

Abstract
Glycogen synthase kinase-3 (GSK-3) has been implicated in numerous pathologies making GSK-3 an attractive therapeutic target. Our group has identified a compound termed COB-187 that is a potent and selective inhibitor of GSK-3. In this study, we probed the mechanism by which COB-187 inhibits GSK-3β. Progress curves, generated via real-time monitoring of kinase activity, indicated that COB-187 inhibition of GSK-3β is time-dependent and subsequent jump dilution assays revealed that COB-187 binding to GSK-3β is reversible. Further, a plot of the kinetic constant (kobs) versus COB-187 concentration suggested that, within the range of concentrations studied, COB-187 binds to GSK-3β via an induced-fit mechanism. There is a critical cysteine residue at the entry to the active site of GSK-3β (Cys-199). We generated a mutant version of GSK-3β wherein Cys-199 was substituted with an alanine. This mutation caused a dramatic decrease in the activity of COB-187; specifically, an IC50 in the nM range for wild type versus >100 μM for the mutant. A screen of COB-187 against 34 kinases that contain a conserved cysteine in their active site revealed that COB-187 is highly selective for GSK-3 indicating that COB-187’s inhibition of GSK-3β via Cys-199 is specific. Combined, these findings suggest that COB-187 inhibits GSK-3β via a specific, reversible, time and Cys-199-dependent mechanism.
Keywords: glycogen synthase kinase 3, cell signaling, Alzheimer’s disease, sepsis, serine/threonine protein kinase, enzyme inhibitor, tideglusib
Graphical Abstract
1. Introduction
Glycogen synthase kinase-3 is a ubiquitously expressed serine/threonine kinase involved in multiple signaling pathways. There are two isoforms of GSK-3, namely GSK-3α (51 kDa) and GSK-3β (47 kDa)1. GSK-3 was first identified as important in glucose metabolism2, and has since been shown to play a significant role in a host of cellular processes3,4. Not surprisingly, the aberrant activity of GSK-3 has been implicated in several human pathologies making GSK-3 an attractive therapeutic target5. To date, the lack of FDA-approved selective and potent inhibitors of GSK-3 has impeded the treatment of GSK-3-implicated diseases.
Most known GSK-3 inhibitors are small molecules; although peptide inhibitors of GSK-3 are being developed6. The majority of the small molecule inhibitors are ATP- competitive [e.g., AR-A0144187] and are highly potent. That said, ATP-competitive inhibitors often suffer from a lack of specificity8 which could lead to high toxicity, off-target effects and failure in clinical trials. Indeed, a significant challenge in the development of kinase inhibitors is designing a compound that is selective towards the kinase of interest amongst the over 500 other kinases. In theory, the specificity of ATP-competitive inhibitors could be improved by designing drugs based on unique features of the GSK-3 ATP-binding pocket9. Perhaps the most advanced ATP-competitive inhibitor is 9-ING-41 which is in phase II clinical trials10. 9-ING-41 appears to be specific as it was reported to be selective for GSK-3 in a screen of 320 related kinases11. This observation bolsters the conjecture that there are features of the ATP-binding pocket that are unique to GSK-3 and this region can be exploited to generate inhibitors that are highly specific for GSK-39.
Several chemical families have been reported whose mechanism of GSK-3 inhibition appears to be distinct from ATP competition12. The most clinically advanced [e.g.13] of these inhibitors is tideglusib. Tideglusib inhibits GSK-3 via an ATP-noncompetitive mechanism14 and was first touted as a GSK-3-specific inhibitor12. However, recent data suggests that tidelgusib does inhibit multiple kinases, in addition to GSK-3, meaning tideglusib is likely only modestly selective towards GSK-315,14. The detailed mechanism of action of tideglusib is complex and still somewhat cloudy. Tideglusib appears to bind irreversibly to GSK-3β, although there is no clear evidence that tideglusib binds covalently to GSK-3β14. Cys-199 in the GSK-3β active site plays a significant role in tideglusib’s activity14.
Our group previously described a selective and potent inhibitor of GSK-3 termed COB-187 [4-hydroxy-4-phenyl-3-(pyridin-4-ylmethyl)thiazolidine-2-thione]15. Molecular assays revealed that COB-187 inhibits both isoforms of GSK-3 with an IC50 in the nanomolar range and, importantly, exhibits high selectivity towards GSK-3 among 404 kinases screened15. In cell-based assays, COB-187 was found to inhibit phosphorylation of canonical GSK-3 substrates indicating that it is a cellular GSK-3 inhibitor15. Recently, we reported that COB-187 dramatically attenuates the cytokine storm induced by LPS in a human macrophage cell line16. Combined, these findings demonstrate that COB-187 is a starting point for the development of novel inhibitors of GSK-3 that have therapeutic potential. To guide the rational and efficient development of such inhibitors, an understanding of the mechanism by which COB-187 inhibits GSK-3 is needed. Thus, in the present study we sought to gain insight into the molecular mechanism by which COB- 187 inhibits GSK-3β.
2. Materials and Methods
2.1. Reagents
The chemical structure of all of the compounds used in this study is given in Fig. 1. COB-187 was prepared as described previously15. Tideglusib, alsterpaullone, AR-A014418, hypothemycin, ethanethiol, tributylphosphine, and THF were purchased from Sigma-Aldrich (St. Louis, MO). Insect-expressed human recombinant full length histidine-tagged GSK-3β (catalog number PR5074A) was from Life Technologies/Thermo Fisher (Waltham, MA). The mutant version of the abovementioned GSK-3β, where the cysteine at position 199 was replaced with an alanine (C199A), was generated by ThermoFisher via a contract agreement. The phospho substrate (catalog number AQT0157) and ATP (catalog number AQT 100XATP) were obtained from AssayQuant Tech (Marlboro, MA). L-Glutathione reduced (GSH) (catalog number: G4251) and Tris(2-carboxyethyl)phosphine hydrochlo (TCEP) (catalog number: C4706) were purchased from Sigma-Aldrich. Dithiotheritol (DTT) (catalog number: AQT1000X DTT) was purchased from AssayQuant Tech. The kinase reaction buffer (AssayQuant Tech; catalog number: AQT10XRB) contained 500 mM HEPES, pH 7.5, 0.1% Brij-35, and 100 mM MgCl2. The enzyme dilution buffer (AssayQuant Tech; catalog number: AQT1XEDB) contained 20 mM HEPES, pH 7.5, 0.01% Brij-35, 5% glycerol, and 1 mg/mL bovine serum albumin.
2.2. Kinase assay
The activity of both versions of GSK-3β (wild type and C199A) was measured with the PhosphoSens® Protein Kinase assay (AssayQuant Tech) which monitors kinase activity in real time. The protocol provided by the manufacturer was followed. The assay was performed in white 96-well plates (Corning). The basis for the assay is a Sox-modified peptide substrate. Upon phosphorylation of the substrate, a phosphate group is “near” the Sox construct. In real time, the assay solution is exposed to 360 nm light and the resulting emission read at 485 nm. The extent of 485 nm emission detected correlates with the extent of peptide phosphorylation17,18,19. All experiments were run at 30 °C. For all of the AssayQuant assays, except the data presented in Figs. 3 and 6, the final concentrations of each reaction component were as follows: 50 mM HEPES, pH 7.5, 14 μM ATP, 0.01% Brij-35, 1 mM EGTA, 10 mM MgCl2, 1% glycerol, 0.2 mg/mL bovine serum albumin, 10 μM peptide substrate, and 0.5 nM GSK-3β or 0.5 nM C199A. For the data presented in Fig. 6, 2 mM of a reducing agent was added to the reaction buffer and % inhibition was determined after 84 minutes of reaction. The progress curves (Fig. 2) were generated by monitoring the kinase reaction and recording data every 3 minutes. The data from each individual experiment was normalized to the 1% DMSO reading at the end point for that experiment. The normalized data was then averaged over the separate experiments to arrive at the data presented in Fig. 2.
