

SUMMARY
In pluripotent cells, a delicate activation-repression balance maintains pro-differentiation genes ready for rapid activation. The identity of transcription factors (TFs) that specifically repress pro-differentiation genes remains obscure. By targeting ~1,700 TFs with CRISPR loss-of-function screen, we found that ZBTB11 and ZFP131 are required for embryonic stem cell (ESC) pluripotency. ESCs without ZBTB11 or ZFP131 lose colony morphology, reduce proliferation rate, and upregulate transcription of genes associated with three germ layers. ZBTB11 and ZFP131 bind proximally to pro-differentiation genes. ZBTB11 or ZFP131 loss leads to an increase in H3K4me3, negative elongation factor (NELF) complex release, and concomitant transcription at associated genes. Together, our results suggest that ZBTB11 and ZFP131 maintain pluripotency by preventing premature expression of pro-differentiation genes and present a generalizable framework to maintain cellular potency.
Graphical abstract
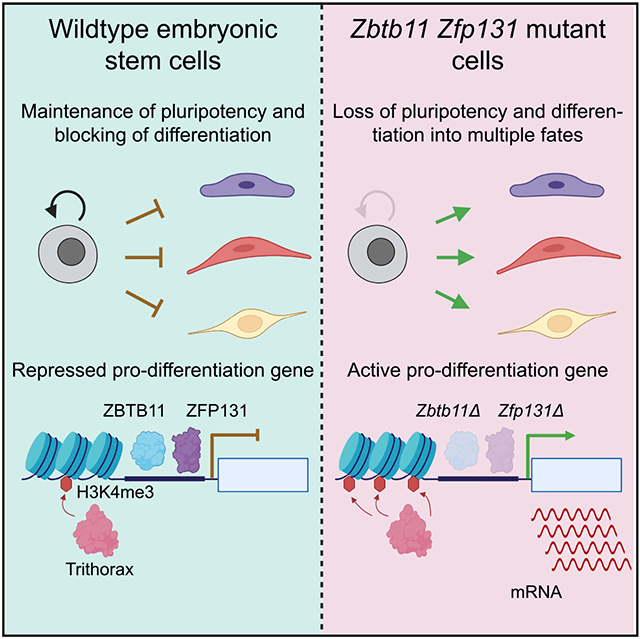
In brief
With a highly sensitive CRISPR loss-of-function screen targeting all TFs in the mouse and human genome, Garipler et al. show that ZBTB11 and ZFP131 TFs are required for embryonic stem cell pluripotency. ZBTB11 and ZFP131 maintain poised pro-differentiation genes in a repressed state to preserve pluripotency.
INTRODUCTION
Early developmental progenitors and stem cells can maintain their fate while being able to differentiate rapidly. Pluripotency is an example of a transient state during early development, which can be recapitulated in vitro with embryonic stem cells (ESCs) isolated from the inner cell mass of blastocyst-stage embryos (Evans and Kaufman, 1981; Ying et al., 2008). ESCs express pluripotency genes while keeping developmentally regulated genes repressed but ready for activation (Bernstein et al., 2006; Mikkelsen et al., 2007; Efroni et al., 2008). The ability to rapidly activate pro-differentiation genes is critical for pluripotent cells to retain potency for lineage commitment yet differentiate rapidly during development. Although essential for understanding pluripotency and differentiation, dedicated transcription factors (TFs) that guide repression mechanisms to specific pro-differentiation genes are still under investigation.
Controlling chromatin states and RNA polymerase II (RNA Pol II) elongation dynamics are two major mechanisms to maintain plasticity for pro-differentiation gene expression. Bivalent genes contain nucleosomes modified by protein complexes associated with active (trithorax group [TrxG]) and repressive (polycomb group [PcG]) transcriptional states (Azuara et al., 2006; Bernstein et al., 2006; Vastenhouw and Schier, 2012). Similarly, reducing other chromatin modifiers, such as histone deacetylase 1/2 (HDAC1/2) complexes, leads to the upregulation of pro-differentiation genes (Kawamura et al., 2005; Karantzali et al., 2008; Dovey et al., 2010; Jamaladdin et al., 2014). Regulating RNA Pol II elongation dynamics by promoter-proximal pausing has emerged as a possible mechanism to repress pro-differentiation genes while preserving the ability to produce full-length transcripts (Stock et al., 2007; Ferrai et al., 2017). However, chromatin modifiers and RNA Pol II lack the sequence specificity to direct them to specific gene sets and must be recruited to a specific set of pro-differentiation genes that must be kept ready for activation.
