

Abstract
Parkinson’s disease is a complex neurodegenerative disorder for which both rare and common genetic variants contribute to disease risk, onset, and progression. Mutations in more than 20 genes have been associated with the disease, most of which are highly penetrant and often cause early onset or atypical symptoms. Although our understanding of the genetic basis of Parkinson’s disease has advanced considerably, much remains to be done. Further disease-related common genetic variability remains to be identified and the work in identifying rare risk alleles has only just begun. To date, genome-wide association studies have identified 90 independent risk-associated variants. However, most of them have been identified in patients of European ancestry and we know relatively little of the genetics of Parkinson’s disease in other populations. We have a limited understanding of the biological functions of the risk alleles that have been identified, although Parkinson’s disease risk variants appear to be in close proximity to known Parkinson’s disease genes and lysosomal-related genes. In the past decade, multiple efforts have been made to investigate the genetic architecture of Parkinson’s disease, and emerging technologies, such as machine learning, single-cell RNA sequencing, and high-throughput screens, will improve our understanding of genetic risk.
Introduction
The burden of Parkinson’s disease is a growing healthcare problem, with a global prevalence that is expected to double from 6·2 million cases in 2015 to 12·9 million cases by 2040.1 Clinically, Parkinson’s disease is defined as a progressive movement disorder, although the presentation of non-motor symptoms can also be severe. Parkinson’s disease has a long prodromal phase with features such as anosmia, constipation, and sleep disturbance.2,3 Pathologically, Parkinson’s disease is characterised by a loss of dopaminergic neurons in the substantia nigra and the presence of Lewy bodies in the midbrain. Lewy bodies are protein aggregates consisting of many proteins, including α-synuclein (encoded by the SNCA gene). Parkinson’s disease is the most common synucleinopathy; other synucleinopathies are multiple system atrophy and dementia with Lewy bodies, and these are clinically, and perhaps genetically, overlapping diseases.4,5
Advancing age is the greatest risk factor for Parkinson’s disease, but both environment and genetics are thought to affect disease risk and progression. Although studying the environmental contribution to disease is complex, potential associations between Parkinson’s disease and several environmental traits have been found, including pesticide exposure, smoking, and caffeine intake.6–8 Genetic contributors to Parkinson’s disease exist across a continuum, ranging from DNA variants that are highly penetrant (ie, causal) to variants that individually exert a small increase in lifetime risk of disease. Genetic risk is often divided into categories: rare DNA variants with high effect sizes, which are typically associated with monogenic or familial Parkinson’s disease; and more common, smaller effect variants, which are usually identified in apparently sporadic Parkinson’s disease.
Rare disease-causing DNA variants were historically identified using linkage studies in large families with Parkinson’s disease, and have more recently been identified with next-generation sequencing techniques, such as exome and genome sequencing. The common genetic components of Parkinson’s disease have mostly been identified using genome-wide association studies (GWASs) and to date 90 independent risk signals have been identified.9 In this Review, we describe current knowledge of Parkinson’s disease genetics, including the most recent developments in the field, and discuss our predictions for the future of Parkinson’s disease genetics.
Current state of Parkinson’s disease genetics
Monogenic Parkinson’s disease
For Parkinson’s disease, the term monogenic is a useful oversimplification. Although several highly penetrant rare variants are linked to Parkinson’s disease (resulting in so-called monogenic Parkinson’s disease), the presentation of the disease is likely to be affected by other factors, including both genetic and non-genetic factors. In some carriers of highly penetrant variants, the disease will not manifest (known as incomplete penetrance), and for those with the disease, the age of onset, clinical presentation, and progression can differ, even among those with the same variant and within families. These observations suggest that additional genetic or environmental factors affect the disease process in addition to the single variant of interest, and highlight the complexity of the causes of Parkinson’s disease. Nevertheless, considerable progress has been made in the identification of rare, highly penetrant variants.
Family studies of Parkinson’s disease genetics first yielded results in 1997, with the discovery of a missense variant in SNCA,10 and family-based studies continue to be a productive line of investigation. To date, rare variants in more than 20 genes have been reported to cause Parkinson’s disease (table). However, the relevance of many of these genes and variants is heavily debated, or replication and functional validation studies have not been done (eg, for LRP10,11–15 TMEM230, and DNAJC1316–18). With large, population-wide sequencing studies such as the UK Biobank19 becoming available, increased efforts are needed to quantify and validate the pathogenicity of variants and nominated pathogenic genes. These efforts will prevent uncertainties in assessing lifetime risk for Parkinson’s disease, both in the clinic and direct-to-consumer genetic testing.
Table:
Mutations that have been reported to cause Parkinson’s disease
| Mutation | Note | Year of discovery | Proposed disease mechanism | Inheritance | Frequency | Nominated by GWAS | Multiple independent families reported* | Functional evidence† | Negative reports published‡ | Confidence as actual PD gene§ | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNCA | Missense or multiplication | Often with dementia | 1997, 2003 | Gain of function or overexpression | Dominant | Very rare | Yes | ++ | ++ | + | Very high |
| PRKN | Missense or loss of function | Often early onset | 1998 | Loss of function | Recessive | Rare | No | ++ | ++ | + | Very high |
| UCHL1 | Missense | .. | 1998 | Loss of function? | Dominant | Unclear | No | − | + | −− | Low |
| PARK7 | Missense | Often early onset | 2003 | Loss of function | Recessive | Very rare | No | ++ | ++ | + | Very high |
| LRRK2 | Missense | .. | 2004 | Gain of function | Dominant | Common | Yes | ++ | ++ | + | Very high |
| PINK1 | Missense or loss of function | Often early onset | 2004 | Loss of function | Recessive | Rare | No | ++ | ++ | + | Very high |
| POLG | Missense or loss of function | Atypical PD | 2004 | Loss of function? | Dominant | Rare | No | ++ | + | + | High |
| HTRA2 | Missense | .. | 2005 | Unclear | Dominant | Unclear | No | − | + | −− | Low |
| ATP13A2 | Missense or loss of function | Atypical PD | 2006 | Loss of function | Recessive | Very rare | No | ++ | ++ | + | Very high |
| FBXO7 | Missense | Often early onset | 2008 | Loss of function | Recessive | Very rare | No | ++ | ++ | + | Very high |
| GIGYF2 | Missense | .. | 2008 | Unclear | Dominant | Unclear | No | + | + | −− | Low |
| GBA | Missense or loss of function | .. | 2009 | Likely loss of function | Dominant (incomplete penetrance) | Common | Yes | ++ | ++ | + | Very high |
| PLA2G6 | Missense or loss of function | Often early onset | 2009 | Loss of function | Recessive | Rare | No | ++ | ++ | + | Very high |
| EIF4G1 | Missense | .. | 2011 | Unclear | Dominant | Unclear | No | − | + | −− | Low |
| VPS35 | Missense | .. | 2011 | Loss of function | Dominant | Very rare | No | ++ | + | + | Very high |
| DNAJC6 | Missense or loss of function | Often early onset | 2012 | Loss of function | Recessive | Very rare | No | ++ | + | + | High |
| SYNJ1 | Missense or loss of function | Often atypical PD | 2013 | Loss of function | Recessive | Very rare | No | ++ | + | + | High |
| DNAJC13 | Missense | Same family as TMEM230 | 2014 | Unclear | Dominant | Unclear | No | + | + | − | Low |
| TMEM230 | Missense | Same family as DNAJC13 | 2016 | Loss of function? | Dominant | Unclear | No | − | + | − | Low |
| VPS13C | Missense or loss of function | .. | 2016 | Loss of function | Recessive | Rare | Yes | ++ | + | + | High |
| LRP10 | Missense or loss of function | .. | 2018 | Loss of function? | Dominant | Unclear | No | − | + | −− | Low |
GWAS=genome-wide association study. PD=Parkinson’s disease.
