

Abstract
Background
Microtubes (MTs), cytoplasmic extensions of glioma cells, are important cell communication structures promoting invasion and treatment resistance through network formation. MTs are abundant in chemoresistant gliomas, in particular, glioblastomas (GBMs), while they are uncommon in chemosensitive IDH-mutant and 1p/19q co-deleted oligodendrogliomas. The aim of this study was to identify potential signaling pathways involved in MT formation.
Methods
Bioinformatics analysis of TCGA was performed to analyze differences between GBM and oligodendroglioma. Patient-derived GBM stem cell lines were used to investigate MT formation under transforming growth factor-beta (TGF-β) stimulation and inhibition in vitro and in vivo in an orthotopic xenograft model. RNA sequencing and proteomics were performed to detect commonalities and differences between GBM cell lines stimulated with TGF-β.
Results
Analysis of TCGA data showed that the TGF-β pathway is highly activated in GBMs compared to oligodendroglial tumors. We demonstrated that TGF-β1 stimulation of GBM cell lines promotes enhanced MT formation and communication via calcium signaling. Inhibition of the TGF-β pathway significantly reduced MT formation and its associated invasion in vitro and in vivo. Downstream of TGF-β, we identified thrombospondin 1 (TSP1) as a potential mediator of MT formation in GBM through SMAD activation. TSP1 was upregulated upon TGF-β stimulation and enhanced MT formation, which was inhibited by TSP1 shRNAs in vitro and in vivo.
Conclusion
TGF-β and its downstream mediator TSP1 are important mediators of the MT network in GBM and blocking this pathway could potentially help to break the complex MT-driven invasion/resistance network.
Keywords: glioblastoma, microtubes, SMAD, TGF-β, Tsp1
Key Points.
TGF-β signaling is upregulated in GBM compared to chemosensitive oligodendroglioma.
TGF-β promotes MT formation in GBM cells and increases calcium exchange and invasion.
SMAD signaling and TSP1 are downstream mediators of TGF-β induced MT formation.
Importance of the Study.
Microtubes (MTs) are important structural elements of GBM cells contributing to invasion and treatment resistance. Thus, identifying molecular pathways that drive MT formation is crucial in developing new targeted treatments. Our data show that TGF-β is a major pathway that distinguishes glioblastoma from chemosensitive oligodendroglioma. In vitro, TGF-β stimulation induced MT formation in GBM cells by activating SMAD signaling and initiating Tsp1 expression. MT formation was inhibited in vivo by blocking either Tsp1 expression or TGF-β signaling. In conclusion, we identified a new function of TGF-β in tumor development and also characterized its specific downstream signaling which might open up new opportunities for GBM treatment.
Microtubes (MTs) are cytoplasmatic extensions of glioblastoma (GBM) cells that form a connective network leading to increased invasion and promoting resistance to radiotherapy.1 The formation of MTs is heterogeneous and more pronounced in a subpopulation of invasive and stem-like GBM cells.1,2 Growth Associated Protein 43 (GAP43), a protein involved in neurite sprouting, was identified as an important mediator involved in the structural development of MTs. Intercellular calcium waves were detected as a way of communication through the MT network and Connexin 43, a major gap junction protein, was found to be functionally involved.1 The MT network was observed extensively in GBM and also in astrocytic tumors in general, however, was scarce in oligodendroglioma. These 2 tumor types can be distinguished at the molecular level by IDH mutation and 1p/19q co-deletion, which are only present in oligodendroglioma.3 In contrast to GBMs and astrocytomas, oligodendroglioma patients show remarkable responses to radio- and chemotherapy and therefore have a much better prognosis.4 Thus, to identify mechanisms of MT formation in GBM and to unravel how MTs promote cell-to-cell communication might ultimately lead to a better understanding of therapeutic resistance and new ways of GBM treatment. Interestingly, a recent study showed that network formation of MTs was enhanced in the resection cavity after neurosurgery in experimental models.5 MT formation in this context might partly explain why recurrent tumors often develop close to the resection site. Recently, it was shown that glioma cells and neurons are connected through so-called neurogliomal synapses that are located on MTs indicating another layer of resistance through connection with normal cells.6 Molecular mediators driving the MT network identified so far such as GAP43 and tweety-homolog 1 (TTYH1)7 are also important in normal physiology of the CNS. Thus, the identification of more tumor-specific pathways that drive MT formation is of great interest in order to develop targeted therapies.
We recently showed by RNA sequencing of patient-derived xenograft (PDX) tissue from laser capture micro-dissected invasive and central tumor areas that the matricellular protein thrombospondin 1 (TSP1 or THBS1) was one of the most upregulated genes in infiltrative areas of GBM.8 TSP1 is a large trimeric calcium-binding molecule, which binds to diverse ligands and receptors.9 In patient biopsies, both TSP1 and TGF-β (transforming growth factor-beta) were expressed at high levels in GBM compared to low-grade gliomas (LGG).10 In addition, TSP1 is an unfavorable prognostic marker for GBM patients.11 The TGF-β18 but not the TGF-β212 canonical pathway transcriptionally regulates TSP1 expression through SMAD3 binding to the THBS1 promoter. TSP1 silencing inhibited cell invasion in vitro and in vivo, also in combination with anti-angiogenic therapy.8 Interestingly, TSP1 was also found to be regulated by calcium flux in several cancers.13
In the present study, we showed that TGF-β1 stimulation of GBM stem-like cell cultures promotes MT formation and that TSP1 is an important mediator of MT formation downstream of TGF-β1.
Materials and Methods
Ethics Statement
Patient material was obtained from surgeries performed at the Haukeland University Hospital (Bergen, Norway). Written consent was obtained from patients with procedures that were approved for the projects (project numbers 013.09 and 151825) by the Regional Ethics Committee (Bergen, Norway). In vivo experiments were conducted in accordance with the European Community for experimental animal use guidelines (L358-86/609EEC) with protocols approved by the Ethical Committee of INSERM (n° MESRI23570).
TCGA Data and GMT Files
For the “The Cancer Genome Atlas” (TCGA) dataset, RNAseqV2 median normalized data (RNA Seq V2 RSEM) and associated clinical data of the TCGA GBM and LGG cohorts were downloaded from cBioPortal (https://www.cbioportal.org/). Details are described in the Supplementary Methods.