2.3. Screen of COB-187 on a panel of kinases
COB-187 was screened on a panel of 34 kinase assays via contract research with Thermo Fisher. Schirmer et al., in a bioinformatic analysis, identified 46 kinases in the human kinome which contain a cysteine residue in their active site20. We screened COB-187 on all of the kinases available from ThermoFisher that have a cysteine residue in their active site. Both Z’-LYTE and LanthaScreen assays were used. The Z’-LYTE assay measures kinase activity via detection of phosphorylation of the peptide while the LanthaScreen assay is based on displacement of the fluorescent analogues of ATP-competitive inhibitors. Both of these assays are endpoint assays and were conducted in the absence of a reducing agent. A peptide, based on human glycogen synthase I containing Ser64114, was the substrate for the GSK-3 kinase assay. The substrate concentration was 2 μM and the concentration of COB-187 was 2 μM. Both kinase and development reactions were carried out for 60 min at room temperature in 384-well plates.
2.4. Jump dilution experiments
Running jump dilution assays21 were performed as follows. Samples of 50 nM GSK-3β in the enzyme dilution buffer were incubated with 10% DMSO or with a concentration of the compounds that results in ~90% enzyme inhibition at this level of enzyme. After a 3-hour incubation, the mixtures were diluted 100-fold with substrate and ATP master mix (supplemented with DMSO) yielding final concentrations of 10 μM, 2 mM, and 1%, for the substrate, ATP and DMSO, respectively. Subsequently, the progress of the reaction was followed and recorded (Fig. 3A and Supp. Fig. S3). To quantify the recovery of GSK-3β pre-treated with COB-187, the activity of GSK-3β, which had been pre-incubated with COB-187, was determined over three different time periods (0-30, 30-60 and 60-90 minutes) since initiation of the jump dilution (Fig. 3B). For each time period, the RFU at the end of the time period was subtracted from the RFU at the beginning of the time period for both the COB-187 and no compound-treated GSK-3β. The resulting values were ratioed (i.e., pre-treated/not pretreated), and the quotient multiplied by 100%. The values from 3 replicate experiments were averaged to arrive at the data presented. For the no-preincubation control for the jump dilution (Fig. 3C), 5 μL of the inhibitors at 10X, i.e., 10 times greater than its final concentration after dilution in the jump dilution assay, was added to the plate. Subsequently, 35 μL of a solution containing substrate and ATP in reaction buffer was added to the corresponding wells. The reaction was then initiated by the addition of 10 μL (5X) of the GSK-3β in the enzyme dilution buffer with the final concentrations of substrate, enzyme, and ATP, 10 μM, 0.5 nM, and 2 mM, respectively. Note that these conditions are identical to the final conditions in the jump dilution assay described above for Fig. 3A.
2.5. ATP and substrate Km values
The ATP Km values were determined by preparing 7-point dilutions of ATP (starting at 448 μM), in the presence of 10 μM of substrate and 0.5 nM of GSK-3β or C199A. The substrate Km values were determined by preparing 8-point dilutions of substrate (starting at 30 μM) in the presence of 250 μM ATP and 0.5 nM of GSK-3β or C199A. The resulting initial velocity data were fit to the Michaelis-Menten model to arrive at the Km.
2.6. IC50 value determination with or without pre-incubation of the inhibitors with enzyme
For the “no preincubation” assays (Fig. 5 and Table 1), 5 μM of the inhibitors at 10X, i.e., 10 times greater than its final concentration, was added to white 96-well plates. Subsequently, 35 μL of a solution containing substrate and ATP in reaction buffer was added to the wells. The reaction was then initiated by the addition of 10 μL (5X) of the GSK-3β in the enzyme dilution buffer. In contrast, for the “preincubation” assays (Fig. 5 and Table 1), 5 μL of the inhibitors at 10X and 10 μL of the enzyme at 5X were mixed for three hours then the reaction was initiated with addition of 35 μL of a solution containing ATP and substrate with the final concentrations the same as for the “no preincubation protocol” stated above. For each condition, the IC50 values were determined by performing serial dilution experiments. For each condition, the pIC50 (the negative logarithm of the IC50) values were determined by fitting dose-response data to the following equation using Prism 8.3.2 (GraphPad Software Inc., San Diego, CA).
| (Eq. 1) |
The input parameters were the inhibitor concentration [I], and the percent inhibition relative to activity in the absence of inhibitor (determined after 84 minutes of reaction). n is the hill coefficient and Min/Max are the lowest and highest asymptotes of the dose-response curve. The no inhibitor samples had the carrier control (1% DMSO for all but the C199A assay where 5% DMSO was utilized to avoid potential precipitation of COB- 187 due to the high levels of COB-187 used). Note that the data generated for hypothemycin did not fit well to Eq. 1 and thus, a simpler 3-parameter equation (i.e., n set to 1) was used.
Statistical analyses –
All statistical analyses were performed using the GraphPad Prism 8.3.2 (GraphPad Software Inc., San Diego, CA). Specific analyses are listed in the figure captions. The following symbols represent the p-values: *p<0.05, **p<0.01, and ***p<0.001.
3. Results
3.1. Analysis of GSK-3β progress curves in the presence of COB-187 reveals a time-dependent velocity
To explore the mechanism of action of COB-187, we used a novel GSK-3β PhosphoSens® Protein Kinase assay that allows real-time monitoring of the reaction. As shown in Supp. Fig. S1 and S2, the substrate Km and ATP Km determined from this assay, ~ 1 μM and ~11 μM respectively, are similar to that reported for other GSK-3 kinase assays [14 and ThermoFisher Z’Lyte assay]. The structures of COB-187 and the other compounds used in this study are provided in Fig. 1. The other compounds were selected due to their complementary mechanisms: hypothemycin, an irreversible inhibitor of GSK-3 likely involving covalent binding to a cys residue in the active site of GSK-314; tideglusib, an irreversible inhibitor of GSK-3 through a mechanism different from hypothemycin’s14 and arguably the most clinically advanced inhibitor of GSK-313; and alsterpaullone, a reversible inhibitor of GSK-314. AR-A014418 was chosen due to the fact that it inhibits GSK-3 in the presence of reducing agents7.
Figure 1. Chemical structure of inhibitors used in this work.