In this column, ++ denotes ≥4 families reported; + denotes ≥2 and <4 families reported; − denotes 1 family reported; −−denotes no reported families,
In this column, ++ denotes ≥4 disease-related reports; + denotes ≥1 and <4 disease-related reports; − denotes no disease-related reports.
Reports that could not replicate the finding that this gene is a PD gene. In this column, + denotes no negative reports; −denotes ≥1 and <4 negative reports; −− denotes ≥4 negative reports.
Sum of the scores in the three preceding columns, with each + adding 1 and each − subtracting 1; very high denotes a score of ≥5; high denotes a score of 4; medium denotes a score of 2 or 3; low denotes a score of ≤1.
The characteristics of the disease-causing variants for monogenic Parkinson’s disease (ie, variants with large risk effects that often result in protein-coding changes or large expression differences) make them amenable to modelling in cellular and animal systems using genetic approaches. The goals of such work are to understand the molecular mechanisms that underlie these monogenic forms of Parkinson’s disease and to evaluate whether these mechanisms are also applicable to the sporadic form of Parkinson’s disease. Studies of patients with disease-causing variants could be done to assess therapies that target a particular genetic cause, and individuals who are non-manifesting carriers could be recruited to assess pre-symptomatic therapies. The rarity of Parkinson’s disease-causing variants means that gathering such cohorts is a challenge, but there are major efforts to do so. Perhaps the most straightforward group to recruit is that of individuals with LRRK2 mutations (both patients with Parkinson’s disease and non-symptomatic carriers), especially Gly2019Ser; this variant is common enough in some populations that well-powered cohorts could be collected.20 Indeed, LRRK2 kinase inhibitors are being developed and tested, suggesting that individuals with LRRK2 mutations will be one of the first precision medicine cohorts for Parkinson’s disease.21 Of note, partial loss of function of LRRK2 seems to lack a phenotype in humans, implying that reducing kinase activity or silencing the harmful allele could be a safe therapeutic strategy.22,23
Given that rare causal variants have been identified and modelled to understand their pathobiology, and that therapeutics are being tested that target this pathobiology in individuals who carry such variants, we might be close to testing the right target in the right patients at the right time. For example, pathogenic variants in PARK7 (also known as DJ-1), PRKN (also known as PARK2), and PINK1 have been implicated in mitochondrial and mitophagy function. Variants in other genes, such as GBA, LRRK2, and VPS35, are likely to be involved in lysosomal and trafficking pathways. However, the insights and therapeutics arising from the study of rare penetrant variants might not be generalisable to the more common, sporadic disease. Aetiological subtypes of Parkinson’s disease are likely to exist, and strategies targeting LRRK2 dysfunction might only be effective for relevant subtypes. Some therapies are unlikely to be effective for disease-associated variants that result in little or no Lewy body pathology (eg, those caused by variants in LRRK2 or PRKN24–26). Therefore, diagnostic and treatment categories need to be considered on the basis of different disease mechanisms. Categorisation for apparently monogenic disease (ie, LRRK2-related or SNCA-related disease) might be straightforward, but for apparently sporadic disease it could be complex; categories might need to be based on the predominant pathomechanism, as indicated by genetics, biomarkers, or both (eg, autophagy-related, mitophagy-related, or lysosomal-related Parkinson’s disease).
Apparently sporadic Parkinson’s disease
Similar to the term monogenic, to describe Parkinson’s disease as sporadic (or idiopathic) is an oversimplification. The implication that typical Parkinson’s disease arises spontaneously or with an unknown cause or origin is inaccurate. We know now that a large proportion of Parkinson’s disease cases are affected by genetic risk factors, and these genetic factors can be detected via GWAS (panel).9 In the early days of Parkinson’s disease research, a genetic component for apparently sporadic disease was not obvious, and for many years, attempts to identify genetic influences in apparently sporadic disease were unsuccessful. With a few notable exceptions, the identification of genetic risk loci for Parkinson’s disease occurred as a direct result of the application of GWASs. GWASs allowed the transition from candidate-based evaluation, in which a single gene, or more commonly a single variant, was tested, to being able to examine the majority of common variations in the human genome at once, in a hypothesis-free manner.