Cell Culture
The patient-derived GBM stem cell (GSC) lines P3,14 GG6 and GG1615 are all derived from IDH wild-type (wt) GBM patients. GG16 was a giant cell glioblastoma.16 Cell lines were cultured in Neurobasal Medium, B27 supplement, Glutamax (NBM; Life Technologies), FGF2 (20 ng/ml), and EGF (20 ng/ml; Peprotech) in non-treated culture flasks (Nunc). U87, U251, and 293T cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained in DMEM supplemented with 10% fetal calf serum (FCS) and 1% glutamine. Medium was changed twice a week. All cell lines were grown at 37°C in a humidified atmosphere of 5% CO2.
In Vitro Experiments
2.5 × 104 cells from GSC lines were seeded on coverslips. Cells were treated with growth factors (10 ng/ml) and or inhibitors (10 µM) for 48 hours. For MT analysis and quantification, 300 cells were counted in random microscopic fields.
Western Blots
Western blot of cultured cells was performed as described previously.8 Details are described in the Supplementary Methods.
Immunofluorescence Staining
Immunofluorescence staining was performed as previously described.8 Details are described in the Supplementary Methods.
Immunohistochemistry
Immunohistochemistry of paraffin-embedded formalin-fixed tissue sections was performed as described previously.8 Details are described in the Supplementary Methods.
RNA Sequencing and Analysis
A total of 3 samples were assigned for each of the control (untreated) and treatment (TGF-β_48 hours) groups. Samples were processed for sequencing at Macrogen Inc, Korea where library preparation, sequencing, and quality control were performed. Details of the sample preparation and data analysis have been reported previously.17
GBM-Brain Organoid Co-Culture Ex Vivo Invasion Assay
For this assay, the preparation and culture of 18-day fetal brain organoids have been described in our previous work.18 For details, see Supplementary Methods.
In Vitro Calcium Imaging
Calcium signal spread across cells was carried out as previously described.19 Fluo-3-AM intensity was determined using the ImageJ z-axis profile function. Mean fluorescence intensity was determined from 20 time lapses per condition.
In Vivo Experiments
Ragγ2C−/− mice were housed with free access to food and water in a 12-hour light/dark cycle. Craniotomy and GBM spheroid implantation were done as previously described.20 LY2109761 was administered orally at 50 mg/kg twice daily (days 1-5 of each week) until the end of observation.
Statistics
All statistical analyses were performed on GraphPad Prism 8.1.2. Data are displayed as mean ± SD. To compare one variable between multiple groups, 1-way ANOVA with Tukey’s post hoc test was used. To compare 2 or more variables between multiple groups, 2-way ANOVA with Tukey’s post hoc test was used. Differences were considered statistically significant when the P-value was below .05.
Results
TGF-β Signaling Is Upregulated in GBM Compared to 1p/19q Co-Deleted Oligodendroglioma
MTs are abundant in GBM, however uncommon in 1p/19q co-deleted oligodendroglioma. To identify new drivers of MT formation, we compared gene expression data from TCGA of IDH-wt tumors, the majority being GBM, to IDH-mutant and 1p/19q co-deleted oligodendroglioma. We found that TGFB1 and TGFB2 were among the significantly differentially expressed genes (Supplementary Table 1). Both TGFB1 and TGFB2 are highly upregulated in IDH-wt tumors compared to IDH-mutant and 1p/19q co-deleted tumors (Figure 1a) and also upregulated in non–co-deleted (IDH-wt and IDH-mutant astrocytoma/GBM) vs co-deleted tumors (Supplementary Figure 1a). MT formation has been previously observed in particular at the invasive front of GBM.1 As TGF-β is a known driver of GBM invasion, we performed more detailed analyses. Interestingly, TGFB1 is located on chromosome 19q, which makes it an attractive candidate for MT formation. The differentially expressed genes of IDH-wt compared to IDH-mutant and 1p/19q co-deleted tumors (1.comparison) were then used to conduct a Gene Ontology (GO) term enrichment analysis. The top regulated pathways were related to extracellular matrix, which are important pathways in tumor invasion (Figure 1b). We performed the same GO term analysis for non–co-deleted vs co-deleted tumors (2.comparison) where extracellular matrix pathways were also found, however, below a number of immune-related pathways (Supplementary Figure 1b). We then analyzed the commonly regulated pathways from these 2 comparisons in a Venn diagram revealing that the top common pathways in both were related to extracellular matrix (Supplementary Figure 1c, d). As MT formation is also highly dependent on extracellular matrix reorganization this could indicate an important role of TGF-β in this process. When focusing on the top 20 GO terms where either TGFB1 or TGFB2 was present in the gene list, the majority of associated genes in these pathways were upregulated in IDH-wt compared to 1p/19q co-deleted tumors (Figure 1c). Next, we identified TGF-β responsive genes from the literature and showed that the majority of these genes are upregulated in IDH-wt compared to 1p/19q co-deleted tumors (Figure 1d). By using connectivity map from the Broad Institute,21 we analyzed drug candidates that would inhibit the expression signature of GBM (top 20 upregulated genes) compared to oligodendroglioma. One of the top drug candidates that came up in this screen were TGF-β inhibitors (Figure 1e) further highlighting an important role of the TGF-β pathway in promoting a GBM gene expression signature, when compared to chemosensitive oligodendroglioma.
Fig. 1.

TGF-β signaling is upregulated in GBM compared to IDH-mutant and 1p/19q co-deleted oligodendroglioma. Analysis of TCGA data comparing IDH-wt GBM and IDH-mutant and 1p/19q co-deleted oligodendroglioma. (a) TGFB1 and TGFB2 are upregulated in IDH-wt tumors. (b) GO term analysis reveals pathways related to extracellular matrix. (c) Heatmap of genes related to the top 20 GO terms where either TGFB1 or TGFB2 is present. (d) TGF-β responsive genes identified from the literature. (e) Analysis of drugs related to gene expression signature by connectivity map reveals that TGF-β inhibitors are one of the top candidate drugs that inhibit IDH-wt GBM signature. Abbreviations: GBM, glioblastoma; GO, Gene Ontology; IDH, isocitrate dehydrogenase; TCGA, The Cancer Genome Atlas; TGF-β, transforming growth factor-beta.
