HCl, pH 7.5, 200 mM NaCl,) overnight at 4°C, followed by the addition of GST-beads to pull down the protein complexes. The beads were then washed three times with protein binding buffer before adding 1 × loading buffer. The protein samples were then analyzed by western blotting.
Immunofluorescence and colocalization
THLE-2 and HeLa cells were transfected on 25 mm coverslips with the indicated plasmid or siRNA using Lipo 8000 transfection reagent and were loaded with 400 μM OA-BSA (BSA, fatty acid-free) complexes for 24 h. The next day, cells were washed with cold PBS and fixed with 4% paraformaldehyde in PBS for 30 min. Cells were then permeabilized with 0.5% Triton X-100 for 5 min and blocked with 5% BSA (MP Biomedicals, USA) for 1 h at RT and stained with primary antibodies for 2 h at RT. The coverslips were washed three times for 30 min with PBS and the secondary antibody was then applied and incubated at RT in the dark for 1 h and stained for 20 min with BODIPY and DAPI. Slides were evaluated and photographed on a Leica TCS SP8 DIVE confocal microscope (Leica Microsystems) using the ×60 oil plan objective.
Liver histology examination
The obtained liver tissue samples were fixed in 4% formaldehyde. The fixed tissues were dehydrated and processed for paraffin embedding, and catted into 5 μm thick slices. The sections were stained with hematoxylin and eosin (H&E). Immunohistochemical (IHC) staining was performed with STX11 antibody (Proteintech, 13301-1-AP, 1:100). Lipid droplets (LDs) numbers and STX11 scores were determined by using ImageJ (US National Institutes of Health).
Live cell imaging
Cells were cultured onto a 35 mm glass-bottom dish (NEST), and live-cell fluorescence imaging was performed using a Leica TCS SP8 DIVE confocal microscope using a 60 × oil 1.4 numerical aperture objective lens. During the live imaging procedure, the culture dish was installed into the living cell workstation to maintain the correct incubation conditions at 37°C and 5% CO2. Two-color time-lapse images were acquired at 30 s intervals for 2 h.
Protein expression and purification
A recombinant BL21 (DE3) E. coli expression system was used to express proteins and subsequent purification. The E. coli were transformed with either pGEX5X-3-STX11, pET30a-MBP-His-ATGL, or pET30a-MBP-His-ATGL-PT and grown in LB medium containing 100 μg/mL ampicillin up to an optical density (OD) of 0.6, followed by induction with 0.5 mM IPTG for 20 h at 16°C. Cells were harvested, resuspended, and then lysed in lysis buffer (20 mM Tris-HCl, pH 8.0, 200 mM NaCl) using sonication in the ice bath. Recombinant proteins were purified with glutathione sepharose beads (GE Healthcare, USA) or with a nickel-nitrilotriacetic acid (Ni-NTA) protein purification system (QIAGEN, Germany) according to the manufacturer’s instructions. Proteins were concentrated and further purified by gel filtration chromatography (Superdex 200 10/300 GL; GE Healthcare, USA) in 20 mM Tris-HCl, pH 8.0, and 200 mM NaCl. Protein concentrations were determined using the one-drop spectrophotometer (OD-1000+).
Transmission electron microscopy
For standard transmission electron microscopy (TEM), THLE-2 cells transfected with siSTX11 were fixed in 4% paraformaldehyde in PBS overnight at 4°C and then fixed in 2% aqueous osmium tetraoxide for 2 h at RT followed by washing three times each for 10 min with deionized water. Samples were then dyed with 2% uranyl acetate for 30 min, and dehydrated through graded alcohols (50%–100%) and 100% acetone each for 10 min. Subsequently, samples were embedded in EPON 812 resin and cured for 24 h. Ultrathin (60 nm) sections were obtained by UC7FC7 ultra-thin slicer machine (Leica) and stained with 2% uranyl acetate and 0.3% lead citrate. Electron microscopy images of the samples were taken using the JEM-2100 transmission electron microscope (JEOL Company).
Quantification and statistical analysis
Data were expressed as mean ± standard error of the mean (SEM). Statistical analyses were performed using either Student’s t-test (two-group comparison) or one-way analysis of variance followed by Student-Newman-Keuls test (more than two group comparison). In all analyses, a probability of less than 0.05 was considered to indicate statistical significance. All statistical analyses were performed using the GraphPad Prism program (V 8.0.2).
Acknowledgments
We are grateful to the staff for providing technical support with using the facility of the Institute of Health Sciences & Technology, Anhui University.
