

Abstract
Background:
Sapoviruses are responsible for sporadic and epidemic acute gastroenteritis worldwide. Sapovirus typing protocols have a success rate as low as 43% and relatively few complete sapovirus genome sequences are available to improve current typing protocols.
Objective/study design:
To increase the number of complete sapovirus genomes to better understand the molecular epidemiology of human sapovirus and to improve the success rate of current sapovirus typing methods, we used deep metagenomics shotgun sequencing to obtain the complete genomes of 68 sapovirus samples from four different countries across the Americas (Guatemala, Nicaragua, Peru and the US).
Results:
VP1 genotyping showed that all sapovirus sequences could be grouped in the four established genogroups (GI (n = 13), GII (n = 30), GIV (n = 23), GV (n = 2)) that infect humans. They include the near-complete genome of a GI.6 virus and a recently reported novel GII.8 virus. Sequences of the complete RNA-dependent RNA polymerase gene could be grouped into three major genetic clusters or polymerase (P) types (GI.P, GII.P and GV.P) with all GIV viruses harboring a GII polymerase. One (GII.P-GII.4) of the new 68 sequences was a recombinant virus with the hotspot between the NS7 and VP1 regions.
Conclusions:
Analyses of this expanded database of near-complete sapovirus sequences showed several mismatches in the genotyping primers, suggesting opportunities to revisit and update current sapovirus typing methods.
Keywords: Viral gastroenteritis, Sapovirus, Genotypes, Next generation sequencing
1. Background
Since the introduction of rotavirus vaccines, norovirus has become the leading cause of medically-attended acute gastroenteritis (AGE) in many countries including the US [1,2]. Of the other viruses associated with AGE, sapoviruses have increasingly been detected in endemic and epidemic AGE [3–6]. Using real-time RT-PCR, studies from low-, middle-, and high-income countries have shown that the prevalence of sapovirus in children < 5 years of age ranges from 3.3 to 17% [7–13]. Although earlier reports described sapovirus infection as one with less severe clinical symptoms than norovirus and rotavirus [14], more recent studies have shown that infections with sapovirus can result in hospitalizations and severe dehydration [9,15]. Sapoviruses were first detected by electron microscopy in children of an infant home with acute gastroenteritis in October 1977 in Japan. The name sapovirus refers to the well-studied prototype strain, Sapporo virus, from another AGE outbreak in the Sapporo prefecture in Japan in 1982. Initially classified as typical caliciviruses or Sapporo-like viruses by electron microscopy, sequencing of the complete genome showed that these non-enveloped 30–35 nm viruses belong to a separate genus Sapovirus in the family Caliciviridae [16]. Sapoviruses have a positive-sense, single stranded RNA genome of 7.3–7.5 kb in length which contains two open reading frames (ORFs). Cleavage of the ORF1 polyprotein by the virus-encoded 3C-like cysteine proteinase yields the mature non-structural (NS) proteins (NS1–NS7) including the RNA-dependent RNA polymerase (RdRp or NS7) as well as the major capsid protein VP1, whereas ORF2 encodes for a minor capsid protein VP2 [17]. The antigenic epitopes are in the hypervariable region of VP1, which is the most diverse region of the genome, likely in the predicted P2 domain [18]. Based on complete VP1 nucleotide sequences, sapoviruses can be divided into up to 19 genogroups (GI–GXIX) [19] of which viruses from 4 (GI, GII, GIV and GV) infect humans while viruses in the other genogroups have been detected in swine (GIII and GV-GXI), sea lions (GV), mink (GXII), dogs (GXIII), bats (GXIV, GXVI-GXIX) and rats (GXV). The human sapoviruses can be further classified into 17 genotypes [17], and an additional proposed GII.8 genotype [20]. GI and GII sapoviruses are the most frequently detected viruses in recent years. Although GIV viruses are relatively rare, in some studies they accounted for up to one third of the cases [21]. They have been detected worldwide in stool samples from young children as early as 1992 in Pakistan [22]. GV viruses were first reported in stool samples from children in Argentina in 1995 [23] but in most studies viruses of this genogroup have been rarely reported [24]. GI.6 and GII.3 viruses became predominant in Japan, but these genotypes gradually disappeared in subsequent years [25]. Several recombinant sapovirus strains have been documented [26] and, like for noroviruses, such strains may have an altered virulence possibly leading to an increased disease burden [27]. Since 2006, most research groups have used the same protocols for the detection [28] and genotyping [29] of human sapoviruses. The real-time RT-PCR assay described by Oka et al. [28] is able to detect viruses from all 4 genogroups. However, reported genotyping success rates for sapovirus range from 43% to 100% [30,31] indicating the current hemi-nested PCR assays do not detect all circulating sapovirus strains.
2. Objective
With protocols for sequencing of the complete genome of enteric viruses directly from stool samples becoming more widely available [32], the aim of our study was to increase the number of complete sapovirus genomes especially for those genotypes for which currently no or only few complete genomic sequences are available in public databases. A larger sequence database will help to develop more broadly-reactive PCR assays with overall improved genotyping success to better understand the molecular epidemiology of human sapoviruses.
3. Study design
3.1. Fecal specimens
Sapovirus positive stool specimens used in this study were obtained from outbreaks or sporadic cases of AGE and collected between 2010 and 2016. CDC’s Internal Program for Research Determination deemed that this study is categorized as public health non-research and that human subject regulations did not apply. Specimens representing rare or uncommon genotypes, or strains for which no complete genomes were available in public databases were selected for whole genome sequencing. In addition, specimens that could not be amplified using the hemi-nested PCR assay were included [29]. All specimens, except four samples collected during an outbreak in a long-term care facility in 2016 in the United States, were collected from children under 5 years of age with sporadic AGE. Sapovirus positive specimens in the US were obtained from two sites (Nashville, Oakland) participating in New Vaccine and Surveillance Network [33] and from an all-age active surveillance study for medically–attended acute gastroenteritis in Oregon. The Peruvian specimens were obtained from children hospitalized at the Instituto Nacional de Salud del Niño in Lima, Peru. The specimens from Nicaragua were from a population-based study in 2010 and had been tested for Shigella, Salmonella, E.coli, Campylobacter, Cryptosporidium, rotavirus, adenovirus, and norovirus [7]. The specimens from Guatemala had been collected as part of an AGE health facility-based surveillance in two Guatemalan departments (Santa Rosa and Quetzaltenango) from April 2010 to February 2016. Table 1 summarizes the number of complete sapovirus genomes (i.e., sequences containing the 5′ UTR, the complete ORF1 and ORF2 and the 3′UTR regions) in public databases (as of April 1st, 2018) and the complete genomes obtained in this study.
Table 1.
Distribution of near-complete sapovirus genomes sequenced in this study compared to complete genomes available in NCBI.
| Genotype | This study | NCBIa |
|---|---|---|
| n | n | |
| GI.1 | 9 | 10 |
| GI.2 | 3 | 3 |
| GI.3 | 0 | 1 |
| GI.4 | 0 | 1 |
| GI.5 | 0 | 1 |
| GI.6 | 1 | 1 |
| GI.7 | 0 | 1 |
| GII.1 | 10 | 2 |
| GII.2 | 4 | 1 |
| GII.3 | 6 | 3 |
| GII.4 | 1 | 1 |
| GII.5 | 5 | 1 |
| GII.6 | 0 | 1 |
| GII.7 | 0 | 1 |
| GII.8b | 4 | 4 |
| GIV.1 | 23 | 5 |
| GV.1 | 2 | 2 |
| GV.2 | 0 | 2 |
| total | 68 | 41 |
NCBI = National Center of Biotechnology Information, complete genomes available on April 1st 2018.
