

Abstract
Juvenile open angle glaucoma (JOAG) is a severe type of glaucoma with onset before age 40 and dominant inheritance. Using exome sequencing we identified 3 independent families from the Philippines with novel EFEMP1 variants (c.238A>T, p.Asn80Tyr; c.1480T>C, p.Ter494Glnext*29; and c.1429C>T, p.Arg477Cys) co-segregating with disease. Affected variant carriers (N= 34) exhibited severe disease with average age of onset of 16 years and with 76% developing blindness. To investigate functional effects, we transfected COS7 cells with vectors expressing the three novel EFEMP1 variants and showed that all three variants found in JOAG patients caused significant intracellular protein aggregation and retention compared to wild type and also compared to EFEMP1 variants associated with other ocular phenotypes including an early-onset form of macular degeneration, Malattia Leventinese/Doyne’s Honeycomb retinal dystrophy. These results suggest that rare EFEMP1 coding variants can cause JOAG through a mechanism involving protein aggregation and retention, and that the extent of intracellular retention correlates with disease phenotype. This is the first report of EFEMP1 variants causing JOAG, expanding the EFEMP1 disease spectrum. Our results suggest that EFEMP1 mutations appear to be a relatively common cause of JOAG in Filipino families, an ethnically diverse population.
Keywords: Glaucoma, Juvenile-onset, EFEMP1, Philippines
INTRODUCTION
Glaucoma is one of the most heritable of all human diseases (Wang et al., 2017) and exhibits both Mendelian and complex inheritance (Wiggs and Pasquale, 2017). Mendelian inheritance is most commonly observed in patients and families with childhood onset of disease (before age 18) although autosomal dominant inheritance is also observed in some families with adult onset disease especially disease caused by MYOC, TBK1 or OPTN mutations (Sears et al., 2019). Most adult-onset cases exhibit complex inheritance with contributions of both genetic and environmental risk factors (Wiggs and Pasquale, 2017). Clinically glaucoma is characterized by irreversible loss of retinal ganglion cells causing permanent damage to the optic nerve (Weinreb et al., 2016). In many patients optic nerve damage is associated with elevation of intraocular pressure (IOP), however retinal ganglion cell degeneration can occur without IOP elevation, a type of glaucoma called normal tension glaucoma or NTG. Increased IOP is caused by impaired drainage of intraocular fluid from the eye through the trabecular meshwork and outflow pathways (Weinreb et al., 2014).
Patients with juvenile-onset glaucoma, defined as diagnosis after age 3 and before the age 40, are typically affected by severe disease characterized by high IOP, and without treatment are likely to become blind over their lifetimes (Papadopoulos et al., 2020). Juvenile-onset primary open-angle glaucoma (JOAG) is one type of glaucoma that can develop during childhood. One gene, MYOC (encoding myocilin), has been identified in JOAG families in populations throughout the world, and accounts for disease in 15–20% of European Caucasian JOAG families (Allen et al. 2015). To further investigate the genetic etiology of JOAG in an ethnically diverse population, we identified and recruited 14 JOAG pedigrees from regions throughout the Philippines.
EFEMP1 (MIM# 601548) (EGF-containing fibulin-like extracellular matrix protein 1) also known as Fibulin 3 is a member of the Fibulin family of extracellular matrix proteins characterized by tandem arrays of EGF (epidermal growth factor)-like domains and a C-terminal fibulin-type module (Figure 1B) (Kobayashi et al., 2007). Among the fibulin family members, EFEMP1 has higher ocular expression (Wagner et al., 2013) and is known to contribute to other ocular phenotypes. Common SNPs near EFEMP1 on chromosome 2p16 have been associated with adult-onset glaucoma (POAG) (Gharahkhani et al., 2021) as well as IOP (Khawaja et al., 2018) and cup-to-disc ratio (CDR) an optic nerve quantitative trait related to POAG risk (Springelkamp et al., 2015). A low frequency EFEMP1 variant c.418C>T, p.Arg140Trp, present in 2 individuals in gnomAD has been identified in 5 affected individuals from an adult-onset POAG African-American family (Mackay et al., 2015) and a stop loss variant (c.1480T>G, Ter494Gluext*29) has been reported in 3 affected members of a Chinese POAG family (Liu et al., 2020). A single EFEMP1 missense allele c.1033C>T, (p.Arg345Trp) is currently the only known cause of Malattia Leventinese (MLVT) (also known as Doyne Honeycomb dystrophy (DHRD; MIM#126600), a rare autosomal dominant retinal degeneration characterized by extracellular deposits (drusen) beneath the retinal pigment epithelium (RPE) (Stone et al., 1999). Recently it has been suggested that the p.Arg345 residue is a mutation hot spot and that founder effects are not likely to account for the predominance of this mutation in MLVT/DHRD patients (Vaclavik et al., 2020). Notably, wildtype EFEMP1 is a secreted protein, while the p.Arg345Trp mutation leads to protein misfolding and reduced secretion (Marmorstein et al., 2002).
Figure 1A. Family pedigrees with juvenile-onset open-angle glaucoma (JOAG) and EFEMP1 mutation status.
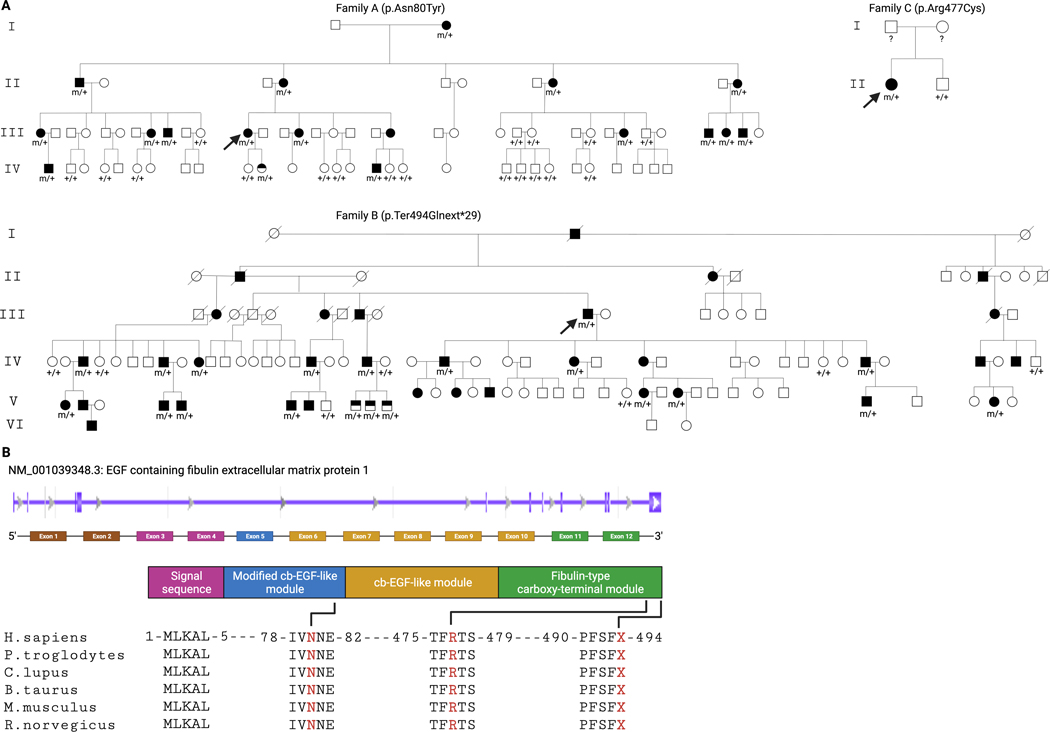
Arrow indicates the proband. Solid symbols indicate subjects diagnosed with JOAG, half-filled symbols indicate subjects who carry the EFEMP1 variant but who had no clinical signs of glaucoma at the time of recruitment, and open symbols with genotype status refer to subjects with no glaucoma and no EFEMP1 variant, open symbols without genotype status refer to subjects with no glaucoma (by history/clinical examination) but for whom DNA was unavailable. Genotypes are heterozygous mutant (m/+) and wild type (+/+). Figure 1B. Schematic diagram of EFEMP1 and evolutionary conservation. EFEMP1 Genbank transcript NM_001039348.3 and color-coded exons corresponding to the protein domain. The novel variants p.Asn80Tyr, p.Arg477Cys, and p.Ter494Glnext*29 are located at highly evolutionarily conserved domains.