TGF-β Promotes MT Formation in GBM Cell Lines In Vitro
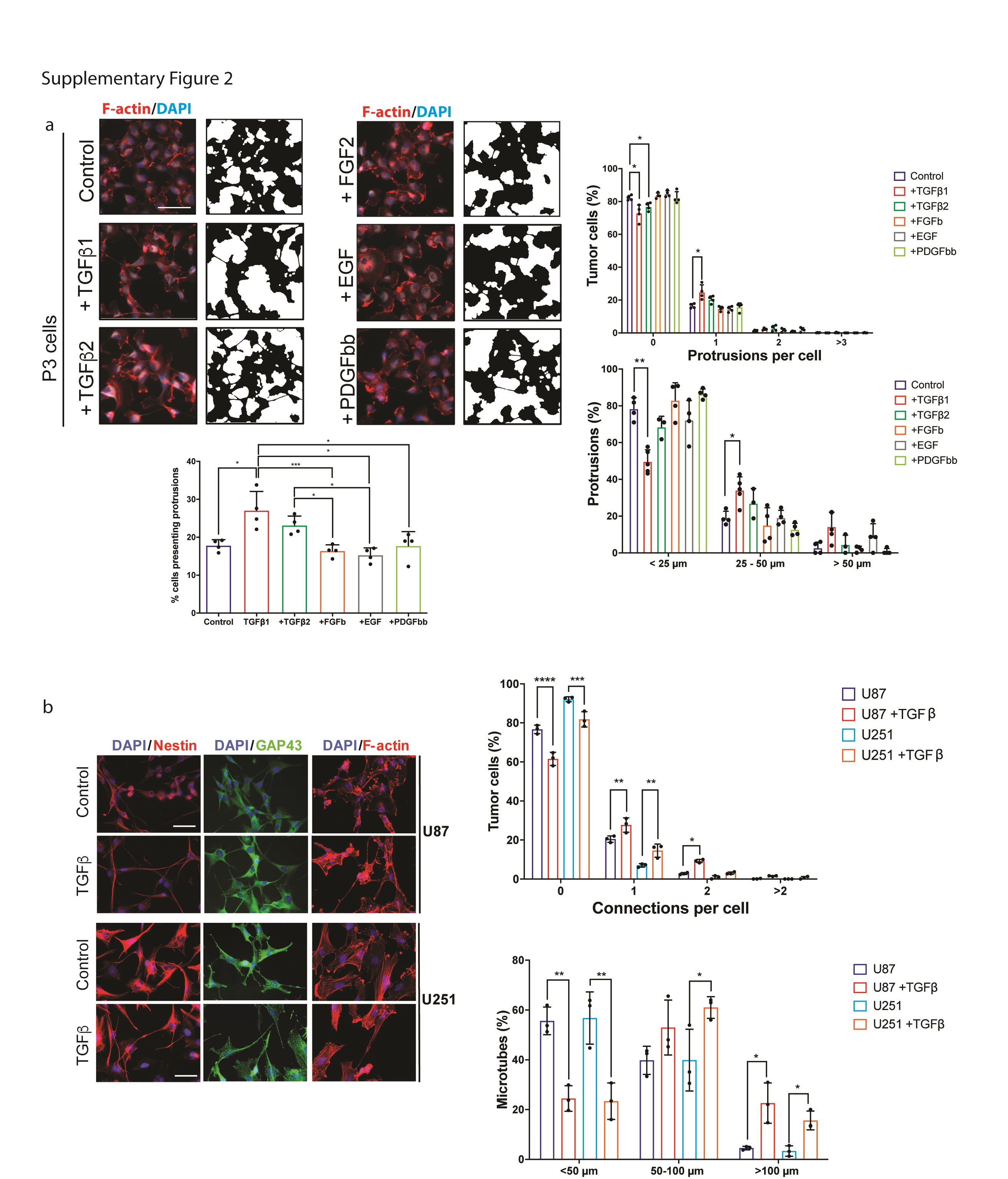
To analyze the ability of different growth factors, including TGF-β, to induce cytoplasmic protrusions (MT-like structures), we stimulated P3 GBM cells with EGF, FGF, PDGFbb, TGF-β1, or TGF-β2. We observed that TGF-β1 was most efficient in promoting cellular protrusions when analyzing the percentage of cells with protrusions, number of protrusions per cell, and length of protrusions (Supplementary Figure 2a). Next, we stimulated 2 different GSC lines (P3 and GG16) and 2 serum cultured GBM cell lines (U87 and U251) with TGF-β1 and analyzed morphological changes of the cells. A clear promotion of cellular protrusions by TGF-β1 was observed in all cell lines (Figure 2a; Supplementary Figure 2b, c). To verify that these protrusions are MTs we stained the cells for GAP43, a known driver for MT formation as well as nestin and actin to visualize the cytoskeleton. The protrusions were rich in GAP43 which colocalized with actin and nestin (Figure 2a; Supplementary Figure 2b). This indicated that the protrusions are MTs as described previously.1,2 To further verify that cells are interconnected through MTs upon TGF-β1 stimulation, we performed Electron microscopy. We observed MTs extending from one cell and inserting into the membrane of a neighboring cell (Figure 2b). We analyzed and quantified the formation of MTs in detail by confocal microscopy using GAP43/nestin or actin stainings. TGF-β1 stimulation increased the number of cells connected through MTs and importantly, also the length of MTs increased significantly (Figure 2c; Supplementary Figure 2b, c). To block TGF-β signaling, we used the TGF-β inhibitor LY2157299, which significantly reduced pSMAD2 phosphorylation in P3 GBM cells (Supplementary Figure 3a). MT formation was significantly inhibited by LY2157299: the number of MTs per cell, the number of cells with MTs, and the length of MTs decreased substantially under treatment with LY2157299 in both P3 and GG16 GBM cells (Figure 2c; Supplementary Figures 2c and 3b). In addition, we used an inducible shRNA to knockdown TGFBRII, as stable knockdown was lethal to the cells (data not shown). The inducible construct confirmed the inhibition of MT formation in P3 cells (Supplementary Figure 3c, d). As GAP43 has been shown to be a major structural element of MTs, we knocked down GAP43 with a shRNA to analyze if MT formation is blocked under TGF-β1 stimulation. As expected, GAP43 knockdown prevented a significant increase in the number and length of MTs under TGF-β1 stimulation (Supplementary Figure 4a, b) verifying that also in our culture system MT formation is dependent on GAP43 as described previously.1 To demonstrate that there is a functional connectivity between the tumor cells through the MT network, we performed calcium imaging using fluorescence indicator of intracellular calcium 3 (Fluo-3).22 The intensity of the calcium wave was calculated for entire frames. Stimulation with TGF-β1 resulted in increased calcium exchange between tumor cells through the MT network which was inhibited by LY2157299 (Figure 2d; Supplementary Figure 5; Movies 1–4).