Proposed new genotype.
3.2. RNA extraction, sapovirus detection by RT-qPCR and VP1 genotyping
Clarified 10% stool suspensions were prepared in phosphate-buffered saline and centrifuged at 10,000 × g for 10 min. Viral nucleic acid was extracted using the MagMAX total nucleic acid isolation Kit (Thermo Fisher Scientific, Carlsbad, CA). Sapovirus RNA was detected by TaqMan-based quantitative RT-PCR as described previously [28]. VP1 subtyping was performed by hemi nested RT-PCR followed by Sanger sequencing of the 2nd round PCR positive products [29].
3.3. Full genome sequencing, de novo assembly, and U50 N50 metrics calculation
Viral metagenomics were performed according to a previously published protocol [34–36]. Briefly, virus particles were filtered using 0.45 μm centrifugal filters (Millipore, Billerica, MA), followed by a nuclease treatment consisting of a cocktail of RNase A and TURBO™ DNase (Thermo Fisher Scientific, Carlsbad, CA), Baseline-ZERO™ (Epicentre, Madison, WI), and Benzonase (Sigma-Aldrich, St. Louis, MO). Viral nucleic acids were extracted using QIAamp® Viral RNA Mini Kit (QIAGEN, Hilden, Germany). Complementary DNA (cDNA) synthesis and random amplification were performed by sequence-independent single primer amplification (SISPA) [35–37]. PCR products were purified using Agencourt® AMPure® XP beads (Beckman Coulter, Brea, CA). An approximate 200-bp fragment library was constructed using the Nextera® XT DNA Library Preparation Kit (Illumina, San Diego, CA). The Nextera® product was purified using Agencourt® AMPure® XP beads (Beckman Coulter, Brea, CA) and quality of the purified library was assessed on an Agilent HS D1000 ScreenTape System (Agilent Technologies, Santa Clara, CA). Library concentration for pooling was determined with a KAPA Library Quantification Kit for Illumina® platforms (Roche, Wilmington, MA). Samples were sequenced on an Illumina MiSeq using MiSeq Reagent Kits v2 (250-cycle paired-end). Full-length sapovirus genomic sequences were generated using a custom bioinformatics pipeline as described previously [38,39]. Briefly, sequences were trimmed/filtered to remove adapters and low quality bases, sequences shorter than 50 nt, and human (host) sequences identified through mapping of reads to the human reference genome hg19 using bowtie2 [40]. Sapovirus were first assembled using SPAdes [41], a de novo assembler, followed by reference mapping and gene annotation using Geneious version 9.1.4 [Biomatters] [42] to verify assembled sequences. For all de novo assembled sequences the new metric U50 was calculated which is circumventing the limitations of N50 by identifying unique, target-specific contigs using a reference sequence as a baseline [43].
3.4. Phylogenetic and genome similarity analyses
Sequences alignment was performed with MUSCLE [44] and phylogenetic trees were constructed using the maximum-likelihood method with 100 bootstraps replications to assess phylogenetic robustness using MEGA version 6 [45]. Using the Sequence Demarcation tool [46] we aligned every unique pair of sequences and calculated the sequence pairwise identity among sequences from this study and published references. Possible recombination events were analyzed using SimPlot [47].
4. Results
Of the 108 sapovirus positive samples that were tested by deep metagenomics shotgun sequencing, we successfully obtained near-complete genome sequences for 68 (63%) strains of which 44 had UG50% values of over 99% indicating that they were successfully obtained during de novo assembly by the bioinformatics pipeline [43] (Table 2). Of the remaining 40 samples, 26 sequences were incomplete and/or had gaps and/or had too few reads by NGS to be confidently assembled and assigned. The other 14 samples had a complete VP1 sequence which allowed genotyping, but since no complete genomes was obtained, they were not further analyzed in this study. All 68 (near-)complete genomes belonged to the four established sapovirus genogroups (GI, GII, GIV and GV) and included 13 GI, 30 GII, 23 GIV and 2 GV sequences were typed based on VP1 sequences. They included 12 sequences from rare sapovirus genotypes, GI.6 [n = 1], GII.3 [n = 6], GII.4 [n = 1] and GII.5 [n = 4]. In addition, based on a pairwise distance cut-off value of ≤0.169 to distinguish different genotypes and ≤0.488 to distinguish different genogroups [17], four sequences could be classified into a recently reported new genotype, GII.8 (Figs. 1 and 2B) [20]. These sequences shared only 75% nucleotide identity with its closest neighbor, GII.7 viruses.
Table 2.
Near-complete genomes of 68 sapovirus strains generated in this study.
| Accession Number | Sample name | Ct | Genotype | Sequence length | U50a | N50 | UG50 | NG50 | UG50% |
|---|---|---|---|---|---|---|---|---|---|
| MG012441 | GI.2_Oakland_6371 | 25 | GI.2 | 7465 | 4100 | 235 | 4100 | 6023 | 54.92% |
| MG012442 | GI.2_Oakland_3023 | 19 | GI.2 | 7457 | 7457 | 1473 | 7457 | 7496 | 99.99% |
| MG012399 | GI.1_Portland_3639 | 24 | GI.1 | 7430 | 7420 | 505 | 7420 | 9503 | 99.87% |
| MG012425 | GIV.1_Portland_3587 | 23 | GIV | 7359 | 5520 | 836 | 5520 | 5520 | 75.01% |
| MG012428 | GIV.1_Portland_3602 | 25 | GIV | 7361 | 1938 | 826 | 1938 | 6497 | 26.13% |
| MG012424 | GIV.1_Portland_3629 | 21 | GIV | 7420 | 7399 | 548 | 7399 | 7399 | 99.69% |
| MG012459 | GIV.1_Portland_3631 | 25 | GIV | 7414 | 7390 | 779 | 7390 | 7392 | 99.68% |
