

SUMMARY
G protein-coupled receptors (GPCRs) in intestinal enteroendocrine cells (EECs) respond to nutritional, neural, and microbial cues and modulate the release of gut hormones. Here we show that Gpr17, an orphan GPCR, is co-expressed in glucagon-like peptide-1 (GLP-1)-expressing EECs in human and rodent intestinal epithelium. Acute genetic ablation of Gpr17 in intestinal epithelium improves glucose tolerance and glucose-stimulated insulin secretion (GSIS). Importantly, inducible knockout (iKO) mice and Gpr17 null intestinal organoids respond to glucose or lipid ingestion with increased secretion of GLP-1, but not the other incretin glucose-dependent insulinotropic polypeptide (GIP). In an in vitro EEC model, overexpression or agonism of Gpr17 reduces voltage-gated calcium currents and decreases cyclic AMP (cAMP) production, and these are two critical factors regulating GLP-1 secretion. Together, our work shows that intestinal Gpr17 signaling functions as an inhibitory pathway for GLP-1 secretion in EECs, suggesting intestinal GPR17 is a potential target for diabetes and obesity intervention.
Graphical abstract
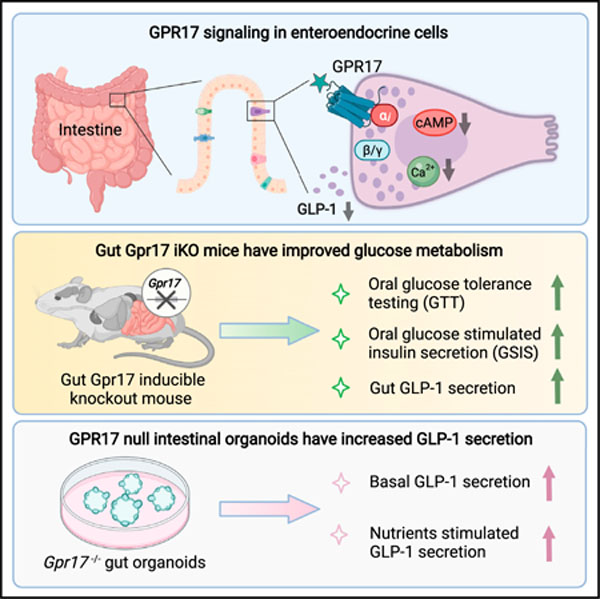
In brief
Yan et al. locate GPR17 expression in the enteroendocrine cells of human and rodent intestinal epithelium. They find that GPR17 signaling inhibits intracellular rise of cAMP and calcium and that loss of intestinal Gpr17 in rodents leads to better glucose tolerance via increased hormone secretion in response to nutrient ingestion.
INTRODUCTION
Enteroendocrine cells (EECs) in the gastrointestinal (GI) tract sense luminal content and release gut hormones, which regulate glucose metabolism and feeding through endocrine and neuronal mechanisms (Gribble and Reimann, 2016; Kim et al., 2018; Schwartz and Zeltser, 2013). The postprandial glucose lowering effect of incretin hormones is often blunted in type 2 diabetes (T2D) and enhanced after bariatric surgery (Douros et al., 2019; Nauck et al., 1986). Glucagon-like peptide-1 (GLP-1) is an incretin hormone secreted by preproglucagon-expressing EECs (traditionally named L cells) and is degraded rapidly by dipeptidylpeptidase 4 (DPP4) (Holst, 2007). Long-acting GLP-1 analogs and DPP4 inhibitors have been developed for the treatment of T2D (Drucker and Nauck, 2006). A potential alternative treatment strategy for T2D is through the modulation of endogenous GLP-1 secretion; however, the factors and pathways involved in the regulation of endogenous GLP-1 secretion remain incompletely understood.
G protein-coupled receptors (GPCRs) regulate GLP-1 secretion in EECs by responding to the rise of nutrients (e.g., lipids, carbohydrates, and peptides), molecules associated with food ingestion (e.g., bile acids), and hormonal and neural factors (e.g., somatostatin, calcium, and endocannabinoids) (Reimann et al., 2012), which in turn activate downstream signaling pathways that regulate gut hormone secretion through second messengers (Ezcurra et al., 2013). As GPCRs are the target of more than a quarter of the Food and Drug Administration-approved drugs (Hauser et al., 2017), understanding the biological actions of GPCRs in EECs will likely provide insight into developing new diabetes therapeutics by promoting endogenous GLP-1 secretion.
GPR17 was initially identified as an orphan GPCR phylogenetically related to purinergic and cysteinyl-leukotriene (CysLT) receptors (Ciana et al., 2006; Parravicini et al., 2008) with an implicated function in oligodendrocyte myelination (Ceruti et al., 2009; Chen et al., 2009; Lecca et al., 2008). We previously identified Gpr17 as the transcriptional target of Forkhead box protein O1 (FoxO1) in hypothalamic neurons and demonstrated its physiological role in maintaining metabolic homeostasis (Reilly et al., 2019; Ren et al., 2015; Ren et al., 2012). In this study, we first made the exciting discovery of GPR17 expression in GLP-1-producing EECs in the GI tract. We further evaluated the metabolic function of intestinal Gpr17 by generating constitutive and inducible intestinal Gpr17 knockout mouse models and characterized Gpr17 downstream signaling pathways in cultured EECs and intestinal organoids. Our results suggest that intestinal GPR17 contributes to the regulation of glucose metabolism by modulating GLP-1 secretion.
RESULTS
GPR17 is expressed in GLP-1-producing cells in human and mouse gut epithelium
Existing data within the Genotype-Tissue Expression (GTEx) portal demonstrated the expression of GPR17 mRNA in human intestinal tissues (https://www.gtexportal.org/home/gene/GPR17#geneExpression). Our qRT-PCR showed human GPR17 expression increased from the proximal to the distal intestine (Figure 1A), similar to the expression pattern of preproglucagon gene GCG (Mojsov et al., 1986). Interestingly, we detected GPR17 expression in flask-shaped cells resembling EECs in the intestinal epithelium by immunohistochemistry (IHC), but not in the submucosa or muscular layers (Figure S1A). To characterize the GPR17-positive cells, we first performed double staining using chromogranin A (CHGA) as a pan-EEC marker (Figure 1B). Our quantified result showed that the majority (66.9%) of GPR17-positive cells were also CHGA positive in human intestine (Figure 1E). Next, we examined if GPR17 was enriched in a subtype of EECs. We found that GPR17 was co-expressed with GLP-1 (Figure 1C), and the majority of GLP-1-producing cells (74.4%) were GPR17 positive (Figure 1F). However, GPR17 was not observed in glucose-dependent insulinotropic polypeptide (GIP)-producing cells in human small intestine (Figure 1D). As antibody specificity is often a concern for GPCR antibodies (Michel et al., 2009), we validated the commercial GPR17 antibody by performing double staining in permeabilized HEK293 cells transiently expressing an HA epitope-tagged human GPR17 construct. Anti-HA and anti-GPR17 antibody staining overlapped in the same cells (Figure S1B). GPR17 antibody staining overlapped with more than 90% of HA antibody staining cells (Figures S1C and S1D) with minimal nonspecific staining (Figure S1E), thereby validating the GPR17 antibody used in our study. Since a substantial fraction of GPR17 staining was detected intracellularly in the permeabilized cells (Figures S1B–S1F), we examined the non-permeabilized cells and found colocalization of anti-HA and anti-GPR17 staining on the cell surface (Figure S1G).
Figure 1. Co-expression of GPR17 and GLP-1 in EECs in human intestine.
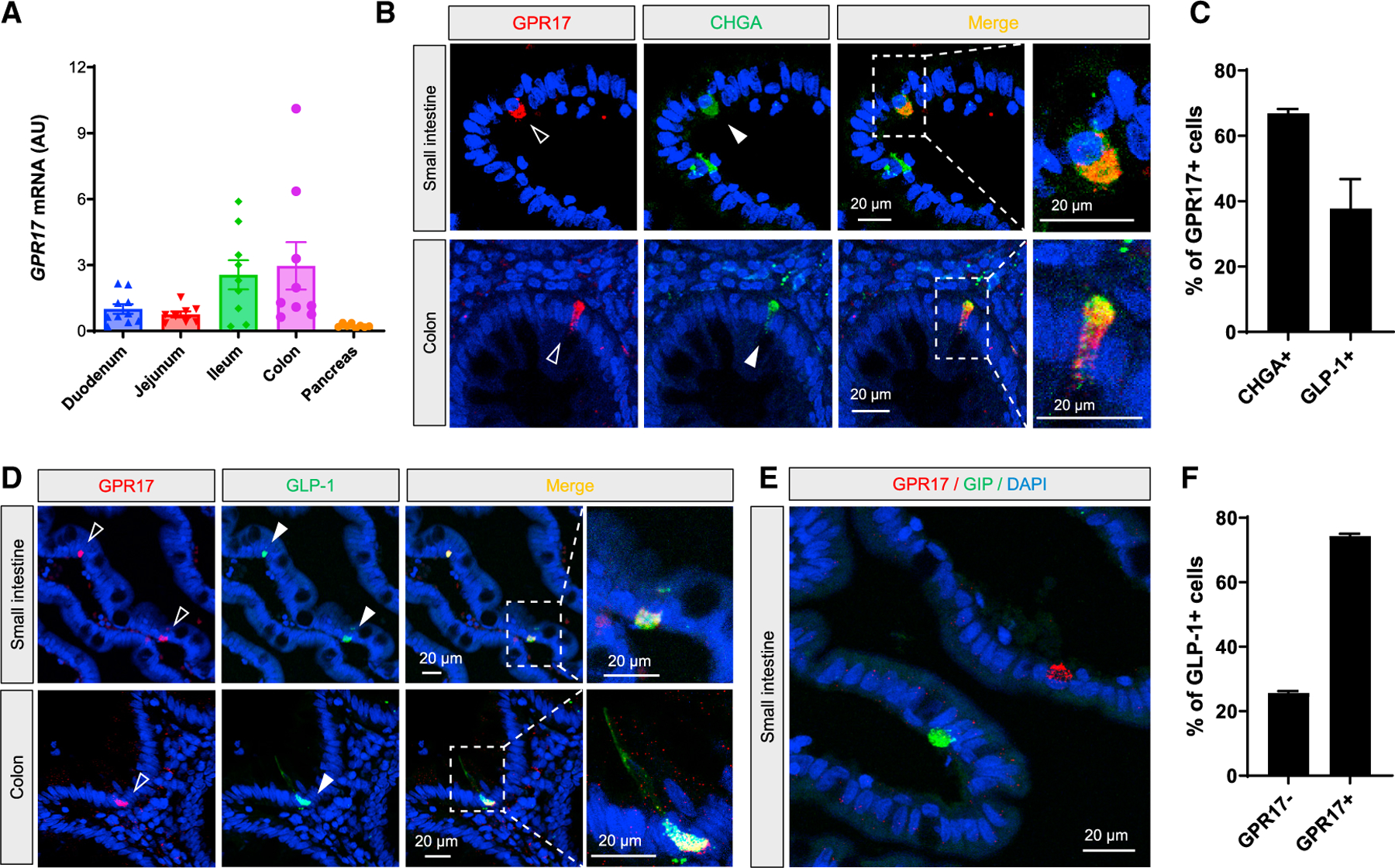
(A) qRT-PCR result of GPR17 mRNA in human gut segments and pancreas (n = 8–10). Gene expression was calculated as 2^-∆∆ct. RPLP0 was used as an internal control.
(B) Co-immunostaining of GPR17 and CHGA, a pan-EEC marker, in human small intestine (top) and colon (bottom).
(C) Co-immunostaining of GPR17 and GLP-1 in human small intestine (top) and colon (bottom).
(D) Co-immunostaining of GPR17 and GIP in human small intestine.
(E) Quantification of cells expressing CHGA in GPR17 + cells (n = 117 cells from two human samples were analyzed), and cells expressing GLP-1 in GPR17 + cells (n = 282 cells were analyzed).
(F) Quantification of cells expressing GPR17 in GLP-1-producing cells (n = 181 cells from two human samples were analyzed). Data are displayed as means ± SEM. See also Figure S1.
We further substantiated our findings using sorted mouse intestinal GLP-1-producing EECs. We crossed Gcg-CreERT mice (Shiota et al., 2017) with Rosa-tdTomato reporter mice to label GLP-1-producing cells. With tamoxifen-induced Cre expression, GLP-1-producing cells were labeled by tdTomato (Figure 2A) and collected by fluorescence-activated cell sorting (FACS) (T+ fraction in Figure 2A). The qRT-PCR result showed that Gpr17 transcripts were enriched in T+ cells (Figure 2B). Consistent with previous reports of transcriptional profiles of GLP-1-producing cells (Habib et al., 2012), we also observed enriched transcripts of Gcg (encoding preproprotein that is eventually cleaved into glucagon-like peptides including GLP-1 and GLP-2 in intestine) and Pyy (encoding peptide YY), but not Gip (Figures 2C–2E). Together, these results indicated that GPR17 was expressed in most GLP-1-producing cells in the gut epithelium.
Figure 2. Enrichment of Gpr17 in GLP-1-producing cells in mouse intestine.
