

Abstract
Human pluripotent stem cells (PSCs) have been utilized as a promising source in regenerative medicine. However, the risk of teratoma formation that comes with residual undifferentiated PSCs in differentiated cell populations is most concerning in the clinical use of PSC derivatives. Here, we report that a monoclonal antibody (mAb) targeting PSCs could distinguish undifferentiated PSCs, with potential teratoma-forming activity, from differentiated PSC progeny. A panel of hybridomas generated from mouse immunization with H9 human embryonic stem cells (hESCs) was screened for ESC-specific binding using flow cytometry. A novel mAb, K312, was selected considering its high stem cell-binding activity, and this mAb could bind to several human induced pluripotent stem cells and PSC lines. Cell-binding activity of K312 was markedly decreased as hESCs were differentiated into embryoid bodies or by retinoic acid treatment. In addition, a cell population negatively isolated from undifferentiated or differentiated H9 hESCs via K312 targeting showed a significantly reduced expression of pluripotency markers, including Oct4 and Nanog. Furthermore, K312-based depletion of pluripotent cells from differentiated PSC progeny completely prevented teratoma formation. Therefore, our findings suggest that K312 is utilizable in improving stem cell transplantation safety by specifically distinguishing residual undifferentiated PSCs.
Keywords: Cell-surface marker, Monoclonal antibody, Pluripotent stem cells, Stem cell differentiation, Teratoma
INTRODUCTION
Human pluripotent stem cells (PSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells, have emerged as promising tools in the fields of regenerative medicine and tissue engineering (1, 2). As PSCs exhibit an unlimited self-renewal capacity and maintain pluripotency, leading to their differentiation into cells of all three embryonic germ layers, they are particularly attractive as well as suitable sources for cell replacement therapy (3, 4). However, a few obstacles hinder the clinical use of PSCs after differentiation induction. In particular, the teratoma risk raised by undifferentiated PSCs is a major limitation in applying PSCs for transplantation (5, 6). Hence, various strategies have been developed to remove undifferentiated PSCs, including treatment with cytotoxic reagents or sorting of undifferentiated cells through flow cytometry (7). Nevertheless, improving the efficiency of undifferentiated cell removal remains essential. Thus, developing an efficient tool to distinguish undifferentiated PSCs with tumorigenic potential from heterogeneous PSC populations is indispensable for the progression of PSC application in clinical settings.
Discovering surface markers specifically expressed on PSCs is a prerequisite to not only assess the pluripotency level of PSCs but also isolate the undifferentiated population using antibody-based separation methods. Several cell-surface markers specific to PSCs, such as stage-specific embryonic antigen (SSEA)-3, SSEA-4, Tra-1-60, and Tra-1-81, have been identified to date and commonly used for detecting PSCs (8). However, the expression of these markers is not completely restricted to PSCs, and hence, for isolating the undifferentiated cell population, an antibody targeting one of these markers to distinguish com-pletely the teratogenic cells would be insufficient. For example, Tang et al. reported that an anti-SSEA-5 monoclonal antibody (mAb) alone reduces the tumorigenic potential, but complete removal of teratoma formation is achieved by targeting three pluripotent cell-surface markers, SSEA-5, CD9, and CD90 (9). Owing to this limitation, identifying a more reasonable cell surface marker to distinguish the teratogenic cell population from heterogeneous PSCs remains necessary.
Previously, we developed a mAb that binds to PSCs and identified L1 cell adhesion molecule (L1CAM) as a target antigen maintaining the pluripotency and self-renewal activity of PSCs (10). In addition, another cell surface molecule, desmoglein-2 (DSG2), has been identified as a novel PSC-specific marker, and DSG2-negative cell population, obtained through anti-DSG2 mAb targeting, exhibits a remarkable suppression of teratoma formation (11).