| MG012423 | GIV.1_Portland_3632 | 28 | GIV | 7358 | 4690 | 1059 | 4690 | 4778 | 63.68% |
| MG012427 | GIV.1_Portland_3633 | 25 | GIV | 7383 | 7339 | 1024 | 7339 | 7352 | 99.15% |
| MG012426 | GIV.1_Portland_3636 | 25 | GIV | 7329 | 7311 | 865 | 7311 | 7311 | 99.75% |
| MG012457 | GIV.1_Portland_3641 | 26 | GIV | 7420 | 7420 | 522 | 7420 | 7420 | 100.00% |
| MG012458 | GIV.1_Portland_3643 | 23 | GIV | 7412 | 7412 | 829 | 7412 | 7420 | 100.00% |
| MG012422 | GIV.1_Portland_3644 | 24 | GIV | 7387 | 7350 | 1077 | 7350 | 7350 | 99.50% |
| MG012456 | GIV.1_Portland_3649 | 23 | GIV | 7413 | 7409 | 747 | 7409 | 7409 | 99.82% |
| MG012455 | GIV.1_Portland_3651 | 21 | GIV | 7426 | 7404 | 1073 | 7404 | 7430 | 99.66% |
| MG012397 | GI.1_Nashville_9427 | 20 | GI.1 | 7356 | 7356 | 491 | 7356 | 7394 | 100.00% |
| MG012400 | GI.1_Nashville_9491 | 22 | GI.1 | 7346 | 6398 | 526 | 6398 | 8745 | 87.10% |
| MG012435 | GI.1_Nashville_9691-1 | 20 | GI.1 | 7425 | 7421 | 800 | 7421 | 7520 | 99.95% |
| MG012437 | GI.1_Nashville_9432 | 19 | GI.1 | 7388 | 7378 | 728 | 7378 | 7411 | 99.86% |
| MG012438 | GI.1_Nashville_9478 | 19 | GI.1 | 7428 | 655 | 540 | 0 | 6151 | 0.00% |
| MG012440 | GI.2_Nashville_9411 | 22 | GI.2 | 7449 | 7449 | 769 | 7449 | 10174 | 99.99% |
| MG012443 | GI.6_Nashville_9367 | 17 | GI.6 | 7430 | 7427 | 575 | 7427 | 10982 | 99.96% |
| MG012444 | GII.1_Nashville_9343 | 15 | GII.1 | 7483 | 2115 | 192 | 2115 | 2253 | 28.26% |
| MG012445 | GII.1_Nashville_9346 | 15 | GII.1 | 7394 | 379 | 269 | 379 | 1856 | 5.13% |
| MG012402 | GII.1_Nashville_9360 | 14 | GII.1 | 7469 | 7469 | 2852 | 7469 | 7592 | 100.00% |
| MG012403 | GII.1_Nashville_9422 | 20 | GII.1 | 7464 | 7464 | 517 | 7464 | 7492 | 100.00% |
| MG012404 | GII.1_Nashville_9353 | 18 | GII.1 | 7363 | 7363 | 385 | 7363 | 7410 | 100.00% |
| MG012405 | GII.1_Nashville_9511 | 21 | GII.1 | 7417 | 7416 | 619 | 7416 | 7457 | 99.99% |
| MG012406 | GII.1_Nashville_9510 | 26 | GII.1 | 7463 | 885 | 403 | 885 | 5949 | 11.86% |
| MG012407 | GII.1_Nashville_9525 | 18 | GII.1 | 7448 | 7446 | 1086 | 7446 | 16746 | 99.97% |
| MG012408 | GII.1_Nashville_9691-2 | 20 | GII.1 | 7148 | 4031 | 800 | 4031 | 7520 | 56.39% |
| MG012409 | GII.1_Nashville_9385 | 14 | GII.1 | 7388 | 7386 | 7553 | 7386 | 7553 | 99.97% |
| MG012410 | GII.2_Nashville_9521 | 22 | GII.2 | 7418 | 4360 | 560 | 4360 | 5162 | 58.78% |
| MG012411 | GII.2_Nashville_9331 | 20 | GII.2 | 7375 | 7374 | 505 | 7374 | 7494 | 99.99% |
| MG012412 | GII.2_Nashville_9393 | 18 | GII.2 | 7429 | 7427 | 655 | 7427 | 7512 | 99.97% |
| MG012413 | GII.2_Nashville_9795 | 22 | GII.2 | 7372 | 7370 | 1386 | 7370 | 7887 | 99.97% |
| MG012415 | GII.3_Nashville_9517 | 27 | GII.3 | 7452 | 485 | 451 | 0 | 2343 | 0.00% |
| MG012418 | GII.3_Nashville_9513 | 16 | GII.3 | 7464 | 7464 | 418 | 7464 | 7544 | 100.00% |
| MG012419 | GII.3_Nashville_9354 | 20 | GII.3 | 7439 | 7424 | 523 | 7424 | 7434 | 99.80% |
| MG012447 | GII.5_Nashville_9349 | 23 | GII.5 | 7383 | 6187 | 533 | 6187 | 6238 | 83.80% |
| MG012449 | GII.5_Nashville_9387 | 20 | GII.5 | 7432 | 7432 | 1373 | 7432 | 7451 | 100.00% |
| MG012430 | GIV.1_Nashville_9327 | 22 | GIV | 7348 | 7348 | 660 | 7348 | 7374 | 100.00% |
| MG012433 | GV.1_Nashville_9424 | 26 | GV.1 | 7455 | 4929 | 573 | 4929 | 5399 | 66.14% |
| MG012434 | GV.1_Nashville_9492 | 25 | GV.1 | 7499 | 7498 | 1045 | 7498 | 7503 | 99.99% |
| MG012421 | GIV.1_Nashville_9489 | 20 | GIV | 7415 | 7415 | 1275 | 7415 | 7568 | 100.00% |
| MG012453 | GII.8_Monterey_6283 | ND | GII.8 | 7404 | 7403 | 714 | 7403 | 7426 | 99.99% |
| MG012452 | GII.8_Monterey_6284 | ND | GII.8 | 7447 | 7446 | 842 | 7446 | 7829 | 99.99% |
| MG012398 | GI.1_Lima_1845 | NA | GI.1 | 7311 | 7311 | 474 | 7311 | 7311 | 100.00% |
| MG012436 | GI.1_Lima_1870 | NA | GI.1 | 7424 | 7421 | 681 | 7421 | 7456 | 99.96% |
| MG012439 | GI.1_Lima_1863 | NA | GI.1 | 7429 | 774 | 254 | 774 | 5139 | 10.42% |
| MG012417 | GII.3_Lima_1856 | NA | GII.3 | 7454 | 7454 | 508 | 7454 | 7454 | 100.00% |
| MG012446 | GII.4_Lima_1873 | NA | GII.4 | 7398 | 253 | 254 | 252 | 3209 | 3.41% |
| MG012451 | GII.5_Lima_1868 | NA | GII.5 | 7434 | 355 | 224 | 355 | 1958 | 4.78% |
| MG674584 | GII.8_Lima_1859 | NA | GII.8 | 7450 | 321 | 221 | 321 | 1150 | 4.31% |
| MG674583 | GII.8_Lima_1867 | NA | GII.8 | 7300 | 4593 | 473 | 4593 | 4610 | 62.91% |
| MG012420 | GIV.1_Lima_1858 | NA | GIV | 7418 | 7298 | 545 | 7298 | 7298 | 98.38% |
| MG012429 | GIV.1_Lima_1861 | NA | GIV | 7389 | 7389 | 514 | 7389 | 7414 | 100.00% |
| MG012431 | GIV.1_Lima_1869 | NA | GIV | 7402 | 7398 | 462 | 7398 | 7402 | 99.99% |
| MG012432 | GIV.1_Lima_1871 | NA | GIV | 7387 | 924 | 462 | 276 | 6039 | 3.74% |
| MG012461 | GIV.1_Lima_1862 | NA | GIV | 7437 | 7437 | 750 | 7437 | 7439 | 100.00% |
| MG012462 | GIV.1_Lima_1864 | NA | GIV | 7415 | 254 | 340 | 245 | 1280 | 3.30% |
| MG012463 | GIV.1_Lima_1865 | NA | GIV | 7437 | 7434 | 644 | 7434 | 7524 | 99.96% |
| MG012414 | GII.3_Leon_6701 | 19 | GII.3 | 7453 | 3135 | 246 | 2165 | 7415 | 29.04% |
| MG012416 | GII.3_Leon_6708 | 20 | GII.3 | 7454 | 7452 | 783 | 7452 | 8083 | 99.97% |
| MG012454 | GIV.1_Leon_1751 | 22 | GIV | 7401 | 7399 | 600 | 7399 | 7469 | 99.97% |
| MG012448 | GII.5_Santa_Rosa_3812 | 21 | GII.5 | 7447 | 7447 | 850 | 7447 | 7574 | 100.00% |
| MG012450 | GII.5_Santa_Rosa_3693 | 21 | GII.5 | 7448 | 499 | 250 | 434 | 4756 | 5.83% |
| MG012460 | GIV.1_Quetzaltenago_3711 | 25 | GIV | 7436 | 7425 | 406 | 7425 | 7577 | 99.85% |
ND: not detected; NA: not available.