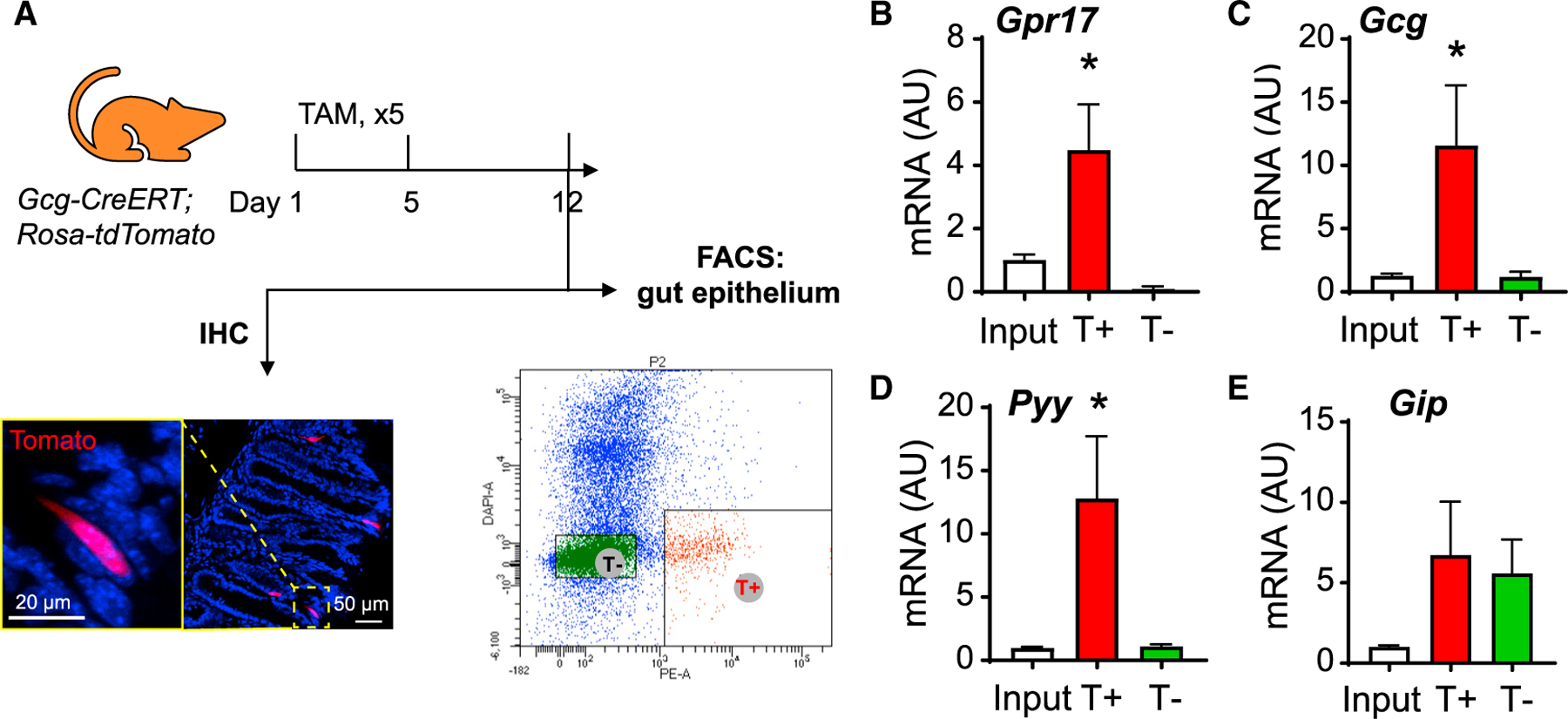
(A) (F) FACS of intestine epithelial cells dissociated from Gcg-CreERT;Rosa-tdTomato mouse post tamoxifen injection. Gcg-expressing cells were labeled by red fluorescence (tdTomato) and sorted by FACS within 1 week after tamoxifen treatment (2 mg/d for 5 days by oral gavage).
(B–E) qRT-PCR analysis of Gpr17 (B), Gcg (C), Pyy (D), and Gip (E) mRNA in sorted cells (n = 7 mice). Gene expression was calculated as 2^-∆∆ct. β-actin was used as an internal control. Gene expression was expressed relative to input fractions. Unpaired two-tailed Student’s t test, *p < 0.05. Data are displayed as means ± SEM.
Inducible intestine-specific Gpr17 knockout mice had improved glucose tolerance
To elucidate the metabolic function of intestinal Gpr17, we generated tamoxifen (TAM)-inducible intestine-specific Gpr17 knockout mice (Gpr17fl/fl; Vil1Cre-ERT2; namely iKO hereafter) (Figure 3A). The Gcg-Cre guided GLP-1-producing cell-specific Gpr17 knockout mice are not suitable for our study. There are two major reasons: (1) Gcg-cre is activated not only in gut but also in any tissues expressing Gcg, such as islets (Figures S2C and S2D) and the nucleus tractus solitarius of the brain stem (Katsurada and Yada, 2016) (data not shown); (2) due to the constant gut epithelial renewal, the Gpr17-deficient GLP-1-producing cells were quickly replaced by the newly regenerated GLP-1-producing cells with intact Gpr17 alleles, once the tamoxifen injection was terminated (Figures S2A and S2B). However, Vil-Cre under the control of the villin promotor targeted epithelial progenitor cells throughout the digestive epithelium and persisted throughout adulthood, despite rapid intestinal cell renewal (el Marjou et al., 2004). Therefore, we bred Gpr17fl/fl mice with Vil1Cre-ERT2 mice to generate inducible gut epithelium-specific Gpr17 knockout mice for our studies.
Figure 3. Inducible intestine-specific Gpr17 knockout mice displayed improved glucose tolerance.
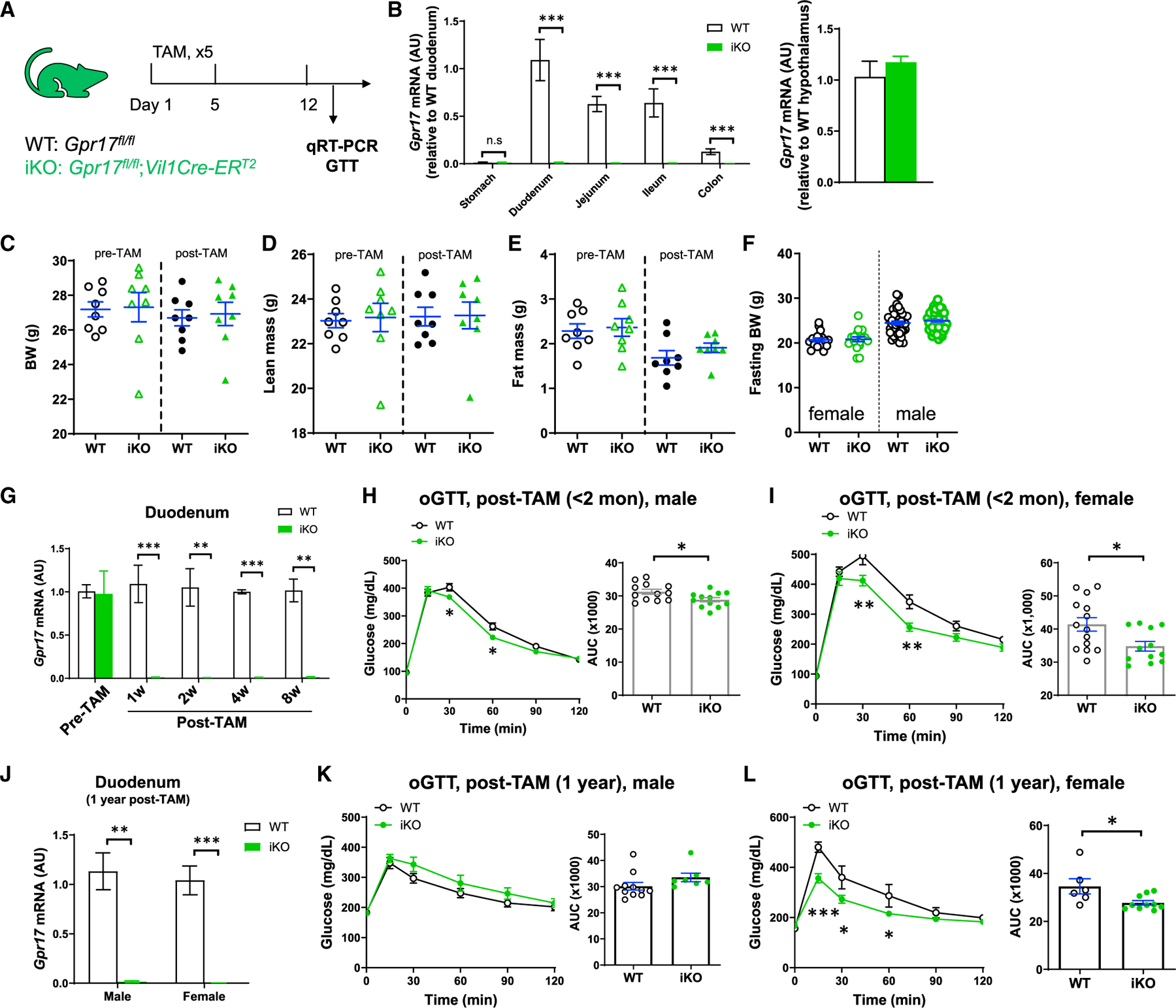
(A) Experimental setup for inducible intestinal Gpr17 knockout mice. Gpr17fl/fl (WT) and Gpr17fl/fl;Vil1Cre-ERT2 (iKO) mice were subjected to experiments at least 1 week after tamoxifen treatment (2 mg/d for 5 days by oral gavage).
(B) Gpr17 mRNA expression in mouse gut and hypothalamus from iKO (n = 9) and WT (n = 6) mice at 1 week after tamoxifen injection. Gene expression was calculated as 2^-∆∆ct. β-actin was used as an internal control. p values were calculated by unpaired two-tailed Student’s t test.
(C–E) Body weight (C), lean mass percent (D), and fat mass percent (E) of male mice before and after tamoxifen injection (n = 8 mice for each group).
(F) Fasting body weight before glucose tolerance tests (n = 17~50 mice for each group).
(G) Time course study measuring Gpr17 mRNA in duodenum following tamoxifen injection (n = 3 mice). p values were calculated with unpaired two-tailed Student’s t test.
(H) Oral glucose tolerance (oGTT, 3 g/kg) in overnight-fasted male iKO (n = 12) and WT (n = 12) mice within 2 months after tamoxifen injection.
(I) oGTT (3 g/kg) in overnight-fasted female iKO (n = 14) and WT (n = 12) mice within 2 months after tamoxifen injection.
(J) Gpr17 mRNA in duodenum in WT and iKO mice 1 year after initial tamoxifen injection.
(K) oGTT (3 g/kg) in 4-h-fasted male iKO (n = 7) and WT (n = 11) mice 1 year after tamoxifen injection.
(L) oGTT (3 g/kg) in 4-h-fasted female iKO (n = 10) and WT (n = 6) mice 1 year after tamoxifen injection.
Two-way ANOVA and unpaired two-tailed Student’s t test were performed for glucose curve and AUC, respectively. *p < 0.05, **p < 0.01, ***p < 0.001. Data are displayed as means ± SEM. See also Figure S2.
After tamoxifen injection, Gpr17 expression was almost completely abolished in the small intestine and colon of iKO mice, but not in the stomach or hypothalamus (Figure 3B). The body weight and composition of iKO mice were comparable to those of the wild-type (WT) control before and after tamoxifen treatments (Figures 3C–3E). There was no difference in fasting body weight before glucose tolerance testing in WT and iKO mice (Figure 3F). The genetic knockout was sustained after the initial tamoxifen injection (Figure 3G). Within 8 weeks after tamoxifen-induced Gpr17 genetic ablation, male iKO mice showed significantly improved oral glucose tolerance (Figure 3H). Similarly, female iKO mice showed even greater significant improvement in glucose tolerance after Gpr17 genetic ablation (Figure 3I). Furthermore, after long-term Gpr17 genetic ablation (1 year post tamoxifen injection) (Figure 3J), female (Figure 3L) but not male mice (Figure 3K) still displayed significantly improved glucose tolerance.
Knocking out intestinal Gpr17 increased nutrient-stimulated insulin and GLP-1 secretion in vivo
To address how intestinal Gpr17 deficiency led to improved glucose metabolism in iKO mice, we examined insulin secretion using the oral glucose-stimulated insulin secretion (GSIS) assay. Both female and male iKO mice responded to oral glucose gavage with increased serum insulin levels (Figures 4A and 4F). Incretin hormones play important roles in regulating insulin secretion in pancreatic islet b-cells (Campbell and Drucker, 2013), and we have observed Gpr17 expression in GLP-1-producing EECs (Figures 1 and 2). We hypothesized that knocking out intestinal Gpr17 increases GLP-1 secretion in response to nutrient ingestion. To test this, we measured incretin (GLP-1 and GIP) secretion in iKO and WT mice during fasting and after nutrient administration. Fasting GLP-1 (0 min, Figures 4B and 4D) and GIP (0 min, Figures 4C and 4E) levels had no difference between male WT and iKO mice. Ten minutes after oral glucose dosing, plasma total GLP-1 levels in iKO male mice were greater than those in WT mice (Figure 4B), while GIP levels were comparable between the two groups (Figure 4C). Meanwhile, iKO males responded to oral corn oil dosing with higher concentrations of total GLP-1 (Figure 4D) and similar GIP levels (Figure 4E). Female iKO mice showed a trend of more GLP-1 secretion after glucose gavage, although this finding was not statistically significant (p = 0.088, Figure 4G). Furthermore, female iKO mice responded to oral corn oil dosing with higher GLP-1 secretion (Figure 4I). Female iKO mice had comparable GIP secretion to WT mice after either oral glucose or corn oil gavage (Figures 4H and 4J). Neither fasting GLP-1 (0 min, Figures 4G and 4I) nor fasting GIP (0 min, Figures 4H and 4J) levels were different in female iKO mice compared with WT mice. In addition, we did not detect any significant changes in Gcg or Gip transcripts in intestinal Gpr17 knockout mice (Figures 4K and 4L).
Figure 4. Gpr17 deficiency enhanced insulin and GLP-1 secretion in response to nutrients in vivo.