Sequence-specific TFs are excellent candidates for recruiting and modulating chromatin complexes to regulate pro-differentiation genes. The pluripotency gene regulatory network (PGRN) relies on a set of interconnected TFs that engage in a positive feedforward loop to activate pluripotency genes (Li and Izpisua Belmonte, 2018). Moreover, the PGRN core TFs, OCT4, NANOG, and SOX2, directly bind to pluripotency and differentiation genes, which must be actively maintained in a repressed state (Boyer et al., 2005). Ad hoc repressive TFs that establish and control poised chromatin states in ESCs were postulated more than a decade ago (Boyer et al., 2005; Creyghton et al., 2008; Surface et al., 2010). However, a full understanding of sequence-specific repressive TFs that regulate the wide variety of genes required for the differentiation into all germ layers had remained elusive.
To rapidly dissect the gene regulatory network of ESCs, we developed a loss-of-function screen with a pooled CRISPR-Cas9 library targeting all annotated TFs in the mouse genome. The screen recovered known PGRN members (OCT4, NANOG, etc.) as well as two non-redundant ZBTB family TFs: ZBTB11 and ZFP131. Inducible knockout lines for ZBTB11 and ZFP131 both demonstrate loss of pluripotency features with abnormal colony morphology, reduced proliferation rate, and mis-expression of pro-differentiation genes. ZBTB11 and ZFP131 co-occupy transcription start sites (TSSs) of pro-differentiation genes along with TrxG, RNA Pol II, and negative elongation factor (NELF). Zbtb11 or Zfp131 knockouts caused a decrease in NELF and an increase in H3K4me3 signal at their binding sites and associated genes. As a result, ZBTB11 or ZFP131 loss causes transcriptomic changes associated with ESC differentiation into all three germ layers. Thus, we propose that ZBTB11 and ZFP131 play non-redundant roles as pluripotency gatekeepers to maintain developmental regulators in a repressed state but ready to be activated.
RESULTS
A CRISPR loss-of-function screen to identify transcription factors required for pluripotency
To functionally identify TFs with essential roles during differentiation or fate maintenance, we designed and constructed a rapid and efficient CRISPR-Cas9 loss-of-function screen targeting all 1,682 annotated TFs in the mouse genome (“CRISPR TF library”; Table S1; Ravasi et al., 2010; Meier et al., 2017). The CRISPR TF library contains 10 single-guide RNAs (sgRNAs) per TF and 1,000 non-targeting sgRNAs (~5% of all sgRNAs) to serve as negative controls. We selected sgRNAs with predicted high on-target activity (Doench et al., 2016), minimal potential for off-target (Hsu et al., 2013), and those that target 5′ exons with annotated functional domains to enrich for loss-of-function mutations (Shi et al., 2015; Figure S1; Table S1).
Because of their rapid cell cycle, bona fide ESCs outcompete differentiating cells or cells with compromised self-renewal abilities, a demonstrated advantage in identifying pluripotency and self-renewal genes (Ivanova et al., 2006). Thus, ESCs transduced with sgRNAs targeting PGRN TFs should differentiate and be outcompeted by ESCs transduced with non-targeting sgRNAs and those targeting TFs that do not affect ESC state. Other Cas9-based screens have been recently performed in mouse and human pluripotent stem cells (Shalem et al., 2014; Yilmaz et al., 2018; Ihry et al., 2019; Shohat and Shifman, 2019). These studies typically provide insights into gene families and signaling pathways required for cell survival and pluripotency. Rather than targeting all protein-coding genes, we focused on a single type of coding genes to gain statistical power and identify phenotype severity by sgRNA depletion scores.