In this study, we generated a novel mAb, K312, which bound to a glycan moiety specifically presented on undifferentiated PSCs. We evaluated the specificity of K312 in separating a cell population with high pluripotency and tumorigenic potential from differentiated PSCs. Our study might provide a new strategy for effectively managing the risk of teratoma formation with stem cell therapy in clinical settings.
RESULTS
Identification of a mAb specific to human PSCs
We generated a panel of hybridomas, which was raised against undifferentiated H9 human ESCs (hESCs). On screening mAbs that bind to H9 hESCs, we identified a novel mAb, K312, and evaluated its selectivity in determining the PSC population. As shown in Fig. 1A, K312 strongly binds to H9 hESCs, similar to mAbs against other cell-surface pluripotency markers, such as Tra-1-60, Tra-1-81, SSEA-3, and SSEA-4. The specific binding of K312 to pluripotent cells was further validated in two induced pluripotent stem cell lines (iPS-fAD and iPS-SPD), as well as two embryonic carcinoma cell lines (NTERA-2 and NCCIT), but K312 does not bind to J1 mouse ESCs (Fig. 1B). We also observed that K312 correlatively binds to H9 hESCs harboring each of the indicated pluripotency markers (Fig. 1C). Subsequently, immunofluorescence analysis of H9 hESCs showed that K312 is co-localized with E-cadherin, a well-known cell-surface pluripotency marker, thereby, verifying the selective binding of K312 to pluripotent cells (Fig. 1D). To identify a target antigen of K312, we performed mass spectrometry analysis of a protein fraction, corresponding to approximately 60 kDa, immunoprecipitated using K312 (Fig. 1E). However, no meaningful candidate protein target was identified as K312 antigen (Supplementary Fig. 1 and 2). As PSC surface markers are occasionally composed of complex glycan structures (12), we hypothesized that K312 might interact with polysaccharide structures of a specific target antigen expressed on PSCs. To assess the ability of K312 to bind glycan, H9 hESC or NCCIT lysates were incubated with PNGase F for deglycosylation and pulled down by K312. Interestingly, the target protein band disappeared after PNGase F treatment, even with 30 min incubation (Fig. 1F). Thus, we could suggest that K312 specifically binds to human, but not mouse, PSCs and recognizes a carbohydrate moiety of the target antigen when it exists in a glycosylated form.
Fig. 1.

Monoclonal antibody K312 specifically binds to human pluripotent stem cells (PSCs). (A) Flow cytometry analysis of the expression of PSC surface markers and the target of K312 on H9 human embryonic stem cells (H9 hESCs). (B) K312 specifically binds to human pluripotent cell lines (iPS-fAD, iPS-SPD, NTERA-2, and NCCIT), but not to J1 mouse embryonic stem cells. (C) Flow cytometry analysis of the correlative expression of PSC markers and the target of K312. H9 hESCs were stained with K312 and the indicated PSC marker-specific antibodies. (D) Immunofluorescence staining shows the co-localization of E-cadherin and the target protein of K312 on H9 hESCs. Scale bar: 25 μm. (E) Immunoprecipitation of H9 hESC lysates with control mouse IgG (mIgG1) or K312; cells were analyzed by immunoblotting. Black arrow indicates a protein band of approximately 60 kDa appearing after immunoprecipitation with K312. (F) Deglycosylation assay. H9 hESC or NCCIT lysates were treated with PNGase F or left untreated for the indicated times, and analyzed by immunoblotting using K312. Cell lysates not treated with PNGase F were used as control (Ctrl).