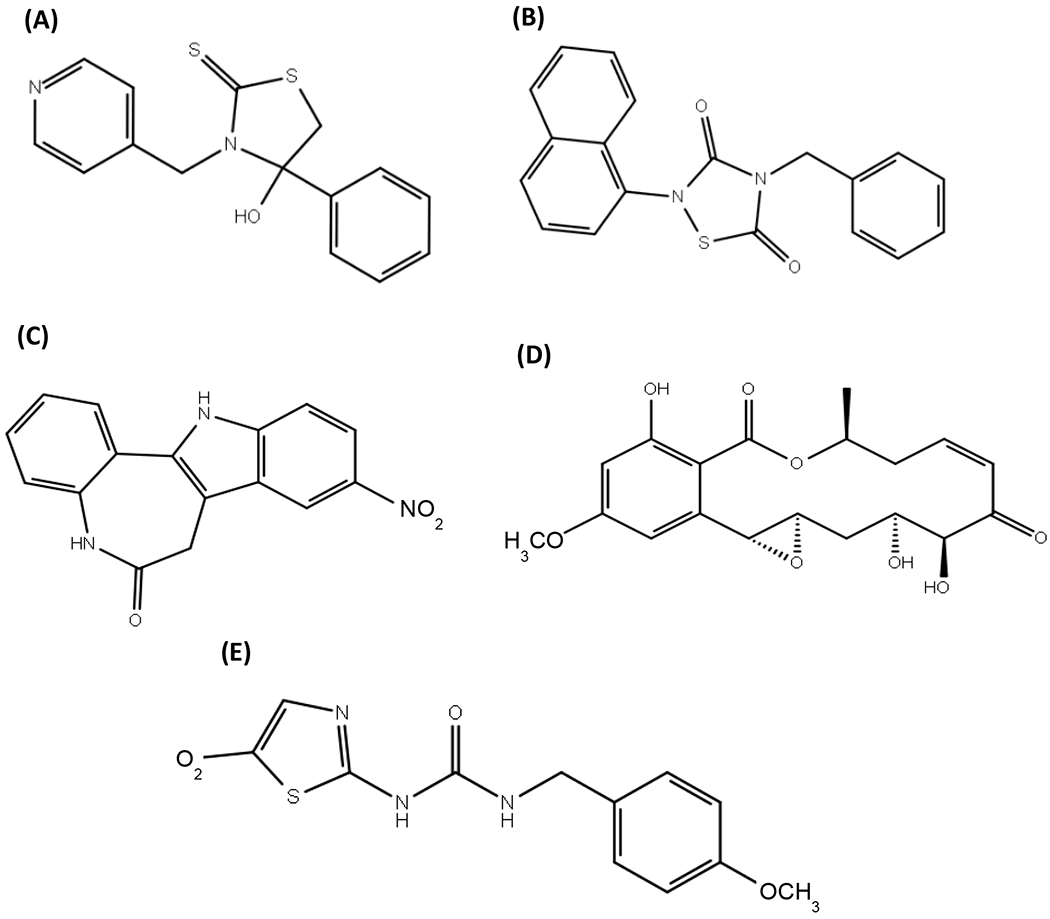
(A) COB-187, (B) Tideglusib, (C) Alsterpaullone, (D) Hypothemycin, and (E) AR-A014418.
Our preliminary studies indicated that COB-187 inhibition of GSK-3β is highly dependent on the ATP concentration. Thus, in the present study, we focused on the condition wherein the peptide substrate concentration was at a saturating level (to achieve a robust signal) and the ATP concentration was near its Km value. We first evaluated the progress curves of the GSK-3β reaction in the presence of COB-187. As shown in Fig. 2, as the concentration of COB-187 increased, the velocity of the GSK-3β reaction decreased and the velocity of the reaction, in the presence of COB-187, appeared to change with time.
Figure 2. Progress curves of GSK-3β activity in the presence of various concentrations of COB-187.
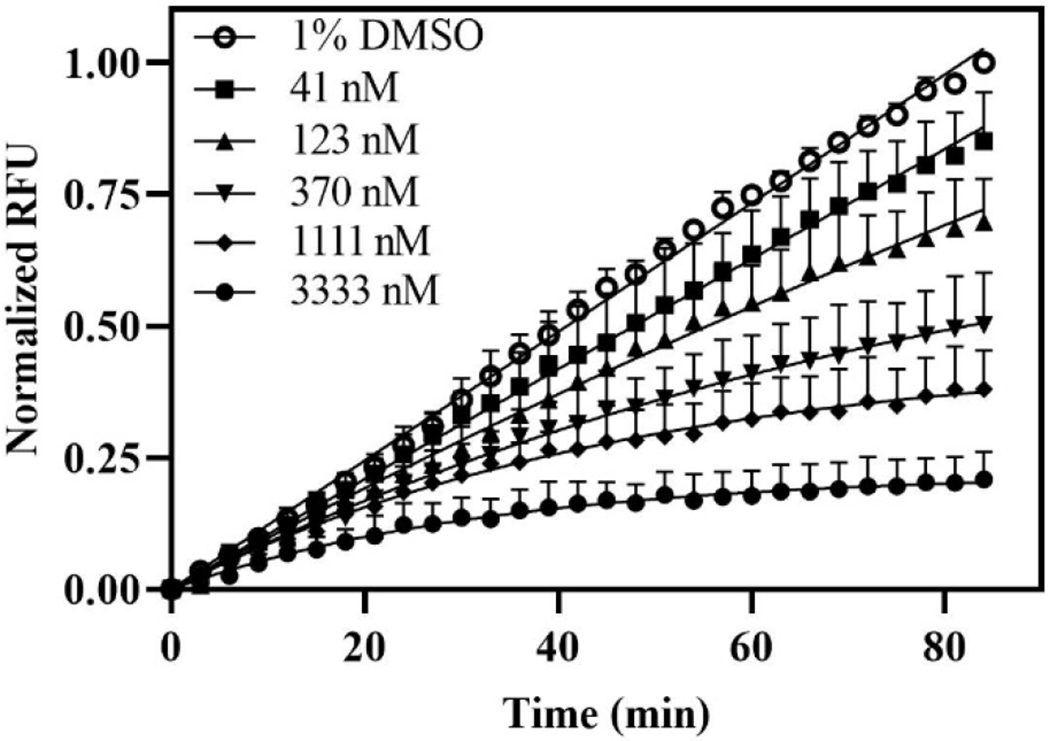
Progress curves were obtained with 0.5 nM GSK-3β at various concentrations of COB-187 provided in the inset; 1% DMSO is carrier control. Normalized RFU (y-axis) correlates with the cumulative enzyme activity while time (x-axis) is the time since the initiation of the reaction. Curve fits shown are to Eq. 4. Error bars represent the SD at that time point; n=5.
To explore this latter observation further, we considered Eq. 6.1 from Copeland22 shown below.
| (Eq. 2) |
This equation models the kinase reaction as consisting of two reaction regimes, an initial regime wherein the velocity of the reaction is V0 and a later regime wherein the reaction velocity is Vs. kobs reflects the “kinetics” of the transition between the two regimes. Note that this is a 3-parameter model; Vo, Vs and kobs. Two simplifications of this model describe other possible mechanisms. First, if the velocity does not change throughout the experiment, Vo = Vs, and Eq. 2 simply becomes a linear model:
| (Eq. 3) |
Alternatively, if the velocity does change with time but Vs is zero, Eq. 2 becomes a 2-parameter exponential decay model:
| (Eq. 4) |
We first compared the fit between the simplest model, i.e., the linear model (Eq. 3), and the next most complicated model, i.e., the 2-parameter model (Eq. 4). This analysis revealed that the progress curves representing GSK-3β activity in the presence of COB-187, with the exception of the lowest concentration analyzed (41 nM), had a better fit to the non-constant velocity model, Eq. 4, than the linear constant velocity model, Eq. 3 (see Supp. Table S1; upper set of data). We next compared the 2-parameter model, Eq. 4, to the 3-parameter model, Eq. 2, and found that the 2-parameter model was preferred (see Supp. Table S1; lower set of data). These observations indicate that there is indeed curvature in the progress curves in the presence of COB-187 that becomes more pronounced with increasing concentrations of COB-187 and reveal that the two-regime velocity model (Eq. 2) provided no better fit than the simpler exponential decay model (Eq. 4).
3.2. COB-187 is a reversible inhibitor of GSK-3β with a low koff
As stated, the progress curves fit well to Eq. 4. This equation can describe irreversible kinetics [see equation 9.1 from Copeland22]. This led us to question whether COB-187 binds to GSK-3β irreversibly. Thus, we next probed the dissociation, or lack thereof, of COB-187 from GSK-3β. For this we performed jump dilution experiments21,22 wherein COB-187 was pre-incubated with GSK-3β at a relatively high concentration of COB-187 and GSK-3β and then the solution was “jump diluted” simultaneously with starting the kinase reaction by adding the substrate and ATP. For comparison, we ran hypothemycin [an irreversible inhibitor14] and tideglusib [reported to be an irreversible inhibitor14] and alsterpaullone [a reversible inhibitor14].