A pilot competition assay using a non-targeting sgRNA versus sgRNAs targeting Oct4 (Pou5f1), Nanog, and Klf5 revealed that cells transduced with sgRNAs targeting PGRN TFs were outcompeted starting by 5 days after transduction and were almost fully depleted by day 12 (Figures S2A and S2B). Therefore, we measured CRISPR TF library sgRNA representation on day 5, day 8, and day 12 after ESC transduction (Figure 1A). As expected, the sgRNA library diversity progressively decreased, as mutant cells for essential and pluripotency TFs were outcompeted over time (Figure 1B; Shannon, 1948). To select candidate TFs, we used three different methods to identify significantly depleted sgRNAs: an empirical false discovery rate (FDR) cutoff derived from the embedded 1,000 non-targeting sgRNAs controls (Chen et al., 2015; Patel et al., 2017), a mixed linear model with random intercepts using normalized sgRNA counts, and model-based analysis of genome-wide CRISPR-Cas9 knockout (MAGeCK) method (Li et al., 2014).
Figure 1. Identification of transcription factors required for pluripotency using the CRISPR TF library.

(A) Transcription factor screen overview. Day −1, CRISPR TF library transduction to growing ESCs; day 0, zeocin selection begins; days 5, 8, and 12, collection days and representation analysis.
(B) Shannon’s diversity index (H) was calculated based on the proportion of individual sgRNAs (pi) in the total number of sgRNAs in the pool (S) for each time point with sgRNA counts. The diversity score decreases as the proportion of essential and pluripotency-targeting sgRNAs decreases in the population.
(C) Log2 fold change of targeting sgRNAs on day 12 compared with the initial library representation. sgRNAs depleted more than the 50th most depleted non-targeting sgRNA are in orange and enriched more than the 50th most enriched non-targeting sgRNA are in blue.
(D) The number of targeting sgRNAs depleted and mean number of sgRNAs with random depletion calculated by 1,000 permutation tests for the different thresholds of sgRNA per gene. TFs with more than or equal to six depleted sgRNAs are unlikely to be false positives.
(E) Venn diagram representation of the number of depleted TFs identified by three different methods: log-fold change, linear modeling, and MAGeCK. All methods picked 196 genes as high-confidence depleted TFs.
(F) The workflow of the secondary screen using Oct4:tdTomato Sox2:Gfp pluripotency reporter ESC line. Pluripotent ESCs are depicted as yellow while differentiating cells lose one or both reporter expression.
(G) Percentage of cells expressing fluorescent reporters when transduced with a single sgRNA that is either non-targeting or targeting a known pluripotency TF or candidate TF. Both fluorescent reporters are expressed by 80% of cells transduced with non-targeting sgRNA. This number decreases in cells transduced with sgRNAs targeting known pluripotency TFs and Zbtb10, Zbtb11, and Zfp131.
See also Figures S1-S6 and Tables S1, S2, and S3.
In the first approach, by measuring the depletion of the nontargeting sgRNAs, we set an empirical FDR cutoff of q < 0.05 and calculated the number of depleted sgRNAs per gene that exceeded this level (Figures 1C, S2C, and S2D). Random permutation simulations demonstrated that at least six depleting or three enriching sgRNAs are required to confidently categorize a gene as depleting or enriching, respectively (Figures 1D and S2E). In total, the empirical FDR method identified 262 depleting candidate TFs (Figure 1E). Although not the focus of this study, using an enrichment analysis, the empirical FDR method also identified 17 genes that increased self-renewal when mutated. These include the tumor suppressor Trp53 (p53) and other genes, which could provide growth advantage in cancers when mutated (Figure S3; Cerami et al., 2012; Gao et al., 2013; Merkle et al., 2017). In the second approach, the mixed linear model identified 218 depleting TFs (Figures 1E and S4A) and, by using the degree of depletion, placed candidate TFs within the PGRN hierarchy (Figure S4A; Table S2; see STAR Methods). Finally, the MAGeCK approach identified 302 TF candidates as significantly depleted using robust-rank aggregation (Figure 1E; Table S2). In total, the three methods identified 196 TFs in common. As expected, this set included positive controls, like essential genes and many known pluripotency factors. After removing these factors from this set, we selected seven high-confidence novel candidate TFs (Ankrd49, Zbtb10, Zbtb11, Zeb1, Zfp131, Zfp638, and Znhit3) with high scores and unknown functions to be tested for their putative role in pluripotency (Table S2).