K312 binding is dependent on the pluripotency of hESCs
We next investigated whether the cell-binding activity of K312 was limited to pluripotent cells. To this end, H9 hESCs were differentiated through embryoid body (EB) formation or retinoic acid (RA) treatment, and then, K312 binding to the cells was evaluated by fluorescence-activated cell sorting (FACS). As shown in Fig. 2A, the binding capacity of K312, as well as of mAbs targeting SSEA-3 and Tra-1-60, gradually decreases as H9 hESCs are differentiated to form EBs until 9 days. In addition, the protein expression levels of various pluripotency markers were evaluated by western blotting. As expected, the expression levels of the K312 target, E-cadherin, EpCAM, Oct4, Nanog, and Sox2 were notably reduced in the differentiated hESCs (Fig. 2B). To validate the loss of binding of K312 on differentiated hESCs, H9 hESCs were treated with RA for 12 days. Consistent with the EB formation assay, RA treatment downregulated the expression of the target of K312 and other pluripotency markers in a time-dependent manner (Fig. 2C, D). Therefore, we demonstrate that K312 specifically binds to the undifferentiated hESCs, and can be used for identifying cells in the pluripotent state.
Fig. 2.
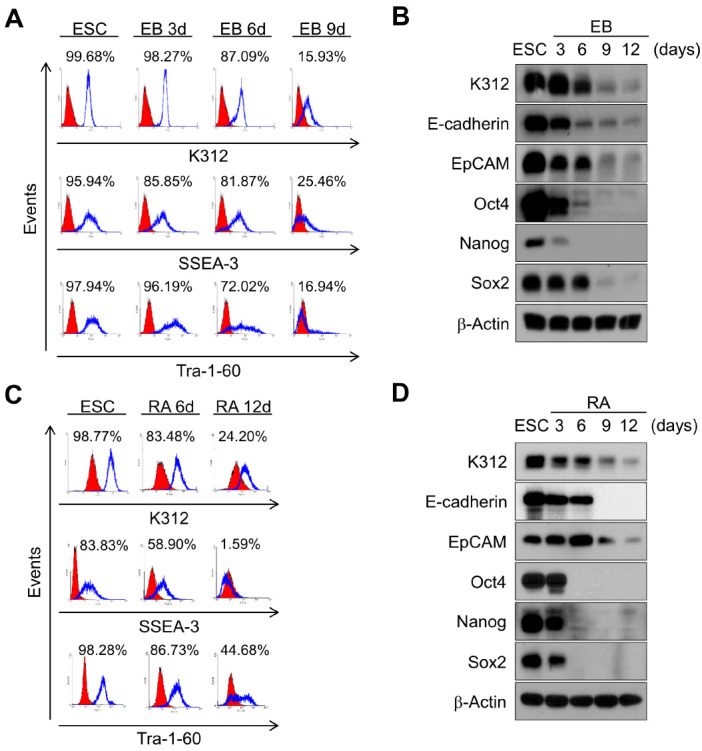
Binding capacity of K312 to H9 hESCs decreases in a differentiation-dependent manner. (A) H9 hESCs were differentiated into embryoid bodies (EBs) for 9 days, and flow cytometry analysis reveals the binding of K312 to cells, in comparison with that of antibodies against stage-specific embryonic antigen (SSEA)-3 and Tra-1-60. (B) Immunoblotting analysis of the expression of the target of K312, PSC surface markers (E-cadherin and EpCAM), and transcription factors (Oct4, Nanog, and Sox2) on the indicated days. (C and D) Downregulation of the target of K312 by retinoic acid (RA)-induced differentiation of H9 hESCs. Binding of K312 to cells and protein expression on each day of differentiation were analyzed by flow cytometry and immunoblotting, respectively.
K312 distinguishes a highly pluripotent ESC population from heterogeneous hESCs
Given the selective binding of K312 to pluripotent hESCs, we sought to examine the ability of K312 in classifying hESCs into two cell populations with high or low pluripotency. Undifferentiated H9 hESCs were gated based on an isotype control antibody and forward scatter, and by probing the cells with K312, we obtained two different cell populations, K312-high and K312-low (Fig. 3A). Next, the expression of pluripotency markers was analyzed by western blotting in these cell subsets. As shown in Fig. 3B, a clear difference exists between K312-high and K312-low cells in the expression of Oct4, Nanog, and E-cadherin, with the expression being higher in K312-high cells than K312-low cells. Furthermore, the clonogenic activity of these cell populations was assessed by colony formation assays. As expected, K312-high cells developed into compact colonies, whereas K312-low cells rarely formed colonies (Fig. 3C, D). These results indicated that K312 successfully distinguishes a cell population with high pluripotency from heterogeneous hESC populations.