Here, we report three independent families from the Philippines with novel EFEMP1 variants segregating with JOAG. Functional studies suggest that these novel JOAG-associated EFEMP1 variants cause protein misfolding, intracellular aggregation and retention. These results expand the spectrum of EFEMP1 associated disease to include childhood glaucoma.
METHODS
Ethical approval for this study was obtained from the Institutional Review Board of the Manila Doctors Hospital and Cebu Velez General Hospital, Philippines and the Mass General Brigham (MGB) IRB. In accordance with the tenets of the Declaration of Helsinki, written informed consent was obtained from the study subjects after explanation of the nature and possible consequences of the study.
Study recruitment
Subjects with juvenile open angle glaucoma (JOAG) were recruited from ophthalmology clinics. When possible, subjects underwent an ophthalmologic exam, which included visual acuity assessment, slit-lamp examination to evaluate the anterior segment, dilated fundus examination to document the structural changes of glaucoma, and gonioscopic examination to evaluate the angle structure.
JOAG was defined as: 1) Age of diagnosis between age 3 and 40; 2) IOP > 21 mmHg; 3) open anterior chamber angle on gonioscopy (grade 3 or 4 of modified Shaeffer classification); 4) characteristic optic disc damage and/or typical visual field loss; and 5) absence of secondary causes of glaucoma.
In total 14 probands were recruited and pedigree information and disease status were ascertained. Symptomatic and asymptomatic family members were invited to participate. A total of 161 subjects from the 14 unrelated families were enrolled in the study and 45 of those underwent whole exome sequencing (WES) (Supp. Table S1). The proband from Family G was initially referred with a diagnosis of JOAG but after further clinical evaluation was determined to be a case of ocular hypertension and aniridia. Genomic DNA was extracted from the blood samples (10cc from venipuncture) with the DNeasy Blood DNA extraction kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions.
Exome sequencing
Whole exome sequencing (WES) was performed at the Massachusetts Eye and Ear Ocular Genomics Institute using Agilent SureSelect Human All Exon v6 with the Illumina HiSeq 2000, or at the Broad Institute of MIT and Harvard using a TWIST Biosciences custom exome bait with the Illumina Novaseq 6000. We used the Ocular Genomics Institute bioinformatics pipeline (Consugar et al., 2015) for alignment and variant calling that includes the Burrows-Wheeler aligner (Li et al., 2010), GATK (https://gatk.broadinstitute.org/), GERP (http://mendel.stanford.edu/SidowLab/downloads/gerp/), Genome Aggregate Database (gnomAD) (gnomAD, https://gnomad.broadinstitute.org/), SIFT (http://sift.jcvi.org/), and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/). A coverage depth cutoff of 10x was applied. Heterozygous was defined as a fraction of a variant base between 0.25–0.75 and homozygous was defined as above 0.75. Average coverage was 105x for 99% of coding sequences.
Variant filtering
Ten genes (MYOC, CYP1B1, LTBP2, PITX2, FOXC1, PAX6, TEK, ANGPT1, LMX1B, CPAMD8) known to cause various forms of early-onset glaucoma were initially screened for pathogenic mutations. If no plausible causative variants were identified, the exome data was examined for novel pathogenic variants. Disease-causing mutations are expected to be rare, disrupt protein function, and have high estimates of pathogenicity. Variants were filtered using: 1) minor allele frequency (MAF) (initially from ExAC and then confirmed from the genome aggregation database (gnomAD; https://gnomad.broadinstitute.org/) (MAF less than 0.001 were retained), 2) tools to score variants according to phylogenetic conservation (GERP) and its effect on protein structure (SIFT, PolyPhen, conserved predicted pathogenic variants were obtained), 3) ocular expression, and any known functional or human disease involvement (ocular expression was prioritized) and 4) examining segregation in all available family members. The following databases were used to filter variants by ocular expression and human disease involvement: Single Cell Portal (SCP) (https://singlecell.broadinstitute.org/single_cell/study/SCP780/cell-atlas-of-aqueous-humor-outflow-pathways-in-eyes-of-humans-and-four-model-species-provides-insights-into-glaucoma-pathogenesis); RetNet, the Retinal Information Network (https://sph.uth.edu/retnet/); Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php). The family A and B pedigree structures suggested dominant inheritance; hence, heterozygous variants were prioritized. Sanger sequencing was used to confirm WES results and to investigate segregation of MYOC (GenBank: NM_000261.2), PAX6 (GenBank: NM_000280.3) variants and EFEMP1 candidate variants in all available family members. The primers for Sanger sequencing are listed in Supp. Table S2.
EFEMP1 expression vector
To evaluate the cellular expression and localization of disease-associated EFEMP1 variants we used a pENTR/D-TOPO entry vector (Invitrogen, Carlsbad, CA) with cloned wild-type EFEMP1 cDNA (provided as a gift from Dr. Rosario Fernandez-Godino, (Garland et al., 2021). The EFEMP1 mutants (p.Asn80Tyr, p.Arg140Trp, p.Arg345Trp, p.Arg477Cys) were created using the QuikChange Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA). To create the p.Ter494Glnext*29 mutation, a gBlocks™ Gene Fragment (Integrated DNA Technologies, Iowa) was designed and cloned in to a pENTR/D-TOPO entry vector. The wild-type and mutant entry clones were verified by sequencing analysis and moved into a Gateway destination expression vector modified to contain an N-terminal V5 epitope tag in-frame (pCAG-V5-IRES-EGFP) (Zhang et al., 2012) by LR recombination. Plasmid DNA was purified using the EndoFree Plasmid Maxi kit (Qiagen, Valencia, CA).
Cell culture
COS-7 cells (ATCC, Manassas, VA) were used to assess EFEMP1 protein subcellular localization. The COS-7 cells were seeded in 6-well plates at a concentration of 100,000 cells per mL and allowed to grow in DMEM + 10% FBS condition media. The cells were transfected with expression clones encoding wild-type or mutant EFEMP1 cDNA using Lipofectamine 3000 (Invitrogen, Carlsbad, CA) 24 hours post-seeding at 80–90% confluence. All intracellular localization experiments and Western blot analyses used this same COS-7 cell line.
Immunocytochemistry
To visualize EFEMP1 intracellular localization, COS-7 cells were seeded on clear coverslips (Neuvitro Corp., Camas, WA). In order to visualize the endoplasmic reticulum, CellLight™ ER-RFP, BacMam 2.0 (Invitrogen, Carlsbad, CA) was added to each well during transfection. The cells were processed for immunocytochemistry 48 hours after transfection. Condition media was aspirated, coverslips washed with PBS, and fixed in 4% PFA. Cells were permeabilized using 0.5% Triton X-100 and blocked in 3% BSA with PBS, followed by incubation in V5 Tag Monoclonal Antibody (Thermofisher, Waltham, MA) overnight. Coverslips were retrieved and incubated in Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 647 (Invitrogen, Carlsbad, CA) for 2 hours and DAPI (Thermofisher, Waltham, MA) for 1 minute. Coverslips were mounted on clear slides with ProLong™ Glass Antifade Mountant (Invitrogen, Carlsbad, CA). Imaging of the transfected cells was done using a confocal laser scanning microscope (SP8, Leica Microsystems, Buffalo Grove, IL).