The performance of a de novo assembly method is measured by N50. U50 is a metric that identifies unique, target-specific non-overlapping contigs by using a reference genome as baseline, aiming at circumventing limitations that are inherent to the N50 metric.
Fig. 1.
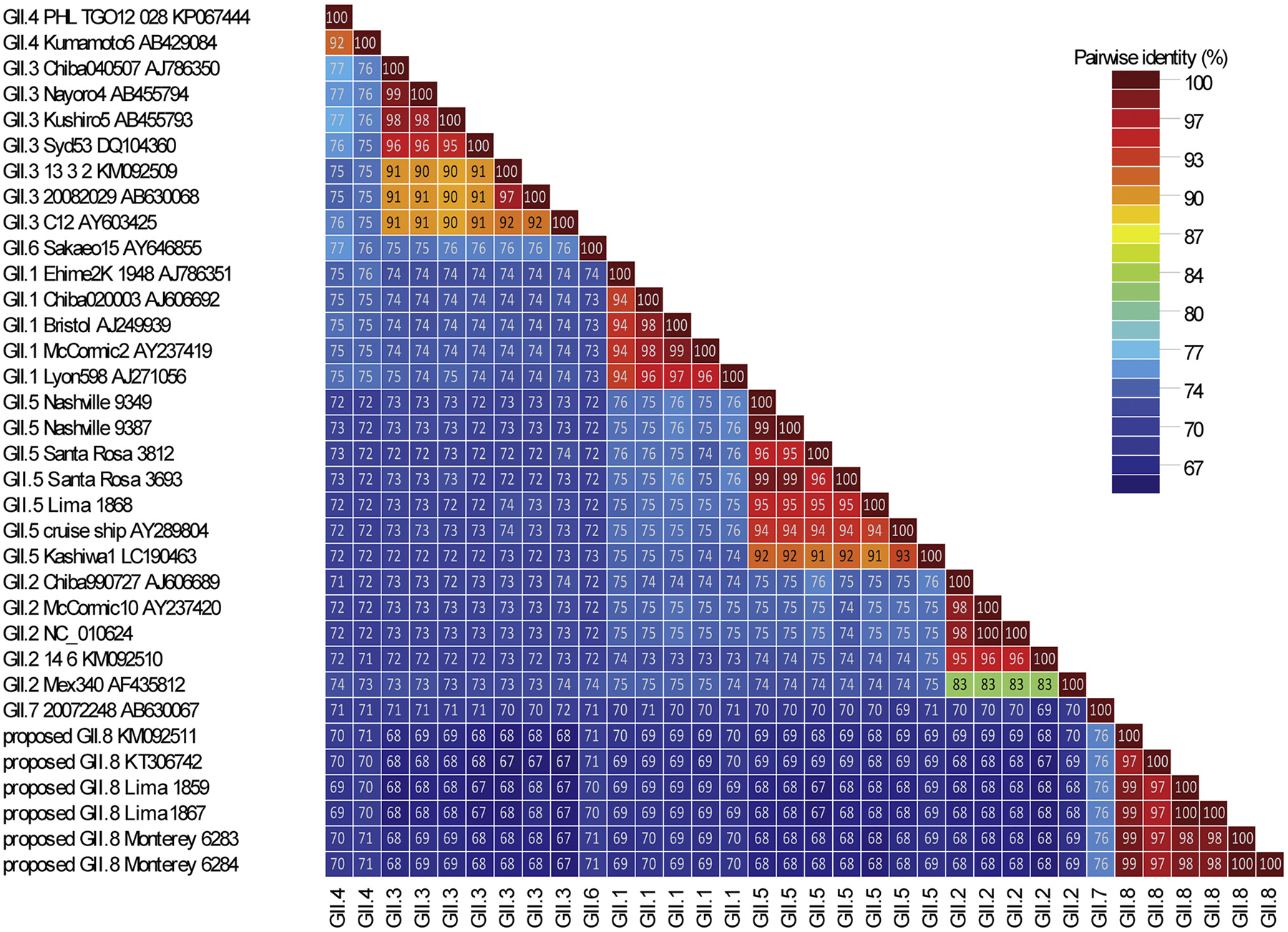
Pairwise sequence comparison of novel sapovirus strains (GII.8 Monterey6283, GII.8 Monterey6284, GII.8 Lima1859, and GII.8 Lima1867) with sapovirus GII.1-GII.7reference sequences. In the color-coded pairwise identity matrix each cell includes the percentage identity among two sequences (horizontally to the left and vertically at the bottom). The colored key indicates the correspondence between pairwise identities and the colors displayed in the matrix.
Fig. 2.