Fig. 3.
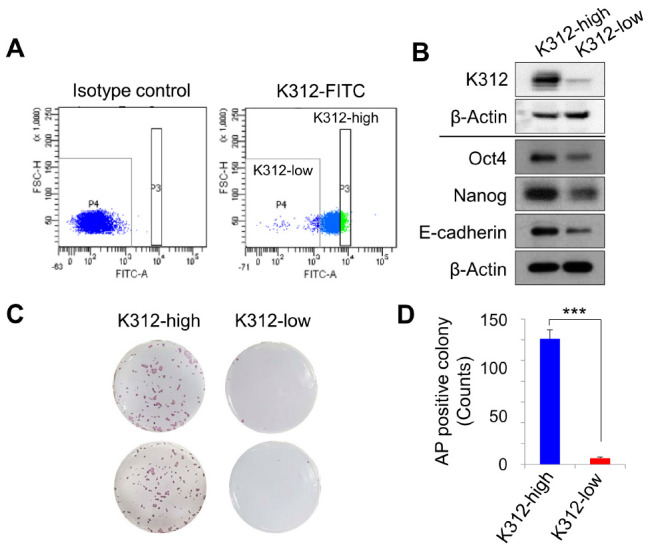
K312 distinguishes highly pluripotent cells from undifferentiated H9 hESCs. (A) H9 hESCs stained with K312 separate into K312-high and -low cell populations by cell gating with an isotype control antibody. (B) Immunoblotting analysis of the expression of the target of K312, Oct4, Nanog, and E-cadherin in K312-high and -low cells. (C and D) Single K312-high and -low cells were cultured for 1 week to form colonies. Colonies were analyzed by alkaline phosphatase staining (C) and counted for quantitative comparison (D). ***P < 0.001, Student’s t-test.
K312 effectively removes teratogenic cells from differentiated PSC progeny
We next investigated whether K312 was utilizable in separating undifferentiated cells from the differentiated progeny. To this end, H9 hESCs were differentiated under RA treatment for 12 days. Under the differentiation conditions, H9 hESCs were converted into a differentiated phenotype characterized by formation of round colonies with well-defined edges and prominent nucleoli in individual cells of a colony (Fig. 4A). Differentiated H9 hESCs were separated into K312-high and -low populations by FACS after labeling the cells with K312 (Fig. 4B). Western blotting revealed that Oct4, Nanog, and the antigen of K312 were only detected in K312-high cells, implying that K312 can be used for distinguishing pluripotent cells from not only undifferentiated hESCs, but also from the differentiated progeny (Fig. 4C).
Fig. 4.

Depletion of pluripotent cells by K312 prevents teratoma formation by differentiated H9 hESCs. H9 hESCs were differentiated by retinoic acid (RA) treatment for 12 days. (A) Morphological differences between undifferentiated and RA-treated hESCs observed using a microscope. (B) Fluorescence-activated cell sorting of K312-high and -low cell populations from RA-treated H9 hESCs. (C) Immunoblotting analysis of the expression of the target of K312, Oct4, and Nanog in K312-high or K312-low cells. (D and E) Teratoma formation by K312-high and -low cells. Control mouse embryonic fibroblasts (MEFs), K312-low cells, or K312-high cells were injected into each side of the mouse testes. (D) Teratoma formation observed 8 weeks after cell transplantation. (E) Differences in the teratoma volume and weight between the groups. Error bars indicate ± SEM. **P < 0.01, Student’s t-test. (F) Representative hematoxylin and eosin (H&E) staining images of the testes sections. (G) RT-PCR analysis of the expression of the representative germ layer markers.