The first GWAS loci for Parkinson’s disease (figure 1) were identified in 2009 with data from approximately 5000 patients and 9000 controls.27 In 2019, 90 independent risk signals have been identified with data from more than 37 000 patients, 17 000 so-called proxy cases (individuals with a parent with Parkinson’s disease) and 1·4 million controls (figure 1).9 Of note, the ancestral alleles that have been identified across loci can be associated with both a higher or lower risk of disease.9 The heritable component of Parkinson’s disease due to common genetic variability is estimated to be around 22%, and the GWAS loci identified to date explain just a fraction of this. Thus, many more risk variants are yet to be discovered. Further calculations estimate that to make major progress in resolving the basis of this heritable component, we need to almost triple the number of cases used in GWASs.9 Efforts to extend sample collection and genotyping have been successful for other diseases, such as cardiovascular disease, and have yielded novel loci, biological insight, and therapeutic implications.28
Figure 1: Timeline of genetic discoveries from GWASs for Parkinson’s disease.
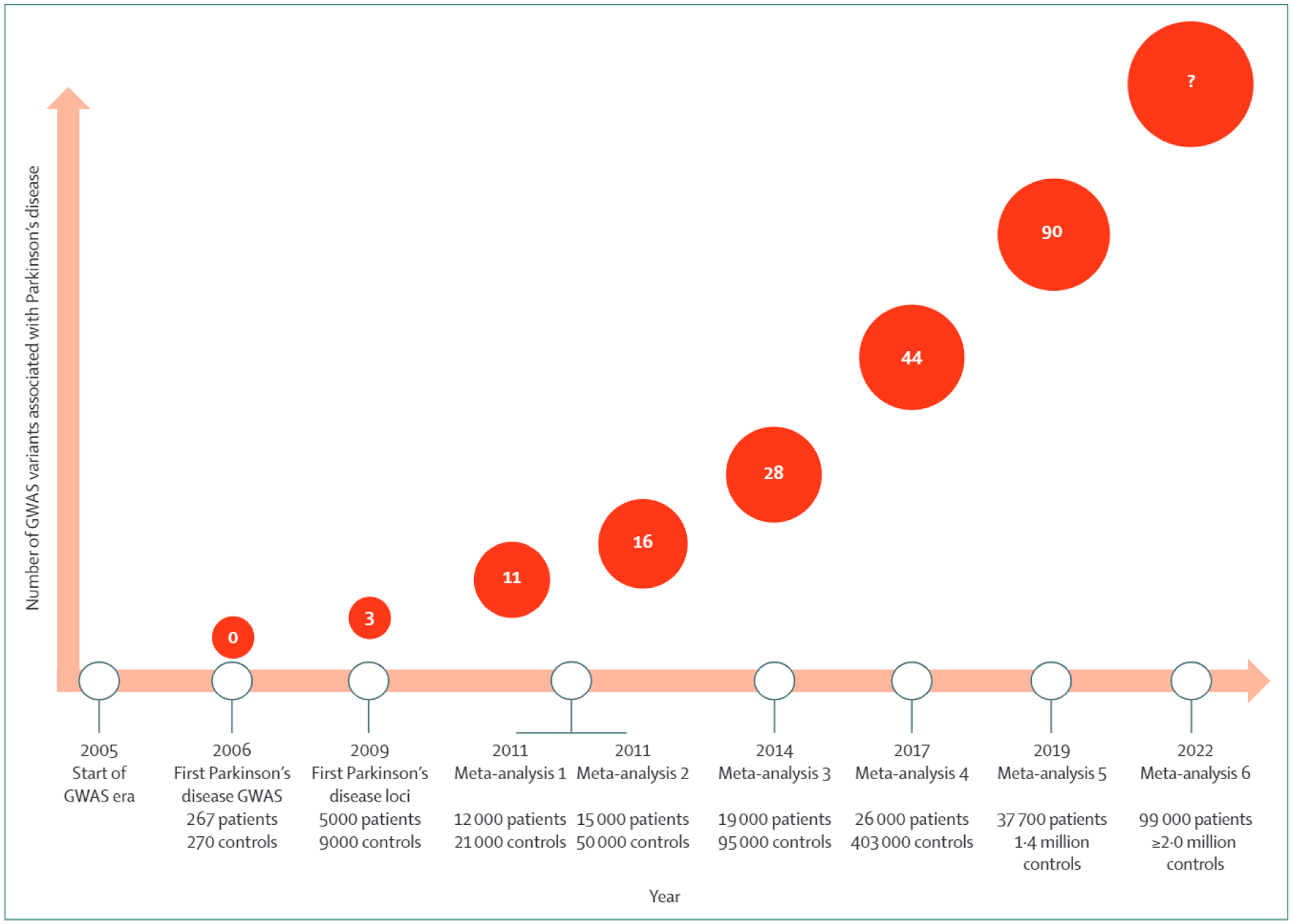
GWAS=genome-wide association study.
As well as discovering more genetic risk loci for Parkinson’s disease, we also need to characterise the biological basis of that risk, which has historically been challenging. The first attempts at such characterisation focused on associations between a risk variant and the level of expression of a gene in the proximity of that variant (known as expression quantitative trait loci [QTLs]; panel) or identifying a damaging coding variant in high linkage disequilibrium (that tends to occur on the same haplotype) with the GWAS signal. Colocalisation analyses between GWAS loci and expression QTLs have provided some causal insight into GWAS loci, but large-scale functional validation of the causality of the expression QTLs has not generally been done. Pathway-based analyses have shown that the genetic risk burden for Parkinson’s disease is high in lysosomal and endocytosis pathways.9,29,30 Mendelian randomisation studies (panel) have also found evidence for causal associations between various traits or exposures and risk of Parkinson’s disease, highlighting the potential overlap between Parkinson’s disease and certain phenotypes.31,32
A large number of the identified GWAS loci are in close proximity to a so-called monogenic Parkinson’s disease gene, such as SNCA, LRRK2, GBA, and VPS13C. These regions are known as pleomorphic risk loci (panel).33 Although rare coding variants (with a high effect size) at these genes can cause Parkinson’s disease, more common variants (with a smaller effect size and often non-coding) can also increase the risk for disease. In addition, some loci are close to genes that cause other diseases, such as MAPT, GRN, NEK1 (associated with frontotemporal dementia and amyotrophic lateral sclerosis34), and NOD1 (associated with Crohn’s disease and Blau syndrome35).