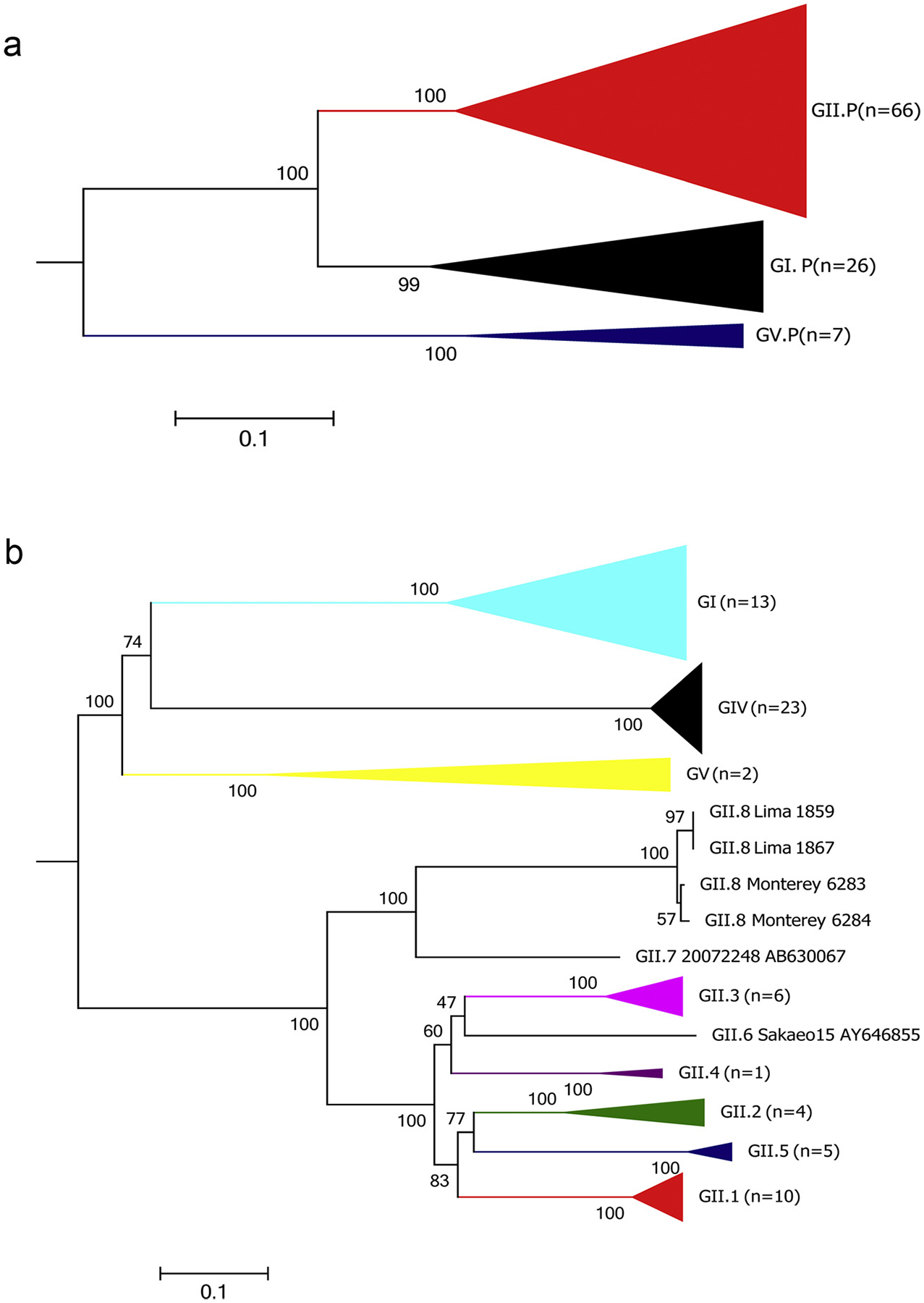
Phylogenetic analysis of 99 sapovirus RdRp (A) and 70 VP1 (B) sequences using Maximum Likelihood method. The tree was inferred by using the Maximum Likelihood method based on the Tamura-Nei model. The trees with the highest log likelihood are shown, on the basis of nucleotide sequence of the complete RdRp (A) and VP1 (B) genes. The trees are drawn to scale with branch lengths measured in the number of substitutions per site. Bootstrap values (100 replicates) are shown next to each branch. Branches have been compressed for clarity and only the GII branch is partially expanded to show how the four samples of GII.8 branch out from the closest GII.7 sapovirus.
Phylogenetic analyses of the complete RdRp gene (approximately 1550 nucleotides) [17] of 99 sapovirus strains, including 68 from this study, showed that they can be grouped into 3 main clusters or polymerase types (P-types: GI.P, GII.P and GV.P) which are distinguished by having less than 43% nucleotide identity (Fig. 2A). GI.P includes all GI viruses based on VP1, GII.P includes all GII and GIV viruses including the new GII.8 and GV.P includes all GV viruses.
The non-structural (NS) protein sequences in ORF1, contained previously recognized conserved motifs in the first five amino acids of NS1 (MASKP) and around the RdRp-VP1 junction region [NS7-VP1] cleavage site (FEME/G, the slash indicates the putative cleavage site by viral protease NS6) in all strains [48]. The rest of non-structural proteins biological functions are not yet experimentally determined, but NS3 and NS5 have typical calicivirus NTPase and VPg motifs, respectively. Conserved amino acids motifs including (G(A/P)PGIGKT) in NS3, (KGKTK and DDEYDE) in NS5, (GDCG) in NS6, (WKGL, KDEL, DYSKDST, GLPSG and YGDD) in NS7 and (PPG and GWS) in VP1 were, with some minor variations, present in all 68 sapovirus sequences obtained in this study. These minor variations were found in the NS3 and NS5 genes. In NS3, the GAPGIGKT motif was present only in GI.1 sequences whereas all other strains, irrespective of genogroup, had the GPPGIGKT motif. In NS5, all sequences had the common KGKTK motif while GI.2 viruses had KGKSK and the novel GII.8 strains had a KGKNK motif.
We identified an RdRp-VP1 recombinant (GII.P-GII.4_Lima_1873) (Fig. 3) with the recombination break point at the RdRp-VP1 junction. No other recombinant viruses were identified throughout ORF1.
Fig. 3.
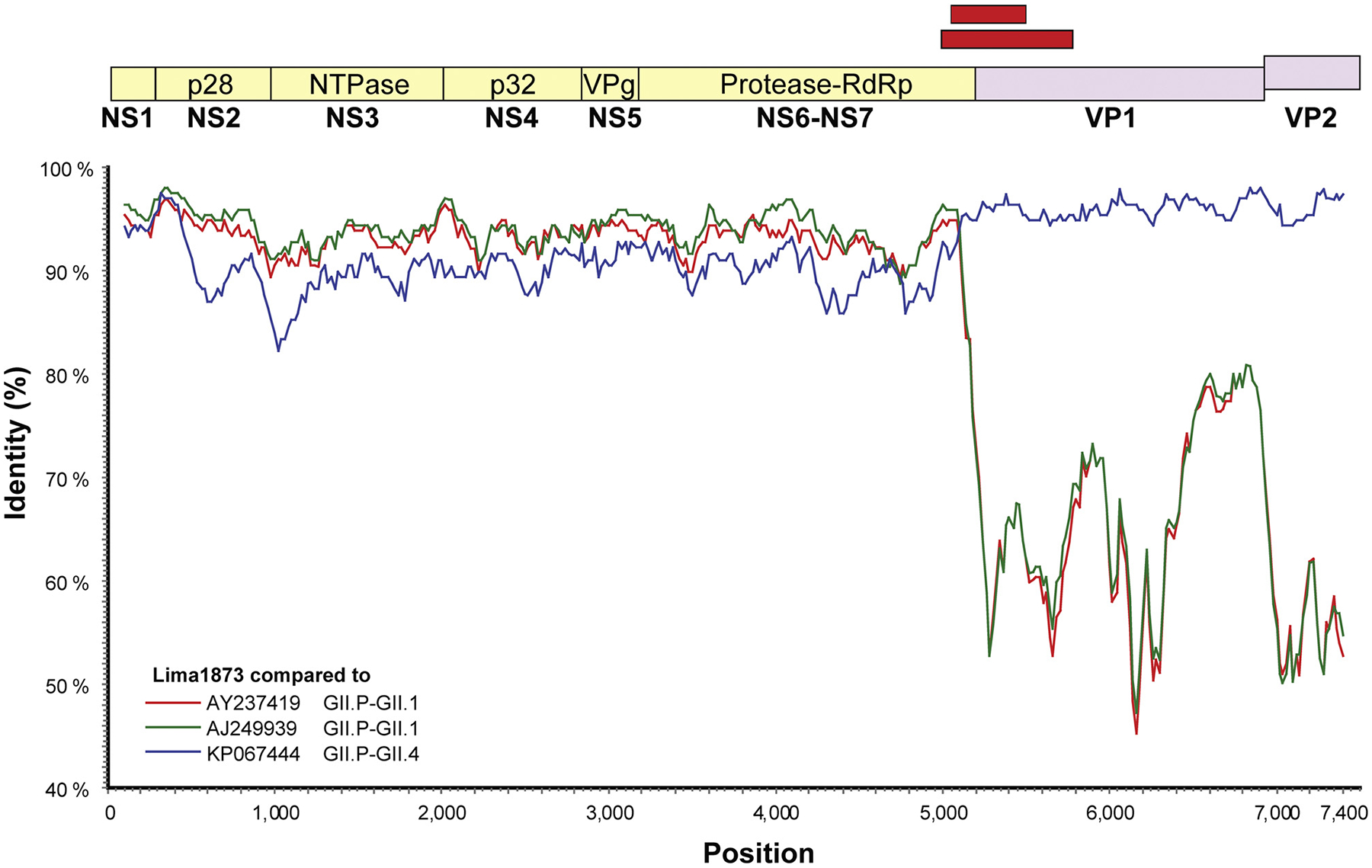
Evidence of recombination for sapovirus GII.4 Lima1873 compared to previous sequenced GII.1 and GII.4 genomes. Sapovirus genomes were analyzed by SimPlot and similarity scores using a 200 nt sliding window are plotted. Three reference strains (A, B and C) were analyzed against the query sequence GII.4_Lima1873. Similarity scores of all three references (two GII.1 and one GII.4) were almost > 85% in the entire NS region, with a notable drop in similarity scores over the capsid region and ORF2 (VP2). The query sequence most closely related to previously described sapovirus recombinant strain with a GII.P polymerase and GII.4 capsid (KP067444). Red boxes indicate the approximate location of the hemi nested genotyping primers [29].