Fig. 2.

TGF-β promotes MT formation and communication via calcium signaling in GBM cells. (a) Cellular protrusions induced by TGF-β1 in P3 GBM cells are identified as MTs due to the expression of GAP43 which co-localizes with the cytoskeleton protein nestin. Scale bar 20 µm. (b) Scanning Electron Microscopy of P3 GBM cells shows that MTs connect 2 neighboring cells through cytoplasmic insertions. Higher magnifications of specific areas are provided as indicated. (c) TGF-β inhibitor LY2157299 inhibits MT formation in P3 and GG16 GBM cells. Immunofluorescence staining for F-actin is shown. Quantification of connections per cell and MT length is presented. Scale bar 10 µm. Statistically significant differences of experimental groups compared to the control are shown on top of the respective bars. *P < .05; **P < .01; ***P < .001; ****P < .0001. (d) Calcium exchange between tumor cells is significantly increased upon TGF-β stimulation and inhibited by LY2157299 in P3 GBM cells. Fluorescence intensity represents the intensity of the calcium signal. The images were taken seconds following the laser injury as indicated. The bar graph represents the intensity at the time point as indicated by the dotted line in the curve diagram. Scale bar 30 µm. *P < .05; **P < .01. Abbreviations: GBM, glioblastoma; MT, microtube; TGF-β, transforming growth factor-beta.
TGF-β Induced MT Formation Is Associated With Invasion
Among the GBM cell lines used in this work, we identified one GSC line, GG6, that did not show increase in MT formation upon TGF-β1 stimulation. GG16, used as a control cell line, confirmed significant MT formation under stimulation (Figure 3a). As MT formation is associated with invasion, we aimed to analyze the invasiveness of the nonresponder cell line GG6 in comparison with the TGF-β1 responding GBM cell line GG16. We used a co-culture system of tumor spheroids with brain organoids from rat fetal brains to mimic the invasive process as described previously.18 Invasion was quantified as indicated in methods. The nonresponding cell line GG6 showed no significant invasion over a time period of 72 hours when co-cultured with brain organoids. In contrast, the TGF-β1 responding cell line GG16 showed substantial invasion into the organoid, which was not further increased by TGF-β1 stimulation, however significantly reduced by TGF-β inhibition with LY2157299, indicating endogenous presence of TGF-β in the organoid microenvironment (Figure 3b). Upon implantation of both cell lines in vivo, we verified a less invasive phenotype of the nonresponder GG6 compared to the responder GG16 and a reduced MT density in nestin immunostained sections (Figure 3c). Importantly, we confirmed a reduced MT density in corresponding patient biopsies from GG6 compared to GG16 stained with nestin antibodies (Figure 3c).
Fig. 3.

TGF-β induced MT formation is associated with invasion. (a) GG6 GBM cells are not responding to TGF-β1 stimulation with increased MT formation in contrast to GG16 GBM cells. Immunofluorescence stainings for nestin are shown. Black and white pictures are presented to better visualize the MT network. Quantification of connections per cell and MT length is presented. Scale bar 20 µm. *P < .05; **P < .01; ***P < .001; ****P < .0001. (b) Invasion of GG16 GBM cells into brain organoids is inhibited by LY2157299. TGF-β stimulation does not significantly increase invasion most likely due to the presence of TGF-β in the microenvironment. In contrast, nonresponder GG6 GBM cells do not show significant invasion into brain organoids. Immunofluorescence pictures of organoids after 72 h showing green fluorescent protein (GFP) signal (GBM cells). Higher magnifications of the invasive areas are provided below each organoid picture. Quantification of invasive cells is provided after 24, 48, and 72 hours. Scale bar 70 µm (zoom 14 µm). Statistically significant differences of experimental groups compared to the control are shown on top of the respective bars. *P < .05; **P < .01; ***P < .001. (c) Immunohistochemical staining of GG6 and GG16 patient and xenograft GBM with nestin antibodies showing an extensive MT network in GG16, which is low to absent in GG6. Quantification of MT density is provided. Scale bar 100 µm (zoom 10 µm). Abbreviations: GBM, glioblastoma; MT, microtube; TGF-β, transforming growth factor-beta.
SMAD Activation Is Important for MT Formation
To identify major signaling mediators in TGF-β1 induced MT formation, we analyzed activation of downstream pathways under stimulation with TGF-β1 in P3, GG6, and GG16 cell lines by double immunofluorescence stainings with nestin (Figure 4a). Canonical (SMAD2/3) signaling correlated with MT formation in P3 and GG16, however, not in GG6 where also pSMAD2/3 activation levels were lower. The non-canonical pathways (MAPK and Pi3K/Akt) did not correlate with MT formation in all 3 cell lines (Figure 4a). Likewise, SMAD inhibition under TGF-β1 stimulation using the SMAD inhibitor SIS3 significantly reduced MT formation (Figure 4b). To further substantiate the role of SMAD2/3 phosphorylation in MT formation, we immunostained consecutive sections from 7 GBM IDH-wt patient biopsies with pSMAD3 and nestin antibodies (Figure 4c; Supplementary Figure 6). Quantification of MT density in randomly selected pSMAD3+ and pSMAD3− areas was performed as described in the methods. For statistical analysis of the data, model selection was performed as indicated in the Supplementary Methods and Supplementary Table 2. The final model indicated a significant association between increased pSMAD3 expression and increased MT density when adjusting for overall cell density (T = 3.80, P = .0002).
Fig. 4.

SMAD activation is important for MT formation. (a) Double immunofluorescence stainings of pSMAD2/nestin, pMAPK/nestin, and pAkt/nestin for P3, GG16, and GG6 cell lines are shown. Arrows indicate cells with MT formation. Arrowheads point to cells without MT formation. Quantification of connections per cell is presented. Scale bar 10 µm. *P < .05; **P < .01; ***P < .001. (b) SMAD inhibitor SIS3 inhibits MT formation under TGF-β1 stimulation. Immunofluorescence staining for F-actin is shown. Quantification of cells with MTs and MT length is presented. Scale bar 10 µm. *P < .05; **P < .01. (c) SMAD3 phosphorylation correlates with MT formation in GBM patient biopsies. Quantification of MT length on consecutive pSMAD3 immunostained and nestin immunostained sections of 1 patient (patient 2) is shown (see the Supplementary Methods for details). Images in the left column show original image data, images in the right column show visualizations of the quantifications. Black lines mark MTs, red outlines denote nuclei, and green outlines pSMAD3-positive nuclei. The graph shows the correlation of pSMAD3 expression with MT length using a linear mixed model. The surface shows the predicted value based on the fixed parameters of the linear mixed model. The results of all patients are included in the graph (each color refers to 1 patient; single graphs are shown in Supplementary Figure 6b). Scale bar 100 µm; P = .0013.