Specifically, in the absence of substrate and ATP, we preincubated 50 nM of GSK- 3β with 10 μM of COB-187, 70 μM of hypothemycin, 20 μM of tideglusib or 400 nM of alsterpaullone. [Note that in preliminary experiments we observed that these concentrations of inhibitors are ≥ the IC90 for each inhibitor when the concentration of GSK-3β is 50 nM.] After the pre-incubation, these solutions were then diluted 100-fold with a master mix solution containing ATP and substrate. The final concentration of GSK-3β in each solution was 0.5 nM and the COB-187, hypothemycin, tideglusib, and alsterpaullone concentrations were 100 nM, 700 nM, 200 nM, and 4 nM, respectively. The ATP and substrate concentrations were 2 mM and 10 μM, respectively, well above the Km values we measured for our system (Supp. Fig. S2).
Representative curves from this analysis are shown in Fig. 3A, where it is revealed that GSK-3β pre-treated with hypothemycin and tideglusib, did not recover activity upon jump dilution. In contrast, GSK-3β pre-treated with alsterpaullone recovered most of its activity upon dilution. The GSK-β activity in the presence of COB-187 was time-dependent. In the first 30 minutes there was limited GSK-3β activity, but after 30 minutes the GSK-3β activity became pronounced and continued to increase (Fig. 3A). Additional replicates of the data presented in Fig. 3A are provided in Supp. Fig. S3.
Figure 3. Activity of inhibitor-treated GSK-3β after jump dilution.
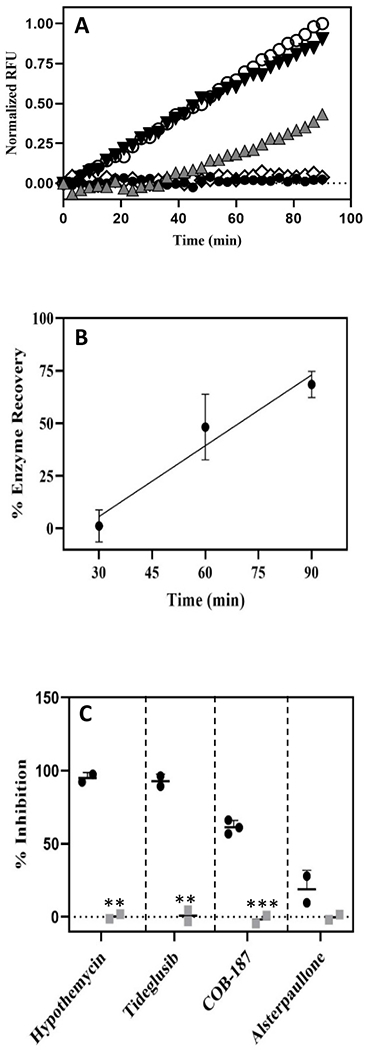
(A) Normalized RFU (y-axis) correlates with the cumulative enzyme activity while time (x-axis) is the time since the initiation of the reaction with the “jump dilution”. Each curve represents a treatment with the DMSO control or an inhibitor: open circles 1% DMSO control; black inverted triangles alsterpaullone; grey upright triangles COB-187; open diamonds tideglusib; and black circles hypothemycin. Curves from separate experiments, each normalized to its own DMSO control, have been plotted on the same axis for comparison (DMSO curve is representative from that set of experiments). Additional replicates provided in Supp. Fig. S3. (B) Recovery of GSK-3β pre-treated with COB-187 over time. The activity of GSK-3β pre-incubated with COB-187, relative to untreated GSK-3β, determined over three different time periods from the initiation of the reaction with the jump dilution, is shown. The activity of GSK-3β pre-treated with COB-187 increased with time from the jump dilution suggesting that the COB-187 was dissociating from GSK-3β. The 95% confidence interval of the slope of the regression line fit to the enzyme recovery data did not span zero indicating that the GSK-3β activity increased with time since the jump dilution. (n=3; error bar ± SD). (C) The percent inhibition of GSK-3β activity for each compound after the jump dilution experiment (black circles) and at the same condition as after the jump dilution but without preincubation of the GSK-3β with the compounds (grey squares) is shown. Error bars denote SD with n ≥2. ***p < 0.001 and **p < 0.01 as determined by t-test comparing the black and the grey symbols for each compound.
Since the RFU values are cumulative, the activity of GSK-3β at a given time point is difficult to discern from the RFU verses time plot. Therefore, we decided to discretize the 1.5-hour time course into three time periods, namely 0 to ≤ 30 min, > 30 to ≤ 60 min and > 60 to ≤ 90 min. Within each time period, we calculated the enzyme recovery and plotted this result versus time (Fig. 3B). As shown, the activity of GSK-3β treated with COB-187 increased over time and exceeds 50% of the untreated value in the last time period.
To further interpret the jump dilution results (Figs. 3A and 3B), we conducted an assay starting at the assay conditions present after the jump dilution (i.e., with no preincubation and at 0.5 nM GSK-3β, 2 mM ATP, 10 μM substrate, 700 nM of hypothemycin, 200 nM of tideglusib, 100 nM of COB-187, and 4 nM of alsterpaullone). As shown in Fig. 3C (grey squares), in stark contrast to that observed in the jump dilution experiment [Figs. 3A and 3C (black circles)], neither hypothemycin, tideglusib or COB-187 showed inhibition under these conditions. This observation (Fig. 3C - grey squares) strongly indicates that it is the pre-incubation step, in the absence of ATP and substrate, that allows hypothemycin, tideglusib and COB-187 to have an inhibitory effect in the jump dilution assay [Figs. 3A and 3C (black circles)], and further suggests that hypothemycin and tideglusib bind irreversibly to GSK-3β, as expected, and that COB-187 binds to GSK-3β through a slowly reversible reaction. Combined, the behavior of COB-187 is not consistent with an entirely irreversible or a reversible compound. Instead, the data in Fig. 3 indicate that COB-187 belongs to a class of compounds known as time-dependent inhibitors with relatively slow koff 22.
3.3. COB-187 appears to bind to GSK-3β via an induced conformation mechanism.
The jump dilution data indicate that there is not an irreversible bond formed between COB-187 and GSK-3β. As outlined by Copeland22, a plot of kobs versus inhibitor concentration allows one to gain insight into whether a “simple reversible slow binding” or “induced-fit mechanism” is operative. These mechanisms correspond to the following reaction schemes22:
| (Eq. 5) |
| (Eq. 6) |
For simple reversible slow binding, the relationship between kobs and the concentration of the inhibitor [I] is linear and is given by the following equation:
| (Eq. 7) |
For the induced-fit mechanism, the relationship between kobs and the concentration of the inhibitor [I] is a rectangular hyperbola and given by the following equation:
| (Eq. 8) |
where Ki app is the apparent Ki value.
Plots of kobs versus the concentration of COB-187 for the five replicate experiments used to generate Fig. 2 are shown in Supp. Fig. S4. Each appears to show curvature. The kobs values from each experiment were averaged and the resulting plot, along with the curve fit to Eqs. 7 and 8, are shown in Fig. 4. Curvature is, again, observed and the data appeared to fit better to Eq. 8 than Eq. 7, suggesting that COB-187 binds to GSK-3β via an induced-fit mechanism for the concentrations of COB-187 investigated. Interestingly, the average k4 determined from curve-fitting each replicate (Supp. Fig. 4) to Eq. 8 and taking the average, was quite low and less than a standard deviation away from zero 2.9 × 10−4 (±4.8 × 10−4 SD) (1/min).
Figure 4. kobs vs concentration of COB-187.