We identified several mismatches between the widely used genotyping primers published a decade ago [29] and 134 VP1 sequences including 68 sequences from this study. Several GII viruses, including the new GII.8, and GIV viruses had multiple mismatches in the primers used in the hemi-nested typing PCR (Fig. 4).
Fig. 4.
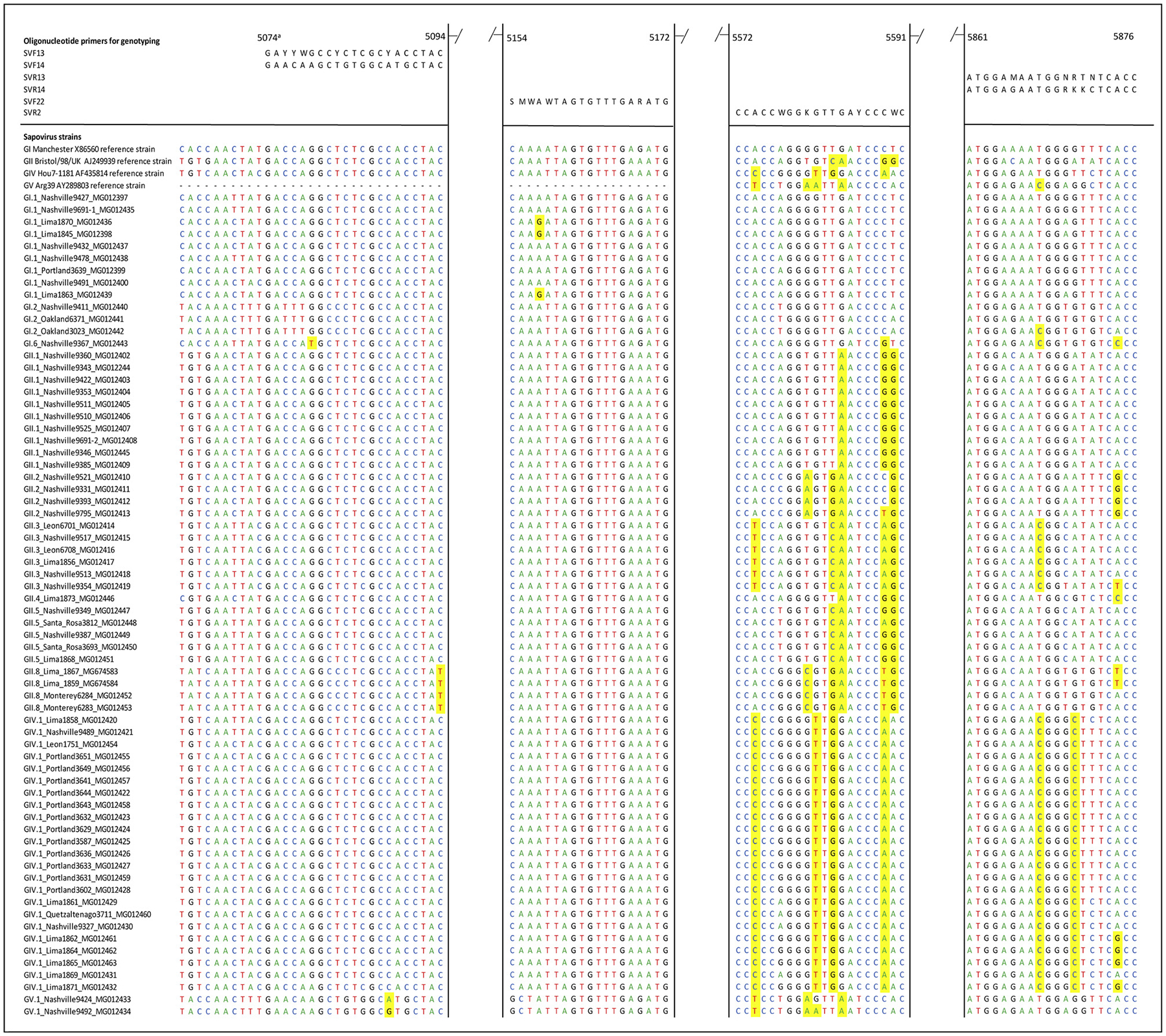
Alignment of 68 near complete genome sequences obtained in this study and the primers used for hemi-nested PCR [29]. Mismatches between individual strains and the primer binding regions are highlighted in yellow.
aNucleotide positions based on Manchester strain (Accesion number X86560).
4.1. Nucleotide sequence accession numbers
The sapovirus nucleotide sequences determined in this study have been deposited in GenBank under the accession numbers MG012397-MG012400; MG012402-MG012463 and MG674583- MG674584.
5. Discussion
To better understand the magnitude of genetic variability of human sapoviruses and to increase the available sequences to improve the success rate of current genotyping protocols, we sequenced (near-) complete genomes of 68 sapovirus strains from four different countries including several rare genotypes. We used the complete VP1 nucleotide sequences to classify strains into established sapovirus genogroups and genotypes [17,23]. Samples from an AGE outbreak in a long-term care facility in the US and from two hospitalized children in Peru could be typed as GII.8, a new genotype which was recently reported [20,49]. Due to the limited number of samples from adults (2 samples from an outbreak in a long term care facility), we cannot conclude if adults are infected by different genotypes then young children which is an important research area to be addressed in future studies. A major contribution of the current study is the addition of 68 near-complete genome sequences to the current 41 complete sapovirus genomes in GenBank. Two types of recombination events have been described including inter-genogroup and intra-genogroup [17]. In the current study one intra-genogroup recombinant (GII.1/GII.4) event was observed among the 68 complete sapovirus genomes analyzed, similar to previously described recombinant viruses in Vietnam [50] and in the Philippines [26]. Other intra-genogroup recombinations have also been also described in sapovirus GI (GI.1/GI.8) in Japan [51] consistently with the breakpoint located between the RdRp and the VP1 genes. Among caliciviruses, RdRp is the most conserved region of the genome and coinfection with multiple sapovirus strains may lead to the emergence of recombinant strains [52,53]. Nevertheless, the frequency of recombination observed in sapovirus is lower than in viruses in the closely related genus Norovirus, in which this phenomenon occurs frequently [27]. This can be partially explained because of the relatively few number of RdRp sequences available for sapovirus compared to norovirus. Thus, the large number of sequences provided in this study allow for a better assessment of the frequency of recombination among sapoviruses.