Western blots
To quantify transfected EFEMP1 protein, COS-7 cells were harvested 48 hours post transfection (average transfection efficiency 75%) (Supp. Table S3). Whole cell lysates were prepared by careful aspiration of condition media and adding cold RIPA buffer (Sigma-Aldrich, St. Louis, MO) and Pierce Protease Inhibitor (Thermofisher, Waltham, MA) to each well and agitating for 30 minutes at 4°C. After incubation, cells were transferred to Eppendorf tubes and centrifuged for 30 minutes at 4°C. The supernatant was retrieved and the protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Thermofisher, Waltham, MA). For each vector group, 5ug of protein was mixed with 4X Protein Sample Loading Buffer (Li-Cor Biosciences, Nebraska) and water for a total volume of 12ul. Each sample was denatured by heating at 95 °C for 3 minutes and loaded in to a 15 well 4–20% Mini-PROTEAN® TGX™ Precast Protein Gel (Bio-Rad, Hercules, CA). Electrophoresis was performed at 120v for approximately 1.5 hours. The gel was retrieved and transferred to a PVDF blotting membrane using the iBlot™ 2 system (Invitrogen, Carlsbad, CA). For normalization of protein loading, total protein staining was done using the Revert™ 700 Total Protein Stain Kit (Li-Cor Biosciences, Nebraska), following the manufacturer’s protocol. After imaging in the 700nm channel using the Odyssey® Imaging System (Li-Cor Biosciences, Nebraska), the membrane was blocked for 1 hour at room temperature using Intercept® Blocking Buffer (Li-Cor Biosciences, Nebraska), then overnight incubation with Anti-Fibulin-3 Antibody (mab3–5): sc-33722 (Santa Cruz Biotechnology, Dallas, TX) diluted with blocking buffer to a final concentration of 1:100. After overnight incubation, the membrane was incubated in IRDye® 800CW secondary goat anti-mouse antibody (Li-Cor Biosciences, Nebraska), at a final concentration of 1:10,000 for 1 hour and then imaged in the 800nm channel. Western blot quantification and analysis was carried out using ImageStudioLite® Software (Li-Cor Biosciences, Nebraska). Relative EFEMP1 abundance was calculated as the ratio between the EFEMP1 signal and the corresponding total protein signal for each sample. Three biological repeats were done for each EFEMP1 condition. Differences between mutant and wildtype EFEMP1 were tested using Student’s t-test with Bonferroni correction.
Statistical Analysis
All the analysis were done using GraphPad Prism software (GraphPad Software, San Diego, CA). Adjusted p value ≤ 0.05 was considered significant.
The dot plot comparing single cell sequencing results for known early-onset glaucoma genes was generated using the Broad Institute single cell sequence portal (https://singlecell.broadinstitute.org/) to visualize gene expression in cells from the aqueous humor pathway. Using single-cell RNA sequencing data from https://singlecell.broadinstitute.org/single_cell/study/SCP780 (van Zyl et al., 2020), the expression of genes in the cells of the anterior segment of the eye were explored using the search bar in the portal. Once the desired genes are selected, a dot plot is produced by the portal, with the size of each dot or circle proportional to the percentage of cells expressing the gene, while the intensity depicts the average normalized transcript count in expressing cells.
RESULTS
Identification of novel coding EFEMP1 variants in three families with JOAG.
Whole exome sequencing was completed for selected affected and unaffected family members, and exome data was filtered to retain rare variants (MAF < 0.1%) that were protein altering and predicted to be pathogenic using in silico programs. Mutations were identified in known disease-causing genes in 4 families (Supp. Table S4; Supp. Figure S1), three with MYOC variants (p.Ter505Trpext*42; p.Gln337Arg; p.Thr438Ile) and one with a PAX6 variant (p.Tyr296*). (The patient with the PAX6 variant was originally referred with a diagnosis of JOAG and after further clinical evaluation the diagnosis of Aniridia was established). Novel variants of interest (Supp. Table S5) were further evaluated for co-segregation with disease in each pedigree using Sanger sequencing. Novel EFEMP1 (MIM# 601548) coding variants (GenBank: NM_001039348.3) were identified in all affected individuals of 3 of the remaining families as follows: (c.238A>T, p.Asn80Tyr) in 16 affected individuals of Family A and not in 18 unaffected family members older than age 18; (c.1480T>C, p.Ter494Glnext*29) was identified in 17 affected family members in family B and not in any unaffected family members older than age 18 (n= 6). Mutations were identified in young family members who were clinically unaffected at the time of sample collection including one Family A member (age 13) and 4 Family B members (ages 7–12). Additionally, a third variant (c.1429C>T, p.Arg477Cys) was identified in a single case (Family C) (Figure 1).
Patients with novel EFEMP1 variants (N= 34 in total for all three families) exhibited severe disease with average age of disease onset of 16 years (range 3–43). Affected individuals had much higher than average IOP (28mmHg) and 76% were blind in at least one eye (Table 1). The majority of patients required surgical treatment for IOP elevation, however not all patients had access to clinical care. While corneal scarring precluded retinal examination for some patients, examination was possible for 19 of 38 individuals who carried EFEMP1 disease-associated variants and none of these exams revealed evidence of subretinal deposits (drusen) characteristic of MLVT/DHRD in affected individuals. A representative fundus image is shown in Figure 2.
Table 1.
Clinical findings in subjects with EFEMP1 novel variants. All clinical data listed were obtained during study recruitment. Normal IOP is less than 21 and visual acuities of 20/200 or greater meet the definition for blindness. NLP, LP, CF, and HM also meet the definition for blindness.
| Family-pedigree position | Sex | Age diagnosed w/ glaucoma | Visual acuity | Intraocular pressure (mmHg) | Vertical cup:disc ratio | Gonioscopy | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| OD | OS | OD | OS | OD | OS | OD | OS | |||
| A-I:2 | F | 11 | NLP | NLP | - | - | - | - | - | - |
| A-II:1 | M | 17 | HM | NLP | 8 | 59 | - | - | Open | Open |
| A-II:4 | F | 12 | NLP | NLP | 30 | 20 | - | - | - | - |
| A-II:8 | F | 28 | NLP | NLP | 54 | 63 | - | - | Open | Open |
| A-II:10 | F | 11 | 20/250 | 20/60 | 46 | 26 | 1 | 1 | Open | Open |
| A-III:1 | F | 22 | 20/25 | NLP | 17 | 13 | 1 | 1 | Open | Open |
| A-III:8 | F | 10 | 20/25 | 20/30 | 12 | 12 | 1 | 1 | Open | Open |
| A-III:9 | M | 31 | 20/25 | 20/25 | 45 | 16 | 0.7 | 0.3 | Open | Open |
| A-III:12 | F | 17 | NLP | NLP | 54 | 2 | 1 | - | Open | Open |
| A-III:15 | M | 18 | 20/60 | LP | 23 | - | 0.9 | - | Open | Open |
| A-III:29 | F | 28 | 20/50 | 20/50 | 23 | 23 | 0.4 | 0.4 | Open | Open |
| A-III:32 | M | 15 | LP | NLP | 43 | - | - | - | Open | - |
| A-III:33 | F | 25 | 20/30 | 20/30 | 13 | 6 | 0.7 | 0.9 | Open | Open |
| A-III:34 | M | 18 | 20/40 | 20/40 | 11 | 16 | 0.9 | 0.8 | Open | Open |
| A-IV:1 | M | 18 | 20/40 | 20/30 | 17 | 46 | 0.7 | 0.6 | Open | Open |
| A-IV:16 | M | 8 | LP | 20/200 | 68 | 20 | - | - | Closed (PAS) | Open |
| B-III:17 | M | 19 | NLP | NLP | - | - | - | - | - | - |
| B-IV:3 | M | 20 | NLP | NLP | - | - | - | - | - | - |
| B-IV:8 | M | 9 | NLP | NLP | - | - | - | - | - | - |
| B-IV:10 | F | 43 | 20/30 | 20/80 | 29 | 65 | 0.8 | 1 | Open | Open |
| B-IV:18 | M | 15 | NLP | NLP | - | - | - | - | - | - |
| B-IV:21 | M | 7 | NLP | 20/30 | - | 12 | - | 0.9 | Open | Open |
| B-IV:24 | M | 7 | NLP | NLP | - | - | - | - | - | - |
| B-IV:28 | F | 8 | NLP | NLP | - | - | - | - | - | - |
| B-IV:38 | M | 18 | NLP | NLP | - | - | - | - | - | - |
| B-V:1 | F | 19 | NLP | NLP | - | - | - | - | - | - |
| B-V:4 | M | 9 | NLP | NLP | - | - | - | - | - | - |
| B-V:5 | M | 10 | 20/20 | 20/20 | 16 | 28 | 0.4 | 0.8 | Open | Open |
| B-V:6 | M | 9 | NLP | NLP | - | - | - | - | - | - |
| B-V:24 | F | 12 | NLP | NLP | - | - | - | - | - | - |
| B-V:26 | F | 11 | NLP | NLP | - | - | - | - | - | - |
| B-V:32 | M | 3 | LP | HM | 20 | 20 | 0.9 | 0.8 | Open | Open |
| B-V:35 | F | 11 | NLP | NLP | - | - | - | - | - | - |
| C-II:1 | F | 18 | CF | CF | 40 | 22 | 1 | 1 | Open | Open |
| Patients with EFEMP1 novel variants but without clinical disease | ||||||||||
| A-IV:11 | F | 13* | 20/20 | 20/20 | 18 | 18 | 0.3 | 0.3 | Open | Open |
| B-V:9 | M | 12* | 20/20 | 20/20 | 15 | 13 | 0.3 | 0.3 | Open | Open |
| B-V:10 | M | 10* | 20/30 | 20/50 | 17 | 18 | 0.3 | 0.3 | Open | Open |
| B-V:11 | M | 7* | 20/50 | 20/40 | 16 | 16 | 0.3 | 0.3 | Open | Open |
For the 4 patients with EFEMP1 variants but without clinical evidence of the disease, the age listed is the age at recruitment. Abbreviations: Single dash (−): exam not performed because of poor view, corneal opacification, or phthisis bulbi; na: not applicable; PAS: peripheral anterior synechiae; NLP: no light perception; LP: light perception; CF: counting fingers; HM: hand movement; M: male; F: female; OD: right eye; OS: left eye.