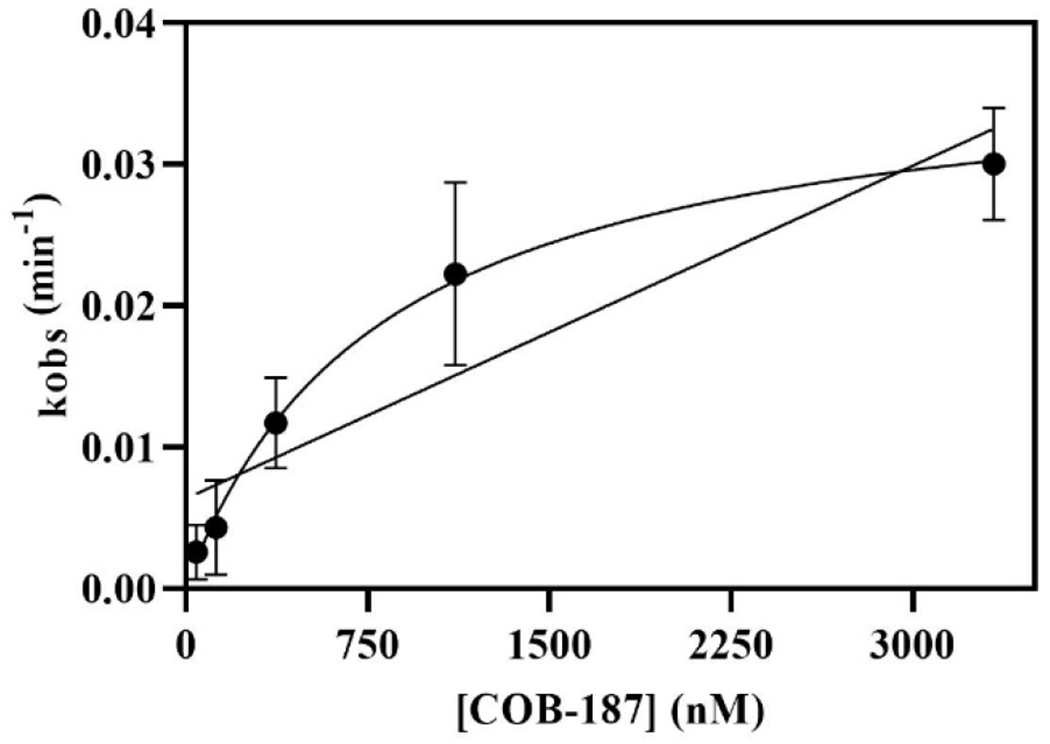
Average kobs, determined from the data in Fig. 2, versus concentration of COB-187. The data were fit to Eq. 7 (linear) and Eq. 8 (rectangular hyperbolic) of the text. The data appears to more closely fit Eq. 8. Replicates used to determine the average kobs are presented in Supp. Fig. S4. Error bars denote ± SD with n=5.
3.4. Preincubation decreases the IC50 value of COB-187 but not as dramatically as that observed for two known irreversible inhibitors
Previous work by Dominguez et al.14 demonstrated that the activity of tideglusib, an apparent irreversible inhibitor, is dramatically influenced by pre-incubation of tideglusib with GSK-3β. This trend was observed for COB-187 in the single dose jump dilution study (Fig. 3). Thus, we investigated the dose-response of COB-187, tideglusib, alsterpaullone, and hypothemycin’s activity on GSK-3β with and without preincubation. In contrast to the jump dilution experiment described in Fig. 3 which included a 100-fold dilution post-incubation, in the present experiment the solution was diluted only 3.3-fold postpreincubation.
As shown in Fig. 5 and Table 1, the potency of hypothemycin increased dramatically (~17-fold) with preincubation. Similar to that reported by Dominguez et al., preincubation caused a dramatic increase (~13-fold) in potency of tideglusib (Fig. 5, Table 1). In contrast, there was no significant increase in the potency of alsterpaullone (Fig. 5, Table 1). Preincubation significantly increased the potency of COB-187 but the shift was not as dramatic (< 4-fold) as that seen for hypothemycin and tideglusib (Fig. 5, Table 1). Thus, as was observed with the jump dilution experiments, COB-187’s behavior is between a reversible and an irreversible behavior.
Figure 5. Dose response curves for inhibition of GSK-3β with and without 3-hour pre-incubation with an inhibitor.
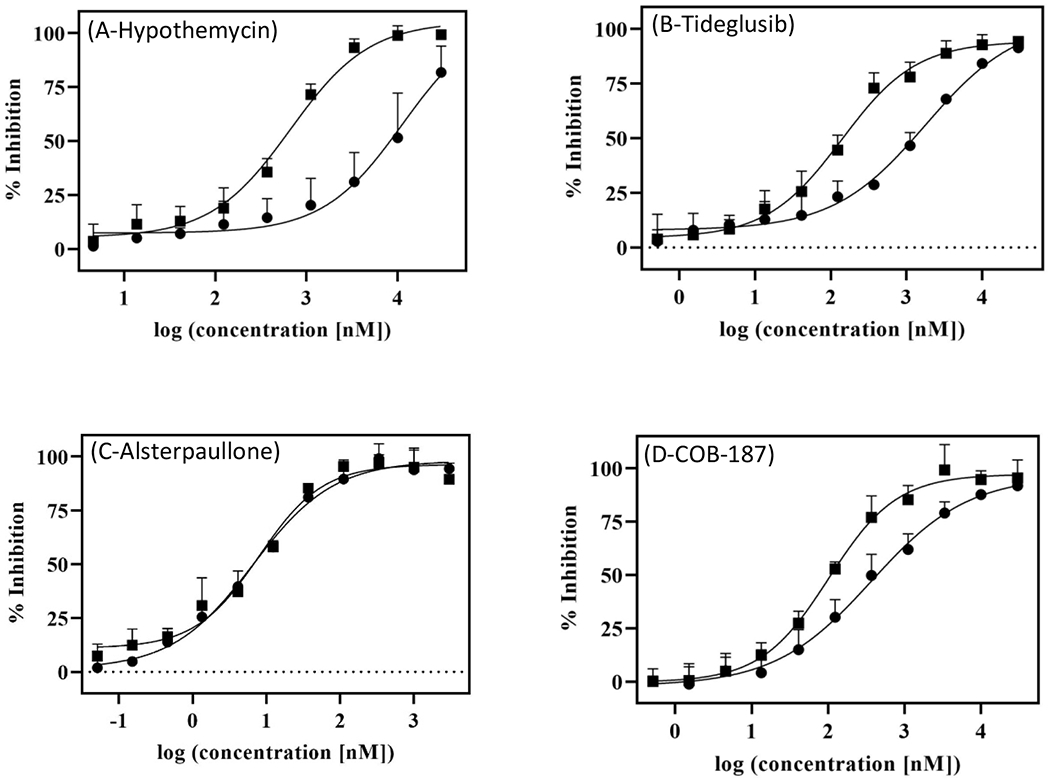
Dose-response curves of (A) Hypothemycin, (B) Tideglusib, (C) Alsterpaullone, and (D) COB-187 on wild type GSK-3β with (∎) or without (●) a 3-hour preincubation of GSK-3β with the compound. Error bars represent SD; n ≥2.
Table 1. IC50 values for inhibition of GSK-3β with and without a 3-hour pre-incubation.
The IC50 values (nM), determined by curve-fitting the data in Fig. 5 to Eq. 1, with and without 3-hour pre-incubation of GSK-3β with the inhibitor. Number in parentheses is the 95% CI;
| Inhibitor | No pre-incubation | 3-hr pre-incubation |
|---|---|---|
| COB-187 | 370 (251 to 592) | 102 (80.3 to 129)* |
| Tideglusib | 1780 (1090 to 4890) | 139 (97.3 to 198)* |
| Hypothemycin | 11300 (3760 to 48800) | 663 (462 to 949)* |
| Alesterpaullone | 6.5 (4.4 to 9.3) | 7.9 (4.7 to 12) |
indicates no overlap of the 95% CIs for the IC50s of the no pre-incubation and the 3-hour pre-incubation conditions for that inhibitor.