Genetic risk scoring based on GWAS loci
In the past five years, interest has grown in predicting diseases or phenotypes using genetic risk scoring (panel) based on GWAS loci. For some phenotypes or traits, a high genetic risk score might be equivalent to a high-risk variant.36 In Alzheimer’s disease, genetic risk scoring is a popular method, with many studies reporting an area under the curve (AUC) of around 0·82 (panel).37,38 However, the majority of the predictive value is driven by the APOE gene (AUC 0·68–0·73 using the APOE locus alone38,39). In Parkinson’s disease, genetic risk scoring has been done in multiple settings, with an AUC ranging from 0·65 to 0·69.9 In comparison with the lowest risk score quartile, the genetic risk for the highest quartile is increased by four times. The main drivers of the genetic risk score are relatively common, high effect size, coding variants in GBA (Glu326Lys and Asn370Ser) and LRRK2 (Gly2019Ser). Using genetics alone, predictions of Parkinson’s disease risk are unlikely to have clinical utility. A better scenario would be if each new patient in the clinic were genetically screened for a broad range of neurological diseases to provide accurate diagnoses. Additionally, population genetic screening efforts can identify subjects at risk, who are interesting candidates for clinical trials. For Parkinson’s disease specifically, combination of the genetic risk score with other observations, such as the smell test and some prodromal features,2,40 will probably be implemented in the near future to help clinicians identify people at prodromal stages. A multimodal predictive model will also help to select prodromal populations for future clinical trials and could help in differentiating synucleinopathies, atypical Parkinson’s disease syndromes, and other diseases from Parkinson’s disease, which would improve trial efficiency.
Additional analyses to assign function to GWAS loci
To gain more information about the biological function of GWAS loci, a promising approach is to do GWASs for additional clinical phenotypes, such as age at onset and longitudinal progression. An age-at-onset GWAS is typically done in a large case series with age as a continuous phenotype. An age-at-onset GWAS from 2019,41 which included more than 28 000 patients with Parkinson’s disease, reported that not all Parkinson’s disease GWAS loci actually contribute to the age at onset. Although risk genes such as GBA, SNCA, and TMEM175 are all associated with younger age at onset, other well-known loci, such as MAPT, RAB29 (also known as RAB7L1), and GCH1 are not associated with age at onset. These findings imply that some pathways can accelerate the underlying disease process and therefore result in earlier onset. The mechanism of earlier onset could be related to SNCA pathology or related pathways, because genes such as TMEM175 and GBA have been associated with increased α-synuclein aggregation,42,43 and an increase of SNCA expression results in increased α-synuclein aggregation.44,45
Progression GWASs are typically performed on longitudinal data and can include many different clinical measurements. In the largest Parkinson’s disease progression GWAS to date, more than 4000 patients with Parkinson’s disease with over 25 000 visits were included, derived from 12 different cohorts.46 Two genome-wide variants were identified, neither of which seem to affect Parkinson’s disease risk. One variant in the intron of SLC44A1 was associated with scores assessed with the Hoehn and Yahr scale,47 a system of measuring Parkinson’s disease progression. The other variant of interest, an intergenic variant and expression QTL for ADRA2A, was associated with the frequency of insomnia at baseline. Additionally, GBA variants were associated with greater motor and cognitive decline over time, confirming previous reports.48,49
Although these age-at-onset and progression GWASs included a very large number of samples, very few significant variants were identified compared with Parkinson’s disease risk GWASs. Two likely explanations are that: 1) these phenotypes are affected by the environment (which is hard to control and test for); and 2) the phenotypes are highly variable, with day-to-day differences, measurement errors, and difficulty in standardising clinical measurements across sites and study designs.
The future of Parkinson’s disease genetics
Diversifying genetics
Genetic diversity is a major challenge in the field of Parkinson’s disease genetics. Similar to many other scientific fields, the majority of research has been done in individuals with mainly European ancestry.50 This lack of diversity means that genetic risk score predictions for disease might not be globally generalisable.51
Several notable findings have been reported in non-European populations; for instance, certain GBA variants and LRRK2 Gly2019Ser are highly enriched in the Ashkenazi Jewish population; MAPT and the GBA GWAS signal were absent in the largest GWAS of Asian patients with Parkinson’s disease to date;52 and population-specific GBA variants have been identified.53,54 These examples are probably representative of much wider diversity in Parkinson’s disease genetics, and many population-specific genetic risk factors are probably yet to be discovered. Future interventional therapies are likely to be targeted at genetic subgroups (eg, GBA-related or LRRK2-related), which could be problematic in regions where carrier prevalence is low, resulting in inadquate commercialisation of the drug in that region. The therapies being developed might not be effective for the majority of patients in some regions. Large collaborative genetic studies, such as the Latin American Research Consortium on the Genetics of Parkinson’s Disease,55 need to be done to collect DNA and phenotype information and provide local education and training for underrepresented populations.
Understanding Parkinson’s disease risk loci
At the genome-wide level, the GWAS loci that have already been identified have provided insight into the causes of Parkinson’s disease, with each novel locus exposing more of the disease-related pathway. However, we are only just beginning to understand the underlying biology. While we continue to dissect the genetic basis of disease, efforts to systematically investigate the functions of these risk loci must run in parallel. To date, only a few Parkinson’s disease GWAS loci have been systematically assessed, such as SNCA and TMEM175.45,56 With use of QTL mapping and the identification of damaging coding variants, we can assign or nominate a gene to a small number of loci. Such QTL attempts have been limited to expression estimates in heterogeneous tissue (eg, postmortem brain regions) with small sample sizes, and because a QTL signal can be cell-specific (and therefore tissue-specific), the tissue in which expression and functional studies should be done is not always known.
The advent of single-cell RNA sequencing and other single-cell techniques, such as assay for transposase accessible chromatin with sequencing (or ATACseq) and HiC sequencing, has modified this approach. With use of expression and epigenomic data that are attributable to specific cell types, the cell type(s) that is most affected by risk variants can be inferred, providing information on the cells or tissue of interest. Studies on the brains and neurons of mice have identified several dopaminergic neuronal subpopulations,57,58 and single-cell RNAseq experiments have been done on the human brain,59 which have already identified multiple cell subpopulations. Although more single-cell reference data are needed, the expression of genes in close proximity to Parkinson’s disease loci seems to be enriched in brain tissues, and specifically in midbrain neurons,9,60 a result that is in contrast with the immune-centred signal observed in Alzheimer’s disease.38 To evaluate these loci, large, unbiased screens and molecular assays need to be done, with resources such as induced pluripotent stem cells (from patients and controls) differentiated into relevant cell types (figure 2). With use of these cells, DNA and RNA molecular assays could be performed to map and analyse these regions in detail. Gene editing tools such as CRISPR could be used to validate the causal variants and genes. Additional studies of genetic perturbations (ie, editing, knockdown, or overexpression of genes) should be done to mimic the aging or stressed state of cells in the human brain. As new single-cell methods are developed and induced pluripotent stem cell datasets expand to include greater numbers of cell lines, more disease-relevant tissues, and epigenetic and proteomic information, the path from genetic finding to biological understanding will become easier and more robust.