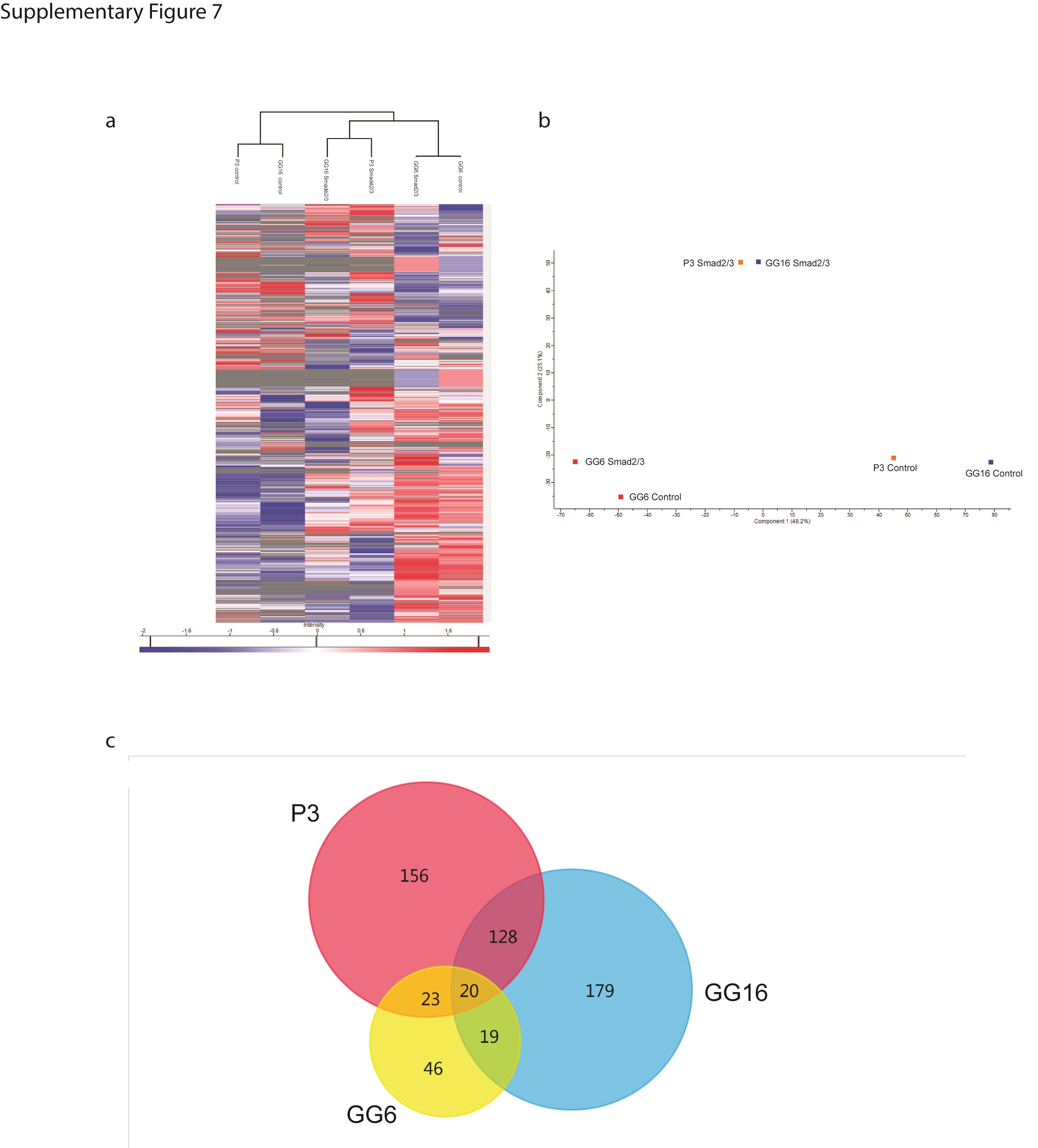
Having demonstrated that pSMAD2/3 signaling is crucial for MT formation downstream of TGF-β, we performed co-immunoprecipitation of pSMAD2/3 signaling complexes in P3, GG6, and GG16 cell lines, followed by mass spectrometry to identify potential binding partners. Interestingly, P3 and GG16 samples clustered together, while GG6 samples were separated (Supplementary Figure 7a). Despite these differences, the proteins in common between all 3 cell lines were related to TGF-β/SMAD signaling as also revealed by KEGG pathway analysis and GO biological function (Supplementary Figure 7, Supplementary Tables 8, 10, and 11). However, the SMAD2/3 proteome was very different between P3/GG16 and GG6. While P3 and GG16 had 128 proteins in common, only 23 and 19 proteins were shared between GG6/P3 and GG6/GG16, respectively (Supplementary Figure 7, Supplementary Tables 3–9). The proteins in common between P3 and GG16 were related to a number of pathways/biological processes (Supplementary Tables 12 and 13), while none could be identified for proteins of GG6. These results further confirm the similarity in TGF-β/SMAD signaling between responders in contrast to the nonresponder GG6.
TSP1 Is a Candidate for MT Formation Downstream of TGF-β1 and SMAD2
To identify targetable proteins involved in MT formation downstream of TGF-β1 and SMAD activation, we performed RNA sequencing of the responder cell lines GG16 and P3 and the nonresponder cell line GG6 with and without TGF-β1 stimulation (48 hours) to identify differences in gene expression profiles that would indicate potential candidates for MT formation downstream of TGF-β1/SMAD2/3 activation. The number of significantly upregulated genes under TGF-β1 stimulation varied substantially between the cell lines (Supplementary Table 14). In particular, GG16 showed only 60 upregulated genes, however, 34 of these genes were shared with P3, while only 1 gene was shared with the nonresponder cell line GG6 (Figure 5a; Supplementary Table 14). 19 genes were commonly upregulated in all 3 cell lines. Gene enrichment analysis showed that in all 3 cell lines, extracellular structure organization and extracellular matrix organization were the top regulated GOs (Figure 5b). When analyzing these GOs for common genes between the responders P3 and GG16, only 4 genes were found, while also 4 genes were in common between all 3 cell lines (Figure 5c; Supplementary Tables 15 and 16). Among the 4 genes in common between the responders P3 and GG16, TSP1 (THBS1) was identified as an interesting candidate because (1) it also showed up in the previous analysis of TCGA data (Figure 1c, d); (2) it is upregulated in invasive tumor areas compared to the core in GBM xenografts,8 and (3)TSP1 is a downstream mediator of TGF-β1 and SMAD3 signaling in GBM and important in tumor cell invasion.8 Analysis of TCGA data also confirmed upregulation of TSP1 in IDH-wt GBM compared to IDH-mutant and 1p/19q co-deleted oligodendroglioma (Figure 5d).