3.5. The ability of COB-187 and tideglusib to inhibit GSK-3β is significantly diminished in the presence of reducing agents
Our previous work15 and the assays described in the above sections, were performed in the absence of reducing agents. As demonstrated by Lee et al.23, the presence of a reducing agent in such assays can have a dramatic effect on the results. Reducing agents are used to limit oxidation of the kinase (e.g., cysteine residues) but can also interfere with, or provide insight into, the mechanism of action of the test compound. Since GSK-3β has a cysteine residue, Cys-199, at the entry to the ATP binding site that has been implicated in tideglusib’s mechanism of inhibition14, we explored the effect of reducing agents on COB-187’s ability to inhibit GSK-3β.
For these studies, we used DTT, TCEP and GSH which are reducing agents frequently used in kinase assays23 and tideglusib, hypothemycin and AR-A014418 for comparison. Note that we chose AR-A014418 since it is an inhibitor that is reported to work in the presence of reducing agents7. As shown in Fig. 6, COB-187, tideglusib and hypothmycin’s potencies were significantly diminished in the presence of the reducing agents with the effect on tideglusib being the most dramatic. In contrast, AR-A014418’s potency was not significantly altered by the presence of DTT or GSH. There was a significant but modest reduction in AR-A014418’s potency in the presence of TCEP. Interestingly, we observed that the AR-A014418 solution has a yellow tint that turns colorless in the presence of TCEP indicating a possible reaction between AR-A014418 and TCEP. Combined, the data presented in Fig. 6 clearly indicate that the inhibitory activities of COB-187, tidegulsib, and hypothemycin are significantly attenuated by the presence of reducing agents.
Fig 6. The effect of reducing agents DTT, GSH, and TCEP on the activity of GSK-3 inhibitors.
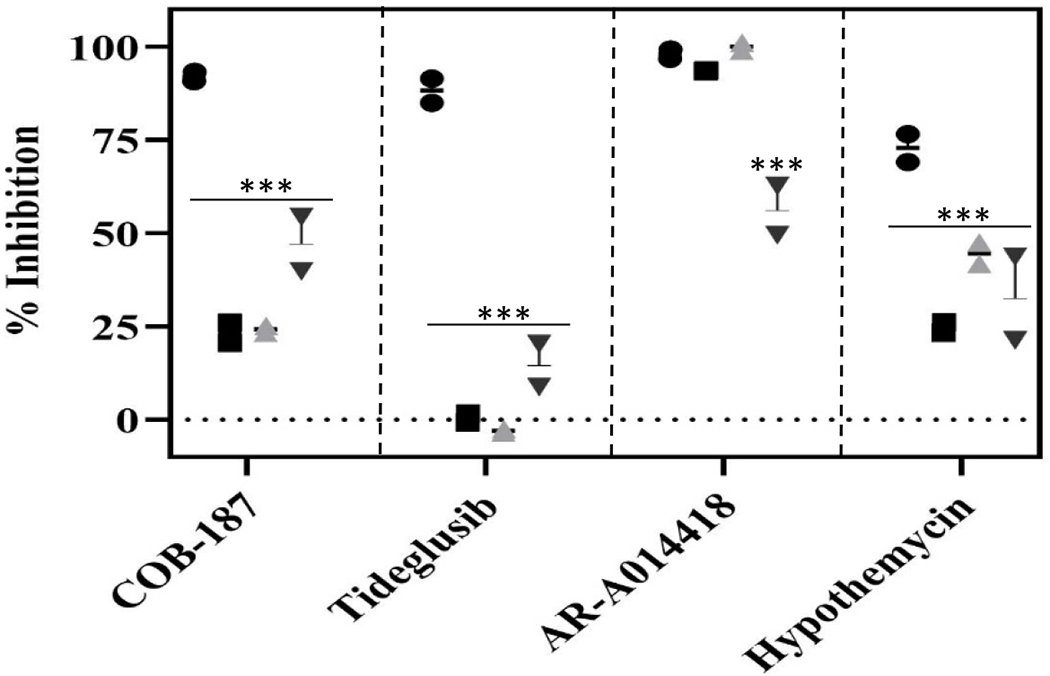
The effect of various inhibitors on GSK-3β activity, in the presence and absence of reducing agents. Circles - no reducing agent; squares - 2 mM DTT; grey upright triangles - 2 mM GSH; and black inverted triangles - 2 mM TECP. Error bars represent SD; n=2. ***p < 0.001 as determined by a Dunnett’s test comparing result with no reducing agent to the result in the presence of DTT, GSH or TCEP within each inhibitor group. Concentration of inhibitors was 10 μM.
3.6. COB-187 does not appear to be a non-specific thiol-reactive compound
As stated, GSK-3β has a cysteine, Cys-199, in its active site14. Thiol-reactive compounds can inhibit kinases which contain a cysteine in their active site by binding covalently to this amino acid 20 Indeed, hypothemycin, which is a Michael acceptor of nucleophiles like cysteine, is reported to covalently bind to kinases that have a conserved cysteine residue in their active site while other kinases, that do not have this residue, appear to be unaffected by hypothemycin14 20. Thus, a fairly straight forward explanation for the data presented in Fig. 6 regarding COB-187, is that (i) COB-187 covalently binds to Cys-199 of GSK-3β in the absence of reducing agents and (ii) COB-187 forms an adduct with the reducing agents rendering it unable to covalently bind to Cys-199 in the presence of reducing agents. That said, there is a key difference between hypothemyicn and COB-187. Specifically, hypothemycin has both an unsaturated α-β carbonyl group and an epoxide both of which are potent electrophiles capable of making a covalent bond with a nucleophile like cysteine20,24. These reactive functional groups of hypothemycin make it primed to react with enzymes containing cysteine or serine residues in their active sites20. In contrast, COB-187 does not have a reactive electrophilic group. Most importantly, the data in Fig. 3 strongly suggest that COB-187 is a reversible inhibitor. Nevertheless, we sought to probe possible interactions between COB-187 and Cys-199.
To understand the potential reaction of COB-187 with thiols such as cysteine or other reducing groups [e.g. DTT, TCEP, tributylphosphine] we attempted reactions between COB-187 and such reagents. COB-187 was stirred with both ethanethiol and tributylphosphine in tetrahydrofuran (THF). Reactions were carried out at several temperatures from room temperature to reflux and were analyzed by HPLC and 1H-NMR. There was no evidence of any reaction and only the starting material (COB-187) was observed.
Next, we screened COB-187 against 34 kinases in the human kinome that contain a conserved cysteine in their active site20,14 As shown in Table 2, in addition to GSK-3α and β, COB-187 only inhibited one kinase [MAPKAPK5(~64%)] other than GSK-3α and GSK-3β at >40%. These results indicate that should COB-187’s mechanism involve the cysteine in the active site of GSK-3β, it is likely not due to a “non-specific” chemical modification. Note that a similar argument was made by Dominguez14 for tideglusib, and COB-187 appears to be more specific for GSK-3 than tideglusib15.
Table 2. COB-187 is selective for GSK-3.