Teratoma assay is commonly used to assess the tumorigenic potential of undifferentiated or differentiated ESCs (13). By transplanting cells into the testis of immunodeficient mice, we further compared the teratoma-forming activity of K312-high and K312-low cells. Surprisingly, teratomas were formed by K312-high cells, but not K312-low cells or control mouse embryonic fibroblasts (MEFs), indicating that the target antigen of K312 is a crucial marker for separating tumorigenic cells from the differentiated progeny (Fig. 4D). Tumor volumes and weights were also significantly different between the K312-high and K312-low groups (Fig. 4E). To evaluate the differentiation of K312-high cells into the three germ layers, the testicles excised from mice were subjected to histological analysis. As shown in Fig. 4F, the neural epithelium, respiratory endothelium, and myxoid tissue, representing the ectoderm, endoderm, and mesoderm, respectively, were developed from K312-high cells, but were not observed in the testicles treated with control MEFs or K312-low cells. In addition to histology, expression of the representative molecular markers of the three germ layers was analyzed by RT-PCR. The markers specific to the endoderm (GATA6, FN1, AMY2A), mesoderm (ACAN, HAND1, MSX1), and ectoderm (MAP2, NESTIN, TUBB3) were markedly expressed in the testicle tissues of mice belonging to the K312-high group but not the K312-low or control group (Fig. 4G). Thus, K312 effectively eliminates the risk of teratoma formation by sorting out teratogenic cells from the differentiated cell progeny after in vitro differentiation of hESCs.
DISCUSSION
Numerous studies have been conducted on the clinical application of PSCs, especially for stem cell transplantation. However, given the plasticity and heterogeneity of PSCs after in vitro differentiation, safety, especially that pertaining to tumorigenicity, is a major concern when differentiated PSCs are transplanted in vivo (14, 15). To address this issue, several antibodies targeting PSC-specific antigens have been developed to isolate fully differentiated PSCs by depleting undifferentiated PSCs (8). In this study, we demonstrate that a novel mAb K312 spec-ifically binding to human PSCs could separate the pluripotent cell population from differentiated PSCs. We found that pluripotent cells are considered to be depleted from the differentiated PSC progeny when K312 fails to bind to the differentiation-induced cells (Fig. 2A, C). This implies that the target antigen of K312 is specifically expressed on PSCs and can be a cell-surface pluripotency marker useful for antibody-based cell sorting approaches. Consistent with the decreased binding of K312, the pluripotency markers were barely detected in the cells negatively isolated using K312 (Fig. 2B, D), indicating that the surface expression of the K312 target is intimately correlated with the pluripotency of cells. Furthermore, K312 clearly distinguished the K312-low and K312-high cell populations from undifferentiated H9 hESCs (Fig. 3A). These results suggest that K312 is also utilizable for assessing whether PSC lines stably maintain their pluripotency.
Well-defined cell-surface pluripotency markers precisely indicate the pluripotent state of PSCs. As is generally known, glycan moieties are incorporated in common cell-surface pluripotency markers, such as glycolipids SSEA-3 and SSEA-4 or keratan sulphate proteoglycan Tra-1-60 and Tra-1-81, and various mAbs binding to glycan in these markers have been developed (8, 16). However, these markers tend to be expressed on tumor cells and are occasionally not suitable to describe the identity of PSCs (17, 18). Thus, it is necessary to discover a valuable surface antigen specific to PSCs. In this study, we determined that the expression of a PSC-specific surface antigen targeted by K312 is well representative of the pluripotent state of various PSCs, with its expression being downregulated in differentiation-induced H9 hESCs, indicating that the antigen could be a novel marker specific for PSCs but not differentiated PSC progeny (Fig. 1B and Fig. 2). Moreover, H9 hESCs expressing SSEA-3, SSEA-4, Tra-1-60, or Tra-1-80 were counterstained with K312, implying that the antigen of K312 is expressed independently of the other markers (Fig. 1C). Although the target of K312 was not identified here, we demonstrated that K312 binds to the glycan moiety of a target specifically expressed on PSCs. Thus, future investigation is warranted to identify the target of K312.