The heminested PCR for typing of sapoviruses was also designed in 2006 based on the limited number of sequences available at that time. Genotypes such as GII.3, and GII.8 have up to 5 mismatches with the SV-R2 reverse primer but design of more broadly-reactive typing primers is compromised by the large genetic variability of GII strains.
In summary, we obtained full and near-complete sapovirus genome sequences of 68 human stool samples from four different countries in the Americas using deep metagenomics shotgun sequencing. Further optimizations of the metagenomics shotgun protocol may be needed to consistently obtain complete genomes from samples with a lower viral load either by using virus-specific RNA baits to enrich sapovirus nucleic acids prior to sequencing or sequence-specific amplification of complete sapovirus genomes. We showed that the current oligonucleotide primers used for genotyping of human sapoviruses [29] have mismatches that are less likely to be successful for amplifying GII sapoviruses, but optimization of amplification conditions of the current protocol may increase the success rate (data not shown). Continued surveillance of sapovirus from different geographic regions using improved detection and typing protocols will help to better determine the disease burden and genotype diversity of these, until recently, underappreciated viral gastrointestinal infections.
Acknowledgments
We thank Nikkail Collins for technical assistance. This work was made possible through support from the Advanced Molecular Detection (AMD) program at the CDC. This research was also supported in part by an appointment to the Research Participation Program at the CDC administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy and the CDC.
Footnotes
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: The findings and conclusions in this article are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. Names of specific vendors, manufacturers, or products are included for public health and informational purposes; inclusion does not imply endorsement of the vendors, manufacturers, or products by the Centers for Disease Control and Prevention or the US Department of Health and Human Services.
Conflict of interest
None.
References
- [1].Payne DC, Boom JA, Staat MA, Edwards KM, Szilagyi PG, Klein EJ, et al. , Effectiveness of pentavalent and monovalent rotavirus vaccines in concurrent use among US children < 5 years of age, 2009–2011, Clin. Infect. Dis 57 (2013) 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hemming M, Rasanen S, Huhti L, Paloniemi M, Salminen M, Vesikari T, Major reduction of rotavirus, but not norovirus, gastroenteritis in children seen in hospital after the introduction of RotaTeq vaccine into the National Immunization Programme in Finland, Eur. J. Pediatr 172 (2013) 739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Svraka S, Vennema H, van der Veer B, Hedlund KO, Thorhagen M, Siebenga J, et al. , Epidemiology and genotype analysis of emerging sapovirus-associated infections across Europe, J. Clin. Microbiol 48 (2010) 2191–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lee LE, Cebelinski EA, Fuller C, Keene WE, Smith K, Vinje J, et al. , Sapovirus outbreaks in long-term care facilities, Oregon and Minnesota, USA, 2002–2009, Emerg. Infect. Dis 18 (2012) 873–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kobayashi S, Fujiwara N, Yasui Y, Yamashita T, Hiramatsu R, Minagawa H, A foodborne outbreak of sapovirus linked to catered box lunches in Japan, Arch. Virol 157 (2012) 1995–1997. [DOI] [PubMed] [Google Scholar]
- [6].Hassan-Rios E, Torres P, Munoz E, Matos C, Hall AJ, Gregoricus N, et al. , Sapovirus gastroenteritis in preschool center, Puerto Rico, 2011, Emerg. Infect. Dis 19 (2013) 174–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Becker-Dreps S, Bucardo F, Vilchez S, Zambrana LE, Liu L, Weber DJ, et al. , Etiology of childhood diarrhea after rotavirus vaccine introduction: a prospective, population-based study in Nicaragua, Pediatr. Infect. Dis. J 33 (2014) 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bucardo F, Carlsson B, Nordgren J, Larson G, Blandon P, Vilchez S, et al. , Susceptibility of children to sapovirus infections, Nicaragua, 2005–2006, Emerg. Infect. Dis 18 (2012) 1875–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bucardo F, Reyes Y, Svensson L, Nordgren J, Predominance of norovirus and sapovirus in Nicaragua after implementation of universal rotavirus vaccination, PLoS One 9 (2014) e98201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liu X, Jahuira H, Gilman RH, Alva A, Cabrera L, Okamoto M, et al. , Etiological role and repeated infections of sapovirus among children aged less than 2 years in a cohort study in a Peri-urban community of Peru, J. Clin. Microbiol 54 (2016) 1598–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chhabra P, Payne DC, Szilagyi PG, Edwards KM, Staat MA, Shirley SH, et al. , Etiology of viral gastroenteritis in children < 5 years of age in the United States, 2008–2009, J. Infect. Dis 208 (2013) 790–800. [DOI] [PubMed] [Google Scholar]
- [12].Grant LR, O’Brien KL, Weatherholtz RC, Reid R, Goklish N, Santosham M, et al. , Norovirus and sapovirus epidemiology and strain characteristics among navajo and apache infants, PLoS One (2017) 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lyman WH, Walsh JF, Kotch JB, Weber DJ, Gunn E, Vinjé J, Prospective study of etiologic agents of acute gastroenteritis outbreaks in child care centers, J. Pediatr 154 (2009) 253–257. [DOI] [PubMed] [Google Scholar]
- [14].Sakai Y, Nakata S, Honma S, Tatsumi M, Numata-Kinoshita K, Chiba S, Clinical severity of Norwalk virus and Sapporo virus gastroenteritis in children in Hokkaido, Japan, Pediatr. Infect. Dis. J 20 (2001) 849–853. [DOI] [PubMed] [Google Scholar]
- [15].Matussek A, Dienus O, Djeneba O, Simpore J, Nitiema L, Nordgren J, Molecular characterization and genetic susceptibility of sapovirus in children with diarrhea in Burkina Faso, Infect. Genet. Evol 32 (2015) 396–400. [DOI] [PubMed] [Google Scholar]
- [16].Nakata S, Honma S, Numata KK, Kogawa K, Ukae S, Morita Y, et al. , Members of the family caliciviridae (Norwalk virus and Sapporo virus) are the most prevalent cause of gastroenteritis outbreaks among infants in Japan, J. Infect. Dis 181 (2000) 2029–2032. [DOI] [PubMed] [Google Scholar]
- [17].Oka T, Wang Q, Katayama K, Saif LJ, Comprehensive review of human sapoviruses, Clin. Microbiol. Rev 28 (2015) 32–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kitamoto N, Oka T, Katayama K, Li TC, Takeda N, Kato Y, et al. , Novel monoclonal antibodies broadly reactive to human recombinant sapovirus-like particles, Microbiol. Immunol 56 (2012) 760–770. [DOI] [PubMed] [Google Scholar]
- [19].Yinda CK, Conceicao-Neto N, Zeller M, Heylen E, Maes P, Ghogomu SM, et al. , Novel highly divergent sapoviruses detected by metagenomics analysis in straw-colored fruit bats in Cameroon, Emerg. Microbes Infect 6 (2017) e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu X, Jahuira H, Gilman RH, Alva A, Cabrera L, Okamoto M, et al. , Etiological role and repeated infections of sapovirus among children aged less than two years in a cohort study in a peri-urban community of Peru, J. Clin. Microbiol 54 (6) (2016) 1598–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Harada S, Okada M, Yahiro S, Nishimura K, Matsuo S, Miyasaka J, et al. , Surveillance of pathogens in outpatients with gastroenteritis and characterization of sapovirus strains between 2002 and 2007 in Kumamoto Prefecture, Japan, J. Med. Virol 81 (2009) 1117–1127. [DOI] [PubMed] [Google Scholar]