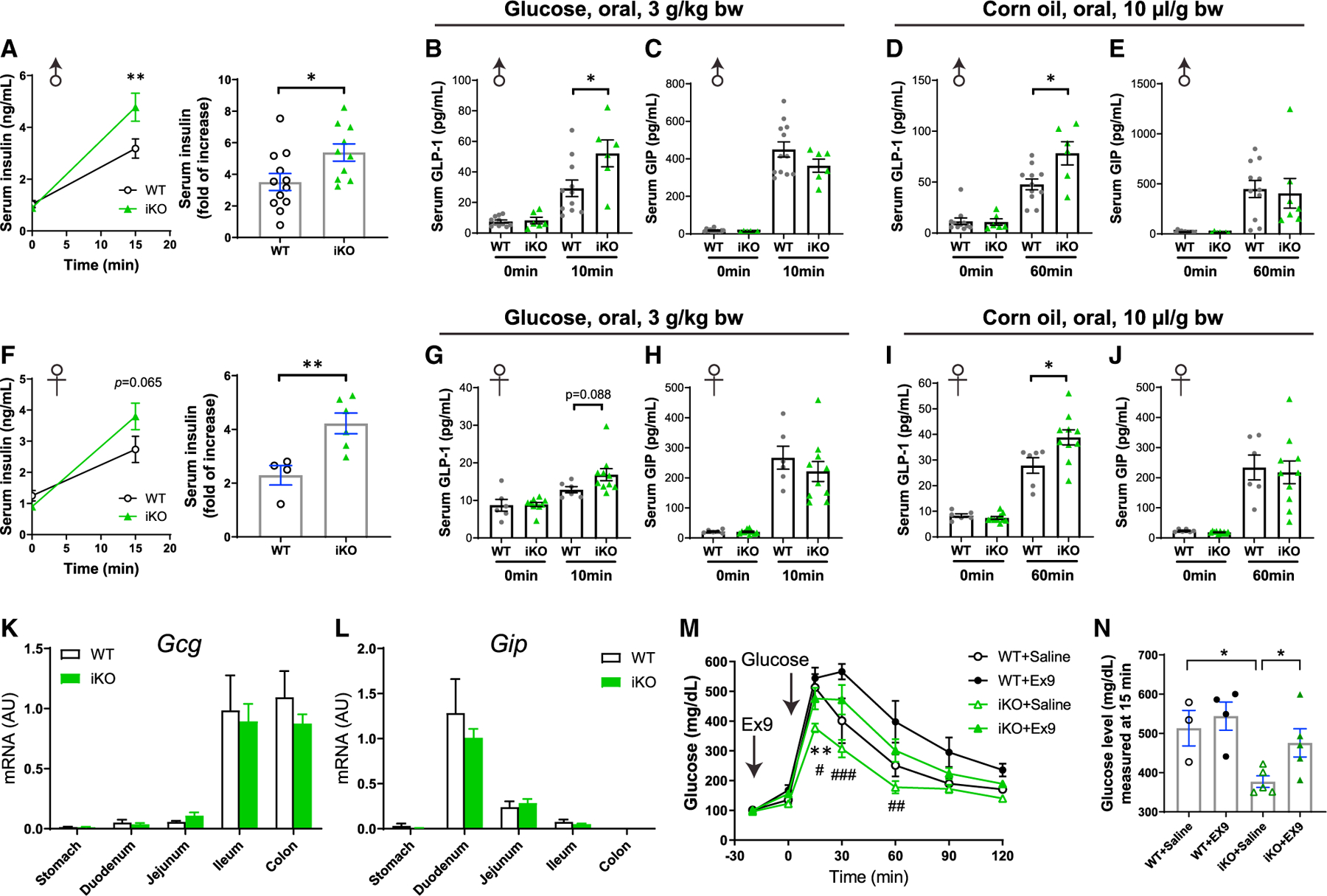
(A) Oral glucose-stimulated insulin secretion (oGSIS) in overnight-fasted male iKO mice versus WT mice after tamoxifen injection (post-TAM). Serum insulin levels were measured before and 15 min after an oral glucose load (3 g/kg) (n = 10–12 mice). Unpaired two-tailed Student’s t test was performed. (B and C) Plasma total GLP-1
(B) and GIP (C) levels in response to an oral glucose load (3 g/kg) in male inducible intestinal Gpr17 deficient mice (iKO, n = 6) and WT mice (n = 11). Tail blood samples were collected at 0 min and 10 min after oral glucose intake in 4~6 h fasted mice. Two-way ANOVA was performed. (D and E) Plasma total GLP-1
(D) and GIP (E) levels in response to an oral corn oil load (10 µL/g) in male inducible intestinal Gpr17 deficient mice (iKO, n = 6) and WT mice (n = 11). Tail blood samples were collected at 0 min and 60 min after oral glucose intake in 4~6 h fasted mice. Two-way ANOVA was performed.
(F) Oral glucose-stimulated insulin secretion (oGSIS) in overnight-fasted female iKO mice versus WT mice after tamoxifen injection (post-TAM). Serum insulin levels were measured before and 15 min after an oral glucose load (3 g/kg) (n = 4~6 mice). Unpaired two-tailed Student’s t test was performed.
(G and H) Plasma total GLP-1 (G) and GIP (H) levels in response to an oral glucose load (3 g/kg) in female iKO mice (n = 10) and WT mice (n = 6). Tail blood samples were collected at 0 min and 10 min after oral glucose intake in 4~6 h fasted mice. Two-way ANOVA was performed.
(I and J) Plasma total GLP-1 (I) and GIP (J) levels in response to an oral corn oil load (10 µL/g) in female iKO mice (iKO, n = 10) and WT mice (n = 6). Tail blood samples were collected at 0 min and 60 min after oral glucose intake in 4~6 h fasted mice. Two-way ANOVA was performed.
(K and L) Gcg (K) and Gip (L) mRNA levels in the gut of inducible Gpr17 deficient mice (iKO) by qRT-PCR (n = 6~9 mice).
(M) ipGTT (3 g/kg) in mice pretreated with GLP-1R antagonist exendin-(9–39) (Ex9, 10µg, [I]p.) (n = 3~5 mice). Two-way ANOVA, *p < 0.05 WT + Saline versus iKO + Saline. #p < 0.05, ##p < 0.01 iKO + Saline vs iKO + Ex9.
(N) Blood glucose levels at 15 min time point in ipGTT. Unpaired two-tailed Student’s t test was performed.
*p < 0.05, **p < 0.01, ***p < 0.001. Data are displayed as means ± SEM.
To further substantiate our finding, we tested whether injection of Ex9, a GLP-1 receptor antagonist, could dampen the improvement of glucose metabolism in iKO mice. We administrated Ex9 at a dose of 10 µg/mouse, which is 20% of the dose that has been previously shown to induce significant glucose intolerance in WT mice (Burmeister et al., 2017; Chambers et al., 2017). As expected, compared with the mild effect on intraperitoneal glucose tolerance test (ipGTT) in WT mice, Ex9 at 10 µg/mouse exerted more profound impact on iKO mice (Figure 4M). Notably, 15 min after glucose dosing, iKO + Ex9 group had significantly higher blood glucose than iKO + saline group (Figure 4N). These data demonstrate that knocking out intestinal Gpr17 improved glucose tolerance through, at least in part, the mechanisms of promoting GLP-1 secretion in response to nutrient ingestion in vivo.
Intestinal Gpr17 deficiency did not affect gut morphology, islets, or gut microbiota
We next determined whether changes in gut morphology, pancreatic islet, and/or gut microbiota may contribute to the glucose phenotype observed in Gpr17 knockout mice. The general morphology of small intestine and colon in adult iKO mice (4 months after TAM injection) was no different compared with WT mice (Figures 5A–5D). The villi and crypt length in iKO small intestine were similar to those of WT (Figures 5B and 5C). The thickness of colonic epithelium (including surface epithelium and crypts) was also comparable between iKO and WT mice (Figure 5E). The size and number of pancreatic islets were not altered in iKO mice compared with WT mice examined by IHC with insulin staining (Figures 5F–5H).
Figure 5. Gpr17 deficiency did not affect gut morphology and islets in intestinal Gpr17 KO mice.
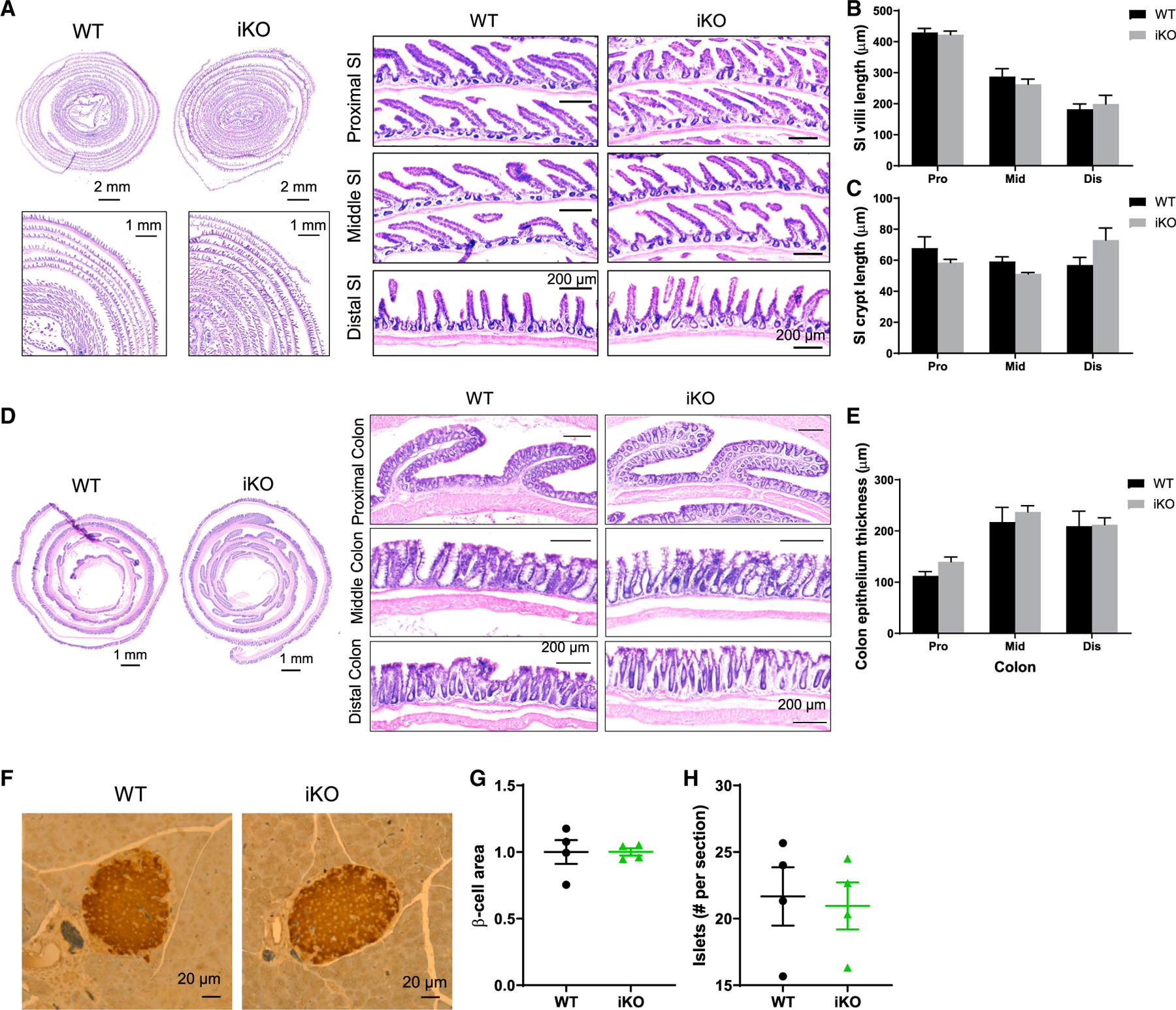
(A–C) H&E staining showing the histology of small intestine (A). Villi (B) and crypt length (C) were quantified; 8~25 villi and crypts for each segment were measured per mouse. Data show the average of three mice for each group.
(D and E) H&E staining showing the histology of colon (D). Colon epithelium thickness (E) (including surface epithelium and crypts) was quantified (n = 3 mice); 9~17 spots for each segment were measured per mouse. Data show the average of three mice for each group.
(F–H) IHC staining of mouse pancreas (WT and iKO) using insulin antibody (n = 4 mice) (F). β-cell size (G) and amount (H) were quantified. Unpaired two-tailed Student’s t tests were performed.
Data are displayed as means ± SEM. See also Figure S3.
The gut microbiome is one of the integral factors for maintaining metabolic homeostasis in vivo (Quigley, 2013). We examined if knocking out intestinal Gpr17 led to changes in gut microbiota composition and found no significant difference in the composition and diversity of gut microbiome (Figures S3A–S3F). Heat-maps arranged using hierarchical clustering methods and principal component analysis of bacteria showed no gene factor-induced difference in WT and iKO fecal samples (Figures S3G and S3H). Therefore, we conclude that acutely knocking out intestinal Gpr17 caused no change in bacterial composition, richness, or diversity of the gut microbiota.
Enhanced GLP-1 secretion in Gpr17 null intestinal organoids
We further investigated the effect of Gpr17 deficiency on incretin secretion using intestinal organoid, a three-dimensional ex vivo mini-gut that retains the architectural complexity and cellular diversity of gut epithelium (Sato et al., 2009). To obtain Gpr17 null organoids, we first generated constitutive intestinal Gpr17 knockout mice (Gpr17fl/fl;Vil1Cre) and then derived the Gpr17 null intestinal organoids from these mice. We detected a significant reduction (approximately 90%) of Gpr17 mRNA in duodenum and negligible levels of Gpr17 mRNA expression in other intestinal segments (jejunum, ileum, and colon) of the knockout mice (Figure 6A). qRT-PCR confirmed the lack of Gpr17 in organoids derived from intestinal Gpr17 knockouts (Figures 6B and 6C), while neither Gcg nor Gip transcription was affected (Figure 6D). Interestingly, Gpr17 null organoids had significantly increased GLP-1 secretion under basal condition with 1 mmol/L glucose, while GIP secretion remained unchanged (Figure 6E). When stimulated with palmitoleic acid (Figure 6F), a monounsaturated fatty acid, and GPBAR-A (Figure 6G), an agonist of bile acid receptor (G-protein-coupled bile acid receptor [GPBAR]), Gpr17 null organoids demonstrated increased GLP-1, but not GIP secretion. When stimulated with forskolin (adenylyl cyclase activator) and 3-Isobutyl-1-methylxanthine (IBMX, phosphodiesterase inhibitor) (Fsk/IBMX), GLP-1 (Figure 6H), but not GIP (Figure 6I), secretion was elevated in Gpr17 null organoids with forskolin at indicated concentrations.
Figure 6. GLP-1 secretion is enhanced in Gpr17-deficient intestinal organoids derived from Gpr17fl/fl;Vil1Cre mice.