Figure 2. Fundus images from a patient from Family B with EFEMP1 variant p.Ter494Glnext*29 (B-V:5).
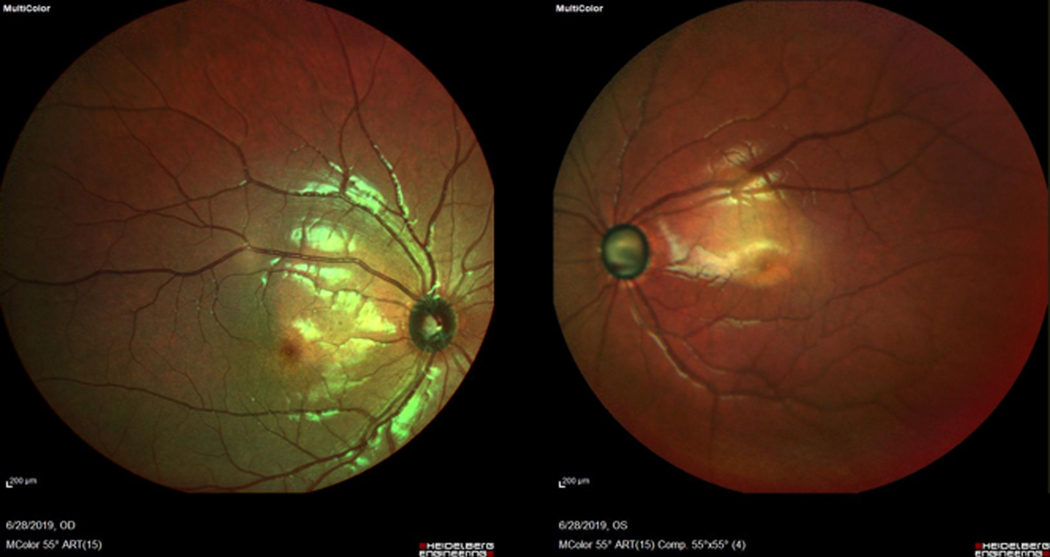
The image shows damage to the optic nerve but no evidence of subretinal deposits (drusen) characteristic of Malattia Leventinese/Doyne Honeycomb dystrophy. This non-mydriatic fundus photo was obtained using Heidelberg Spectralis® MultiColor Imaging (Heidelberg Engineering, Heidelberg, Germany).
The three variants, p.Asn80Tyr, p.Ter494Glnext*29, and p.Arg477Cys, identified in affected individuals are not present in population databases including gnomAD and TOPMed and have not been previously reported in any individuals. For the two missense alleles, in silico programs SIFT and Polyphen2 predict p.Arg477Cys to be damaging and probably damaging respectively while p.Asn80Tyr is predicted to be tolerated and possibly damaging (Supp. Table S6). p.Asn80Tyr is located within the first EGF-domain, while p.Arg477Cys and p.Ter494Glnext*29 are within the terminal fibulin-like carboxy terminal domain. p.Ter494Glnext*29 results from replacement of the wild type stop codon with a glutamine residue and the addition of 29 amino acid residues to the polypeptide before a stop codon is encountered (Figure 1).
Expression and localization of EFEMP1 wild-type and mutant proteins
EFEMP1 is expressed in multiple ocular anterior segment cell types especially the trabecular meshwork beam cells and juxtacanilicular matrix cells (Figure 3) (van Zyl et al., 2020), two structures necessary for outflow of intraocular fluid (aqueous humor) and maintenance of normal IOP levels. EFEMP1 ocular expression is similar to Myocilin (MYOC) also an extracellular matrix protein involved in JOAG. MYOC mutations cause protein misfolding and endoplasmic reticulum aggregation, and efforts to reduce mutation-related misfolding can result in lower IOP in animal models (Orwig et al., 2014; Jain et al., 2017). The MLVT/DHRD causing EFEMP1 variant, p.Arg345Trp, also causes protein misfolding and both intracellular retention (Marmostein et al., 2002) and extracellular protein deposition (Fernandez-Godino et al., 2015).
Figure 3. Single cell RNA sequencing EFEMP1 expression and known early onset glaucoma genes from the human aqueous humor outflow pathway.
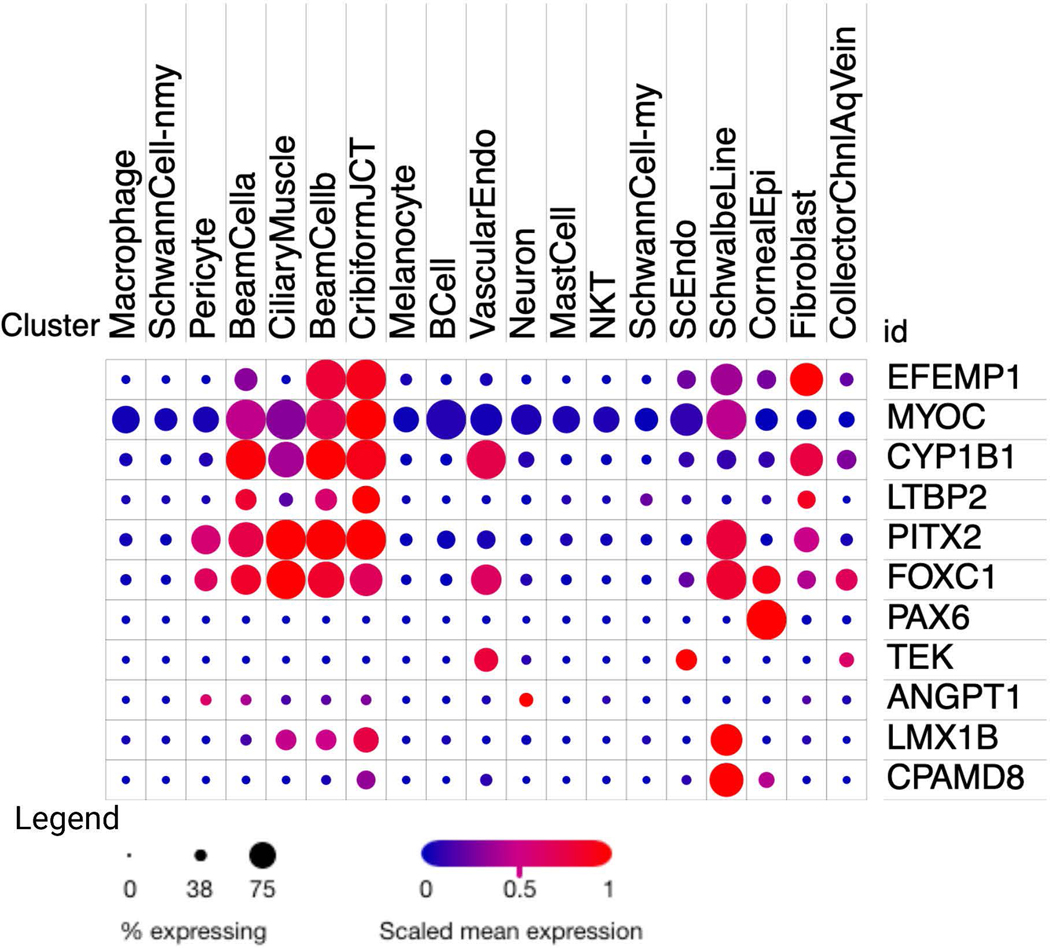
Comparative expression of EFEMP1 and genes known to cause various types of early onset glaucoma. This plot was generated from the Broad Institute of MIT and Harvard’s Single Cell Portal using data from “Cell atlas of aqueous humor outflow pathways in eyes of humans and four model species provides insight into glaucoma pathogenesis” by van Zyl et al, 2019. The dot plot is produced by the portal, with the size of each dot or circle proportional to the percentage of cells expressing the gene, and the intensity depicts the average normalized transcript count in expressing cells.