This work was supported by grants from Natural Science Foundation of China (No. 31771310 to X.Y. and No. 41871210 to L.W.), Anhui Province Natural Science Foundation (No. 1708085MC67 to X.Y.), Anhui Province Higher Education Natural Science Foundation (Z010162002 to X.Y. and KJ2019A0041 to F.W.), the Undergraduate Training Program for Innovation and Entrepreneurship of Anhui University (201610357007) and the Recruitment Program for leading Talent Team of Anhui Province (2019-16).
Author contributions
Conceptualization, X.Y., H.G., and G.Z.; Methodology, G.Z., J.H., and Z.Y.; Software, G.Z., J.H., and L.W.; Validation, M.Q., H.Z., and X.G.Y.; Formal Analysis, G.Z., X.Y., and F.W.; Investigation, G.Z. and X.G.Y.; Resources, X.Y., F.W., and L.W.; Data Curation, G.Z. and J.H.; Writing – Original Draft Preparation, G.Z.; Writing – Review & Editing, H.B., J.H, and F.W.; Visualization, G.Z., M.Q., and H.Z.; Supervision, X.Y.; Project Administration, X.Y.
Declaration of interests
The authors declare no competing interests.
Published: April 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104085.
Contributor Information
Honghua Ge, Email: gehonghua@gmail.com.
Xingyuan Yang, Email: xingyuan@ahu.edu.cn.
Supplemental information
Data and code availability
-
•
This study did not generate/analyze datasets or computer code/algorithm.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.
References
- Bhatt-Wessel B., Jordan T.W., Miller J.H., Peng L. Role of DGAT enzymes in triacylglycerol metabolism. Arch. Biochem. Biophys. 2018;655:1–11. doi: 10.1016/j.abb.2018.08.001. [DOI] [PubMed] [Google Scholar]
- Bolsoni-Lopes A., Alonso-Vale M.I. Lipolysis and lipases in white adipose tissue - an update. Arch. Endocrinol. Metab. 2015;59:335–342. doi: 10.1590/2359-3997000000067. [DOI] [PubMed] [Google Scholar]
- Cerk I.K., Wechselberger L., Oberer M. Adipose triglyceride lipase regulation: an overview. Curr. Protein Pept. Sci. 2018;19:221–233. doi: 10.2174/1389203718666170918160110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrazhynetskaya A., Ma J., Guerreiro-Cacais A.O., Arany Z., Rudd E., Henter J.I., Karre K., Levitskaya J., Levitsky V. Syntaxin 11 marks a distinct intracellular compartment recruited to the immunological synapse of NK cells to colocalize with cytotoxic granules. J. Cell. Mol. Med. 2012;16:129–141. doi: 10.1111/j.1582-4934.2011.01280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy E., Subramaniam S., Murphy R.C., Nishijima M., Raetz C.R., Shimizu T., Spener F., van Meer G., Wakelam M.J., Dennis E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009;50:S9–S14. doi: 10.1194/jlr.R800095-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman J.G., Moore H.P., Krishnamoorthy R., Rathod M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl) J. Biol. Chem. 2009;284:34538–34544. doi: 10.1074/jbc.M109.068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross D.A., Silver D.L. Cytosolic lipid droplets: from mechanisms of fat storage to disease. Crit. Rev. Biochem. Mol. Biol. 2014;49:304–326. doi: 10.3109/10409238.2014.931337. [DOI] [PubMed] [Google Scholar]
- Harriff M.J., Karamooz E., Burr A., Grant W.F., Canfield E.T., Sorensen M.L., Moita L.F., Lewinsohn D.M. Endosomal MR1 trafficking plays a key role in presentation of Mycobacterium tuberculosis ligands to MAIT cells. PLoS Pathog. 2016;12:e1005524. doi: 10.1371/journal.ppat.1005524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsuzawa K., Hirose H., Tani K., Yamamoto A., Scheller R.H., Tagaya M. Syntaxin 18, a SNAP receptor that functions in the endoplasmic reticulum, intermediate compartment, and cis-golgi vesicle trafficking. J. Biol. Chem. 2000;275:13713–13720. doi: 10.1074/jbc.275.18.13713. [DOI] [PubMed] [Google Scholar]
- Hellewell A.L., Foresti O., Gover N., Porter M.Y., Hewitt E.W. Analysis of familial hemophagocytic lymphohistiocytosis type 4 (FHL-4) mutant proteins reveals that S-acylation is required for the function of syntaxin 11 in natural killer cells. PLoS One. 2014;9:e98900. doi: 10.1371/journal.pone.0098900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W., Lev S. Tethering the assembly of SNARE complexes. Trends Cell Biol. 2014;24:35–43. doi: 10.1016/j.tcb.2013.09.006. [DOI] [PubMed] [Google Scholar]