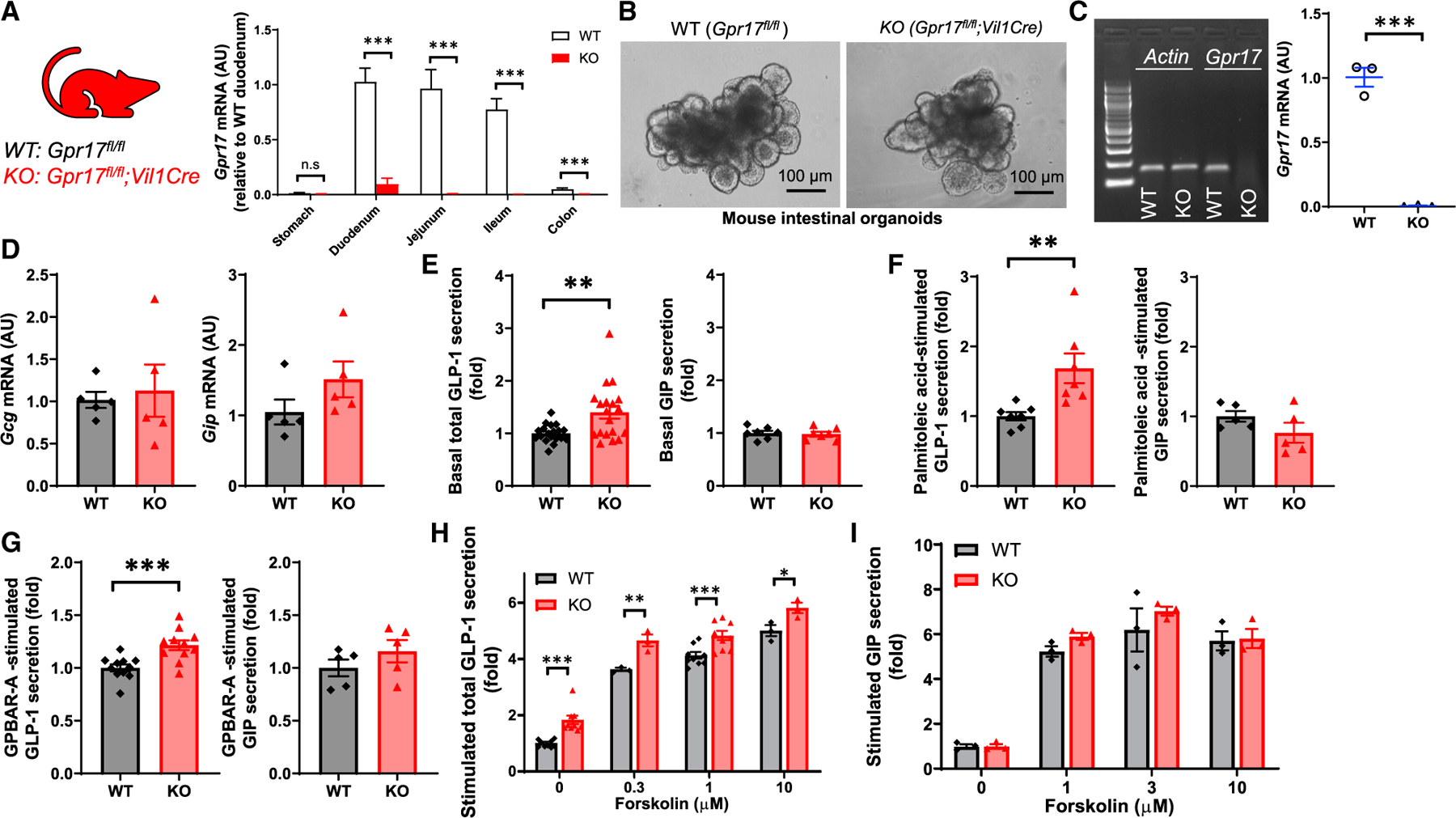
(A) Gpr17 mRNA expression in the gut of Gpr17fl/fl (WT) and Gpr17fl/fl;Vil1Cre (KO) mice. Gene expression was calculated as 2^-∆∆ct. β-actin was used as an internal control. Gene expression was expressed relative to duodenum (n = 5 mice for WT, n = 10 mice for KO). Unpaired t tests were performed for statistical analysis.
(B) Bright-field images of intestinal organoids derived from adult WT and constitutive Gpr17 KO mice.
(C) qRT-PCR of Gpr17 mRNA in organoids (n = 3 mice). Gene expression was normalized to β-actin. Representative gel electrophoresis image of qRT-PCR products was shown. Unpaired two-tailed Student’s t test was performed.
(D) qRT-PCR of Gcg and Gip mRNA in organoids (n = 5 mice).
(E) GLP-1 (n = 19 replicates for each group) and GIP (n = 7 replicates for each group) secretion from intestinal organoids under basal conditions (1 mmol/L glucose) for 2 h. Unpaired two-tailed Student’s t test was performed.
(F) Gpr17 deficiency enhanced fatty acid-induced GLP-1 secretion (n = 7 replicates for each group), but not GIP (n = 5 replicates for each group). Organoids were incubated with palmitoleic acid, a monounsaturated fatty acid, at 40 µmol/L for 2 h.
(G) Gpr17 deficiency enhanced bile acid receptor agonist-induced GLP-1 secretion (n = 11 replicates for each group), but not GIP (n = 5 replicates for each group). Organoids were incubated with GPBAR-A, an agonist of bile acid receptor (TGR5), at 20 µmol/L for 2 h.
(H and I) Gpr17 deficiency enhanced cAMP-elevating agents-induced GLP-1 secretion (n = 3~13 replicates) (H), but not GIP (n = 3 replicates) (I). Organoids were incubated with cAMP-elevating agents, forskolin (0, 0.3, 1, 3, and 10 µmol/L) and 10 µmol/L IBMX for 2 h. p values were calculated using two-way ANOVA test. Percentage of GLP-1 secretion was calculated by measuring GLP-1 levels in the supernatants and cell lysates and normalized to WT organoids in parallel on the same day. Data were obtained from at least three independent experiments.
*p < 0.05, **p < 0.01, ***p < 0.001. Data are displayed as means ± SEM.
Gpr17 deficiency enhanced GLP-1 secretion in cultured GLUTag cells through the Ca2+ and cAMP signaling pathways
Our data showed that intestinal Gpr17 knockout led to better glucose metabolism via increased GLP-1 and insulin secretion in vivo. However, the exact molecular mechanisms are still unclear. To answer this question, we used in vitro cultured GLUTag cells, an established murine intestinal EEC line that secretes GLP-1 in a regulated manner (Brubaker et al., 1998; Drucker et al., 1994). The rise of intracellular calcium triggers exocytosis in various cell types, including neurons (Pang and Südhof, 2010), pancreatic beta cells (Satin, 2000), and EECs (Kuhre et al., 2015). We used whole-cell patch clamp to monitor the currents of voltage-dependent calcium channels (VDCCs) (Figures 7A and 7B). Overexpressing Gpr17 (GFP + cells) in GLUTag cells resulted in reduced amplitude of Ca2+ current triggered by membrane depolarization with I/V curve shown in Figure 7C and area under the curve (AUC) shown in Figure 7D. No difference regarding cell capacitance between transfected cell and non-transfected cells was observed (Figure 7E). Reduced amplitude of Ca2+ current indicated less Ca2+ flux into intracellular space when Gpr17-expressing EECs responded to nutrient stimulation. The cAMP signaling pathway is well known to potentiate Ca2+-induced exocytosis in a variety of secretory cells including EECs (Ezcurra et al., 2013; Seino and Shibasaki, 2005). We measured cAMP levels in live GLUTag cells cotransfected with GloSensor, a cAMP biosensor (Buccioni et al., 2011), and mouse Gpr17 (mGpr17) to determine whether Gpr17 regulates cAMP generation in EECs. The cAMP level was lower in cells expressing mGpr17 as compared with empty vector, suggesting constitutive activity consistent with Gαi coupling (Figure 7F). Treatment with the synthetic GPR17 agonist, MDL29,951, had concentration-dependent effects on the cAMP response in cells expressing mGpr17 (Figure 7F). Specifically, a biphasic pattern was observed where inhibition of cAMP was detected for MDL29,951 concentrations between ~10 nM and 300 nM, but stimulation of cAMP was observed at MDL29,951 concentrations ≥ ~30 µM. GloSensor cAMP signal differences were also observed between empty vector and mGpr17-expressing cells upon addition of 3 µM forskolin (Figure 7G), suggesting constitutive receptor activity suppressing cAMP levels in these cells. MDL29,951 further inhibited the forskolin-stimulated GloSensor cAMP response in a concentration-dependent manner (Figure 7G). However, MDL29,951 had no effect on GloSensor cAMP responses in empty vector-transfected cells (Figure 7G), suggesting that the effects of MDL29,951 are mediated by mGpr17. Time course recording showed that MDL29,951 inhibited forskolin-stimulated induction of luminescence in GLUTag cells expressing mGpr17 (Figures 7H and 7I). Taken together, these experiments suggest that mGpr17 expression constitutively inhibits cAMP levels in GLUTag cells through Gαi -coupling. Furthermore, agonist activation of mGpr17 had biphasic effects on basal cAMP levels that were dependent on MDL29,951 concentration, but exclusively inhibitory effects on forskolin-stimulated cAMP.
Figure 7. Gpr17 regulates GLP-1 secretion through Ca2+ and cAMP signaling.
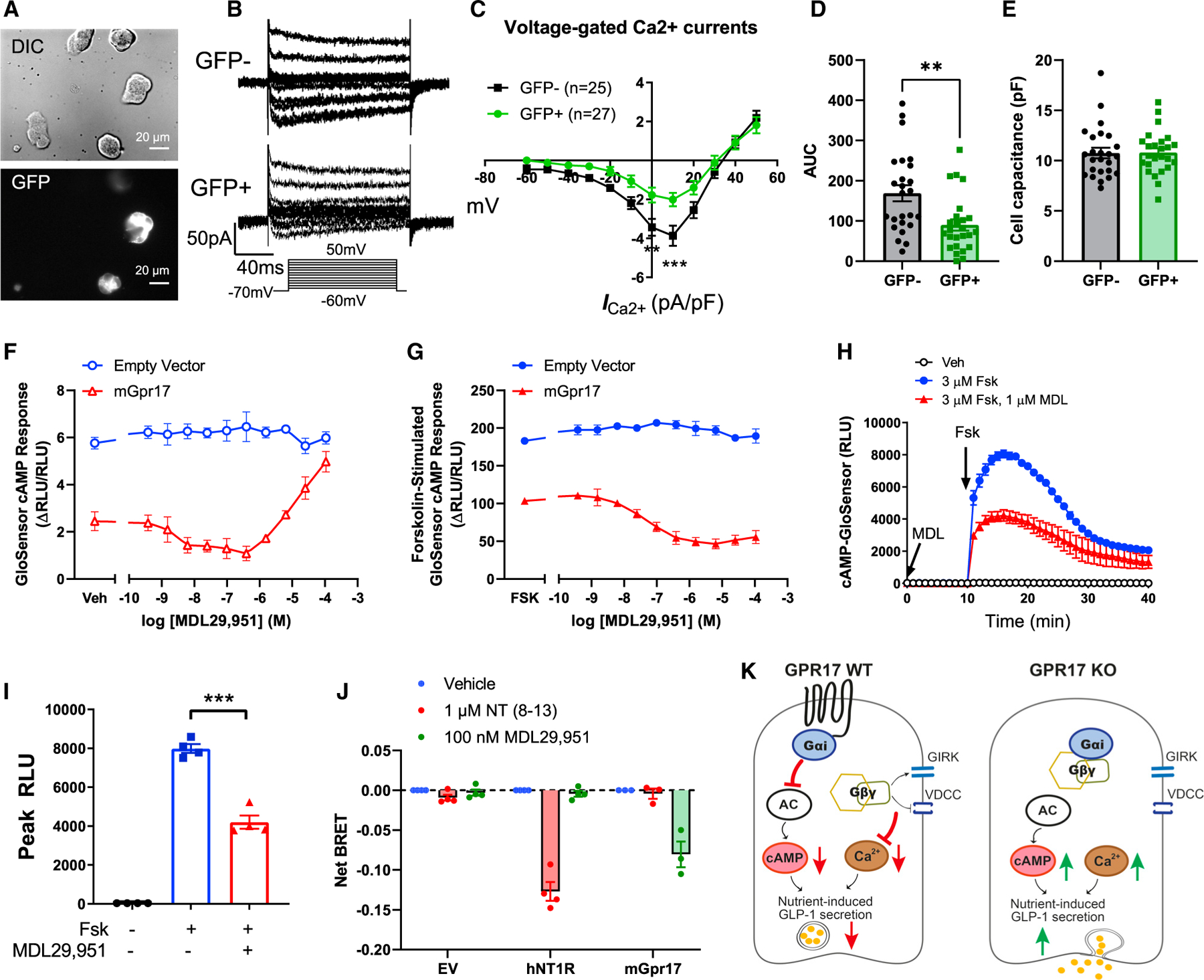
(A) Whole-cell patch clamp recording voltage-gated Ca2+ currents in GLUTag cells that were transiently transfected with pcDNA3.1-mGpr17-GFP. All cells were patched under IR-DIC optics and GFP-positive cells were identified by using an upright microscope fitted with fluorescence optics.
(B) Ca2+ current responses to 150 ms voltage steps of 10 mV increments from a holding potential of −70 mV. The inset shows the voltage pulse protocol. Tetrodotoxin (TTX) at 0.3 µmol/L was added to block Na+ currents.
(C) Current–voltage relationship of the voltage-gated Ca2+ currents recorded in Gpr17-overexpressed GLUTag cells (GFP+; n = 27 cells) and nontransfected cells (GFP−; n = 25 cells). Statistical comparisons between groups in were performed using two-way ANOVA and Sidak’s post hoc tests.
(D) Averaged AUC for voltage-current curve of Ca2+ currents.
(E) Cell capacitance were comparable between Gpr17-overexpressed GLUTag cells (GFP+) and nontransfected cells (GFP−).
(F) Gpr17 expression and agonist stimulation, MDL29,951, inhibited GloSensor cAMP response in GLUTag cells without forskolin addition.
(G) Gpr17 expression and MDL29,951 inhibited forskolin-stimulated GloSensor cAMP response in a concentration-dependent manner in GLUTag cells. Data for (F) and (G) were represented as the mean ± SEM of two independent experiments performed with duplicate wells.
(H and I) Time course recording showing MDL29,951 inhibited forskolin-stimulated cAMP response in mouse Gpr17-overexpressing GLUTag cells (n = 4 replicates) (H). Peak RLU were quantified (I). Vehicle (Veh) or MDL29,951 (1 µmol/L) were incubated for 10 min before forskolin addition. Unpaired two-tailed Student’s t test was performed.
(J) GPR17-Gαi/Gβγ coupling is identified by TRUPATH in HEK293 cells transiently transfected with either pcDNA3.1 (−) empty vector, pcDNA3.1 (+)-hNT1R, or pcDNA3-HA-mGpr17 together with pcDNA5/FRT/TO-Gαi1-Rluc8, pcDNA3.1-Gβ3, and pcDNA3.1-Gγ9-GFP2. The net BRET response represents the BRET ratio for each well subtracted by the mean BRET ratio response of the vehicle-treated wells for each receptor transfection condition. hNT1R, human neurotensin receptor type 1. Data are represented as the mean ± SEM of three or four independent experiments performed with triplicate wells.