Fig. 5.

TSP1 is a candidate for MT formation downstream of TGF-β1 and SMAD2/3. RNA sequencing data of P3, GG6, and GG16 GBM cells unstimulated and stimulated with TGF-β1 for 48 hours. (a) Venn diagram showing number of common and unique upregulated genes upon TGF-β1 stimulation. Hypergeometric test for all overlaps, P < .0001. (b) Gene enrichment analysis showing upregulation of pathways related to extracellular matrix/structure in all 3 cell lines. (c) Venn diagram showing the number of common and unique regulated genes among the 3 cell lines from extracellular matrix and extracellular structure GOs. Hypergeometric test for GG6/GG16 P < .05, for all other overlaps, P < .0001. (d) Comparison of TSP1 expression in TCGA data between IDH-wt and IDH-mutant, 1p/19q co-deleted tumors. Abbreviations: GBM, glioblastoma; GO, Gene Ontology; IDH, isocitrate dehydrogenase; MT, microtube; TCGA, The Cancer Genome Atlas; TGF-β, transforming growth factor-beta; TSP1, thrombospondin 1.
TSP1 Is Activated by TGF-β1 and Mediates MT Formation
As described previously,8 Tsp1 expression is upregulated by TGF-β signaling through activation of the Tsp1 promoter by pSMAD3 (Figure 6a). Here, we confirmed that TSP1 protein expression was induced upon TGF-β1 stimulation in the responder cell lines GG16 and P3, while it was absent in the nonresponder cell line GG6 (Figure 6b; Supplementary Figure 8). We used a shRNA to knock down expression of TSP1 and analyzed MT formation under TGF-β1 stimulation. ShTsp1 significantly reduced MT formation in P3 cells (connections per cell and MT length) when compared to scrambled control (Figure 6c). Likewise, overexpression of TSP1 in P3 cells showed increase in MT formation compared to control cells (Supplementary Figure 9).
Fig. 6.

Inhibition of TSP1 reduces MT formation in vitro and in vivo. (a) Schematic figure showing upregulation of Tsp1 expression by TGF-β signaling through binding of pSMAD3 to the Tsp1 promoter. Tsp1 expression can be inhibited by LY2157299 or shRNA Tsp1. (b) Western blot of Tsp1 in P3 shControl and P3 shTsp1 with and without TGF-β1 stimulation. (c) MT formation is inhibited by shTsp1. Immunofluorescence staining for F-actin is shown. Quantification of connections per cell and MT length is presented. Scale bar 10 µm. *P < .05; **P < .01; ***P < .001. (d) Formation of MT network in vivo in the orthotopic P3-GFP GBM xenograft model was analyzed by intravital imaging. ShTsp1 and LY2157299 inhibit MT formation compared to shcontrol. Immunofluorescence pictures of intravital imaging are shown. Quantification of MT number per tumor cell is presented. Scale bar 150 µm. *P < .05; **P < .01; ***P < .001. Abbreviations: MT, microtube; TGF-β, transforming growth factor-beta; TSP1, thrombospondin 1.
To verify that TGF-β and TSP1 are important players of MT formation in vivo, we performed intravital imaging of P3 cells implanted orthotopically in NOD/SCID mice. After 7-70 days, both inhibition of TGF-β by LY2157299 and knockdown of TSP1 by shRNA reduced the number of MTs per tumor cell in vivo compared to controls (Figure 6d).
Discussion
In the present study, we identified a new molecular mechanism of MT formation in GBM. We showed that TGF-β1, a multifunctional cytokine, promotes MT formation in vitro and in vivo. MTs have been defined as major drivers of GBM invasion and resistance to standard treatments such as radiation and chemotherapy.1 Since this hallmark study, where the GAP43 protein was identified as a major structural protein of MTs, new and fundamental insights into detailed molecular mechanisms of MT formation are still lacking. In search of molecular drivers of MT formation, we performed a bioinformatics analysis comparing IDH-mutant 1p/19q co-deleted vs IDH-wt tumors. We chose this starting point because MT formation is abundant in IDH-wt tumors while it is low to absent in IDH-mutant and co-deleted tumors.1 This analysis brought our attention to TGF-β. Among other gene candidates, TGFB1 and TGFB2 are expressed at significantly lower levels in co-deleted tumors. Importantly, TGFB1 is located on chromosome 19q and very important in GBM invasion,23 which are additional justifications for its potential role in MT formation. TGF-β is a cytokine with multiple functions which are dependent on the cell type and its associated microenvironment.24 In GBM, TGF-β is well characterized as a cytokine that promotes invasion, angiogenesis as well as suppression of the immune system.25 Thus, our results add an additional function of TGF-β to its divergent roles in GBM development. We demonstrated experimentally in different ways that TGF-β1 is important in MT formation: (1) by stimulation with TGF-β1; (2) by blocking of TGF-β activity using the inhibitor LY2157299 and an inducible shRNA. As our study is among the first to analyze MT formation in vitro, we had to verify that the structures we analyzed are corresponding to the MTs which have been shown in vivo.1 We identified MTs in our culture system by demonstrating co-expression of major structural proteins of MTs such as nestin, actin, and GAP43. Nestin, a protein related to cellular stemness, has recently been identified as an important protein expressed in the GBM MT network in vivo.2 Thus, we used this marker in our study to identify and quantify MTs. Knockdown of GAP43, which significantly reduced MT formation under TGF-β1 stimulation, further corroborated that the structures we analyzed in vitro are very similar to the MTs formed in vivo.