To assess the functional effects of the JOAG-related EFEMP1 variants, we hypothesized that these variants could cause protein misfolding, aggregation and intracellular retention similar to that observed for the p.Arg345Trp MLVT/DHRD variant and for JOAG-related MYOC mutations. To test this hypothesis we transfected COS7 cells with Gateway destination expression vectors (pCAG-V5-IRES-EGFP) (Zhang et al., 2012) for wildtype EFEMP1 cDNA, the three JOAG-related EFEMP1 variants, and also the MLVT/DHRD variant (p.Arg345Trp) and the POAG-related variant (p.Arg140Trp). Imaging transfected cells showed that wild type EFEMP1 was distributed throughout the cell, while significant intracellular aggregation was observed for each JOAG-related variant (Figure 4). Co-localization with an endoplasmic reticulum (ER) marker, CellLight™ ER-RFP, BacMam 2.0 (Invitrogen, Carlsbad, CA), suggested that EFEMP1 protein aggregates are formed in the vicinity of the ER. In contrast, although both the p.Arg345Trp and p.Arg140Trp variants showed increased intracellular retention compared to wildtype, both variants exhibited more diffuse cellular distribution compared to the variants found in JOAG patients.
Figure 4. Variant and wildtype EFEMP1 subcellular localization in COS-7 cells.
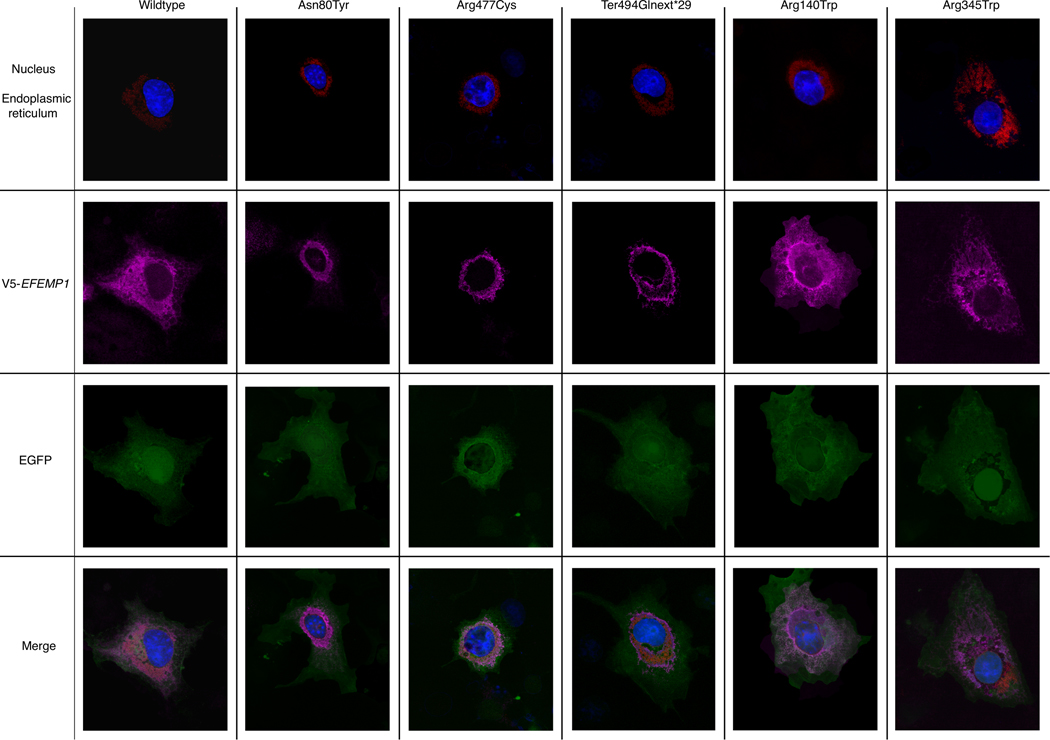
Cultured COS-7 cells were transfected with wildtype and variant EFEMP1 and processed with immunocytochemistry and the following stains: DAPI (nucleus), CellLight ER-RFP (endoplasmic reticulum), Alexa 647 (EFEMP1 protein), GFP (GFP expression reporter). Imaged using Leica SP8 confocal microscope using 63x objective (glycerol immersion), digital zoom 2x.
To measure intracellular protein retention, we collected an equal number of transfected cells from experiments using the wild type vector, the 3 JOAG-related vectors and the p.Arg345Trp and p.Arg140Trp vectors. Using Western blot assays (Figure 5A, Supp. Figure S2) we determined the fraction of EFEMP1 protein retained within the cell in comparison to overall protein and relative to wild type protein (Figure 5B). Compared to wild type cells intracellular EFEMP1 protein was significantly increased for all three of the JOAG-related variants (P< 0.05). JOAG-related intracellular protein was also increased compared to the MLVT/DHRD variant p.Arg345Trp and the POAG-related p.Arg140Trp.
Figure 5. EFEMP1 expression in COS7 cells.
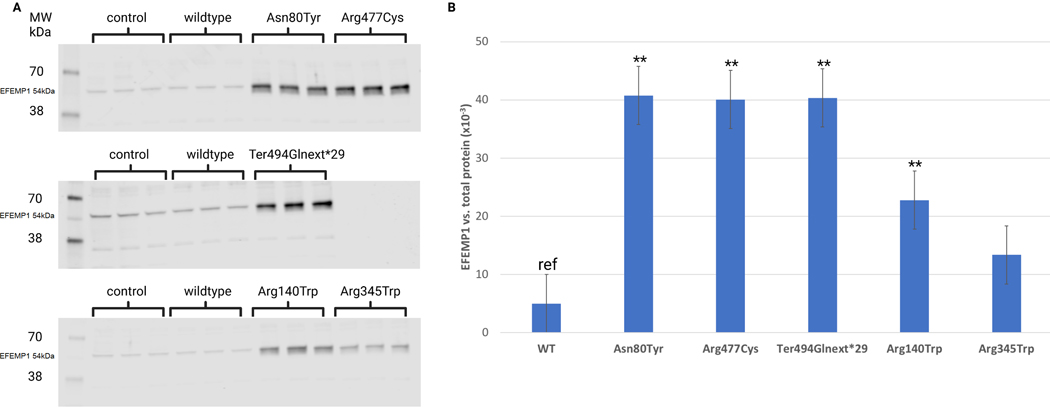
A. Western blot of wild-type (WT) and mutant EFEMP1 proteins. Transfected COS7 cell-lysates showing expression of WT and mutant EFEMP1. EFEMP1 (54 kDa) is indicated on each blot. The identity of the faint lower bands is not known but only the EFEMP1 band intensity was used to calculate the EFEMP1 signal. The control group consists of cell-lysates from non-transfected COS7 cells. B. The ratio of intracellular EFEMP1 protein to total protein for wild type and each variant. Relative EFEMP1 abundance was calculated as the ratio between the EFEMP1 signal and the corresponding total protein signal in each sample measured using Revert 700 total protein stain. Three biological repeats were done for each EFEMP1 condition. Differences between mutant EFEMP1 and wildtype EFEMP1 were tested using Student’s t-test with Bonferroni correction. Error bars indicate the standard error. *p<0.05, **p<0.001.