Figure 2:
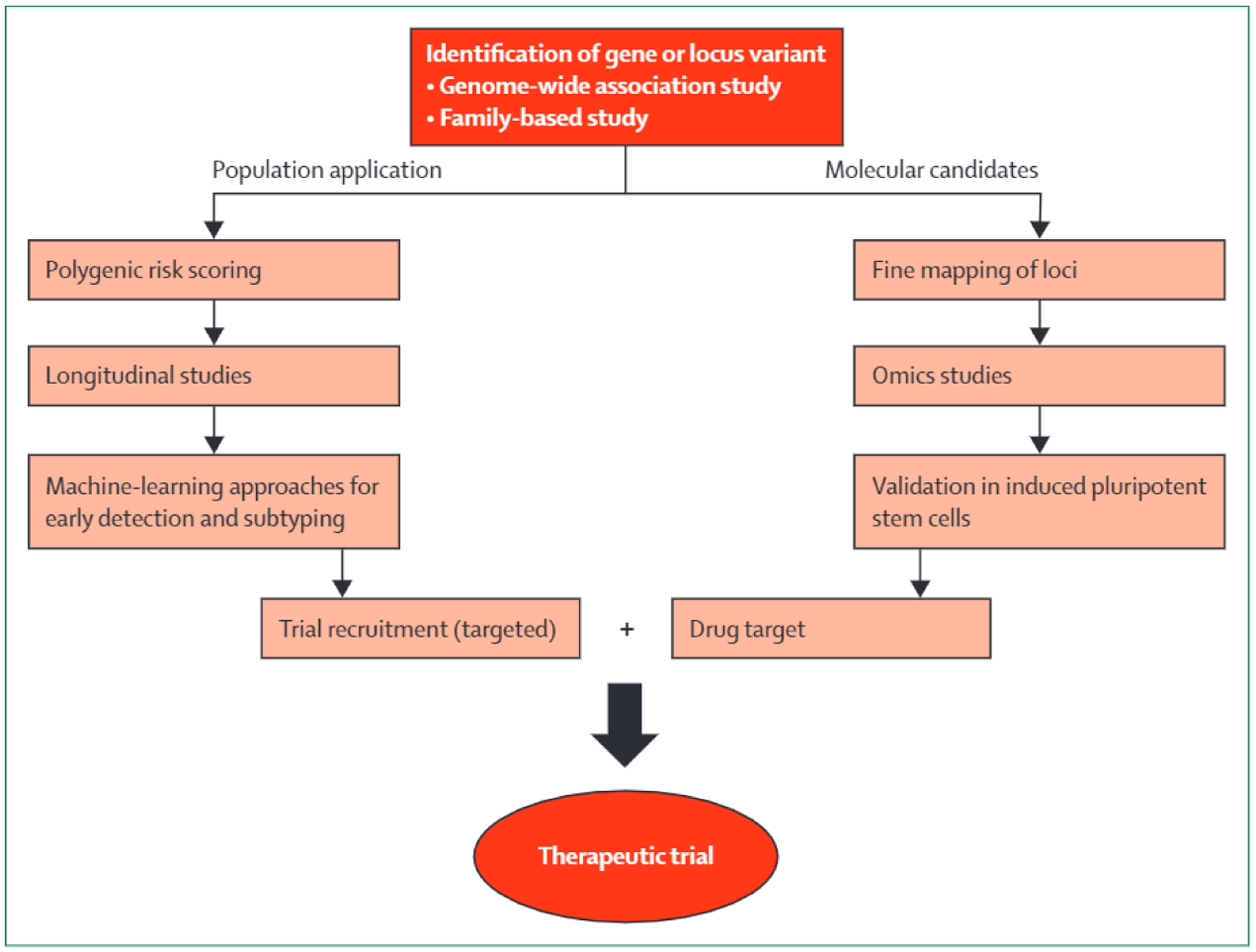
Schematic for genetics-driven drug discovery
Improving prediction of disease risk and progression
Prediction of Parkinson’s disease risk and disease progression has been restricted by a scarcity of harmonised and deeply phenotyped cohorts. To date, the vast majority of the clinical trials for neurodegenerative diseases have failed, and did not result in approved therapies. Common problems include recruiting patients at the wrong time in their disease course, small group sizes, and no genetic screening or grouping before enrolment. For Parkinson’s disease in particular, an absence of genetic screening or grouping prior to enrolment could have a considerable effect on the outcome because of the potential genetic subtypes of the disease, as discussed earlier.61 A universal therapy for Parkinson’s disease is unlikely to be found; therefore, specific clinical trials are underway for GBA-related Parkinson’s disease (NCT02906020) and LRRK2-related Parkinson’s disease (NCT3710707).
Early detection is likely to be crucial for neurodegenerative disease interventions. Predictive analytics, using polygenic risk scores and multimodal machine-learning studies, will drive the identification of high-risk individuals, and these people can be selected for interventional therapy. Additionally, data-driven efforts to subtype disease could help in designing more efficient trials and more accurately quantifying efficacy.62 A large number of phenotypes (eg, imaging results, loss of smell, and other clinical observations)2,63,64 that are associated with Parkinson’s disease could also be included in prediction and progression models. Typically, prediction studies can only be done on a small scale, with a limited scope.2,40,46,65 Similarly, autopsy-confirmed studies assessing Parkinson’s disease risk, the spread of Lewy body pathology, or other features often have very small sample sizes. If performed on a larger scale, these studies could be informative in defining the genetic basis and spread of pathology.66–68
An ongoing problem in many fields of biomedical research is that data can be deep (a lot of data on a small number of patients) or wide (a large sample size), but not both. To some extent, this issue is being addressed by the Accelerating Medicines Partnership and its ongoing efforts to aggregate data from public and private organisations on disease biomarkers and therapeutic targets.69 Longitudinal studies, such as the Parkinson’s Progression Markers Initiative and the Parkinson’s Disease Biomarkers Program, have substantially increased our understanding of Parkinson’s disease and are valuable resources for researchers in the field.