A secondary screen identifies ZBTB-domain-containing TFs as novel candidates for PGRN
Disrupting pluripotency or self-renewal decreases the ESCs’ proliferation rate. To discriminate between effects on pluripotency and self-renewal, we utilized a dual-color pluripotency reporter ESC line (Figure 1F). We generated an Oct4:: tdTomato and Sox2::Gfp pluripotency reporter line. When transduced with non-targeting sgRNAs, 85% of ESCs expressed both GFP and tdTOMATO (Figure 1G). In contrast, only 45%–65% of cells transduced with sgRNAs targeting Klf5, Nanog, or Oct4 expressed both fluorescent proteins under these culture and infection conditions (Figure 1G). We transduced the pluripotency dual-color reporter ESC line with two sgRNAs targeting each candidate TF (Figure 1F). To ensure robust, independent validation, we made sure that at least one sgRNA for each gene was not found in our original set of 10 sgRNAs per TF (Table S3). Transduction with sgRNAs targeting Zbtb10, Zbtb11, and Zfp131 reduced OCT4 and SOX2 reporters similarly to the known pluripotency TFs (Figure 1G). In contrast, sgRNAs targeting Zfp638, Ankrd49, Znhit3, and Zeb1 did not significantly reduce OCT4 and SOX2 reporter expression. We did not continue our investigation on these TFs, since they are likely to control self-renewal or other ESC features, but not pluripotency.
A sgRNA competition assay between Zbtb10, Zbtb11, and Zfp131 targeting sgRNAs and non-targeting sgRNA (Figure S5A) demonstrated that Zbtb11 and Zfp131 sgRNAs depleted as fast as the core PRGN Oct4 and Nanog sgRNAs (Figures S5B, S2A, and S2B). Zbtb10 depleted like Klf5, slower than the other candidate and core TFs (Figures S5B, S2A, and S2B). The comparative depletion rate in the competition assay is in line with the original screen (Figure S4). Moreover, we performed a similar CRISPR TF screen using a library targeting all human TFs with 10 sgRNAs per gene and found that sgRNAs targeting the human homologs of Zbtb10, Zbtb11, and Zfp131 are also depleted in human pluripotent stem cells (Figures S6A-S6C). Thus, a rapid TF-wide screen combined in mouse and human ESC with a small-scale secondary screen using dual fluorescent reporter line was sufficient to identify novel candidate TFs for PGRN.
Zbtb11 and Zfp131 are required for pluripotency
Zbtb10, Zbtb11, and Zfp131 are C2H2 zinc finger BTB domain TFs expressed in ESCs and during early mouse and human development and downregulated upon differentiation (Boroviak et al., 2018; Nowotschin et al., 2019). To better characterize their role in pluripotency, we generated clonal ESC knockout lines carrying null alleles for each TF. We introduced frameshift deletions in both endogenous alleles while supplying an exogenous doxycycline-inducible hemagglutinin (HA)-tagged rescue copy (Figure S5C). Doxycycline removal causes the extinction of the rescue construct, and consequently, ESCs only express the knockout alleles for each candidate TF (Zbtb10Δ, Zbtb11Δ, or Zfp131Δ thereafter). The addition of the HA tag to exogenous allele does not disturb ESC morphology or Oct4 expression (Figures S5D and S5E). To validate the competitive disadvantage seen during the screen, we repeated the competition assay at the same time point (day 5) with H2B-GFP-labeled wild-type (WT) ESCs plated in equal proportions with Zbtb10Δ, Zbtb11Δ, or Zfp131Δ ESCs (Figure S6D). H2B-GFP-labeled WT ESCs significantly outcompeted Zbtb11Δ and Zfp131Δ cells (Figure S6E). However, the H2B-GFP-labeled WT ESCs did not significantly outcompete the Zbtb10Δ ESCs (Figure S6E).