Prior to the emergence of various efforts for the clinical application of PSCs, the tumorigenic potential of differentiated PSCs should be assessed in vivo. In this regard, the teratoma assay is the most reliable method because it directly monitors the differential development of PSCs into a wide range of tissues (13). In terms of assessing the risk of teratoma formation with PSCs differentiated in vitro, several studies have shown that an antibody targeting a PSC-specific marker, such as SSEA-5 or claudin-6, can sort teratogenic cells from differentiated progeny, and thereby manage the teratoma risk accompanied with differentiation-induced PSCs (9, 19). Although these studies demonstrate the substantial utilization of antibody-based depletion of tumorigenic PSCs, complete elimination of teratoma formation is observed when the tumorigenic cells are immuno-depleted using antibodies against SSEA-5 and two additional pluripotency markers (CD9 and CD30) or are killed by claudin-6-targeting cytotoxic drugs. Interestingly, we observed that the K312-low cell population did not completely result in develop-ing a teratoma, same as the control MEFs, which indicates that managing the risk of teratoma formation is possible just by K312-based depletion of teratogenic cells (Fig. 4D, E). These data also imply that the target of K312 is essential for maintaining the self-renewal capacity and tumorigenic activity of PSCs.
In conclusion, K312, which specifically targeted PSCs, not only identified the pluripotent state of PSCs, but also separated undifferentiated tumorigenic PSCs from differentiated PSCs, suggesting that it is valuable to further the clinical application of stem cell-based therapy by eliminating the risk of teratoma formation.
MATERIALS AND METHODS
Cell culture
H9 hESCs, iPS-fAD and iPS-SPD, were cultured in DMEM/F12 supplied with 20% serum replacement and 10 ng/ml basic fibroblast growth factor (bFGF). For culturing J1 mouse ESCs, leukemia inhibitory factor was added instead of bFGF in the medium as described above. These feeder-based cell lines were cultured on irradiated MEFs. Embryonic carcinoma cell lines, NTERA-2 and NCCIT, were cultured as previously described (11). For feeder-free culture, H9 hESC clumps were transferred onto Matrigel (BD Bioscience)-coated plates and cultured with mTeSR medium (STEMCELL Technologies). EB formation assay was performed using an AggreWell plate (STEMCELL Technologies) as described previously (11). For in vitro differentiation, H9 hESCs were cultured in the medium not containing bFGF and treated with 10 μM RA (Sigma-Aldrich).
Hybridoma generation and mAb purification
BALB/c mice were immunized with H9 hESCs as described previously (10). Hybridomas were generated and screened to select mAbs binding to H9 hESCs via flow cytometry. mAbs secreted from hybridoma cells were purified using Protein G-Sepharose column chromatography as described previously (10).
FACS analysis
Cells were dissociated using Accutase (STEMCELL Technologies) for 10 min and washed using 3% bovine serum albumin in PBS. Cells (1 × 106) were incubated with 1 μg of the indicated antibody for 1 h at room temperature. K312 was detected using a FITC-labeled secondary antibody, and phycoerythrin-labeled antibodies against SSEA-3, SSEA-4, Tra-1-60, and Tra-1-81 (BioLegend) were used for double staining. Cell sorting was performed using a FACSAria Cell sorter (BD Bioscience).
Immunofluorescence staining
H9 hESCs were incubated with K312 and anti-E-cadherin (Cell Signaling Technology) antibodies for 16 h at 4°C after fixing. Secondary antibodies labeled with FITC and phycoerythrin were used to detect K312 and anti-E-cadherin antibodies, respectively. Fluorescence images were captured using a Zeiss 510LSM META laser-scanning microscope (Carl Zeiss).