(K) Schematic diagram illustrates the molecular mechanisms of how GPR17 negatively regulates nutrient-induced GLP-1 secretion in EECs. GPR17 signals through Gαi pathway to reduce cAMP production in enteroendocrine cells, in turn dampening nutrient-stimulated cAMP rise and Ca2+-stimulated exocytosis. Gβγ subunits released from Gαi reduce Ca2+ influx upon nutrient stimulation through activation of G-Protein-Coupled Inwardly Rectifying Potassium (GIRK) channels and inhibition of VDCCs. On the other hand, GPR17 deficiency increases cAMP and intracellular Ca2+ rise, thereby potentiating nutrient-induced GLP-1 secretion.
*p < 0.05, **p < 0.01, ***p < 0.001. Data are displayed as means ± SEM.
Activation of several GPCRs could directly inhibit VDCCs (Ruiz-Velasco and Ikeda, 2000; Sun et al., 2001) and cAMP production (Robichaux and Cheng, 2018) via Gβγ and Gαi subunits, respectively. To directly prove the Gαi/Gβγ coupling of GPR17, we performed bioluminescence resonance energy transfer (BRET) assay in HEK293 cells transiently expressing TRUPATH Gαβγ biosensor (Olsen et al., 2020). BRET was reduced in mGpr17-expressing cells, but not in empty vector- or human neurotensin receptor type 1 (NT1R)-expressing cells, when treated with 100 nM MDL29,951 (Figure 7J), suggesting GPR17 was coupled to Gαi and its activation could induce Gαi/Gβγ dissociation.
In summary, we showed that GPR17 acts as a brake to negatively regulate GLP-1 secretion in EECs by inhibiting Ca2+ signaling and cAMP generation in response to nutrient stimulation (Figure 7K). Inhibition of GPR17 signaling could potentiate nutrient-stimulated GLP-1 secretion, and consequently improve glycemic control in vivo.
DISCUSSION
GLP-1 and GIP are incretin hormones secreted by enteroendocrine cells in the intestinal epithelial lining within minutes after meal ingestion (Ezcurra et al., 2013; Holst, 2007). GLP-1 and GIP regulate glucose homeostasis and appetite via endocrine and neural mechanisms (Kim et al., 2018; Steinert et al., 2017). In T2D, the overall incretin effect is reduced (Nauck et al., 1986). The cellular mechanisms and intestinal luminal cues governing endogenous GLP-1 secretion are subjects of intense investigation yet remain poorly understood. Here we demonstrated that an orphan GPCR, GPR17, was expressed in GLP-1-producing enteroendocrine cells in the human and rodent intestinal epithelium using complementary approaches (IHC- and FACS-based gene expression characterization). The presence of GPR17 in intestine and its colocalization with GLP-1 led us to further evaluate its previously unrecognized physiological roles in glucose metabolism. We generated the acute inducible (iKO) intestine-specific Gpr17 loss-of-function mouse models. Both female and male iKO mice showed improved glucose tolerance with increased oral GSIS. Furthermore, iKO mice also responded to oral nutrient (glucose or lipid) ingestion with increased GLP-1 secretion. Finally, our in vitro studies showed that Gpr17 signaling in cultured EECs inhibited Ca2+ influx and cAMP production via coupling with Gαi. Gpr17 null intestinal organoids also showed increased GLP-1 secretion. Collectively, these findings demonstrated that inhibiting intestinal Gpr17 signaling improved glucose metabolism via potentiating nutrient-induced GLP-1 secretion from enteroendocrine cells.
GPCRs constitute a large family of proteins that engage in regulating various physiological responses by sensing a diverse array of ligands and coupling with discrete downstream cellular signaling cascades. For instance, GPCRs expressed in EECs can be activated by luminal nutrients, microbiome metabolites, and basolateral stimulations from the enteric nervous system or the circulation (Hauser et al., 2017; Husted et al., 2017; Mace et al., 2015). Efforts have been made to identify EEC-specific GPCRs (Haber et al., 2017; Panaro et al., 2014), though sequencing depth and library preparation often impose limitations to detect GPCR transcripts with low abundance in single-cell RNA sequencing (scRNAseq) studies (Mawla and Huising, 2019; Wang and Kaestner, 2019). We took direct experimental approaches and found Gpr17 mRNA and protein expression in the GLP-1-producing EECs of the rodent and human gut epithelium. Our in vivo physiological and in vitro molecular mechanism studies supported our hypothesis that Gpr17 intestinal knockout improves glucose homeostasis by promoting GLP-1 secretion. In addition to demonstrating increased GLP-1 secretion in iKO mice and organoids, we unbiasedly tested and subsequently ruled out the following alternative hypotheses. First, the improved glucose tolerance phenotype is not secondary to defects in nutrient intake, as Gpr17 intestinal iKO mice exhibited normal weight, body composition, and gut morphology with ad libitum feeding on normal chow diet. Second, we performed extensive profiling and found no difference in the gut microbiota composition of Gpr17 intestinal iKO mice. Last, the number and size of the pancreatic islets in the acute Gpr17 intestinal iKO mice were comparable to the measurements of the WT mice. Taken together, the results from these control experiments further strengthen our initial hypothesis that intestinal Gpr17 plays an important role in glucose homeostasis by modulating GLP-1 secretion.
We previously identified Gpr17 as an effector of FoxO1 orexigenic signals in the brain (Ren et al., 2012). Loss of Gpr17 in the brain confers increased hypothalamic neuronal sensitivity to insulin and leptin, leading to reduced ad libitum feeding and increased relative energy expenditure (Ren et al., 2015). Other groups reported that loss of Gpr17 mitigates brain damage through a FoxO-dependent mechanism (Lecca et al., 2008; Marschallinger et al., 2015). In the intestine, we reason that FoxO1 is less likely to mediate the effects GPR17, since FoxO1 expression was restricted in the chromogranin A-positive endocrine cells and serotonin (5HT)-positive cells, but not GLP-1-producing cells (Bouchi et al., 2014). Moreover, knocking out Gpr17 is less likely to affect the transcription factor upstream. Indeed, in our inducible Gpr17 knockout mice, Gpr17 ablation did not cause a compensatory increase of FoxO1 transcripts in the intestine (data not shown).
As a result of physiological compensation and developmental adaption, germ-line knockout models of established metabolic factors often showed lack of body weight phenotype or surprisingly milder glucose phenotype (Qian et al., 2002; Scrocchi et al., 1996; Scrocchi and Drucker, 1998), which limits the interpretation of negative data as a true absence of function (Chambers et al., 2013). This could be the case for the lack of phenotype in one Gpr17 germ-line knockout model (Mastaitis et al., 2015), although another study reported beneficial metabolic effects (Ou et al., 2019). Being aware of the potential confounding factors of germ-line knockout approaches, we tested the inducible tissue-specific knockouts in our studies. Our inducible intestine-specific knockout mice yielded largely consistent phenotypes for both sexes and revealed metabolic functions of Gpr17 signaling in the gut that are distinct from our previously reported role in the brain. Therefore, we speculate that inhibiting Gpr17 signaling in brain and gut yields concerted improvement of metabolic outcomes through different cellular mechanisms and complementary endocrine pathways.
In gut EECs, our data revealed that GLP-1 secretion was regulated by GPR17 and the downstream signaling pathways mediated via G protein subunits: Gαi and Gβγ (Figure 7K). Previous Gpr17 signaling studies using the heterologous expression models have yielded complicated and sometimes even paradoxical results (Ciana et al., 2006; Hennen et al., 2013; Simon et al., 2016). Adding to the complexity of the issue, GPCRs in metabolic organs have the capacity to signal through multiple pathways in cell-type-specific manners, and it is often not clear which pathway is associated with a given biological effect for a particular receptor (Husted et al., 2017). To better understand the physiological function of GPR17, we used physiologically relevant models, including inducible tissue-specific knockout animals, intestinal organoids, and cultured EECs. Our results indicate that GPR17 engaged Gαi signaling pathway is physiologically relevant for regulating incretin secretion in the enteroendocrine cells. First, Gαi subunits released from the coupled GPCR directly inhibit adenylyl cyclase activity and consequently reduce cAMP production. Second, Gβγ subunits released from Gαi modulate membrane potential through activation of G protein-gated inwardly rectifying K+ channels (GIRKs) and inhibition of voltage-dependent Ca2+ channels (VDCCs) (Lüscher and Slesinger, 2010; Smrcka, 2008; Sun et al., 2001). In the voltage-clamp setting, which would bypass the modulation of Ca2+-influx through GIRK-channel, we found GPR17 overexpression inhibited voltage-gated Ca2+ channels, suggesting the constitutive activation of GPR17 could directly inhibit VDCCs. The activation of Gαi-coupled GPCR causes voltage-dependent inhibition of Ca2+ channels (Bean, 1989; Ikeda and Dunlap, 1999) mediated by Gβγ subunits (Ruiz-Velasco and Ikeda, 2000), a process that has been well documented in neuronal cells (Dunlap et al., 1995). Given the many shared features of neuronal transmitter release in neurons and gut hormone secretion in EECs, we hypothesized that activation of GPR17 could directly modulate voltage-gated Ca2+ channels through Gβγ subunits in EECs. Gpr17 signaling through the Gαi/Gβγ pathway has been reported in several articles (Ciana et al., 2006; Hennen et al., 2013; Inoue et al., 2019; Merten et al., 2018; Simon et al., 2016). Furthermore, by application of Gabg TRUPATH biosensor (Olsen et al., 2020), we directly proved that GPR17 is coupled to Gαi, and its activation could dissociate Gβγ from Gαi. Therefore, we conclude that GPR17 functions as a brake to negatively modulate nutrient-stimulated GLP-1 secretion by inhibiting cAMP and Ca2+ signaling through Gαi and Gβγ subunits.
Limitations of the study
There are limitations with our current studies. First, the nature of the endogenous ligand(s) for GPR17 is still unclear. Several groups have performed initial studies on defining GPR17 ligands. Candidates include uracil nucleotides, cysteinyl leukotrienes (CysLTs), and other synthetic compounds (Ciana et al., 2006; Hennen et al., 2013; Parravicini et al., 2010; Parravicini et al., 2008; Pugliese et al., 2009). Although results were inconsistent among these studies (Simon et al., 2017), cysteinyl-leukotriene receptor (CysLTR) inhibitors did consistently show antagonistic activity for GPR17 signaling (Hennen et al., 2013; Merten et al., 2018), which raises the possibility of endogenous GPR17 agonists sharing structural similarity with cysteinyl leukotrienes, a class of inflammatory lipids. Although beyond the scope of our current study, identifying the endogenous Gpr17 ligand will provide valuable insights to Gpr17 biology and warrants future investigation. Second, not all GPR17-positive cells were stained positive for GLP-1, suggesting GPR17 may regulate other gut hormone secretion in different types of EECs. It remains to be determined whether or how Gpr17 signaling impacts the secretion of other gut hormones in EECs. We acknowledge that the increased GLP-1 secretion in knockout animals in vivo and intestinal organoid cultures in vitro may be one of the mechanisms leading to better glucose homeostasis. Other enteroendocrine mechanisms may also contribute to the glucose phenotype, which warrants future investigation. Third, we observed sexual dimorphism in glucose tolerance after longterm knockout. Sex differences in metabolic regulation have been well documented in animal and human studies (Gannon et al., 2018; Tramunt et al., 2020), such as the landmark paper revealing the essential role of incretin hormones in the double incretin receptor knockout (DIRKO) mice (Hansotia et al., 2004). Sexual dimorphism was observed in three aspects of glucose metabolism dependent or independent of the incretin receptors genetic knockout. First, female and male single and double incretin receptor knockout mice displayed different degrees of impaired glucose tolerance during the oral and intraperitoneal glucose tolerance tests. Second, islets isolated from female and male DIRKO mice showed different degrees of impaired glucose-stimulated first and second phase insulin secretion during the perifusion experiment. Third, islets isolated from female mice had half of the islet insulin content of the male counterparts, regardless of the genotypic differences. Likewise, human studies reported women exhibited more insulin secretion for a given glucose load (Basu et al., 2017; Basu et al., 2006; Horie et al., 2018). At the molecular level, sex hormones have been reported to regulate GLP-1 secretion. Estradiol positively regulates proglucagon-derived peptide secretion in mouse and human alpha and L cells (Handgraaf et al., 2018). Testosterone binding to androgen receptor enhances cAMP production and the incretin effect on insulin secretion in islet beta cells (Navarro et al., 2016). Though we cannot pinpoint the exact mechanism for the sex differences we observed after longterm Gpr17 genetic ablation in this study, we will address the potential interaction of Gpr17 and gonadal hormones in molecular signaling pathways and its implication for cellular physiological function in gut and islet tissues in future studies.
In conclusion, our results demonstrated the role of GPR17 signaling in the gut epithelium related to incretin secretion and the regulation of glucose metabolism. Additional insights into GPR17 biology may lead to more targeted therapies for T2D and obesity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hongxia Ren (renh@iu.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Mouse fecal microbiome data have been deposited at NCBI Bioproject and are publicly available as of the date of publication. Accession number is listed in the key resources table. IHC raw images and quantified data have been deposited at IU DataWorks and are publicly available as of the date of publication. DOI is listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human intestinal samples
Human gut full thickness biopsies were obtained from two deceased donors (male, aged 26 and 30 years old) by transplant surgeon at IU School of Medicine during organ procurements. Fresh samples were maintained and transported in cold DMEM/F12 medium (Invitrogen, Carlsbad, CA); and processed for qRT-PCR within 24 h after resection. Human jejunum and colon were fixed in 4% PFA for 24 h and sectioned for immunohistochemistry staining. Consent for research was obtained from the next of kin of deceased donors by Indiana Donor Network. The study was reviewed and Indiana University Human Research Protection Program (IU HRPP) staff determined the project is not human subjects research and does not require further IU Institutional Review Boards (IRB) review.
Animals
B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (#007909, Rosa-tdTomato), B6.Cg-Tg(Vil1-cre)997Gum/J (#004586, Vil1-Cre), and B6; 129S-Gcgtm1.1(cre/ERT2)Gkg/J (#030681, Gcg-CreERT) mice were purchased from the Jackson Laboratory (JAX). Vil1Cre-ERT2 mice were kindly shared by Dr. Lori Sussel. Gpr17fl/fl mice were generated as described before (Ren et al., 2015) and were bred with Vil1-Cre mice (Madison et al., 2002) to generate control WT (Gpr17fl/fl) and constitutive intestine-specific Gpr17 knockouts (Gpr17fl/fl;Vil1Cre). Inducible intestine-specific Gpr17 knockouts (iKO) were generated by mating Vil1Cre-ERT2 mice (el el Marjou et al., 2004) with Gpr17fl/fl mice. Gcg expressing-dependent tdTomato reporter mice were generated by mating Rosa-tdTomato with Gcg-CreERT mice. Cre recombinase was activated by oral gavage of 10 mg/mL tamoxifen (Sigma-Aldrich, St. Louis, MO)/corn oil solution (200 µL/mouse/day) for 5 consecutive days. Experiments were conducted using 3~16-month-old male and female mice as indicated in the figure legends. All animals were bred and housed under specific pathogen-free conditions and handled in accordance with the guidelines for animal care of Indiana University School of Medicine Animal Care and Use Committee.