- Jenkins C.M., Mancuso D.J., Yan W., Sims H.F., Gibson B., Gross R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004;279:48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- Kim S.-J., Tang T., Abbott M., Viscarra J.A., Wang Y., Sul H.S. AMPK phosphorylates desnutrin/ATGL and hormone-sensitive lipase to regulate lipolysis and fatty acid oxidation within adipose tissue. Mol. Cell Biol. 2016;36:1961–1976. doi: 10.1128/MCB.00244-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogl T., Muller J., Jessen B., Schmitt-Graeff A., Janka G., Ehl S., zur Stadt U., Aichele P. Hemophagocytic lymphohistiocytosis in syntaxin-11-deficient mice: T-cell exhaustion limits fatal disease. Blood. 2013;121:604–613. doi: 10.1182/blood-2012-07-441139. [DOI] [PubMed] [Google Scholar]
- Kohjima M., Enjoji M., Higuchi N., Kato M., Kotoh K., Yoshimoto T., Fujino T., Yada M., Yada R., Harada N., et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007;20:351–358. [PubMed] [Google Scholar]
- Lass A., Zimmermann R., Haemmerle G., Riederer M., Schoiswohl G., Schweiger M., Kienesberger P., Strauss J.G., Gorkiewicz G., Zechner R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman syndrome. Cell Metab. 2006;3:309–319. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Martinez-Lopez N., Garcia-Macia M., Sahu S., Athonvarangkul D., Liebling E., Merlo P., Cecconi F., Schwartz G.J., Singh R. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the Brown adipose tissue and liver. Cell Metab. 2016;23:113–127. doi: 10.1016/j.cmet.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashek D.G. Hepatic lipid droplets: a balancing act between energy storage and metabolic dysfunction in NAFLD. Mol. Metab. 2020;50:101115. doi: 10.1016/j.molmet.2020.101115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P., Leray V., Diez M., Serisier S., Le Bloc'h J., Siliart B., Dumon H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008;92:272–283. doi: 10.1111/j.1439-0396.2007.00752.x. [DOI] [PubMed] [Google Scholar]
- Notari L., Baladron V., Aroca-Aguilar J.D., Balko N., Heredia R., Meyer C., Notario P.M., Saravanamuthu S., Nueda M.L., Sanchez-Sanchez F., et al. Identification of a lipase-linked cell membrane receptor for pigment epithelium-derived factor. J. Biol. Chem. 2006;281:38022–38037. doi: 10.1074/jbc.M600353200. [DOI] [PubMed] [Google Scholar]
- Offenhauser C., Lei N., Roy S., Collins B.M., Stow J.L., Murray R.Z. Syntaxin 11 binds Vti1b and regulates late endosome to lysosome fusion in macrophages. Traffic. 2011;12:762–773. doi: 10.1111/j.1600-0854.2011.01189.x. [DOI] [PubMed] [Google Scholar]
- Olzmann J.A., Carvalho P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019;20:137–155. doi: 10.1038/s41580-018-0085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong K.T., Mashek M.T., Bu S.Y., Greenberg A.S., Mashek D.G. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology. 2011;53:116–126. doi: 10.1002/hep.24006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabha Das K.M., Wechselberger L., Liziczai M., De la Rosa Rodriguez M., Grabner G.F., Heier C., Viertlmayr R., Radler C., Lichtenegger J., Zimmermann R., et al. Hypoxia-inducible lipid droplet-associated protein inhibits adipose triglyceride lipase. J. Lipid Res. 2018;59:531–541. doi: 10.1194/jlr.M082388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlemuter G., Bigorgne A., Cassard-Doulcier A.M., Naveau S. Nonalcoholic fatty liver disease: from pathogenesis to patient care. Nat. Clin. Pract. Endocrinol. Metab. 2007;3:458–469. doi: 10.1038/ncpendmet0505. [DOI] [PubMed] [Google Scholar]
- Sathyanarayan A., Mashek M.T., Mashek D.G. ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Rep. 2017;19:1–9. doi: 10.1016/j.celrep.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R., Xie H., Schweiger M. Of mice and men: the physiological role of adipose triglyceride lipase (ATGL) Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2019;1864:880–899. doi: 10.1016/j.bbalip.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze R.J., Schott M.B., Casey C.A., Tuma P.L., McNiven M.A. The cell biology of the hepatocyte: a membrane trafficking machine. J. Cell Biol. 2019;218:2096–2112. doi: 10.1083/jcb.201903090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwerbel K., Kamitz A., Krahmer N., Hallahan N., Jahnert M., Gottmann P., Lebek S., Schallschmidt T., Arends D., Schumacher F., et al. Immunity-related GTPase induces lipophagy to prevent excess hepatic lipid accumulation. J. Hepatol. 