DISCUSSION
Collectively, these data support the hypothesis that EFFMP1 coding variants can cause JOAG by a mechanism that involves intracellular protein aggregation and retention. Similarly, JOAG-causing MYOC mutations also promote formation of intracellular protein aggregates and the extent of protein aggregation appears to correlate with disease severity (Patterson-Orazem et al., 2019). Misfolded myocilin is hypothesized to cause ER stress leading to cellular dysfunction and potentially apoptosis (Yam et al., 2007), however the specific cells that are impacted and the underlying molecular events are not known. Studies of human and mouse MYOC knockouts (Kim et al., 2001; Pang et al., 2002; Wiggs et al., 2001) and mouse knockins (Kim et al., 2001); Zode et al., 2011) indicate that MYOC mutations are gain-of-function. The observation that the retina and optic nerves of EFEMP1 knockout mice are anatomically normal (McLaughlin et al., 2007; Stanton et al., 2017; Daniel et al., 2020) suggests that EFEMP1 loss of function also does not underlie glaucoma development. MYOC and EFEMP1 are among a group of proteins expressed in ocular extracellular matrix that also includes Thrombospondin1 (THBS1), and Angiopoietin-like 7 (ANGPTL7) among other proteins with potential glaucoma involvement (Wirtz et al., 2021; Tanigawa et al., 2020). Although the specific mechanisms underlying the contribution of ECM proteins to glaucoma is not known, preventing mutant MYOC expression (Jain et al., 2017) or encouraging secretion of misfolded myocilin can reduce intraocular pressure in mice (Zode et al., 2011; Zode et al., 2021), and similar approaches may also be therapeutically useful for patients with EFEMP1 mutations.
In this study we showed that the JOAG associated variants demonstrate increased intracellular protein retention compared with the MLVT/DHRD associated variant p.Arg345Trp. EFEMP1 p.Arg345Trp has recently been shown to effect cholesterol efflux (Tsai et al., 2021) which may potentially impact the contribution of this mutation to the characteristic extracellular protein aggregation observed in this disease (Fu et al., 2007). While some POAG genomic loci include genes that may influence cholesterol efflux (ABCA1, ARHGEF12, CAV1/2) (Gharahkhani et al., 2021; Springelkamp et al., 2015; Jacobo-Albavera et al., 2021; Wang et al., 2014; Okuhira et al., 2010), the role of cholesterol metabolism in glaucoma is not clear and given the involvement of EFEMP1 in JOAG could be interesting to explore further. It is also of interest that only the single missense allele p.Arg345Trp is known to cause MLVT/DHRD, yet at least 3 protein variants can cause JOAG and including the 2 variants described in adult-onset POAG families, 5 variants in total may contribute to glaucoma. It is possible that EFEMP1 missense alleles are more likely to affect cells of the ocular outflow pathway than the retinal pigment epithelial cells involved in MVLT/DHRB. Interestingly, patients with MVLT/DHRB are not known to be at increased risk of glaucoma, and the JOAG patients with EFEMP1 mutations do not have any evidence of the MVLT/DHRB retinal dystrophy (Figure 2).
Additionally, we show that the JOAG EFEMP1 variants demonstrate increased intracellular retention compared to a possible POAG associated variant, p.Arg140Trp found in a family with 5 members affected by adult-onset POAG (Mackay et al., 2015), a finding that may be related to the less severe and genetically complex adult-onset POAG. Interestingly, the p.Arg140Trp variant exhibited increased intracellular retention compared to wild type but decreased retention compared to the 3 JOAG variants supporting a possible genotype phenotype relationship. Our results could support a contribution of p.Arg140Trp to POAG in the reported family, which is interestingly of African American ancestry. Exome-based studies of African American POAG cases or families have not yet been completed and further investigation in this population could reveal additional EFEMP1 disease-related variants. A stop lost variant (c.1480T>G, p.Ter464Gluext*29) similar to the mutation affecting JOAG family B (c.11480 T>C, p.Ter464Glnext*29) has recently been described in 3 members of a Chinese adult POAG family (Liu et al., 2020). While individuals in this family are described as affected by POAG, the age of disease onset is the mid-20s which would meet our diagnostic criteria for JOAG and is consistent with our results.
It is interesting that two EFEMP1 variants altering the stop codon have been identified in glaucoma families (p.Ter464Gluext*29; Liu et al., 2020 and p.Ter464Glnext*29, Family B in this study). Single-nucleotide variants that abolish the stop codon (“nonstop” or ‘stop-lost’ alterations) cause read-through of the wild type stop codon resulting in C-terminal extension of the protein polypeptide chain. In humans functional characterization has suggested that stop-lost variants can cause disease by loss of protein function through activation of proteasome degradation pathway (Shibata et al., 2015) or by a gain of function mechanism involving protein aggregation (Bock et al., 2018). Interestingly, a REEP1 stop-lost mutation revealed a 3’ untranslated region that promotes protein aggregation causing peripheral neuropathy in patients with autosomal dominant Charcot-Marie-Tooth disease type 2 (Bock et al., 2018). Our results are consistent with the hypothesis that the 29 amino acids added to the EFEMP1 C-terminus by the glaucoma-related stop-lost variants promote protein aggregation.
EFEMP1 is located on chromosome 2p16 within a genomic region that was initially identified as a POAG genomic locus (GLC1H) in a linkage study of a Jamaican family and European Caucasian families from the UK (Suriyapperuma et al., 2007). Subsequently linkage to this region has also been observed in four Chinese families, including two with disease onset before age 40 (Liu et al., 2012; Liu et al., 2008). POAG candidate association studies have also implicated SNPs in the 2p16 region in cases and controls from Barbados (Jiao et al., 2009), an African-American cohort (Liu et al., 2010), Chinese (Chen et al., 2012) and South Indian cohorts (Balasubbu et al., 2021). Together with our results, these studies support EFEMP1 as the GLC1H gene and also could suggest that EFEMP1 is more commonly associated with glaucoma in non-white populations.
Our findings also suggest a phenotype-genotype spectrum that correlates the extent of intracellular protein retention with disease phenotype and extend the EFEMP1 phenotypic spectrum to include severe childhood-onset glaucoma. The mutations causing JOAG cause more severe intracellular protein retention compared to the MLVT/DHRD EFEMP1 variant and also to a variant found in an adult-onset glaucoma family. Further study will be required to determine the mechanism underlying intracellular protein retention as well as the molecular events leading to elevated IOP and glaucoma.
This study investigates the etiology of JOAG in a collection of affected families from the Philippines, a population with diverse ancestry including Asian, African and European Caucasian origins (Larena et al., 2021). Among the pedigrees that we have collected we find that EFEMP1 variation is a relatively common cause of juvenile glaucoma (20%), equal to MYOC in this population. Interestingly, EFEMP1 variants have not been observed in any prior studies of childhood glaucoma which have focused primarily of families with European Caucasian or Asian ancestry (Allen et al., 2015; Huang et al., 2018). As well, MYOC and other currently known childhood gene mutations have infrequently been identified in patients with African ancestry (Liu et al., 2012). These results suggest that investigation of diverse populations such as this Filipino cohort will be necessary to develop a more comprehensive set of childhood glaucoma genes.
A limitation of our study is the absence of an ancestry matched population allowing for assessing population specific allele frequencies. The variants we identified in this study are absent from the largest sequence database (gnomAD), however Filipino individuals are not specifically included in these datasets making it difficult to confirm that these variants are novel in the Filipino population. Further confirmation would require future study using a sufficiently sized Filipino sequence dataset.
The discovery of genes causing childhood glaucoma makes it possible to use genetic testing to inform genetic counseling for affected families. Treatment initiated at early stages of disease can delay irreversible optic nerve degeneration and provide the best chance that an affected child will maintain useful sight throughout their lifetime. Informed genetic counseling makes it possible to create surveillance and treatment plans for individuals with disease-causing mutations and alleviates the burden of screening family members without disease-causing mutations. Currently however a molecular diagnosis can only be achieved for approximately 20–25% of cases based on known genes (Allen et al., 2015). Discovery of novel disease-causing genes such as EFEMP1 is needed to improve the overall diagnostic yield and effectiveness of childhood glaucoma genetic testing.