Cell lines
Murine enteroendocrine cells, GLUTag cells, were kindly made available to the community by Dr. Daniel J. Drucker (Toronto, Canada) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 1 g/L glucose, as described previously (Ghanem et al., 2016). HEK293 cells and HeLa cells were cultured in Minimum Essential Medium (MEM), 10% fetal bovine serum (FBS), and 1x Penicillin-Streptomycin solution and maintained at 37°C and 5% CO2.
METHOD DETAILS
qRT-PCR
Gut segment dissection was performed as previously described (Panaro et al., 2014). In brief, gut was removed from the stomach to the rectum. Segments measuring 1 cm were removed from the glandular stomach (distal end), duodenum (adjacent to the pyloric sphincter), jejunum (halfway between the pyloric sphincter and the ileocecal valve), ileum (3 cm above the ileocecal valve), and colon (halfway between the cecocolic junction and anus). Total RNA for gut segments were extracted by Trizol (Invitrogen) and reverse transcribed with Superscript II reverse transcriptase (Invitrogen). We performed quantitative real-time PCR (qRT-PCR) in CFX Connect Real-Time system (Bio-Rad, Hercules, CA) using primers spanning introns. Primer sequences are listed in Table S1.
Immunohistochemistry staining
Immunohistochemistry staining of human small intestine (jejunum) and colon were performed by using antibodies for GPR17 (NLS4229; Novus, Centennial, CO), Chromogranin A (SC-393941; Santa Cruz Biotechnology, Dallas, Texas), GIP (ab30679; abcam, Cambridge, MA) and GLP-1 (NBP1–50697; Novus). Images and quantified results were deposited in IU DataWorks (Yan et al., 2021). Insulin and glucagon staining in pancreatic sections were performed by CDMD islets core facility using methods described previously (Tong et al., 2016).
GPR17 antibody was validated in HEK293 cells and HeLa cells transiently transfected by pcDNA3.1-HA-hGPR17 plasmids. Cells were transfected with pcDNA3.1-HA-hGPR17 plasmids for overnight. GPR17 (NLS4229; Novus) and HA (#901513; Biolegend, San Diego, CA) antibodies were used for immunostaining. Cells were permeabilized by 0.1% Triton X-100 for total GPR17 staining or non-permeabilized for cell-surface GPR17 staining. Chamber slides were imaged under LSM-700 confocal microscope (ZEISS, Oberkochen, Germany). 10–22 randomly selected cells per image and total 10 images were quantified for staining intensity by using Image J (NIH).
Fluorescence-activated cell sorting (FACS) of Gcg-expressing cells in mouse gut epithelium
The whole colon and the last quarter of the small intestine from Gcg-CreERT;Rosa-tdTomato mice were isolated and rinsed in cold PBS. Intestine epithelium tissues were collected by scraping and resuspended in TrypLE Express (Thermo Fisher Scientific, Waltham, MA) at 37°C for 20 min to dissociate into single cell suspension. Cells were re-suspended in 1mL 1X PBS containing 1% BSA and 10 µM Y27632 (STEMCELL Technologies, Cambridge, MA). Cells were stained by DAPI before subjected to flow cytometry sorting by FACSAria 2 SORP (Becton Dickinson, Franklin Lakes, NJ) and data were analyzed on FACSDiva 8.0.1 software (Becton Dickinson). Forward scatter and size scatter were used to get single cells based on granularity and height. DAPI negative gate was used to get live cells. tdTomato positive cells with a fluorescence signal measured above 103 were gated to collect the Gcg-CreERT positive (i.e. GLP-1-producing) cells. We collected the tdTomato-positive (T+), and tdTomato-negative (T-) live cells for RNA extraction with Trizol LS reagent (Invitrogen).
oGSIS in vivo
Adult mice were fasted overnight. Insulin levels (#EZRMI-13K; Millipore, Burlington, MA) were measured in tail serum samples collected before and 15 min after oral glucose (3 g/kg) administration.
Glucose tolerance tests
Adult mice were fasted for overnight or 4–6 h for glucose tolerance tests. Tail blood glucose levels were measured by AlphaTRAK2 glucose meter at 0, 15, 30, 60, 90, 120 min after glucose administration via intraperitoneal (i.p.) injection or oral gavage. The dose of glucose for each experiment was noted in the figure legends.
Small intestine and colon histology
The mouse entire small intestine (SI), from the gastric pylorus to the ileocecal valve, and the colon, from cecocolic junction to anus, were dissected from anaesthetized mice (3–4 months old). The SI and colon were opened longitudinally and wrapped as a ‘‘Swiss roll’’ (Moolenbeek and Ruitenberg, 1981), respectively. Swiss rolls were fixed in 4% paraformaldehyde (PFA) for 24–48 h, cryoprotected with 30% sucrose (w/v PBS), and sectioned (10 mm) for H&E staining.
Gut incretins secretion in vivo
Adult mice were given glucose (3 g/kg) via oral gavage after being deprived of food for 4–6 h. Tail blood was collected from tail at 0min and 10 min after glucose administration. Blood was collected and mixed with an inhibitor cocktail (Aprotinin, A6279, Sigma-Aldrich; DPP-IV inhibitor, DPP4–010, Millipore; 0.5M EDTA, AM9261, Invitrogen. 1:1:1, 10 mL mixture for each sample.). On another occasion, tail blood was collected at 0 min and 60 min after 100% corn oil (10 µL/g BW) via oral gavage after 4~6 h fasting. Total GIP (#EZRMGIP-55K; Millipore) and total GLP-1 (#K1503PD-1; MSD, Rockville, MD) measurements were measured in plasma samples.
Mouse intestinal organoids culture
Mouse intestinal organoids were cultured in mouse IntesiCultTM Organoid Growth Medium (STEMCELL Technologies, Cambridge, MA) by following the manufacturer’s instruction. 20 cm of small intestine proximal to stomach were harvested for culturing intestinal organoids, as this region allows optimal efficiency for generating organoids (Zietek et al., 2015). Organoids were used for experiments after two passages from primary culture or frozen.
GLP-1 and GIP secretion studies in intestinal organoids
Intestinal organoids were cultured in 24-well plates with a density of 200–300 organoids per well for 7–10 days. Secretion experiments were performed by incubating the organoids with test regents in treated solution (138 mM NaCl, 4.5 mM KCl, 4.2 mM NaHCO3, 1.2 mM NaH2PO4, 2.5 mM CaCl2, 1.2 mM MgCl2, and 10 mM HEPES supplemented with 0.1% (wt/vol) fatty acid-free BSA and 1% DPP-IV inhibitor, pH 7) for 2 h at 37°C. At the end of the incubation period, supernatant was collected, and the organoids were lysed in RIPA buffer supplemented with protease inhibitor and DPP-IV inhibitor. Incretins in supernatant and lysates were assayed using ELISA kit for GIP (#EZRMGIP-55K; Millipore) and total GLP-1 (#K1503PD-1; MSD). Secretion was first calculated as the percentage of secreted GLP-1 (supernatant) in total GLP1 (both supernatant and cell lysates) and then normalized to the basal secretion in WT organoids in parallel on the same day.
Whole-cell patch clamp
The voltage-gated Ca2+ currents in GLUTag cells were recorded by the standard whole-cell patch clamp with a procedure adapted from previous reports (Reimann et al., 2005; Rogers et al., 2011). Cells and plasmid (pcDNA3.1-mGpr17-GFP) mixture were cultured onto glass cover slips in 12-well plate. After overnight transfection, the medium was replaced with fresh growth medium, and the cells were subjected to patch clamp 1 day after that. Single cells and well-defined cells in small clusters were patched. GFP positive or negative cells were identified by using an upright microscope (E600FN, Nikon, Japan) fitted with fluorescence optics. Microelectrodes were pulled from borosilicate glass capillary (1B150F, WPI, Sarasota, FL) by a Micropipette Puller (P-1000, Sutter Instrument, Novato, CA) and had 4~6 MΩ when filled with pipette solution. Recordings were acquired with an MultiClamp 700B (Molecular Devices, San Jose, CA), a DigiData 1322A (Molecular Devices) and analyzed with pClamp10 software (Molecular Devices). The bath solution contained (mmol/L): 4.5 CsCl, 118 NaCl, 4.2 NaHCO3, 1.2 NaH2PO4, 2.6 CaCl2, 1.2 MgCl2, 10 HEPES, 20 TEA, 1 glucose (pH7.4). The pipette solution contained (mmol/L): 107 CsCl, 1 CaCl2, 7MgCl2, 11 EGTA, 10 HEPES, 5 Na2ATP (pH7.4). Tetrodotoxin (TTX, 0.3 µmol/L) was added into the bath solution to block Na+ currents.
GloSensor cAMP assay
GLUTag cells were transiently transfected with pGloSensor-cAMP-22F (Promega, Madison, WI) together with either pcDNA3.1(−) empty vector or pcDNA3-HA-mGpr17 (mGpr17) plasmids and cultured in poly-D-lysine coated 96-well plates. Transfection mix was replaced with fresh growth media the next day. The following day, growth media was removed, cells were washed with CO2-independent media (Gibco, Life Technologies Corporation, Grand Island, NY), and cells were loaded with equilibration buffer (CO2-independent media containing 2% GloSensor cAMP reagent) (Promega, Madison, WI) for 2 h at 37°C and 5% CO2. Cells were then allowed to equilibrate to ambient room temperature for 30 min. Luminescence was read at ambient room temperature every 2 minutes using a SpectraMax iD5 plate reader (Molecular Devices, San Jose, CA). Initial baseline luminescence was measured for 8 minutes. Subsequently, drug treatments were added to wells, followed by luminescence measurements. Specifically, luminescence was measured for 10 min following each of IBMX and MDL29,951 additions. Finally, forskolin was added and luminescence was measured for 30 min.
Data were expressed as the difference in RLU form the average initial baseline, divided by the average initial baseline for each individual well (∆RLU/RLU) and represent the Mean ± SEM of two independent experiments performed with duplicate wells. Concentration response curves for MDL29,951 were from the 4 minute time point after MDL29,951 addition. Concentration response curves for MDL29,951 in the presence of 3 µM forskolin represent the average of the 16–30 min time points following forskolin addition.
Bioluminescence resonance energy transfer (BRET) assay
BRET assay was performed using TRUPATH biosensor (Addgene, Watertown, MA) as described previously (Olsen et al., 2020). HEK293 cells were seeded into 6-well plates at a density of 600,000 cells/well. The following day, cells were transiently transfected with 1:1:1:1 ratio of either pcDNA3.1(−) empty vector, pcDNA3.1(+)-hNT1R, or pcDNA3-HA-mGpr17 together with pcDNA5/FRT/TO- Gαi 1-Rluc8, pcDNA3.1-Gβ3, and pcDNA3.1-Gγ9-GFP2 using Lipofectamine 3000. 24 h post-transfection, cells were dissociated from the plate with 0.05% trypsin-EDTA, resuspended in MEM containing 1% FBS and seeded into poly-D-lysine-coated white, opaque, 96-well plates at 30,000 cells/well. The next day, cells were washed with Hank’s balanced salt solution (HBSS)-based assay buffer (HBSS, 20 mM HEPES, pH 7.2), followed by addition of 80 µL/well assay buffer, and then loaded with 10 µL/well 50 µM coelenterazine 400a diluted in assay buffer for 5 min at ambient room temperature. 10 µL/well of the indicated treatments were added in triplicate and incubated for 5 min at room temperature before filtered luminescence was read at 410 nm and 515 nm on a SpectraMax iD5 plate reader. BRET ratios for each well were calculated as the ratio of luminescence emission at 515 nm divided by 410 nm. The net BRET response represents the BRET ratio for each well subtracted by the mean BRET ratio response of the vehicle-treated wells for each receptor transfection condition. Data were represented as the Mean ± SEM of three or four independent experiments performed with triplicate wells.
Microbiome analysis
Fresh fecal samples were collected in the morning during ad libitum feeding. Frozen fecal samples were sent to the University of Missouri for DNA isolation and 16S rRNA gene V4 sequencing using the Illumina MiSeq platform as previously described (Ericsson et al., 2018). Briefly, DNA was extracted using PowerFecal kits (Qiagen) according to the manufacturer’s instructions, with the exception that samples were homogenized in the provided bead tubes using a TissueLyser II (Qiagen, Venlo, Netherlands) for 3 min at 30/s, rather than performing the initial homogenization of samples using the vortex adapter described in the protocol, before proceeding according to the protocol and eluting in 100 µL of elution buffer (Qiagen). DNA yields were quantified via fluorometry (Qubit 2.0, Invitrogen, Carlsbad, CA) using quant-iT BR dsDNA reagent kits (Invitrogen) and normalized to a uniform concentration and volume.
Extracted fecal DNA was processed at the University of Missouri DNA Core Facility. Bacterial 16S rRNA amplicons were constructed via amplification of the V4 region of the 16S rRNA gene with universal primers (U515F/806R) previously developed against the V4 region, flanked by Illumina standard adapter sequences (Caporaso et al., 2011; Walters et al., 2011). Oligonucleotide sequences are available at proBase (Loy et al., 2007). Dual-indexed F and R primers were used in all reactions. PCR was performed in 50 µL reactions containing 100 ng metagenomic DNA, primers (0.2 µM each), dNTPs (200 µM each), and Phusion high-fidelity DNA polymerase (1U, Thermo Fisher). Amplification parameters were 98°C(3min) + [98°C(15sec) + 50°C(30sec) + 72°C(30sec)] x 25 cycles +72°C(7min). Amplicon pools (5 µL/reaction) were combined, mixed, and then purified by addition of Axygen™ Axyprep MagPCR clean-up beads to an equal volume of 50 µL of amplicons and incubated for 15 min at room temperature (RT). Products were then washed multiple times with 80% ethanol and the dried pellet was resuspended in 32.5 µL EB buffer (Qiagen), incubated for 2 min at RT, and then placed on a magnetic stand for 5 min. The final amplicon pools were evaluated using an Advanced Analytical Fragment Analyzer automated electrophoresis system, quantified using quant-iT HS dsDNA kits, and diluted according to Illumina’s standard protocol for sequencing as 23250 bp paired-end reads on the MiSeq instrument.