Mouse ESCs grow in compact colonies and are typically cultured with LIF and with or without 2i compounds (MEK and GSK3 inhibitors; Ying et al., 2008). In this work, we examined cells in both media (+2i + LIF and −2i + LIF) to capture phenotypes in these common culture conditions. All comparisons between knockout and WT cells are within each culture condition. When cultured in the absence of doxycycline for 3 days, Zbtb11Δ and Zfp131Δ cells lost ESC morphology both with and without 2i (Figure 2A), and the number of OCT4-expressing cells decreased in Zbtb11Δ and Zfp131Δ (Figures S6F and S6G). On the other hand, Zbtb10Δ cells looked similar to WT cells (Figure 2A) and the number of OCT4-positive cells was mildly reduced in the Zbtb10Δ line (Figure S6G). Thus, ZBTB10 is not required for ESCs however might have self-renewal or accessory roles. Together, the characterization of clonal knockout lines confirms that ESCs require ZBTB11 and ZFP131.
Figure 2. Zbtb11Δ and Zfp131Δ knockout cells express genes associated with multiple germ layers.

(A) Zbtb10Δ, Zbtb11Δ, and Zfp131Δ colony morphology in +2i + LIF and −2i + LIF conditions 3 days after doxycycline removal. Zbtb11Δ and Zfp131Δ cells lose typical ESC colony formation ability and adopt morphologies associated with differentiation. Scale bar, 200 μm.
(B) Volcano plots of log2 fold change of transcripts expressed in Zbtb10Δ, Zbtb11Δ, and Zfp131Δ cells versus wild-type (WT) cells grown in the absence of doxycycline for 3 days in +2i + LIF media (n = 3). Significant changes (*p < 0.05) were marked in color for each genotype. Zbtb11 and Zfp131 mutations in ESCs induce aberrant gene expression.
(C and D) GO-enrichment analysis of significantly upregulated genes in (C) Zbtb11Δ (693) and (D) Zfp131Δ (916) in +2i + LIF media (n = 3). Zbtb11 and Zfp131 mutations induce ESCs to express genes with a strong developmental signature.
(E) The log2 fold change of developmentally regulated TFs obtained from bulkRNA-seq experiment in Zbtb11Δ and Zfp131Δ cells versus WT in +2i + LIF and −2 + LIF conditions. Zbtb11 and Zfp131 mutations induce the expression of TFs associated with all three embryonic layers while downregulating pluripotency TFs.
See also Figures S6 and S7.
Loss of Zbtb11 and Zfp131 compromises pluripotent stem cell fate
Morphological changes of knockouts described above led us to hypothesize that Zbtb11Δ and Zfp131Δ ESC would induce genes associated with cell differentiation. To test this hypothesis, we performed bulk RNA sequencing (RNA-seq) on knockout cells from cells grown in LIF with and without 2i. In line with phenotypic characterization, the Zbtb10Δ transcriptome was similar to WT ESCs (Figures 2B and S7A). On the other hand, Zbtb11 and Zfp131 knockout ESCs had dissimilar transcriptomes when compared with WT ESCs (Figures 2B and S7A). Zbtb11Δ cells downregulated 475 and upregulated 996 genes, whereas Zfp131Δ cells downregulated 835 and upregulated 1,359 genes with the 2i compounds. In summary, because of the slight growth disadvantage and the relatively small transcriptomic differences, we speculate that Zbtb10 is not a central factor required for pluripotency and decided not to pursue it further. On the other hand, we conclude that Zbtb11 and Zfp131 are required for pluripotency because of the strong morphological and transcriptional changes in Zbtb11Δ and Zfp131Δ cells, even in strong pluripotency-maintaining conditions.
To understand ZBTB11’s and ZFP131’s role in maintaining pluripotency, we characterized the set of dysregulated genes in their respective knockout lines. Notably, both knockout lines induced a strong differentiation signature in culture conditions with and without 2i compounds. Upregulated genes in Zbtb11Δ and Zfp131Δ cells are associated with cell differentiation Gene Ontology (GO) terms. Importantly, those include cell fate commitment, morphogenesis, and tissue development (Figures 2C, 2D, S7B, and S7C). We also detected downregulated mitochondrial and other cellular biosynthetic and metabolic processes genes in Zbtb11Δ and Zfp131Δ ESCs (Figures S7D-S7F; Wilson et al., 2020). Thus, Zbtb11 and Zfp131 loss caused profound transcriptional changes indicative of cell differentiation in the two most common ESC culture conditions.