In conclusion we have identified 3 different EFEMP1 coding variants that segregate with a severe form of glaucoma affecting children in 3 independent families from the Philippines. Our results suggest that disease-associated variants cause significant intracellular EFEMP1 aggregation and retention and that the extent of intracellular retention appears to be correlated with EFEMP1-related disease phenotypes. This study further supports a role for EFEMP1 in ocular extracellular matrix and in regulation of intraocular fluid dynamics and IOP and provides new opportunities for genetic testing and therapeutic intervention.
Supplementary Material
Acknowledgments
Supported by the ARVO Foundation David L. Epstein award (ERAC) and NIH/NEI R01 EY031830 (JLW) and P30EY014104 (JLW)
Funding information: Supported by the ARVO Foundation David L. Epstein award (ERAC) and NIH/NEI R01 EY031830 (JLW) and P30EY014104 (JLW)
Footnotes
Declaration of Interests
JLW has received research support from Aerpio pharmaceuticals and is a consultant for Aerpio, Allergan, Maze, Editas, Regenxbio and Avellino.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article or supplementary information. All variants identified in the present study have been submitted to ClinVar (https://www.ncbi.nih.gov/clinvar/) and have received the following accession numbers: VCV001294422, VCV001294423, VCV001294421.
Web Resources
CADD, https://cadd.gs.washington.edu/snv
GATK (https://gatk.broadinstitute.org/)
Genbank, https://www.ncbi.nlm.nih.gov/genbank/
GERP (http://mendel.stanford.edu/SidowLab/downloads/gerp/)
gnomAD, https://gnomad.broadinstitute.org/
Human Gene Mutation Database (HGMD), http://www.hgmd.cf.ac.uk/ac/index.php
OMIM, https://www.omim.org/
Polyphen2, http://genetics.bwh.harvard.edu/pph2/
RetNet, the Retinal Information Network, https://sph.uth.edu/retnet/
Single Cell Portal (SCP), https://singlecell.broadinstitute.org
References
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, … MacArthur DG (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen KF, Gaier ED, Wiggs JL. (2015) Genetics of Primary Inherited Disorders of the Optic Nerve: Clinical Applications. Cold Spring Harb Perspect Med. 5, a017277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubbu S, Krishnadas SR, Jiao X, Hejtmancik JF, Sundaresan P (2012) Evaluation of SNPs on chromosome 2p with primary open angle glaucoma in the South Indian cohort. Invest Ophthalmol Vis Sci 53: 1861–1864. [DOI] [PubMed] [Google Scholar]
- Bock AS, Günther S, Mohr J, Goldberg LV, Jahic A, Klisch C, Hübner CA, Biskup S, Beetz C. (2018). A nonstop variant in REEP1 causes peripheral neuropathy by unmasking a 3’UTR-encoded, aggregation-inducing motif. Hum Mutat. 39, 193–196. [DOI] [PubMed] [Google Scholar]
- Chen LJ, Tam PO, Leung DY, Fan AH, Zhang M, Tham CC, et al. (2012) SNP rs1533428 at 2p16.3 as a marker for late-onset primary open-angle glaucoma. Mol Vis 18: 1629–1639. [PMC free article] [PubMed] [Google Scholar]
- Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, Wang DY, Au ED, Sims KB, Sweetser DA, Fulton AB, Liu Q, Wiggs JL, Gai X, Pierce EA (2015). Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med. 17:253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel S, Renwick M, Chau VQ, Datta S, Maddineni P, Zode G, Wade EM, Robertson SP, Petroll WM, Hulleman JD (2020) Fibulin-3 knockout mice demonstrate corneal dysfunction but maintain normal retinal integrity. J Mol Med (Berl). 98, 1639–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Godino R, Garland DL, Pierce EA (2015) A local complement response by RPE causes early-stage macular degeneration. Hum Mol Genet. 24, 5555–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Garland D, Yang Z, Shukla D, Rajendran A, Pearson E, Stone EM, Zhang K, Pierce EA (2007) The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum. Mol. Genet. 16, 2411–2422. [DOI] [PubMed] [Google Scholar]
- Garland DL, Pierce EA, Fernandez-Godino R. (2021). Complement C5 is not critical for the formation of sub-RPE deposits in Efemp1 mutant mice. Sci Rep. 11:10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharahkhani P, Jorgenson E, Hysi P, Khawaja AP, Pendergrass S, Han X, Ong JS, Hewitt AW, Segrè AV, Rouhana JM, et al. (2021) Genome-wide meta-analysis identifies 127 open-angle glaucoma loci with consistent effect across ancestries. Nat. Commun. 12, 1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Xie L, Wu Z, Cao Y, Zheng Y, Pang CP, Zhang M. (2018) Detection of mutations in MYOC, OPTN, NTF4, WDR36 and CYP1B1 in Chinese juvenile onset open-angle glaucoma using exome sequencing. Sci Rep. 8, 4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobo-Albavera L, Domínguez-Pérez M, Medina-Leyte DJ, González-Garrido A, Villarreal-Molina T. (2021) The Role of the ATP-Binding Cassette A1 (ABCA1) in Human Disease. Int J Mol Sci. 22, 1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Zode G, Kasetti RB, Ran FA, Yan W, Sharma TP, Bugge K, Searby CC, Fingert JH, Zhang F, et al. (2017) CRISPR-Cas9-based treatment of myocilin-associated glaucoma. Proc. Natl. Acad. Sci. U. S. A. 2017 Oct 17;114(42):11199–11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Yang Z, Yang X, Chen Y, Tong Z, Zhao C, et al. (2009) Common variants on chromosome 2 and risk of primary open-angle glaucoma in the Afro-Caribbean population of Barbados. Proc Natl Acad Sci U S A 106: 17105–17110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khawaja AP, Cooke Bailey JN, Wareham NJ, Scott RA, Simcoe M, Igo RP Jr, Song YE, Wojciechowski R, Cheng CY, Khaw PT, et al. (2018) Genome-wide analyses identify 68 new loci associated with intraocular pressure and improve risk prediction for primary open-angle glaucoma. Nat. Genet. 50, 778–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BS, Savinova OV, Reedy MV, Martin J, Lun Y, Gan L, Smith RS, Tomarev SI, John SW, Johnson RL (2001) Targeted Disruption of the Myocilin Gene (Myoc) Suggests that Human Glaucoma-Causing Mutations Are Gain of Function Mol Cell Biol 21, 7707–7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Kostka G, Garbe JHO, Keene DR, Bächinger HP, Hanisch F-G, Markova D, Tsuda T, Timpl R, Chu M-L, Sasaki T. (2007) A comparative analysis of the fibulin protein family. Biochemical characterization, binding interactions, and tissue localization. J. Biol. Chem. 282, 11805–11816. [DOI] [PubMed] [Google Scholar]
- Larena M, Sanchez-Quinto F, Sjödin P, McKenna J, Ebeo C, Reyes R. Casel O, Huang J-Y, Hagada KP, Guilay D. (2021). Multiple migrations to the Philippines during the last 50,000 years. Proc. Natl. Acad. Sci. U S A 118, e2026132118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Liu T, Li J, Yang J, Du Q, Wang J, et al. (2008) A genome-wide scan maps a novel autosomal dominant juvenile-onset open-angle glaucoma locus to 2p15–16. Mol Vis 14: 739–744. [PMC free article] [PubMed] [Google Scholar]
- Liu T, Xie L, Ye J, Liu Y, He X (2012) Screening of candidate genes for primary open angle glaucoma. Mol Vis 18: 2119–2126. [PMC free article] [PubMed] [Google Scholar]
- Liu T, Tang C, Shi X. (2020) Analysis of variants in Chinese individuals with primary open-angle glaucoma using molecular inversion probe (MIP)-based panel sequencing. Mol. Vis. 26, 378–391. [PMC free article] [PubMed] [Google Scholar]