A beneficial step forward would be to combine the available data for Parkinson’s disease into a single progression or risk score that could be implemented in a practical and understandable manner in the clinic. Novel machine-learning approaches could offer a way of combining genetic data, imaging, and other clinical phenotypes to improve the prediction of Parkinson’s disease progression and Parkinson’s disease risk.62,70 However, at present, these efforts are limited by the scarcity of deep datasets.
Conclusion
Understanding the genetic factors that influence Parkinson’s disease risk, onset, and progression is crucial to developing treatments that might slow or stop disease progression. To date, many genes and GWAS loci have been identified that contribute to the development of Parkinson’s disease. We need to continue this search for genetic risk factors, while also making a concerted, coordinated effort to understand the consequences of these discoveries at the molecular and biological levels. This understanding of both genetic and functional information will be key to the identification of cause-related therapies.
The application of cause-related therapies will require us to be able to identify the prominent causal factors in each patient, preferably before clinical signs and symptoms. Longitudinal studies, with high-quality data and large samples, will be crucial to improving our prediction of Parkinson’s disease risk and disease progression. The ultimate goal of Parkinson’s disease research is to treat patients with the right therapy at the right time; although much research remains to be done, we believe that the path to this goal is now clear.
Search strategy and selection criteria.
References for this Review were identified by searches of PubMed and bioRxiv, from database inception to April 1, 2019, and of the reference lists of relevant articles. The search terms used were: “Parkinson” or “Parkinson’s disease” in combination with “genetics”, “gene”, “genome-wide”, “meta-analysis”, or “GWAS”. No language restrictions were applied. The final reference list was generated on the basis of relevance to the topics covered in this Review.
Declaration of interests
MAN is supported by a consulting contract between Data Tecnica International and the National Institute on Aging, National Institutes of Health. MAN also consults for Lysosomal Therapies and Neuron23. CB and ABS are employed by the National Institutes of Health.
Panel: Glossary of terms
- Area under the curve
The area under the curve (AUC) refers to the area under the receiver operating curve, which is a plot of true positive rate against false positive rate. The AUC is commonly used to evaluate the predictive value of a model, such as a genetic risk score. In practice, an AUC measures how good a model is at correctly identifying cases as cases and controls as controls. An AUC of 1 means the model is always right and an AUC of 0·5 means that the model is equivalent to random guessing.
- Genetic heritability
Heritability has historically been estimated from twin studies, and in the past decade has also been applied to GWAS and sequencing data from large datasets. Genetic heritability is a measure of the extent to which genetics is involved in a given trait or phenotype. Heritability is often represented as an H2 value between 0 (no genetic contribution) and 1 (complete genetic contribution). Complex diseases usually have intermediate heritability values, implying that both genetics and other factors (eg, environmental factors) contribute to disease risk.
- Genetic risk score
A genetic risk score, also known as a polygenic risk score, is calculated by aggregating the allelic status of multiple variants and their effect sizes, typically derived from a GWAS. If a trait or phenotype has high genetic heritability, the genetic risk score can be a good prediction tool.
- Genome-wide association study
A genome-wide association study (GWAS) is a hypothesis-free method to investigate whether any genetic variants are associated with a particular trait or phenotype. The phenotype being evaluated can be a case-control phenotype (eg, Parkinson’s disease vs control), a continuous phenotype (eg, body-mass index), or time-to-event (eg, incident stroke). For example, GWAS identified a common variant, rs356182 in the SNCA locus, that is associated with an increased risk for Parkinson’s disease. The frequency of the minor allele (G) in the general European population is around 34%, whereas in the Parkinson’s disease population it is around 40%; the odds ratio of the G allele is around 1·3.
- GWAS locus
A GWAS locus is a region in the genome that is associated with a certain trait or phenotype. In general, several variants within the locus will tend to be inherited together on a haplotype; these variants are considered to be in linkage disequilibrium. Therefore, although a variant might be associated with a disease, it might not be causal. Identifying causal variants and genes remains challenging because a GWAS locus can contain hundreds of significant variants spanning many different genes in the same region. Of note, GWAS reports often name risk loci by their closest gene. This gene is often misinterpreted to also be the causal gene.
- Mendelian randomisation
Mendelian randomisation is a framework for inferring a potential causal association between an exposure and an outcome using genetic data. Within the GWAS context, Mendelian randomisation usually involves comparing two sets of independent GWAS summary statistics, one representing the exposure and the other representing the outcome of interest. This comparison provides some insight into the causal effect of the exposure on the outcome. Mendelian randomisation is often used to tie together QTLs and GWASs, by providing a possible molecular context for the association signals at GWAS loci.
- Pleomorphic risk locus
A pleomorphic risk locus is a region, often identified by GWAS, at which both rare (typically high effect size or causal) and common (typically small effect) variants contribute to a specific trait. For example, some missense variants in SNCA or multiplications of SNCA clearly cause Parkinson’s disease. With use of GWAS, several other variants in the SNCA locus have been identified to moderately increase Parkinson’s disease risk, probably by increasing SNCA expression. Another example is the GBA gene, for which some variants (encoding lysosomal acid glucosylceramidase) cause Gaucher’s disease (a lysosomal storage disorder) in a homozygous state and are risk factors for Parkinson’s disease in a heterozygous state.
- Quantitative trait locus
Within the GWAS context, a quantitative trait locus (QTL) is usually a region in the genome that is associated with a molecular trait, such as gene expression or methylation. For instance, a variant within a QTL might disrupt the local binding motif of a transcription factor, leading to reduced gene transcription. The effect of a QTL can be either local (or cis), typically defined as when the affected gene is within 1 megabase of the variant, or distant (or trans), when it is further away. In general, cis effects are stronger and easier to detect than trans effects.