DNA sequences were assembled and annotated at the MU Informatics Research Core Facility. Primers were designed to match the 5′ ends of the forward and reverse reads. Cutadapt (Kechin et al., 2017) (version 2.6; https://github.com/marcelm/cutadapt) was used to remove the primer from the 5′ end of the forward read. If found, the reverse complement of the primer to the reverse read was then removed from the forward read as were all bases downstream. Thus, a forward read could be trimmed at both ends if the insert was shorter than the amplicon length. The same approach was used on the reverse read, but with the primers in the opposite roles. Read pairs were rejected if one read or the other did not match a 5′ primer, and an error-rate of 0.1 was allowed. Two passes were made over each read to ensure removal of the second primer. A minimal overlap of three bp with the 3′ end of the primer sequence was required for removal. The QIIME2 (Bolyen et al., 2019) DADA2 (Callahan et al., 2016) plugin (version 1.10.0) was used to denoise, de-replicate, and count ASVs (amplicon sequence variants), incorporating the following parameters: 1) forward and reverse reads were truncated to 150 bases, 2) forward and reverse reads with number of expected errors higher than 2.0 were discarded, and 3) Chimeras were detected using the “consensus” method and removed. R version 3.5.1 and Biom version 2.1.7 were used in QIIME2. Taxonomies were assigned to final sequences using the Silva.v132 (Pruesse et al., 2007) database, using the classifysklearn procedure.
QUANTIFICATION AND STATISTICAL ANALYSIS
We analyzed data with Student’s t–test or one–way or two-way ANOVA using GraphPad Prism software. All of the statistical details of experiments can be found in the figure legends. No method was used to determine whether the data met assumptions of the statistical approach. We used the customary threshold of p < 0.05 to declare statistical significance.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-GPR17 (1:200) | Novus | Cat#NLS4229; RRID: AB_2232678 |
| Mouse monoclonal anti-Chromogranin A (1:200) | Santa Cruz Biotechnology | SC-393941; RRID: AB_2801371 |
| Mouse monoclonal anti-GIP (1:100) | Abcam | Cat# ab30679, RRID: AB_2109682 |
| Mouse monoclonal anti-GLP1 (1:50) | Novus | Cat# NBP1–50697, RRID: AB_11016296 |
| Mouse monoclonal anti-HA (1:1000) | BioLegend | Cat# 901513, RRID: AB_2565335 |
| Goat anti-Rabbit IgG (H+L) (1:500) | Thermo Fisher Scientific | Cat# A-21428, RRID: AB_2535849 |
| Goat anti-Mouse IgG (H+L) (1:500) | Thermo Fisher Scientific | Cat# 35502, RRID: AB_844397 |
|
Biological samples | ||
| Human gut tissue | IU Health Methodist Hospital | NA |
|
Chemicals, peptides, and recombinant proteins | ||
| Aprotinin | Sigma-Aldrich | A6279; CAS: 9087–70-1 |
| DPP-IV inhibitor | Millipore | Cat#DPP4–010 |
| Exendin-3 (9–39) amide (Ex9) | Tocris | 2081; CAS: 133514–43-9 |
| Forskolin | Tocris | 1099; CAS: 66575–29-9 |
| IBMX | Sigma-Aldrich | I7018; CAS: 28822–58-4 |
| MDL29,951 | Laboratory of Donald Landry and Andrew Wasmuth | NA |
| Pertussis toxin (PTX) | Sigma-Aldrich | P2980; CAS: 70323–44-3 |
| Tamoxifen | Sigma-Aldrich | T5648; CAS: 10540–29-1 |
| Tetrodotoxin (TTX) | Tocris | 1069; CAS: 18660–81-6 |
| Y27632 | STEMCELL Technologies | Cat#72302 |
| NT(8–13) | Sigma-Aldrich | N5266; CAS: 60482–95-3 |
|
Critical commercial assays | ||
| GLP-1 ELISA kit | Meso Scale Discovery | Cat#K1503PD-1 |
| GIP ELISA kit | Millipore | Cat#EZRMGIP-55K |
| Insulin ELISA kit | Millipore | Cat#EZRMI-13K |
| TRUPATH kit | Gift from Bryan Roth | Addgene#1000000163 |
|
Deposited data | ||
| Mouse Fecal Microbiome Data | This paper | SRA: PRJNA756295 |
| IHC raw images and quantified data | This paper | https://doi.org/10.7912/D2/22 |
|
Experimental models: Cell lines | ||
| HEK293 | ATCC | CRL-1573 |
| GLUTag | Gift from Sonia M. Najjar | NA |
| HELA | ATCC | CCL-2 |
|
Experimental models: Organisms/strains | ||
| Mouse: B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | The Jackson Laboratory | CAT#007909 |
| Mouse: B6.Cg-Tg(Vil1-cre)997Gum/J | The Jackson Laboratory | CAT#004586 |
| Mouse: B6;129S-Gcgtm1.1(cre/ERT2)Gkg/J | The Jackson Laboratory | CAT#030681 |
| Mouse: B6.Cg-Tg(Vil1-cre/ERT2)23Syr/J | The Jackson Laboratory | CAT#020282 |
| Mouse: Gpr17fl/fl | Columbia University Transgenic Mouse Core Facility | NA |
|
Oligonucleotides | ||
| See Table S1 for a list of all primers and oligonucleotides | This paper | NA |
|
Recombinant DNA | ||
| pcDNA3.1 (2021(−) | (Conley et al., 2021) | NA |
| pcDNA3.1-mGpr17-GFP | (Conley et al., 2021) | NA |
| pcDNA3.1-HA-hGPR17 | (Conley et al., 2021) | NA |
| pcDNA3.1(+)-hNT1R | CDNA | NTSR100000 |
| pcDNA3-HA-mGPR17 | This paper | NA |
| pGloSensor™-22F | Promega | E2301 |
|
Software and algorithms | ||
| pClamp 10.7 | Molecular Devices | NA |
| Prism 9 | GraphPad | NA |
Highlights.
Gpr17 is expressed in GLP-1-producing EECs in human and rodent gut epithelium
Loss of gut Gpr17 increases insulin release and improves glucose tolerance in mice
Gpr17 deficiency increases GLP-1 secretion in mice and intestinal organoids
Gpr17-Gαi/βγ signaling regulates GLP-1 secretion by inhibiting cAMP and VDCCs
ACKNOWLEDGMENTS
We thank Dr. Daniel J. Drucker (Toronto, Canada) for sharing GLUTag cells and Dr. Sonia M. Najjar (Ohio University) for shipping GLUTag cells to us. We thank Drs. Wasmuth and Landry at the Organic Chemistry Collaborative Center and Dr. Rebecca A. Haeusler at Columbia University College of Physicians and Surgeons for synthesizing MDL29,951 and sharing the human cDNA samples, respectively. We thank Dr. Gary Schwartz (Albert Einstein College of Medicine) for insightful discussions during project development and comments on the manuscript. We thank the support from the islets core, histology core, and flow cytometry core at Indiana University School of Medicine. Dr. Ren’s group was supported by funding from NIH (R01DK120772, R00DK098294, R03TR003350, P30DK097512, T32DK064466, and UL1TR002529).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110179.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Basu A, Dube S, and Basu R (2017). Men are from mars, women are from venus: Sex differences in insulin action and secretion. Adv. Exp. Med. Biol 1043, 53–64. 10.1007/978-3-319-70178-3_4. [DOI] [PubMed] [Google Scholar]
- Basu R, Dalla Man C, Campioni M, Basu A, Klee G, Toffolo G, Cobelli C, and Rizza RA (2006). Effects of age and sex on postprandial glucose metabolism: Differences in glucose turnover, insulin secretion, insulin action, and hepatic insulin extraction. Diabetes 55, 2001–2014. 10.2337/db05-1692. [DOI] [PubMed] [Google Scholar]
- Bean BP (1989). Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature 340, 153–156. 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol 37, 852–857. 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchi R, Foo KS, Hua H, Tsuchiya K, Ohmura Y, Sandoval PR, Ratner LE, Egli D, Leibel RL, and Accili D (2014). FOXO1 inhibition yields functional insulin-producing cells in human gut organoid cultures. Nat. Commun 5, 4242. 10.1038/ncomms5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker PL, Schloos J, and Drucker DJ (1998). Regulation of glucagon-like peptide-1 synthesis and secretion in the GLUTag enteroendocrine cell line. Endocrinology 139, 4108–4114. 10.1210/endo.139.10.6228. [DOI] [PubMed] [Google Scholar]
- Buccioni M, Marucci G, Dal Ben D, Giacobbe D, Lambertucci C, Soverchia L, Thomas A, Volpini R, and Cristalli G (2011). Innovative functional cAMP assay for studying G protein-coupled receptors: Application to the pharmacological characterization of GPR17. Purinergic Signal 7, 463–468. 10.1007/s11302-011-9245-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister MA, Ayala JE, Smouse H, Landivar-Rocha A, Brown JD, Drucker DJ, Stoffers DA, Sandoval DA, Seeley RJ, and Ayala JE (2017). The Hypothalamic glucagon-like peptide 1 receptor is sufficient but not necessary for the regulation of energy balance and glucose homeostasis in mice. Diabetes 66, 372–384. 10.2337/db16-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016). DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 13, 581–583. 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JE, and Drucker DJ (2013). Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab 17, 819–837. 10.1016/j.cmet.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, and Knight R (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. United States America 108, 4516–4522. 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceruti S, Villa G, Genovese T, Mazzon E, Longhi R, Rosa P, Bramanti P, Cuzzocrea S, and Abbracchio MP (2009). The P2Y-like receptor GPR17 as a sensor of damage and a new potential target in spinal cord injury. Brain : A J. Neurol 132, 2206–2218. 10.1093/brain/awp147. [DOI] [PubMed] [Google Scholar]
- Chambers AP, Sandoval DA, and Seeley RJ (2013). Integration of satiety signals by the central nervous system. Curr. Biol 23, R379–R388. 10.1016/j.cub.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim KS, Gutierrez-Aguilar R, Li B, Drucker DJ, D’Alessio DA, et al. (2017). The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab 25, 927–934.e923. 10.1016/j.cmet.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wu H, Wang S, Koito H, Li J, Ye F, Hoang J, Escobar SS, Gow A, Arnett HA, et al. (2009). The oligodendrocyte-specific G protein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat. Neurosci 12, 1398–1406. 10.1038/nn.2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciana P, Fumagalli M, Trincavelli ML, Verderio C, Rosa P, Lecca D, Ferrario S, Parravicini C, Capra V, Gelosa P, et al. (2006). The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J 25, 4615–4627. 10.1038/sj.emboj.7601341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley Jason, Sun Hongmao, Ayers Kristin, Zhu Hu, Chen Rong, Shen Min, Hall Matthew, and Ren Hongxia (2021). Human GPR17 missense variants identified in metabolic disease patients have distinct downstream signaling profiles. Journal of Biological Chemistry 297 (1), 100881. 10.1016/j.jbc.2021.100881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douros JD, Tong J, and D’Alessio DA (2019). The effects of bariatric surgery on islet function, insulin secretion, and glucose control. Endocr. Rev 40, 1394–1423. 10.1210/er.2018-00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker DJ, Jin T, Asa SL, Young TA, and Brubaker PL (1994). Activation of proglucagon gene transcription by protein kinase-A in a novel mouse enteroendocrine cell line. Mol. Endocrinol 8, 1646–1655. 10.1210/mend.8.12.7535893. [DOI] [PubMed] [Google Scholar]
- Drucker DJ, and Nauck MA (2006). The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 368, 1696–1705. 10.1016/s0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI, and Turner TJ (1995). Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci 18, 89–98. [PubMed] [Google Scholar]
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, and Robine S (2004). Tissue-specific and inducible cre-mediated recombination in the gut epithelium. Genesis 39, 186–193. 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- Ericsson AC, Gagliardi J, Bouhan D, Spollen WG, Givan SA, and Franklin CL (2018). The influence of caging, bedding, and diet on the composition of the microbiota in different regions of the mouse gut. Scientific Rep 8, 4065. 10.1038/s41598-018-21986-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezcurra M, Reimann F, Gribble FM, and Emery E (2013). Molecular mechanisms of incretin hormone secretion. Curr. Opin. Pharmacol 13, 922–927. 10.1016/j.coph.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon M, Kulkarni RN, Tse HM, and Mauvais-Jarvis F (2018). Sex differences underlying pancreatic islet biology and its dysfunction. Mol. Metab 15, 82–91. 10.1016/j.molmet.2018.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem SS, Heinrich G, Lester SG, Pfeiffer V, Bhattacharya S, Patel PR, DeAngelis AM, Dai T, Ramakrishnan SK, Smiley ZN, et al. (2016). Increased glucose-induced secretion of glucagon-like peptide-1 in mice lacking the carcinoembryonic antigen-related cell adhesion molecule 2 (CEACAM2). J. Biol. Chem 291, 980–988. 10.1074/jbc.M115.692582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, and Reimann F (2016). Enteroendocrine cells: Chemosensors in the intestinal epithelium. Annu. Rev. Physiol 78, 277–299. 10.1146/annurev-physiol-021115-105439. [DOI] [PubMed] [Google Scholar]
- Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y, et al. (2017). A single-cell survey of the small intestinal epithelium. Nature 551, 333–339. 10.1038/nature24489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib AM, Richards P, Cairns LS, Rogers GJ, Bannon CA, Parker HE, Morley TC, Yeo GS, Reimann F, and Gribble FM (2012). Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology 153, 3054–3065. 10.1210/en.2011-2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handgraaf S, Dusaulcy R, Visentin F, Philippe J, and Gosmain Y (2018). 17-b Estradiol regulates proglucagon-derived peptide secretion in mouse and human a- and L cells. JCI Insight 3. 10.1172/jci.insight.98569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansotia T, Baggio LL, Delmeire D, Hinke SA, Yamada Y, Tsukiyama K, Seino Y, Holst JJ, Schuit F, and Drucker DJ (2004). Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of DPP-IV inhibitors. Diabetes 53, 1326–1335. 10.2337/diabetes.53.5.1326. [DOI] [PubMed] [Google Scholar]
- Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, and Gloriam DE (2017). Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov 16, 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennen S, Wang H, Peters L, Merten N, Simon K, Spinrath A, Blattermann S, Akkari R, Schrage R, Schroder R, et al. (2013). Decoding signaling and function of the orphan G protein-coupled receptor GPR17 with a small-molecule agonist. Sci. Signal 6, ra93. 10.1126/scisignal.2004350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst JJ (2007). The physiology of glucagon-like peptide 1. Physiol. Rev 87, 1409–1439. 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- Horie I, Abiru N, Eto M, Sako A, Akeshima J, Nakao T, Nakashima Y, Niri T, Ito A, Nozaki A, et al. (2018). Sex differences in insulin and glucagon responses for glucose homeostasis in young healthy Japanese adults. J. Diabetes Investig 9, 1283–1287. 10.1111/jdi.12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husted AS, Trauelsen M, Rudenko O, Hjorth SA, and Schwartz TW (2017). GPCR-mediated signaling of metabolites. Cell Metab 25, 777–796. 10.1016/j.cmet.2017.03.008. [DOI] [PubMed] [Google Scholar]