- Liu W, Liu Y, Challa P, Herndon LW, Wiggs JL, Girkin CA, Allingham RR, Hauser MA. (2012) Low prevalence of myocilin mutations in an African American population with primary open-angle glaucoma. Mol. Vis. 18, 2241–2246. [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Qin X, Schmidt S, Allingham RR, Hauser MA (2010) Association between chromosome 2p16.3 variants and glaucoma in populations of African descent. Proc Natl Acad Sci U S A 107: E61; author reply E62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DS, Bennett TM, Shiels A. (2015) Exome Sequencing Identifies a Missense Variant in EFEMP1 Co-Segregating in a Family with Autosomal Dominant Primary Open-Angle Glaucoma. PLoS One. 10, e0132529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein LY, Munier FL, Arsenijevic Y, Schorderet DF, McLaughlin PJ, Chung D, Traboulsi E, Marmorstein AD (2002) Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc. Natl. Acad. Sci. U. S. A. 99, 13067–13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin PJ, Bakall B, Choi J, Liu Z, Sasaki T, Davis EC, Marmorstein AD, Marmorstein LY (2007) Lack of fibulin-3 causes early aging and herniation, but not macular degeneration in mice. Hum. Mol. Genet. 16, 3059–3070. [DOI] [PubMed] [Google Scholar]
- Okuhira K, Fitzgerald ML, Tamehiro N, Ohoka N, Suzuki K, Sawada J-I, Naito M, Nishimaki-Mogami T. (2010) Binding of PDZ-RhoGEF to ATP-binding cassette transporter A1 (ABCA1) induces cholesterol efflux through RhoA activation and prevention of transporter degradation J. Biol. Chem. 285, 16369–16377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orwig SD, Chi PV, Du Y, Hill SE, Cavitt MA, Suntharalingam A, Turnage KC, Dickey CA, France S, Fu H, Lieberman RL (2014) Ligands for glaucoma-associated myocilin discovered by a generic binding assay. ACS Chem. Biol. 9, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang CP, et al. (2002) TIGR/MYOC gene sequence alterations in individuals with and without primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci. 43, 3231–3235. [PubMed] [Google Scholar]
- Papadopoulos M, Vanner EA, Grajewski AL, International Study of Childhood Glaucoma – Childhood Glaucoma Research Network Study Group. (2020) International Study of Childhood Glaucoma. Ophthalmol. Glaucoma. 3(2), 145–157. [DOI] [PubMed] [Google Scholar]
- Patterson-Orazem AC, Hill SE, Wang Y, Dominic IM, Hall CK, Lieberman RL (2019) Differential Misfolding Properties of Glaucoma-Associated Olfactomedin Domains from Humans and Mice. Biochemistry. 58, 1718–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears NC, Boese EA, Miller MA, Fingert JH (2019) Mendelian genes in primary open angle glaucoma. Exp Eye Res. 186:107702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata N, Ohoka N, Sugaki Y, Onodera C, Inoue M, Sakuraba Y, Takakura D, Hashii N, Kawasaki N, Gondo Y, Naito M. (2015) Degradation of Stop Codon Read-through Mutant Proteins via the Ubiquitin-Proteasome System Causes Hereditary Disorders. J Biol Chem. 290, 28428–28437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springelkamp H, Iglesias AI, Cuellar-Partida G, Amin N, Burdon KP, van Leeuwen EM, Gharahkhani P, Mishra A, van der Lee SJ, Hewitt AW, et al. (2015) ARHGEF12 influences the risk of glaucoma by increasing intraocular pressure. Hum Mol Genet. 24, 2689–2699. [DOI] [PubMed] [Google Scholar]
- Springelkamp H, Mishra A, Hysi PG, Gharahkhani P, Höhn R, Khor CC, Cooke Bailey JN, Luo X, Ramdas WD, Vithana E, et al. (2015) Meta-analysis of Genome-Wide Association Studies Identifies Novel Loci Associated With Optic Disc Morphology. Genet. Epidemiol. 39, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton JB, Marmorstein AD, Zhang Y, Marmorstein LY (2017) Deletion of Efemp1 Is Protective Against the Development of Sub-RPE Deposits in Mouse Eyes. Invest Ophthalmol Vis Sci. 58, 1455–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EM, Lotery AJ, Munier FL, Héon E, Piguet B, Guymer RH, Vandenburgh K, Cousin P, Nishimura D, Swiderski RE, et al. (1999) A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat. Genet. 22, 199–202. [DOI] [PubMed] [Google Scholar]
- Suriyapperuma SP, Child A, Desai T, Brice G, Kerr A, Crick RP, et al. (2007) A new locus (GLC1H) for adult-onset primary open-angle glaucoma maps to the 2p15-p16 region. Arch Ophthalmol 125: 86–92. [DOI] [PubMed] [Google Scholar]
- Tanigawa Y, Wainberg M, Karjalainen J, Kiiskinen T, Venkataraman G, Lemmelä S, Turunen JA, Graham RR, et al. (2020) Rare protein-altering variants in ANGPTL7 lower intraocular pressure and protect against glaucoma. PLoS Genet. 16, e1008682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YT, Li Y, Ryu J, Su PY, Cheng CH, Wu WH, Li YS, Quinn PMJ, Leong KW, Tsang SH (2021) Impaired cholesterol efflux in retinal pigment epithelium of individuals with juvenile macular degeneration. Am. J. Hum. Genet. 108, 903–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaclavik V, Tran HV, Schorderet DF (2020) Malattia Leventinese: EFEMP1 R345W Variant Is a Hot Spot Mutation, Not a Founder Mutation. Ophthalmol Retina. 4, 1023. [DOI] [PubMed] [Google Scholar]
- van Zyl T, Yan W, McAdams A, Peng Y-R., Shekhar K, Regev A, Juric D, Sanes JR (2020) Cell atlas of aqueous humor outflow pathways in eyes of humans and four model species provides insight into glaucoma pathogenesis. Proc Natl Acad Sci U S A 117, 10339–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AH, Anand VN, Wang WH, Chatterton JE, Sun D, Shepard AR, Jacobson N, Pang IH, Deluca AP, Casavant TL, Scheetz TE, Mullins RF, Braun TA, Clark AF (2013) Exon-level expression profiling of ocular tissues. Exp Eye Res. 111:105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Gu HM, Zhang DW. (2014) Caveolin-1 and ATP binding cassette transporter A1 and G1-mediated cholesterol efflux. Cardiovasc Hematol Disord Drug Targets. 2014;14(2):142–8. [DOI] [PubMed] [Google Scholar]
- Wang K, Gaitsch H, Poon H, Cox NJ, Rzhetsky A. (2017) Classification of common human diseases derived from shared genetic and environmental determinants. Nat. Genet. 49, 1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb RN, Aung T, Medeiros FA (2014) The pathophysiology and treatment of glaucoma: A review. JAMA 311, 1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb RN, Leung CK, Crowston JG, Medeiros FA, Friedman DS, Wiggs JL, Martin KR (2016) Primary open-angle glaucoma. Nat. Rev. Dis. Primers. 2, 16067. [DOI] [PubMed] [Google Scholar]
- Wiggs JL, Pasquale LR (2017) Genetics of glaucoma. Hum. Mol. Genet. 26, R21–R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggs JL, Vollrath D. (2001) Molecular and clinical evaluation of a patient hemizygous for TIGR/MYOC. Arch. Ophthalmol. 119, 1674–1678. [DOI] [PubMed] [Google Scholar]
- Wirtz MK, Sykes R, Samples J, Edmunds B, Choi D, Keene DR, Tufa SF, Sun YY, Keller KE (2021) Identification of missense extracellular matrix gene variants in a large glaucoma pedigree and investigation of the N700S thrombospondin-1 variant in normal and glaucomatous trabecular meshwork cells. Curr. Eye. Res. 2021 Jun 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam GH, Gaplovska-Kysela K, Zuber C, Roth J. (2007) Aggregated myocilin induces Russell bodies and causes apoptosis: Implications for the pathogenesis of myocilin-caused primary open-angle glaucoma. Am. J. Pathol. 170, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Liu Q, Austin C, Drummond I, Pierce EA (2012). Knockdown of ttc26 disrupts ciliogenesis of the photoreceptor cells and the pronephros in zebrafish. Mol. Biol. Cell. 23,3069–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zode GS, Bugge KE, Mohan K, Grozdanic SD, Peters JC, Koehn DR, Anderson MG, Kardon RH, Stone EM, Sheffield VC. Topical ocular sodium 4-phenylbutyrate rescues glaucoma in a myocilin mouse model of primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2012. Mar 21;53(3):1557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zode GS, Kuehn MH, Nishimura DY, Searby CC, Mohan K, Grozdanic SD, Bugge K, Anderson MG, Clark AF, Stone EM, Sheffield VC. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J Clin Invest. 2011. Sep;121(9):3542–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.