COB-187 was screened against a panel of recombinant human kinases that have a cysteine in their active site. Assay - Z’-LYTE (Z) or Lantha (L); ATP - level of ATP used in the Z’-LYTE assay; Right column presents the % inhibition in the presence of COB-187 ±SD (n=2). Bold face text - inhibition ≥ 80%; underlined text - inhibition between 79% and 40%; and plain text- inhibition < 40%.
| Kinases with Cys | Assay | ATP | Inhibition |
|---|---|---|---|
| AAK1 | L | - | 11 ± 1.5 |
| CDKL5 | Z | Km | 2 ± 3.2 |
| FLT1 (VEGFR1) | Z | Km | 0 ± 1.3 |
| FLT3 | z | Km | 2 ± 1.0 |
| FLT4 (VEGFR3) | z | Km | 1 ± 1.9 |
| GAK | L | - | 24 ± 5.9 |
| GSK3A (GSK3 alpha) | Z | Km | 91 ± 2.2 |
| GSK3B (GSK3 beta) | Z | Km | 89 ± 4.7 |
| KDR (VEGFR2) | Z | Km | 8 ± 8.2 |
| KIT | Z | Km | 4 ± 1.9 |
| MAP2K1 (MEK1) | Z | 100 | 6 ± 0.3 |
| MAP2K1 (MEK1) | L | - | 5 ± 0.4 |
| MAP2K2 (MEK2) | Z | 100 μM | 7 ± 0.8 |
| MAP2K2 (MEK2) | L | - | 4 ±0.5 |
| MAP2K3 (MEK3) | L | - | 7 ± 1.8 |
| MAP2K4 (MEK4) | L | - | 12 ± 0.3 |
| MAP2K5 (MEK5) | L | - | 1 ± 0.9 |
| MAP3K7/MAP3K7IP1 (TAK1-TAB1) | L | - | 21 ± 0.6 |
| MAPK1 (ERK2) | Z | Km | 29 ± 0.5 |
| MAPK15 (ERK7)* | L | - | 26 ± 0.9 |
| MAPK3 (ERK1) | Z | Km | 26 ± 3.4 |
| MAPKAPK5 (PRAK) | Z | Km | 60 ± 5 |
| MKNK1 (MNK1) | Z | Km | −2 ± 3.5 |
| MKNK2 (MNK2) | L | - | 1 ± 0.7 |
| NLK | L | - | 24 ± 1.2 |
| PDGFRA (PDGFR alpha) | Z | Km | −2 ± 0.5 |
| PDGFRB (PDGFR beta) | Z | Km | −2 ± 1.3 |
| PRKCN (PKD3) | Z | Km | 10 ± 0.1 |
| PRKD2 (PKD2) | Z | Km | 11 ± 1.8 |
| RPS6KA1 (RSK1) | Z | Km | 6 ± 1.1 |
| RPS6KA2 (RSK3) | Z | Km | 9 ± 3.4 |
| RPS6KA3 (RSK2) | Z | Km | 11 ± 0.8 |
| RPS6KA6 (RSK4) | Z | Km | 18 ± 2.0 |
| TGFBR2 | L | - | 20 ± 2.2 |
| ZAK | L | - | 9 ± 0.2 |
Note that in a previous screen (Noori et al.15), we reported inhibition of MAPK15 (ERK7) above 40% (specifically 42%) whereas here we found it to be < 40% (specifically 26%). The average of the two screens for MAPK15 is < 40%.
3.7. The mechanism of COB-187 inhibition of GSK-3β involves Cys-199
Previous studies have shown that Cys-199 is one of the key residues in the active site of GSK-314. While COB-187 does not appear to make a covalent bond with GSK-3β, the possibility remains that COB-187’s mechanism of inhibition involves Cys-199 or even binds to Cys-199 via a noncovalent bond. Thus, to further probe the mechanism by which COB-187 inhibits GSK-3β, we generated a mutated form of GSK-3β wherein Cys-199 was replaced with an alanine. This mutant is termed C199A. Prior to using C199A with COB-187, its substrate and ATP Km values were measured. This analysis revealed that replacement of Cys-199 with alanine significantly increases the ATP and substrate Km values; a 3.4- and 2.4-fold increase, respectively (Supp. Fig. S2). A comparison of the kinase activity of wild type GSK-3β to C199A in the presence of COB-187 revealed a dramatic (> 300-fold) decrease in the potency of COB-187 for the mutant; IC50 370 nM for wild type and 121,000 nM for C199A (n ≥ 4), Table 3.
Table 3. The IC50 for COB-187 is dramatically increased for the GSK-3β C199A mutant compared to wild type GSK-3β.
The IC50 values (nM) were determined from a curve fit to Eq. 1. Number in parentheses is the 95% CI;
| Inhibitor | Wild type | C199A |
|---|---|---|
| COB-187 | 370 (251 to 592) | 121,000 (88,100 to 187,000)* |
indicates no overlap of the 95% CIs for the IC50s between mutant and wild type. n ≥4.
4. Discussion
Our previous studies utilizing molecular assays demonstrated that COB-187 is a selective and potent inhibitor of GSK-3β15. In this study, we investigated the mechanism of inhibition. Using a molecular assay that allows real-time observation of GSK-3β activity, we observed that the rate of product formation in the presence of COB-187 was significantly reduced (Fig. 2). Curve fitting of the progress curves revealed that a model wherein the reaction velocity changes with time, i.e., Eq. 4, as compared to a model wherein the reaction velocity is non-variant with time, i.e., Eq. 3, provided a better fit to the data. This effect was most distinct at the higher levels of COB-187.
An important observation from this analysis is that the more complex time-dependent velocity model, i.e., Eq. 2, did not provide a better fit to the data compared to the simpler, time-dependent velocity model, i.e., Eq. 4. As stated in the results section, Eq. 4 can be used to describe irreversible kinetics [9.1 from Copeland22]. Thus, it could be argued that either (i) COB-187 is binding irreversibly to GSK-3β or (ii) COB-187 binds reversibly to GSK-3β but this reversible interaction is “functionally irreversible” in the context of the conducted experiments. Additional observations made in this paper suggest that the latter scenario is operative. First, a jump dilution analysis (Fig. 3; and Supp. Fig. S3) clearly reveals that GSK-3β recovers its activity after being exposed to a relatively high level of COB-187 that is subsequently diluted. Second, a likely mechanism of COB-187 irreversible binding would be via a covalent bond between the Cys residue in the active site of GSK-3β (Cys-199) and COB-187. In this case, it would be expected that COB-187 would inhibit multiple kinases that have a Cys residue in their active site. However, a screen of COB-187 against a panel of over 30 such kinases revealed that COB-187 is quite specific for GSK-3 (Table 2). Thus, COB-187 appears to be a specific reversible inhibitor of GSK-3β that behaves in an irreversible manner in the context of the present experiments.
The above considerations also provide insight into the kinetics of the reaction and suggest that COB-187 belongs to the class of compounds that exhibit relatively slow kinetics22. As discussed in Copeland22, a plot of kobs vs. the concentration of inhibitor provides insight into the mechanism of the slow binding. Our analysis (Supp. Fig. S4 and Fig. 4) suggests that, for the range of COB-187 concentrations analyzed, COB-187 inhibits GSK-3β via an induced-fit mechanism. Interestingly, the koff values determined from these experiments were quite low and not statistically different from zero. These later observations combined with Fig. 3 again support the argument that COB-187 does not form an irreversible bond with GSK-3β, but behaves, under the conditions used in the progress curve data collection, as a functionally irreversible inhibitor of GSK-3β. Finally, the fact that pre-incubation does appear to enhance the inhibitory effect of COB-187 (Fig. 5) may reflect the induced-fit nature of the binding.
Mutating Cys-199 of GSK-3β to an alanine had a dramatic attenuating (>300-fold) effect on the potency of COB-187 clearly indicating that Cys-199 is involved in the mechanism of inhibition. Interestingly, the Km value for both ATP and the substrate is significantly increased with the mutant compared to the wild type GSK-3β, but this increase is only ~3-fold and ~2-fold respectively. In contrast, the increase in the IC50 of COB-187 is > 300-fold when going from the wild type to C199A. This observation is consistent with the hypothesis that an interaction between Cys-199 and COB-187 is a key part of its mechanism of inhibition.