MTs connect tumor cells with each other forming a communicating network. To show that network formation is promoted by TGF-β1, we performed calcium imaging. As expected, calcium exchange increased upon TGF-β1 stimulation and was inhibited by LY2157299. Network formation and increase in calcium exchange in GBM cells in vitro was also observed by da Silva et al using ROCK inhibition.19
The majority of our cell lines responded to TGF-β1 with increase in MT formation. Interestingly, we identified one cell line (GG6), which did not respond and also showed very limited invasive capacity upon TGF-β stimulation using ex vivo brain organoid co-cultures and in vivo xenografts. These results highlight that MT formation and invasion are highly connected as also demonstrated previously.1 Inhibition of TGF-β activity in GBM in a clinical setting has been unsuccessful so far.26,27 Reasons for these disappointing results may be the dose limitations due to the important physiological roles of TGF-β in the body and also the BBB which often impairs drug penetration.28 Thus, we aimed to further characterize downstream signaling of TGF-β inducing MT formation to reveal more cancer-specific downstream signaling. First, we identified the canonical SMAD2/3 signaling as being the most important downstream pathway and also showed in a patient setting that SMAD3 activation correlates with MT length in patient biopsies. Previously, SMAD2/3 signaling was shown to impact glioma invasion,15,29 whereas SMAD3 was defined as the main transcription factor regulating invasion and TSP1 expression in GBM cells.8 To further identify more specific downstream targets of TGF-β and SMAD signaling, we performed RNA sequencing and bioinformatics analysis of cell lines P3 and GG16 (responding to TGF-β with MT formation) and compared the data to the nonresponder cell line GG6. TSP1 came up as an interesting candidate which was absent in the nonresponder GG6 as also verified by western blotting. Interestingly, we have previously shown that TSP1 is activated by TGF-β signaling and specifically by SMAD3 through a SMAD3 binding site in the TSP1 promoter.8 We also demonstrated in this study that TSP1 promotes GBM invasion downstream of TGF-β and SMAD. Thus, as invasion and MT formation are linked to each other, we demonstrated that knockdown of TSP1 inhibited invasion and MT formation both in vitro and in vivo. TSP1 has a substantial interactome that is still not fully explored consisting of extracellular matrix components, matricellular proteins, different receptors, proteases, and growth factors.8,30 Thus, TSP1 likely involves a potential network of additional proteins in MT formation/invasion, which need to be identified in future studies.
In conclusion, we identified TGF-β as a new mediator of MT formation through SMAD and Tsp1 signaling. Blocking this pathway might be an attractive strategy to treat GBM in the future.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to thank B. Nordanger and H. S. Sdik for expert technical assistance and the Molecular Imaging Center (MIC) in Bergen, Norway for technical support.
Contributor Information
Justin V Joseph, Department of Clinical Medicine, University of Aarhus, Aarhus, Denmark; Department of Biomedicine, University of Bergen, Bergen, Norway.
Capucine R Magaut, INSERM, LAMC, U1029, University of Bordeaux, Pessac, France.
Simon Storevik, Department of Biomedicine, University of Bergen, Bergen, Norway; Department of Pathology, Haukeland University Hospital, Bergen, Norway.
Luiz H Geraldo, INSERM U970, Paris Cardiovascular Research Center, Paris, France.
Thomas Mathivet, INSERM U970, Paris Cardiovascular Research Center, Paris, France.
Md Abdul Latif, Department of Biomedicine, University of Bergen, Bergen, Norway; Department of Clinical Science, University of Bergen, Bergen, Norway.
Justine Rudewicz, Department of Biomedicine, University of Bergen, Bergen, Norway.
Joris Guyon, INSERM, LAMC, U1029, University of Bordeaux, Pessac, France.
Matteo Gambaretti, INSERM, LAMC, U1029, University of Bordeaux, Pessac, France.
Frida Haukas, Department of Biomedicine, University of Bergen, Bergen, Norway.
Amalie Trones, Department of Biomedicine, University of Bergen, Bergen, Norway.
Lars A Rømo Ystaas, Department of Biomedicine, University of Bergen, Bergen, Norway.
Jubayer A Hossain, Department of Biomedicine, University of Bergen, Bergen, Norway; Department of Pathology, Haukeland University Hospital, Bergen, Norway.
Sandra Ninzima, Department of Biomedicine, University of Bergen, Bergen, Norway; Department of Biological Sciences, University of Bergen, Bergen, Norway.
Sylvain Cuvellier, CNRS, IBGC, UMR5095, University of Bordeaux, Bord, eaux, France.
Wenjing Zhou, Department of Biomedicine, University of Bergen, Bergen, Norway; Department of Blood Transfusion, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, P.R. China; Department of Neurosurgery, Qilu Hospital of Shandong University and Institute of Brain and Brain-Inspired Science, Shandong University; Shandong Key Laboratory of Brain Function Remodeling, Jinan, Shandong, P.R. China.
Tushar Tomar, PamGene International B.V., BJ ‘s-Hertogenbosch, the Netherlands.
Barbara Klink, Department of Biomedicine, University of Bergen, Bergen, Norway; National Center of Genetics (NCG), Laboratoire national de santé (LNS), Dudelange, Luxembourg; Department of Oncology, Luxembourg Institute of Health (LIH), Strassen, Luxembourg.
Lalit Rane, Department of Clinical Science, University of Bergen, Bergen, Norway.
Bronwyn K Irving, School of Medicine, University of Leeds, Leeds, UK.
Joanne Marrison, Department of Biology, University of York, York, UK.
Peter O’Toole, Department of Biology, University of York, York, UK.
Heiko Wurdak, School of Medicine, University of Leeds, Leeds, UK.
Jian Wang, Department of Biomedicine, University of Bergen, Bergen, Norway; Department of Neurosurgery, Qilu Hospital of Shandong University and Institute of Brain and Brain-Inspired Science, Shandong University; Shandong Key Laboratory of Brain Function Remodeling, Jinan, Shandong, P.R. China.
Zhang Di, Department of Neurosurgery, Qilu Hospital of Shandong University and Institute of Brain and Brain-Inspired Science, Shandong University; Shandong Key Laboratory of Brain Function Remodeling, Jinan, Shandong, P.R. China.
Even Birkeland, Department of Biomedicine, University of Bergen, Bergen, Norway.
Frode S Berven, Department of Biomedicine, University of Bergen, Bergen, Norway.
Frank Winkler, Neurology Clinic and National Center for Tumor Diseases, University of Heidelberg, Heidelberg, Germany.
Frank A E Kruyt, Department of Medical Oncology, University of Groningen, University Medical Centre Groningen, Groningen, Netherlands.
Andreas Bikfalvi, INSERM, LAMC, U1029, University of Bordeaux, Pessac, France.