- Ikeda SR, and Dunlap K (1999). Voltage-dependent modulation of N-type calcium channels: Role of G protein subunits. Adv. Second Messenger Phosphoprotein Res 33, 131–151. 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- Inoue A, Raimondi F, Kadji FMN, Singh G, Kishi T, Uwamizu A, Ono Y, Shinjo Y, Ishida S, Arang N, et al. (2019). Illuminating G-protein-coupling selectivity of GPCRs. Cell 177, 1933–1947.e1925. 10.1016/j.cell.2019.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsurada K, and Yada T (2016). Neural effects of gut- and brain-derived glucagon-like peptide-1 and its receptor agonist. J. Diabetes Investig 7, 64–69. 10.1111/jdi.12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kechin A, Boyarskikh U, Kel A, and Filipenko M (2017). cutPrimers: A new tool for accurate cutting of primers from reads of targeted next generation sequencing. J. Comput. Biol 24, 1138–1143. 10.1089/cmb.2017.0096. [DOI] [PubMed] [Google Scholar]
- Kim KS, Seeley RJ, and Sandoval DA (2018). Signalling from the periphery to the brain that regulates energy homeostasis. Nat. Rev. Neurosci 19, 185–196. 10.1038/nrn.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhre RE, Frost CR, Svendsen B, and Holst JJ (2015). Molecular mechanisms of glucose-stimulated GLP-1 secretion from perfused rat small intestine. Diabetes 64, 370–382. 10.2337/db14-0807. [DOI] [PubMed] [Google Scholar]
- Lecca D, Trincavelli ML, Gelosa P, Sironi L, Ciana P, Fumagalli M, Villa G, Verderio C, Grumelli C, Guerrini U, et al. (2008). The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PLoS one 3, e3579. 10.1371/journal.pone.0003579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loy A, Maixner F, Wagner M, and Horn M (2007). probeBase–an online resource for rRNA-targeted oligonucleotide probes: New features 2007. Nucleic Acids Res 35, D800–D804. 10.1093/nar/gkl856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, and Slesinger PA (2010). Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci 11, 301–315. 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace OJ, Tehan B, and Marshall F (2015). Pharmacology and physiology of gastrointestinal enteroendocrine cells. Pharmacol. Res. Perspect 3, e00155. 10.1002/prp2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, and Gumucio DL (2002). Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem 277, 33275–33283. 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- Marschallinger J, Schäffner I, Klein B, Gelfert R, Rivera FJ, Illes S, Grassner L, Janssen M, Rotheneichner P, Schmuckermair C, et al. (2015). Structural and functional rejuvenation of the aged brain by an approved anti-asthmatic drug. Nat. Commun 6, 8466. 10.1038/ncomms9466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastaitis J, Min S, Elvert R, Kannt A, Xin Y, Ochoa F, Gale NW, Valenzuela DM, Murphy AJ, Yancopoulos GD, and Gromada J (2015). GPR17 gene disruption does not alter food intake or glucose homeostasis in mice. Proc. Natl. Acad. Sci. United States America 112, 1845–1849. 10.1073/pnas.1424968112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawla AM, and Huising MO (2019). Navigating the depths and avoiding the shallows of pancreatic islet cell transcriptomes. Diabetes 68, 1380–1393. 10.2337/dbi18-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merten N, Fischer J, Simon K, Zhang L, Schröder R, Peters L, Letombe AG, Hennen S, Schrage R, Bödefeld T, et al. (2018). Repurposing HAMI3379 to block GPR17 and promote rodent and human oligodendrocyte differentiation. Cell Chem Biol 25, 775–786.e775. 10.1016/j.chembiol.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MC, Wieland T, and Tsujimoto G (2009). How reliable are G-protein-coupled receptor antibodies? Naunyn Schmiedebergs Arch. Pharmacol 379, 385–388. 10.1007/s00210-009-0395-y. [DOI] [PubMed] [Google Scholar]
- Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, and Habener JF (1986). Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J. Biol. Chem 261, 11880–11889. [PubMed] [Google Scholar]
- Moolenbeek C, and Ruitenberg EJ (1981). The “Swiss roll”: A simple technique for histological studies of the rodent intestine. Lab Anim 15, 57–59. [DOI] [PubMed] [Google Scholar]
- Nauck M, Stockmann F, Ebert R, and Creutzfeldt W (1986). Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 29, 46–52. 10.1007/BF02427280. [DOI] [PubMed] [Google Scholar]
- Navarro G, Xu W, Jacobson DA, Wicksteed B, Allard C, Zhang G, De Gendt K, Kim SH, Wu H, Zhang H, et al. (2016). Extranuclear actions of the androgen receptor enhance glucose-stimulated insulin secretion in the male. Cell Metab 23, 837–851. 10.1016/j.cmet.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RHJ, DiBerto JF, English JG, Glaudin AM, Krumm BE, Slocum ST, Che T, Gavin AC, McCorvy JD, Roth BL, and Strachan RT (2020). TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol 16, 841–849. 10.1038/s41589-020-0535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Z, Ma Y, Sun Y, Zheng G, Wang S, Xing R, Chen X, Han Y, Wang J, Lu QR, et al. (2019). A GPR17-cAMP-lactate signaling axis in oligodendrocytes regulates whole-body metabolism. Cell Rep 26, 2984–2997.e2984. 10.1016/j.celrep.2019.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaro BL, Tough IR, Engelstoft MS, Matthews RT, Digby GJ, Moller CL, Svendsen B, Gribble F, Reimann F, Holst JJ, et al. (2014). The melanocortin-4 receptor is expressed in enteroendocrine L cells and regulates the release of peptide YY and glucagon-like peptide 1 in vivo. Cell Metab 20, 1018–1029. 10.1016/j.cmet.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, and Südhof TC (2010). Cell biology of Ca2+-triggered exocytosis. Curr. Opin. Cell Biol 22, 496–505. 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parravicini C, Abbracchio MP, Fantucci P, and Ranghino G (2010). Forced unbinding of GPR17 ligands from wild type and R255I mutant receptor models through a computational approach. BMC Struct. Biol 10. 10.1186/1472-6807-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parravicini C, Ranghino G, Abbracchio MP, and Fantucci P (2008). GPR17: Molecular modeling and dynamics studies of the 3-D structure and purinergic ligand binding features in comparison with P2Y receptors. BMC Bioinformatics 9, 263. 10.1186/1471-2105-9-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, and Glockner FO (2007). SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35, 7188–7196. 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese AM, Trincavelli ML, Lecca D, Coppi E, Fumagalli M, Ferrario S, Failli P, Daniele S, Martini C, Pedata F, and Abbracchio MP (2009). Functional characterization of two isoforms of the P2Y-like receptor GPR17: [35S]GTPgammaS binding and electrophysiological studies in 1321N1 cells. Am. J. Physiol. Cell Physiol 297, C1028–C1040. 10.1152/ajp-cell.00658.2008. [DOI] [PubMed] [Google Scholar]
- Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, Yu H, Shen Z, Feng Y, Frazier E, et al. (2002). Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol. Cell Biol 22, 5027–5035. 10.1128/mcb.22.14.5027-5035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley EM (2013). Gut bacteria in health and disease. Gastroenterol. Hepatol. (N Y) 9, 560–569. [PMC free article] [PubMed] [Google Scholar]
- Reilly AM, Zhou S, Panigrahi SK, Yan S, Conley JM, Sheets PL, Wardlaw SL, and Ren H (2019). Gpr17 deficiency in POMC neurons ameliorates the metabolic derangements caused by long-term high-fat diet feeding. Nutr. Diabetes 9, 29. 10.1038/s41387-019-0096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann F, Maziarz M, Flock G, Habib AM, Drucker DJ, and Gribble FM (2005). Characterization and functional role of voltage gated cation conductances in the glucagon-like peptide-1 secreting GLUTag cell line. J. Physiol 563, 161–175. 10.1113/jphysiol.2004.076414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann F, Tolhurst G, and Gribble FM (2012). G-protein-coupled receptors in intestinal chemosensation. Cell Metab 15, 421–431. 10.1016/j.cmet.2011.12.019. [DOI] [PubMed] [Google Scholar]
- Ren H, Cook JR, Kon N, and Accili D (2015). Gpr17 in AgRP Neurons regulates feeding and sensitivity to insulin and leptin. Diabetes 64, 3670–3679. 10.2337/db15-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Orozco IJ, Su Y, Suyama S, Gutierrez-Juarez R, Horvath TL, Wardlaw SL, Plum L, Arancio O, and Accili D (2012). FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell 149, 1314–1326, S0092–8674(12)00577–6 [pii]. 10.1016/j.cell.2012.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaux WG 3rd, and Cheng X (2018). Intracellular cAMP sensor EPAC: Physiology, pathophysiology, and therapeutics development. Physiol. Rev 98, 919–1053. 10.1152/physrev.00025.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GJ, Tolhurst G, Ramzan A, Habib AM, Parker HE, Gribble FM, and Reimann F (2011). Electrical activity-triggered glucagon-like peptide-1 secretion from primary murine L-cells. J. Physiol 589, 1081–1093. 10.1113/jphysiol.2010.198069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Velasco V, and Ikeda SR (2000). Multiple G-protein betagamma combinations produce voltage-dependent inhibition of N-type calcium channels in rat superior cervical ganglion neurons. J. Neurosci. : Official J. Soc. Neurosci 20, 2183–2191. 10.1523/jneurosci.20-06-02183.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satin LS (2000). Localized calcium influx in pancreatic beta-cells: its significance for Ca2+-dependent insulin secretion from the islets of Langerhans. Endocrine 13, 251–262. 10.1385/ENDO:13:3:251. [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, and Clevers H (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265. 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, and Zeltser LM (2013). Functional organization of neuronal and humoral signals regulating feeding behavior. Annu. Rev. Nutr 33, 1–21. 10.1146/annurev-nutr-071812-161125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrocchi LA, Brown TJ, MaClusky N, Brubaker PL, Auerbach AB, Joyner AL, and Drucker DJ (1996). Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat. Med 2, 1254–1258. 10.1038/nm1196-1254. [DOI] [PubMed] [Google Scholar]
- Scrocchi LA, and Drucker DJ (1998). Effects of aging and a high fat diet on body weight and glucose tolerance in glucagon-like peptide-1 receptor −/− mice. Endocrinology 139, 3127–3132. 10.1210/endo.139.7.6092. [DOI] [PubMed] [Google Scholar]
- Seino S, and Shibasaki T (2005). PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol. Rev 85, 1303–1342. 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- Shiota C, Prasadan K, Guo P, Fusco J, Xiao X, and Gittes GK (2017). Gcg (CreERT2) knockin mice as a tool for genetic manipulation in pancreatic alpha cells. Diabetologia 60, 2399–2408. 10.1007/s00125-017-4425-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon K, Hennen S, Merten N, Blattermann S, Gillard M, Kostenis E, and Gomeza J (2016). The orphan G protein-coupled receptor GPR17 negatively regulates oligodendrocyte differentiation via Galphai/o and its down-stream effector molecules. J. Biol. Chem 291, 705–718. 10.1074/jbc.M115.683953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon K, Merten N, Schroder R, Hennen S, Preis P, Schmitt NK, Peters L, Schrage R, Vermeiren C, Gillard M, et al. (2017). The orphan receptor GPR17 is unresponsive to uracil nucleotides and cysteinyl leukotrienes. Mol. Pharmacol 91, 518–532. 10.1124/mol.116.107904. [DOI] [PubMed] [Google Scholar]
- Smrcka AV (2008). G protein βγ subunits: Central mediators of G protein-coupled receptor signaling. Cell Mol. Life Sci. : CMLS 65, 2191–2214. 10.1007/s00018-008-8006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert RE, Feinle-Bisset C, Asarian L, Horowitz M, Beglinger C, and Geary N (2017). Ghrelin, CCK, GLP-1, and PYY(3–36): Secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. Physiol. Rev 97, 411–463. 10.1152/physrev.00031.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QQ, Huguenard JR, and Prince DA (2001). Neuropeptide Y receptors differentially modulate G-protein-activated inwardly rectifying K+ channels and high-voltage-activated Ca2+ channels in rat thalamic neurons. J. Physiol 531, 67–79. 10.1111/j.1469-7793.2001.0067j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Kono T, Anderson-Baucum EK, Yamamoto W, Gilon P, Lebeche D, Day RN, Shull GE, and Evans-Molina C (2016). SERCA2 deficiency impairs pancreatic b-cell function in response to diet-induced obesity. Diabetes 65, 3039–3052. 10.2337/db16-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramunt B, Smati S, Grandgeorge N, Lenfant F, Arnal JF, Montagner A, and Gourdy P (2020). Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 63, 453–461. 10.1007/s00125-019-05040-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters WA, Caporaso JG, Lauber CL, Berg-Lyons D, Fierer N, and Knight R (2011). PrimerProspector: de novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 27, 1159–1161. 10.1093/bioinformatics/btr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, and Kaestner KH (2019). Single-Cell RNA-Seq of the pancreatic islets–a promise not yet fulfilled? Cell Metab 29, 539–544. 10.1016/j.cmet.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Kubal C, and Ren H (2021). Dataset for Confocal Study of G Protein-Coupled Receptors 17 (GPR17), Chromogranin A (CHGA), Glucagon-like Peptide-1 (GLP-1) and Glucose-dependent Insulinotropic Polypeptide (GIP) in Human Intestine (IUPUI University Library) 10.7912/D2/22. [DOI]
- Zietek T, Rath E, Haller D, and Daniel H (2015). Intestinal organoids for assessing nutrient transport, sensing and incretin secretion. Scientific Rep 5, 16831. 10.1038/srep16831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mouse fecal microbiome data have been deposited at NCBI Bioproject and are publicly available as of the date of publication. Accession number is listed in the key resources table. IHC raw images and quantified data have been deposited at IU DataWorks and are publicly available as of the date of publication. DOI is listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.