- [22].Phan TG, Okame M, Nguyen TA, Maneekarn N, Nishio O, Okitsu S, et al. , Human astrovirus, norovirus (GI, GII), and sapovirus infections in Pakistani children with diarrhea, J. Med. Virol 73 (2004) 256–261. [DOI] [PubMed] [Google Scholar]
- [23].Farkas T, Zhong WM, Jing Y, Huang PW, Espinosa SM, Martinez N, et al. , Genetic diversity among sapoviruses, Arch. Virol 149 (2004) 1309–1323. [DOI] [PubMed] [Google Scholar]
- [24].Nakamura N, Kobayashi S, Minagawa H, Matsushita T, Sugiura W, Iwatani Y, Molecular epidemiology of enteric viruses in patients with acute gastroenteritis in Aichi prefecture, Japan, 2008/09–2013/14, J. Med. Virol 88 (2016) 1180–1186. [DOI] [PubMed] [Google Scholar]
- [25].Harada S, Oka T, Tokuoka E, Kiyota N, Nishimura K, Shimada Y, et al. , A confirmation of sapovirus re-infection gastroenteritis cases with different genogroups and genetic shifts in the evolving sapovirus genotypes, 2002–2011, Arch. Virol 157 (2012) 1999–2003. [DOI] [PubMed] [Google Scholar]
- [26].Liu X, Yamamoto D, Saito M, Imagawa T, Ablola A, Tandoc AO 3rd et al. , Molecular detection and characterization of sapovirus in hospitalized children with acute gastroenteritis in the Philippines, J. Clin. Virol 68 (2015) 83–88. [DOI] [PubMed] [Google Scholar]
- [27].Cannon JL, Barclay L, Collins NR, Wikswo ME, Castro CJ, Magana LC, et al. , Genetic and epidemiologic trends of norovirus outbreaks in the US demonstrated emergence of novel GII.4 recombinant viruses, 2013–2016, J. Clin. Microbiol (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Oka T, Katayama K, Hansman GS, Kageyama T, Ogawa S, Wu FT, et al. , Detection of human sapovirus by real-time reverse transcription-polymerase chain reaction, J. Med. Virol 78 (2006) 1347–1353. [DOI] [PubMed] [Google Scholar]
- [29].Okada M, Yamashita Y, Oseto M, Shinozaki K, The detection of human sapoviruses with universal and genogroup-specific primers, Arch. Virol 151 (2006) 2503–2509. [DOI] [PubMed] [Google Scholar]
- [30].Varela MF, Polo D, Romalde JL, Prevalence and genetic diversity of human sapoviruses in shellfish from commercial production areas in Galicia, Spain, Appl. Environ. Microbiol 82 (2016) 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Murray TY, Nadan S, Page NA, Taylor MB, Diverse sapovirus genotypes identified in children hospitalised with gastroenteritis in selected regions of South Africa, J. Clin. Virol 76 (2016) 24–29. [DOI] [PubMed] [Google Scholar]
- [32].Ng TF, Zhang W, Sachsenroder J, Kondov NO, da Costa AC, Vega E, et al. , A diverse group of small circular ssDNA viral genomes in human and non-human primate stools, Virus Evol. 1 (2015) vev017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Payne DC, Staat MA, Edwards KM, Szilagyi PG, Gentsch JR, Stockman LJ, et al. , Active, population-based surveillance for severe rotavirus gastroenteritis in children in the United States, Pediatrics 122 (2008) 1235–1243. [DOI] [PubMed] [Google Scholar]
- [34].Ng TFF, Marine R, Wang C, Simmonds P, Kapusinszky B, Bodhidatta L, et al. , High variety of known and new RNA and DNA viruses of diverse origins in untreated sewage, J. Virol 86 (2012) 12161–12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ng TF, Mesquita JR, Nascimento MS, Kondov NO, Wong W, Reuter G, et al. , Feline fecal virome reveals novel and prevalent enteric viruses, Vet. Microbiol 171 (2014) 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ng TF, Kondov NO, Deng X, Van Eenennaam A, Neibergs HL, Delwart E, A metagenomics and case-control study to identify viruses associated with bovine respiratory disease, J. Virol 89 (2015) 5340–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ng TF, Marine R, Wang C, Simmonds P, Kapusinszky B, Bodhidatta L, et al. , High variety of known and new RNA and DNA viruses of diverse origins in untreated sewage, J. Virol 86 (2012) 12161–12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Deng X, Naccache SN, Ng T, Federman S, Li L, Chiu CY, et al. , An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data, Nucleic Acids Res. 43 (2015) e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Montmayeur AM, Ng TF, Schmidt A, Zhao K, Magana L, Iber J, et al. , High-throughput next-generation sequencing of polioviruses, J. Clin. Microbiol 55 (2017) 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Langmead B, Salzberg SL, Fast gapped-read alignment with bowtie 2, Nat. Methods 9 (2012) 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. , SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing, J. Comput. Biol 19 (2012) 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. , Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data, Bioinformatics 28 (2012) 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Castro CJ, Ng TFF. U50: a new metric for measuring assembly output based on non-overlapping, target-specific contigs, J. Comput. Biol (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Edgar RC, MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Res. 32 (2004) 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tamura K, Stecher G, Peterson D, Filipski A, Kumar S, MEGA6: molecular evolutionary genetics analysis version 6.0, Mol. Biol. Evol 30 (2013) 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Muhire BM, Varsani A, Martin DP, SDT: a virus classification tool based on pairwise sequence alignment and identity calculation, PLoS One 9 (2014) e108277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, et al. , Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination, J. Virol 73 (1999) 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Oka T, Lu Z, Phan T, Delwart EL, Saif LJ, Wang Q, Genetic characterization and classification of human and animal sapoviruses, PLoS One (2016) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kagning Tsinda E, Malasao R, Furuse Y, Gilman RH, Liu X, Apaza S, et al. , Complete coding genome sequences of uncommon GII.8 Sapovirus strains identified in diarrhea samples collected from peruvian children, Genome Announc. (2017) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nguyen TA, Hoang L, Pham le D, Hoang KT, Okitsu S, Mizuguchi M, et al. , Norovirus and sapovirus infections among children with acute gastroenteritis in Ho Chi Minh City during 2005–2006, J. Trop. Pediatr 54 (2008) 102–113. [DOI] [PubMed] [Google Scholar]
- [51].Phan TG, Trinh QD, Yagyu F, Sugita K, Okitsu S, Muller WE, et al. , Outbreak of sapovirus infection among infants and children with acute gastroenteritis in Osaka City, Japan during 2004–2005, J. Med. Virol 78 (2006) 839–846. [DOI] [PubMed] [Google Scholar]
- [52].Bull RA, Norovirus recombination in ORF1/ORF2 overlap, Emerg. Infect. Dis 11 (2005) 1079–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Simmonds P, Karakasiliotis I, Bailey D, Chaudhry Y, Evans DJ, Goodfellow IG, Bioinformatic and functional analysis of RNA secondary structure elements among different genera of human and animal caliciviruses, Nucleic Acids Res. 36 (2008) 2530–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]