These findings impact the overall process of developing novel GSK-3 inhibitors based on COB-187 in several ways. First, the results bolster the hypothesis that the COB-187 mechanism of inhibition is distinct from the most clinically advanced GSK-3 inhibitor tideglusib. Specifically: (i) we have previously provided evidence that COB-187 is significantly more selective than tideglusib amongst over 400 kinases screened15; (ii) tideglusib has been reported to be an irreversible inhibitor of GSK-3β14, a finding supported by the results presented here (Fig. 3A), while COB-187 appears to be a reversible inhibitor (Fig. 3); and (iii) preincubation of tideglusib with GSK-3β causes a dramatic drop in the IC50 value (~13-fold) while preincubation of COB-187 with GSK-3β causes only a modest decrease in the IC50 value (< 4 fold). Second, these findings facilitate the rational and efficient development of a library of COB-187-like compounds that have an improved, compared to COB-187, inhibition profile in molecular/cell-based assays. Indeed, we are currently using docking studies and molecular simulations in our lead optimization process. The mechanistic findings revealed in the present study complement this effort (e.g., the finding that the Cys-199 residue is critical to the mechanism provides a locus for the docking and simulation studies). Finally, the majority of this study was done with the GSK-3β isoform. While the two GSK-3 isoforms have significant similarity in their kinase domains3, there is increasing interest in GSK-3α specific inhibitors25. Thus, a series of studies with GSK-3α and a family of COB-187-like molecules, similar to the studies conducted here with COB-187 and GSK-3β, are clearly warranted and will provide further insight into the mechanism of COB-187 inhibition of GSK-3 and, perhaps, guide the development of novel isoform specific inhibitors.
In summary, COB-187 appears to be a specific, reversible, time-dependent inhibitor of GSK-3β and the mechanism involves Cys-199.
Supplementary Material
Acknowledgments
DJG would like to thank Elaine R. Goetz for useful discussions.
Funding
This work was supported by the National Institutes of Health [R15GM110602].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
Ohio University owns a patent on COB-187. SCB and DJG are inventors on the patent.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References.
- 1.Woodgett JR Molecular cloning and expression of glycogen synthase kinase-3/Factor A. EMBO J vol. 9, 2431–2438 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Embi N, Rylatt DB & Cohen P Glycogen synthase kinase-3 from rabbit skeletal muscle separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochein vol. 107, 519–27 (1980). [PubMed] [Google Scholar]
- 3.Woodgett JR & Cormier KW Recent advances in understanding the cellular roles of GSK-3. F1000Research vol. 6, 167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kauffman MR, Nazemidashtarjandi S, Ghazanfari D, Allen AE, Reynolds NM, Faik A, Burdick MM, McCall KD, Goetz DJ Evidence that knock down of GSK-3β in Chronic Myelogenous Leukemia cells augments IFN-γ-induced apoptosis. Leuk. Res vol. 99, 106464 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jope RS, Yuskaitis CJ & Beurel E Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochemical Research vol. 32, 577–595 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eldar-Finkelman H, Licht-Murava A, Pietrokovski S & Eisenstein M Substrate competitive GSK-3 inhibitors - strategy and implications. Biochimica et Biophysics Acta - Proteins and Proteomics vol. 1804, 598–603 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Bhat R, Xue Y, Berg S, Hellberg S, Ormö M, Nilsson Y, Radesäter AC, Jerning E, Markgren PO, Borgegård T, Nylöf M, Giménez-Cassina A, Hernández F, Lucas JJ, Díaz-Nido J, and Avila J. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem vol. 278, 45937–45945 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Anastassiadis T, Deacon SW, Devarajan K, Ma H & Peterson JR Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol vol. 29, 1039–1045 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eldar-Finkelman H & Martinez A GSK-3 inhibitors: preclinical and clinical focus on CNS. Front. Mol. Neurosci vol. 4, article 32, 1–18 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anraku T, Kuroki H, Kazama A, Bilim V, Tasaki M, Schmitt D, Mazar A, Giles FJ, Ugolkov A, and Tomita Y. Clinically relevant GSK-3β inhibitor 9-ING-41 is active as a single agent and in combination with other antitumor therapies in human renal cancer. Int. J. Mol. Med vol. 45, 315–323 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuroki H, Anraku T, Kazama A, Bilim V, Tasaki M, Schmitt D, Mazar AP, Giles FJ, A. 9-ING-41, a small molecule inhibitor of GSK-3beta, potentiates the effects of anticancer therapeutics in bladder cancer. Sci. Rep vol. 9, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez A, Alonso M, Castro A, Dorronsoro I, Gelpí JL, Luque FJ, Pérez C, and Moreno FJ. SAR and 3D-QSAR studies on thiadiazolidinone derivatives: Exploration of structural requirements for glycogen synthase kinase 3 inhibitors. J. Med. Chem vol. 48, 7103–7112 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Lovestone S, Boada M, Dubois B, Hüll M, Rinne JO, Huppertz HJ, Calero M, Andrés MV, Gómez-Carrillo B, León T, and Del Ser T. A phase II trial of tideglusib in alzheimer’s disease. J. Alzheimer’s Dis vol. 45, 75–88 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Domínguez JM, Fuertes A, Orozco L, Del Monte-Millán M, Delgado E, and 28 M. Medina. Evidence for irreversible inhibition of glycogen synthase kinase-3β by tideglusib. J. Biol. Chem vol. 287, 893–904 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noori MS, Bhatt PM, Courreges MC, Ghazanfari D, Cuckler C, Orac CM, McMills MC, Schwartz FL, Deosarkar SP, Bergmeier SC, McCall KD, and Goetz DJ. Identification of a novel selective and potent inhibitor of glycogen synthase kinase-3. Am. J. Physiol. - Cell Physiol vol. 317, 1289–1303 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noori MS, Courreges MC, Bergmeier SC, McCall KD & Goetz DJ Modulation of LPS-induced inflammatory cytokine production by a novel glycogen synthase kinase-3 inhibitor. Eur. J. Pharmacol vol. 883, 173340 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shults MD, Janes KA, Lauffenburger DA & Imperiali BA multiplexed homogeneous fluorescence-based assay for protein kinase activity in cell lysates. Nat. Methods vol. 2, 277–283 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Luković E, González-Vera JA & Imperiali B Recognition-domain focused chemosensors: Versatile and efficient reporters of protein kinase activity. J. Am. Chem. Soc vol. 130, 12821–12827 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shults MD & Imperiali B Versatile fluorescence probes of protein kinase activity. J. Am. Chem. Soc vol. 125, 14248–14249 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Schirmer A, Kennedy J, Murli S, Reid R & Santi DV Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. Proc. Natl. Acad. Sci. U. S. A vol. 103, 4234–4239 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Copeland RA, Basavapathruni A, Moyer M & Scott MP Impact of enzyme concentration and residence time on apparent activity recovery in jump dilution analysis. Anal. Biochem vol. 416, 206–210 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Copeland RA Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists: Second Edition (Wiley, 2013). doi: 10.1002/9781118540398. [DOI] [PubMed] [Google Scholar]
- 23.Lee H, Torres J, Truong L, Chaudhuri R, Mittal A, and Johnson ME. Reducing agents affect inhibitory activities of compounds: Results from multiple drug targets. Anal. Biochem vol. 423, 46–53 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnan S, Miller RM, Tian B, Mullins RD, Jacobson MP, and Taunton J. Design of reversible, cysteine-targeted michael acceptors guided by kinetic and computational analysis. J. Am. Chem. Soc vol. 136, 12624–12630 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benajiba L Wanger F Ross L Galinsky I DeAngelo D, Gale J Pan J Zhang Y Sacher J Weiwer M Stone R, Holson E Stegmaier K Identification of a first in class GSK3-alpha selective inhibitor as a new differentiation therapy for AML. Blood vol. 126, 870 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.