Rolf Bjerkvig, Department of Biomedicine, University of Bergen, Bergen, Norway; Department of Oncology, Luxembourg Institute of Health (LIH), Strassen, Luxembourg.
Thomas Daubon, Department of Biomedicine, University of Bergen, Bergen, Norway; INSERM, LAMC, U1029, University of Bordeaux, Pessac, France; CNRS, IBGC, UMR5095, University of Bordeaux, Bord, eaux, France.
Hrvoje Miletic, Department of Biomedicine, University of Bergen, Bergen, Norway; Department of Pathology, Haukeland University Hospital, Bergen, Norway.
Funding
S.S. was supported by a fellowship from Helse Vest (grant nr. 912251). This work was supported by the Norwegian Cancer Society (grant nr. 197933), Fondation ARC, Ligue Contre le Cancer, and ARTC.
Conflict of interest statement. The authors declare that they have no conflict of interest.
Authorship statement. Performed experiments and analyzed data: J.V.J., C.R.M., S.S., L.H.G., T.M., J.G., M.G., F.H., A.T., L.A.R.Y., J.A.H., S.N., S.C., W.Z., T.T., B.K., L.R., B.K.I., J.M., Z.D., E.B., and T.D. Analyzed data: P.O., H.W., J.W., F.W., A.B., F.S.B., T.D., and H.M. Analyzed sequencing data: M.D.A.L. and J.R. Wrote the manuscript: R.B., T.D., and H.M.
References
- 1. Osswald M, Jung E, Sahm F, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528(7580):93–98. [DOI] [PubMed] [Google Scholar]
- 2. Xie R, Kessler T, Grosch J, et al. Tumor cell network integration in glioma represents a stemness feature. Neuro Oncol. 2020;23(5):757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 4. van den Bent MJ. New perspectives for the diagnosis and treatment of oligodendroglioma. Expert Rev Anticancer Ther. 2001;1(3):348–356. [DOI] [PubMed] [Google Scholar]
- 5. Weil S, Osswald M, Solecki G, et al. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 2017;19(10):1316–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Venkataramani V, Tanev DI, Strahle C, et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. 2019;573(7775):532–538. [DOI] [PubMed] [Google Scholar]
- 7. Jung E, Osswald M, Blaes J, et al. Tweety-homolog 1 drives brain colonization of gliomas. J Neurosci. 2017;37(29):6837–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Daubon T, Léon C, Clarke K, et al. Deciphering the complex role of thrombospondin-1 in glioblastoma development. Nat Commun. 2019;10(1):1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adams JC, Lawler J. The thrombospondins. Cold Spring Harb Perspect Biol. 2011;3(10):a009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kawataki T, Naganuma H, Sasaki A, Yoshikawa H, Tasaka K, Nukui H. Correlation of thrombospondin-1 and transforming growth factor-beta expression with malignancy of glioma. Neuropathology. 2000;20(3):161–169. [DOI] [PubMed] [Google Scholar]
- 11. Qi C, Lei L, Hu J, Wang G, Liu J, Ou S. Thrombospondin-1 is a prognostic biomarker and is correlated with tumor immune microenvironment in glioblastoma. Oncol Lett. 2021;21(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Naganuma H, Satoh E, Kawataki T, Amagasaki K, Satoh H, Nukui H. Cell density regulates thrombospondin-1 production in malignant glioma cells. J Neurooncol. 2003;63(2):147–153. [DOI] [PubMed] [Google Scholar]
- 13. Firlej V, Mathieu JR, Gilbert C, et al. Thrombospondin-1 triggers cell migration and development of advanced prostate tumors. Cancer Res. 2011;71(24):7649–7658. [DOI] [PubMed] [Google Scholar]
- 14. Eskilsson E, Rosland GV, Talasila KM, et al. EGFRvIII mutations can emerge as late and heterogenous events in glioblastoma development and promote angiogenesis through Src activation. Neuro Oncol. 2016;18(12):1644–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joseph JV, Conroy S, Tomar T, et al. TGF-β is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 2014;5:e1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roesch S, Rapp C, Dettling S, Herold-Mende C. When immune cells turn bad-tumor-associated microglia/macrophages in glioma. Int J Mol Sci. 2018;19(2):436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hossain JA, Latif MA, Ystaas LAR, et al. Long-term treatment with valganciclovir improves lentiviral suicide gene therapy of glioblastoma. Neuro Oncol. 2019;21(7):890–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bjerkvig R, Laerum OD, Mella O. Glioma cell interactions with fetal rat brain aggregates in vitro and with brain tissue in vivo. Cancer Res. 1986;46(8):4071–4079. [PubMed] [Google Scholar]
- 19. da Silva B, Irving BK, Polson ES, et al. Chemically induced neurite-like outgrowth reveals a multicellular network function in patient-derived glioblastoma cells. J Cell Sci. 2019;132(19):jcs228452. [DOI] [PubMed] [Google Scholar]
- 20. Mathivet T, Bouleti C, Van Woensel M, et al. Dynamic stroma reorganization drives blood vessel dysmorphia during glioma growth. EMBO Mol Med. 2017;9(12):1629–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. [DOI] [PubMed] [Google Scholar]
- 22. Minta A, Kao JP, Tsien RY. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J Biol Chem. 1989;264(14):8171–8178. [PubMed] [Google Scholar]
- 23. Caja L, Bellomo C, Moustakas A. Transforming growth factor β and bone morphogenetic protein actions in brain tumors. FEBS Lett. 2015;589(14):1588–1597. [DOI] [PubMed] [Google Scholar]
- 24. Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity. 2019;50(4):924–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Birch JL, Coull BJ, Spender LC, et al. Multifaceted transforming growth factor-beta (TGFβ) signalling in glioblastoma. Cell Signal. 2020;72:109638. [DOI] [PubMed] [Google Scholar]
- 26. Brandes AA, Carpentier AF, Kesari S, et al. A phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016;18(8):1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wick A, Desjardins A, Suarez C, et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Invest New Drugs. 2020;38(5):1570–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pandit R, Chen L, Götz J. The blood-brain barrier: physiology and strategies for drug delivery. Adv Drug Deliv Rev. 2020;165–166:1–14. [DOI] [PubMed] [Google Scholar]
- 29. Liu Z, Kuang W, Zhou Q, Zhang Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int J Mol Med. 2018;42(6):3395–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Resovi A, Pinessi D, Chiorino G, Taraboletti G. Current understanding of the thrombospondin-1 interactome. Matrix Biol. 2014;37:83–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.