Footnotes
For the Parkinson’s Progression Markers Initiative see https://www.ppmi-info.org
For the Parkinson’s Disease Biomarkers Program see https://pdbp.ninds.nih.gov
References
- 1.Dorsey ER, Bloem BR. The Parkinson pandemic: a call to action. JAMA Neurol 2018; 75: 9–10. [DOI] [PubMed] [Google Scholar]
- 2.Schrag A, Anastasiou Z, Ambler G, Noyce A, Walters K. Predicting diagnosis of Parkinson’s disease: a risk algorithm based on primary care presentations. Mov Disord 2019; 34: 480–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noyce AJ, Lees AJ, Schrag AE. The prediagnostic phase of Parkinson’s disease. J Neurol Neurosurg Psychiatry 2016; 87: 871–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guerreiro R, Ross OA, Kun-Rodrigues C, et al. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol 2018; 17: 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sailer A, Scholz SW, Nalls MA, et al. A genome-wide association study in multiple system atrophy. Neurology 2016; 87:1591–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kieburtz K, Wunderle KB. Parkinson’s disease: evidence for environmental risk factors. Mov Disord 2013; 28: 8–13. [DOI] [PubMed] [Google Scholar]
- 7.Noyce AJ, Bestwick JP, Silveira-Moriyama L, et al. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann Neurol 2012; 72: 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalia LV, Lang AE. Parkinson’s disease. Lancet 2015; 386: 896–912. [DOI] [PubMed] [Google Scholar]
- 9.Nalls MA, Blauwendraat C, Vallerga CL, et al. Expanding Parkinson’s disease genetics: novel risk loci, genomic context, causal insights and heritable risk. bioRxiv 2019; published online March 4. DOI: 10.1101/388165 (preprint). [DOI] [Google Scholar]
- 10.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997; 276: 2045–47. [DOI] [PubMed] [Google Scholar]
- 11.Quadri M, Mandemakers W, Kuipers D, Breedveld GJ, Bonifati V. LRP10 in α-synucleinopathies: authors’ reply. Lancet Neurol 2018; 17:1035–36. [DOI] [PubMed] [Google Scholar]
- 12.Shi CH, Luo HY, Fan Y, Li YS, Xu YM. LRP10 in α-synucleinopathies. Lancet Neurol 2018; 17:1034–35. [DOI] [PubMed] [Google Scholar]
- 13.Tesson C, Brefel-Courbon C, Corvol JC, Lesage S, Brice A, French Parkinson’s Disease Genetics Study Group. LRP10 in α-synucleinopathies. Lancet Neurol 2018; 17:1034. [DOI] [PubMed] [Google Scholar]
- 14.Guerreiro R, Orme T, Neto JL, Bras J, International DLB Genetics Consortium. LRP10 in α-synucleinopathies. Lancet Neurol 2018; 17:1032–33. [DOI] [PubMed] [Google Scholar]
- 15.Kia DA, Sabir MS, Ahmed S, Trinh J, Bandres-Ciga S, International Parkinson’s Disease Genomics Consortium. LRP10 in α-synucleinopathies. Lancet Neurol 2018; 17:1032. [DOI] [PubMed] [Google Scholar]
- 16.Deng HX, Pericak-Vance MA, Siddique T. Reply to ‘TMEM230 variants in Parkinson’s disease’ and ‘Doubts about TMEM230 as a gene for parkinsonism’. Nat Genet 2019; 51: 369–71. [DOI] [PubMed] [Google Scholar]
- 17.Iqbal Z, Toft M. TMEM230 variants in Parkinson’s disease. Nat Genet 2019; 51: 366. [DOI] [PubMed] [Google Scholar]
- 18.Farrer MJ. Doubts about TMEM230 as a gene for parkinsonism. Nat Genet 2019; 51: 367–68. [DOI] [PubMed] [Google Scholar]
- 19.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018; 562: 203–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Correia Guedes L, Ferreira JJ, Rosa MM, Coelho M, Bonifati V, Sampaio C. Worldwide frequency of G2019S LRRK2 mutation in Parkinson’s disease: a systematic review. Parkinsonism Relat Disord 2010; 16: 237–42. [DOI] [PubMed] [Google Scholar]
- 21.West AB. Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp Neurol 2017; 298: 236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blauwendraat C, Reed X, Kia DA, et al. Frequency of loss of function variants in LRRK2 in Parkinson disease. JAMA Neurol 2018; 75:1416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whiffin N, Armean IM, Kleinman A, et al. Human loss-of-function variants suggest that partial LRRK2 inhibition is a safe therapeutic strategy for Parkinsons disease. bioRxiv 2019; published online Feb 27. DOI: 10.1101/561472 (preprint). [DOI] [Google Scholar]
- 24.Sanchez-Contreras M, Heckman MG, Tacik P, et al. Study of LRRK2 variation in tauopathy: progressive supranuclear palsy and corticobasal degeneration. Mov Disord 2017; 32:115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blauwendraat C, Pletnikova O, Geiger JT, et al. Genetic analysis of neurodegenerative diseases in a pathology cohort. Neurobiol Aging 2019; 76: 214.e1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson’s disease. Mov Disord 2012; 27: 831–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simón-Sánchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 2009; 41:1308–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bovijn J, Krebs K, Chen C-Y, et al. Lifelong genetically lowered sclerostin and risk of cardiovascular disease. bioRxiv 2019; published online Feb 1. DOI: 10.1101/531004 (preprint). [DOI] [Google Scholar]
- 29.Robak LA, Jansen IE, van Rooij J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017; 140: 3191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bandres-Ciga S, Saez-Atienzar S, Bonet-Ponce L, et al. The endocytic membrane trafficking pathway plays a major role in the risk of Parkinson’s disease. Mov Disord 2019; 34:460–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noyce AJ, Kia DA, Hemani G, et al. Estimating the causal influence of body mass index on risk of Parkinson disease: a Mendelian randomisation study. PLoS Med 2017; 14: e1002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noyce AJ, Bandres-Ciga S, Kim J, et al. The Parkinson’s disease Mendelian randomization research portal. bioRxiv 2019; published online April 10. DOI: 10.1101/604033 (preprint). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singleton A, Hardy J. A generalizable hypothesis for the genetic architecture of disease: pleomorphic risk loci. Hum Mol Genet 2011; 20: R158–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen HP, Van Broeckhoven C, van der Zee J. ALS genes in the genomic era and their implications for FTD. Trends Genet 2018; 34: 404–23. [DOI] [PubMed] [Google Scholar]