Immunoblotting
Cells were dissolved in RIPA buffer (50 mM Tris-HCl at pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS, and 0.5% sodium deoxycholate) with protease and phosphatase inhibitors to isolate cell lysates. Cell lysates (20 μg) were mixed with SDS sample buffer, heated for 10 min, and analyzed by SDS-PAGE. Separated proteins were transferred onto a PVDF membrane and blocked with 3% skim milk in PBS for 1 h. The membrane was incubated with K312 or primary antibodies against pluripotency markers for 16 h at 4°C. After incubating the membrane with appropriate secondary antibodies conjugated with horse-radish peroxidase, ECL solution (GE Healthcare) was added and the immunoreactive bands were visualized by FUSION SOLO S chemiluminescence system (Vilver).
Immunoprecipitation
Cell lysates, as prepared for immunoblotting, were precleared of protein G beads for 1 h. The bead-unbound fractions were incubated with K312 for 4 h at 4°C, and then, K312 was precipitated by protein G beads for 12 h. Target protein-antibody reactants were released by heating, and the samples were centrifuged to remove the beads. Collected supernatant was analyzed by SDS-PAGE, followed by immunoblotting.
Deglycosylation assay
H9 hESCs or NCCIT cells were lysed in RIPA buffer, and 10 μg of lysate was incubated with 500 units of PNGase F (NEB) at 37°C for 0.5, 1, 2, and 4 h. Samples reacting or not reacting with PNGase F were immunoblotted using K312 as described earlier (Immunoblotting section).
Alkaline phosphatase staining
Colony-forming cells were fixed in 10% formalin solution and stained using an alkaline phosphatase staining kit (Sigma-Aldrich), as recommended by the manufacturer. Alkaline phosphatase-positive colonies were manually counted under light microscope and images were captured using HP Scanjet (Hewlett-Packard).
Teratoma assay
Six-week-old NSG mice (Jackson Laboratory) were cared for, following the guidelines of the Animal Care Committee of the Korea Research Institute of Bioscience and Biotechnology. The teratoma assay was performed as described previously (11). Briefly, control irradiated MEFs, K312-low cells, or K312-high cells (5 × 105) were injected into each side of the mouse testes using a 31-gauge Ultra-FineTM syringe (BD Bioscience). Teratomas were excised 8 weeks from injection, and their weights and sizes were measured. For histology, tissue fixation, paraffin processing, embedding, sectioning, and hematoxylin and eosin staining were performed as described previously (20).
Reverse transcription PCR (RT-PCR)
Total RNA was extracted from mice testicle tissues using TRIzol reagent (iNtRON Biotechnology) and cDNA was synthesized as described previously (20). For RT-PCR, primers to detect GATA6, FN1, AMY2A, HAND1, MSX1, MAP2, NESTIN, TUBB3, and GAPDH gene expression were used with primer sequences obtained from a previous study (19). Primer sequences used to analyze ACAN gene expression were 5’-TCTGTAACCCAGGC TCCAAC-3’ (sense) and 5’-CTGGCAAAATCCCCACTAAA-3’ (antisense).