2020;73:771–782. doi: 10.1016/j.jhep.2020.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A.M., Czaja M.J. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.M., Lee Y.H., Kim J.W., Ham D.S., Kang E.S., Cha B.S., Lee H.C., Lee B.W. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy. 2015;11:46–59. doi: 10.4161/15548627.2014.984271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugaya Y., Satoh H. Liver-specific G0/G1 switch gene 2 (G0s2) expression promotes hepatic insulin resistance by exacerbating hepatic steatosis in male Wistar rats. J. Diabetes. 2017;9:754–763. doi: 10.1111/1753-0407.12482. [DOI] [PubMed] [Google Scholar]
- Tang B.L., Low D.Y., Hong W. Syntaxin 11: a member of the syntaxin family without a carboxyl terminal transmembrane domain. Biochem. Biophys. Res. Commun. 1998;245:627–632. doi: 10.1006/bbrc.1998.8490. [DOI] [PubMed] [Google Scholar]
- Tian X., Teng J., Chen J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy. 2021;17:2680–2688. doi: 10.1080/15548627.2020.1823124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez A.C., Cabaniols J.P., Brown M.J., Roche P.A. Syntaxin 11 is associated with SNAP-23 on late endosomes and the trans-golgi network. J. Cell Sci. 1999;112:845–854. doi: 10.1242/jcs.112.6.845. [DOI] [PubMed] [Google Scholar]
- Villena J.A., Roy S., Sarkadi-Nagy E., Kim K.H., Sul H.S. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem. 2004;279:47066–47075. doi: 10.1074/jbc.M403855200. [DOI] [PubMed] [Google Scholar]
- Ward C., Martinez-Lopez N., Otten E.G., Carroll B., Maetzel D., Singh R., Sarkar S., Korolchuk V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta. 2016;1861:269–284. doi: 10.1016/j.bbalip.2016.01.006. [DOI] [PubMed] [Google Scholar]
- Wilfling F., Thiam A.R., Olarte M.J., Wang J., Beck R., Gould T.J., Allgeyer E.S., Pincet F., Bewersdorf J., Farese R.V., Jr., et al. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife. 2014;3:e01607. doi: 10.7554/eLife.01607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F., Wang H., Haas J.T., Krahmer N., Gould T.J., Uchida A., Cheng J.X., Graham M., Christiano R., Frohlich F., et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell. 2013;24:384–399. doi: 10.1016/j.devcel.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A., Mottillo E.P. Adipocyte lipolysis: from molecular mechanisms of regulation to disease and therapeutics. Biochem. J. 2020;477:985–1008. doi: 10.1042/BCJ20190468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Lu X., Lombes M., Rha G.B., Chi Y.I., Guerin T.M., Smart E.J., Liu J. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010;11:194–205. doi: 10.1016/j.cmet.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Ding Y., Chen Y., Zhang S., Huo C., Wang Y., Yu J., Zhang P., Na H., Zhang H., et al. The proteomics of lipid droplets: structure, dynamics, and functions of the organelle conserved from bacteria to humans. J. Lipid Res. 2012;53:1245–1253. doi: 10.1194/jlr.R024117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R., Madeo F., Kratky D. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017;18:671–684. doi: 10.1038/nrm.2017.76. [DOI] [PubMed] [Google Scholar]
- Zimmermann R., Strauss J.G., Haemmerle G., Schoiswohl G., Birner-Gruenberger R., Riederer M., Lass A., Neuberger G., Eisenhaber F., Hermetter A., et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- Zimmermann R., Lass A., Haemmerle G., Zechner R. Fate of fat: the role of adipose triglyceride lipase in lipolysis. Biochim. Biophys. Acta. 2009;1791:494–500. doi: 10.1016/j.bbalip.2008.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HeLa cells transfected with STX11-EGFP and ATGL-mcherry and then treated with isoproterenol (1 μM). Images were taken at 0.5 min intervals for 2 h using a Leica confocal microscope. Bar, 10 μm.
HeLa cells transfected with ATGL-mcherry and then treated with isoproterenol (1 μM). Images were taken at 0.5 min intervals for 2 h using a Leica confocal microscope. Bar, 10 μm.
Data Availability Statement
-
•
This study did not generate/analyze datasets or computer code/algorithm.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.