- 35.Caruso R, Warner N, Inohara N, Núñez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 2014; 41: 898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018; 50:1219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Escott-Price V, Myers A, Huentelman M, Shoai M, Hardy J. Polygenic risk score analysis of Alzheimer’s disease in cases without APOE4 or APOE2 alleles. J Prev Alzheimers Dis 2019; 6:16–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 2019; 51: 404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Escott-Price V, Sims R, Bannister C, et al. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 2015; 138: 3673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nalls MA, McLean CY, Rick J, et al. Diagnosis of Parkinson’s disease on the basis of clinical and genetic classification: a population-based modelling study. Lancet Neurol 2015; 14:1002–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blauwendraat C, Heilbron K, Vallerga CL, et al. Parkinson’s disease age at onset genome-wide association study: defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov Disord 2019; 34: 866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du TT, Wang L, Duan CL, et al. GBA deficiency promotes SNCA/α-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015; 11:1803–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jinn S, Drolet RE, Cramer PE, et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc Natl Acad Sci USA 2017; 114: 2389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pihlstrøm L, Blauwendraat C, Cappelletti C, et al. A comprehensive analysis of SNCA-related genetic risk in sporadic Parkinson disease. Ann Neurol 2018; 84: 117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soldner F, Stelzer Y, Shivalila CS, et al. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016; 533: 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iwaki H, Blauwendraat C, Leonard HL, et al. Genome-wide association study of Parkinson’s disease progression biomarkers in 12 longitudinal patients’ cohorts. bioRxiv 2019; published online March 27. DOI: 10.1101/585836 (preprint). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goetz CG, Poewe W, Rascol O, et al. Movement Disorder Society Task Force report on the Hoehn and Yahr staging scale: status and recommendations. Mov Disord 2004; 19:1020–28. [DOI] [PubMed] [Google Scholar]
- 48.Davis MY, Johnson CO, Leverenz JB, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol 2016; 73:1217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu G, Boot B, Locascio JJ, et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol 2016; 80: 674–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morales J, Welter D, Bowler EH, et al. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol 2018; 19: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 2019; 51: 584–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foo JN, Tan LC, Irwan ID, et al. Genome-wide association study of Parkinson’s disease in East Asians. Hum Mol Genet 2017; 26: 226–32. [DOI] [PubMed] [Google Scholar]
- 53.Velez-Pardo C, Lorenzo-Betancor O, Jimenez-Del-Rio M, et al. The distribution and risk effect of GBA variants in a large cohort of PD patients from Colombia and Peru. Parkinsonism Relat Disord 2019; published online Feb 4. DOI: 10.1016/j.parkreldis.2019.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Shu L, Sun Q, et al. Integrated genetic analysis of racial differences of common variants in Parkinson’s disease: a meta-analysis. Front Mol Neurosci 2018; 11: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zabetian CP, Mata IF, Latin American Research Consortium on the Genetics of PD (LARGE-PD). LARGE-PD: examining the genetics of Parkinson’s disease in Latin America. Mov Disord 2017; 32:1330–31. [DOI] [PubMed] [Google Scholar]
- 56.Jinn S, Blauwendraat C, Toolan D, et al. Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Hum Mol Genet 2019; published online July 2. DOI: 10.1093/hmg/ddz136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saunders A, Macosko EZ, Wysoker A, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 2018; 174: 1015–30.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hook PW, McClymont SA, Cannon GH, et al. Single-cell RNA-seq of mouse dopaminergic neurons informs candidate gene selection for sporadic Parkinson disease. Am J Hum Genet 2018; 102: 427–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhong S, Zhang S, Fan X, et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 2018; 555: 524–28. [DOI] [PubMed] [Google Scholar]
- 60.Bryois J, Skene NG, Hansen TF, et al. Genetic identification of cell types underlying brain complex traits yields novel insights into the etiology of Parkinson’s disease. bioRxiv 2019; published online Jan 23. DOI: 10.1101/528463 (preprint). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leonard H, Blauwendraat C, Krohn L, et al. Genetic variability and potential effects on clinical trial outcomes: perspectives in Parkinson’s disease. bioRxiv 2018; published online Oct 30. DOI: 10.1101/427385 (preprint). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Faghri F, Hashemi SH, Leonard H, et al. Predicting onset, progression, and clinical subtypes of Parkinson disease using machine learning. bioRxiv 2019; published online June 5. DOI: 10.1101/338913 (preprint). [DOI] [Google Scholar]
- 63.Heilbron K, Noyce AJ, Fontanillas P, et al. The Parkinson’s phenome-traits associated with Parkinson’s disease in a broadly phenotyped cohort. NPJ Parkinsons Dis 2019; 5: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saeed U, Compagnone J, Aviv RI, et al. Imaging biomarkers in Parkinson’s disease and Parkinsonian syndromes: current and emerging concepts. Transl Neurodegener 2017; 6: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu G, Locascio JJ, Corvol J-C, et al. Prediction of cognition in Parkinson’s disease with a clinical-genetic score: a longitudinal analysis of nine cohorts. Lancet Neurol 2017; 16: 620–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Pablo-Fernandez E, Lees AJ, Holton JL, Warner TT. Prognosis and neuropathologic correlation of clinical subtypes of Parkinson disease. JAMA Neurol 2019; 76: 470–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heckman MG, Kasanuki K, Diehl NN, et al. Parkinson’s disease susceptibility variants and severity of Lewy body pathology. Parkinsonism Relat Disord 2017; 44: 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beecham GW, Dickson DW, Scott WK, et al. PARK10 is a major locus for sporadic neuropathologically confirmed Parkinson disease. Neurology 2015; 84: 972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.National Institutes of Health. Accelerating medicines partnership (AMP). 2018. https://www.nih.gov/research-training/accelerating-medicines-partnership-amp/parkinsons-disease (accessed March 15, 2019).
- 70.Latourelle JC, Beste MT, Hadzi TC, et al. Large-scale identification of clinical and genetic predictors of motor progression in patients with newly diagnosed Parkinson’s disease: a longitudinal cohort study and validation. Lancet Neurol 2017; 16: 908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]