SUPPLEMENTARY MATERIALS
ACKNOWLEDGEMENTS
This research was supported by the Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program (KGM5272221) and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2021R1I1A2057698).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
References
- 1.Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat Rev Mol Cell Biol. 2016;17:194–200. doi: 10.1038/nrm.2016.10. [DOI] [PubMed] [Google Scholar]
- 2.Park J, Lee Y, Shin J, et al. Mitochondrial genome mutations in mesenchymal stem cell derived from human dental induced pluripotent stem cells. BMB Rep. 2019;52:689–694. doi: 10.5483/BMBRep.2019.52.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graf T, Stadtfeld M. Heterogeneity of embryonic and adult stem cells. Cell Stem Cell. 2008;3:480–483. doi: 10.1016/j.stem.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Yamanaka S. Pluripotent stem cell-based cell therapy-promise and challenges. Cell Stem Cell. 2020;27:523–531. doi: 10.1016/j.stem.2020.09.014. [DOI] [PubMed] [Google Scholar]
- 5.Blum B, Benvenisty N. The tumorigenicity of human embryonic stem cells. Adv Cancer Res. 2008;100:133–158. doi: 10.1016/S0065-230X(08)00005-5. [DOI] [PubMed] [Google Scholar]
- 6.Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer. 2011;11:268–277. doi: 10.1038/nrc3034. [DOI] [PubMed] [Google Scholar]
- 7.Wuputra K, Ku CC, Wu DC, Lin YC, Saito S, Yokoyama KK. Prevention of tumor risk associated with the reprogramming of human pluripotent stem cells. J Exp Clin Cancer Res. 2020;39:100. doi: 10.1186/s13046-020-01584-0.bbea83d8a3f846d884cf96409bb4ee49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi HS, Kim WT, Ryu CJ. Antibody approaches to prepare clinically transplantable cells from human embryonic stem cells: identification of human embryonic stem cell surface markers by monoclonal antibodies. Biotechnol J. 2014;9:915–920. doi: 10.1002/biot.201300495. [DOI] [PubMed] [Google Scholar]
- 9.Tang C, Lee AS, Volkmer JP, et al. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat Biotechnol. 2011;29:829–834. doi: 10.1038/nbt.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Son YS, Seong RH, Ryu CJ, et al. Brief report: L1 cell adhesion molecule, a novel surface molecule of human embryonic stem cells, is essential for self-renewal and pluripotency. Stem Cells. 2011;29:2094–2099. doi: 10.1002/stem.754. [DOI] [PubMed] [Google Scholar]
- 11.Park J, Son Y, Lee NG, et al. DSG2 is a functional cell surface marker for identification and isolation of human pluripotent stem cells. Stem Cell Rep. 2018;11:115–127. doi: 10.1016/j.stemcr.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Satomaa T, Heiskanen A, Mikkola M, et al. The N-glycome of human embryonic stem cells. BMC Cell Biol. 2009;10:42. doi: 10.1186/1471-2121-10-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wesselschmidt RL. The teratoma assay: an in vivo assessment of pluripotency. Methods Mol Biol. 2011;767:231–241. doi: 10.1007/978-1-61779-201-4_17. [DOI] [PubMed] [Google Scholar]
- 14.Narsinh KH, Sun N, Sanchez-Freire V, et al. Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. J Clin Invest. 2011;121:1217–1221. doi: 10.1172/JCI44635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nussbaum J, Minami E, Laflamme MA, et al. Transplantation of undifferentiated murine embryonic stem cells in the heart: teratoma formation and immune response. FASEB J. 2007;21:1345–1357. doi: 10.1096/fj.06-6769com. [DOI] [PubMed] [Google Scholar]
- 16.Lanctot PM, Gage FH, Varki AP. The glycans of stem cells. Curr Opin Chem Biol. 2007;11:373–380. doi: 10.1016/j.cbpa.2007.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schopperle WM, DeWolf WC. The TRA-1-60 and TRA-1-81 human pluripotent stem cell markers are expressed on podocalyxin in embryonal carcinoma. Stem Cells. 2007;25:723–730. doi: 10.1634/stemcells.2005-0597. [DOI] [PubMed] [Google Scholar]
- 18.Brimble SN, Sherrer ES, Uhl EW, et al. The cell sur-face glycosphingolipids SSEA-3 and SSEA-4 are not essen-tial for human ESC pluripotency. Stem Cells. 2007;25:54–62. doi: 10.1634/stemcells.2006-0232. [DOI] [PubMed] [Google Scholar]
- 19.Ben-David U, Nudel N, Benvenisty N. Immunologic and chemical targeting of the tight-junction protein Claudin-6 eliminates tumorigenic human pluripotent stem cells. Nat Commun. 2013;4:1992. doi: 10.1038/ncomms2992. [DOI] [PubMed] [Google Scholar]
- 20.Park J, Lee NG, Oh M, et al. Selective elimination of human pluripotent stem cells by anti-Dsg2 antibody-doxorubicin conjugates. Biomaterials. 2020;259:120265. doi: 10.1016/j.biomaterials.2020.120265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.