Zbtb11 and Zfp131 loss induced ESCs to express genes associated with all three germ layers
We next asked whether ZBTB11 and ZFP131 repress a unique differentiation trajectory each or whether they repress the expression of genes associated with all three germ layers. Both knockouts significantly downregulated pluripotency TFs and upregulated TFs associated with mesoderm, endoderm, ectoderm, and trophectoderm development compared with WT ESCs (Figure 2E). We did not detect any strong upregulation of genes associated with germline and primordial germ cells (Figure S7G). These results indicated that ZBTB11 and ZFP131 TFs are required for pluripotency maintenance by repressing pro-differentiation genes.
The observed upregulation of developmental genes in bulk RNA-seq offered two alternative scenarios: knockout cells differentiate into multiple germ layer trajectories or each cell simultaneously expresses genes of various germ layers. To discriminate between these two alternatives, we performed single-cell RNA-seq (scRNA-seq). We mixed Zbtb11Δ or Zfp131Δ cells with H2B-GFP-expressing WT ESCs to landmark control ESC expression in each experiment. H2B-GFP-expressing WT naive stem cells cluster away in both analyses with a high expression of stem cell markers (cluster 6 in Zbtb11Δ, Figure 3A, and cluster 4 in Zfp131Δ, Figure 3D). Zbtb11Δ and Zfp131Δ each formed distinct clusters. Zbtb11Δ cells were grouped in clusters with different germ layer markers (Figures 3B and 3C). For example, cluster 3 contained cells expressing mesoderm markers, while cluster 4 was associated with markers of ectoderm fate (Figures 3B, 3C, and S8A). Cluster 5 included cells expressing genes related to endoderm (Gata4, Gata6, and Sox7). Zfp131Δ clusters express genes associated with different germ layers with endoderm in cluster 6 and mesoderm markers in cluster 7 (Figures 3E, 3F, and S8B). Zfp131Δ cells upregulated ectodermal markers (Otx2, Neurod1, and Id2) across several clusters (Figures 3F and S8B). Although Zbtb11Δ and Zfp131Δ clusters separated from ESCs, a portion of cells retained the expression of the pluripotency factors Sox2 and Nanog (Figures S8A and S8B). This co-expression can be due to either capturing an early differentiation phase or aberrant expression. To better characterize differentiation trajectory, we constructed pseudotime trajectories (Figures S8C and S8D; Trapnell et al., 2014; Qiu et al., 2017; Cao et al., 2019). Although, with some aberrant expression patterns, pseudotime trajectories highlight the differentiation of Zbtb11Δ and Zfp131Δ cells to multiple lineages, together, neither Zbtb11Δ nor Zfp131Δ cells were grouped with WT ESCs nor formed a single cluster with each cell co-expressing genes from all germ layers. From the bulk and scRNA-seq analysis, we conclude that ZBTB11 and ZFP131 maintain the pluripotent state by actively repressing multiple developmental regulatory programs (with each forming its own cluster), as opposed to one specific differentiation trajectory.
Figure 3. Zbtb11Δ and Zfp131Δ knockout cells differentiate into multiple lineages.

(A) Uniform manifold approximation and projection (UMAP) plot of Zbtb11Δ and H2b:Gfp (internal control) cells single-cell RNA-seq. Knockout cells were grown in −2i + LIF media (n = 1).
(B) Violin plots of genes expressed in each cluster in Zbtb11Δ and H2b:Gfp cells. ESC, mesoderm, endoderm, and ectoderm markers are expressed in distinct clusters.
(C) Expression of genes associated with pluripotency and germ layers in Zbtb11Δ cells. Zbtb11Δ cells segregate in different clusters compared with H2b::Gfp and express developmentally regulated genes.
(D) UMAP plot of Zfp131Δ and H2b:Gfp (internal control) cells single-cell RNA-seq. Knockout cells were grown in −2i + LIF media (n = 1).
(E) Violin plots of genes expressed in each cluster in Zfp131Δ and H2b:Gfp cells. ESC, mesoderm, endoderm, and ectoderm markers are expressed in distinct clusters.
(F) Expression of genes associated with pluripotency and germ layers in Zfp131Δ and H2b:Gfp UMAP plots. Zfp131Δ cells segregate in different clusters compared with H2b::Gfp and express developmentally regulated genes.
See also Figure S8.
ZBTB11 and ZFP131 bind to sites with active chromatin features
To gain insights into how ZBTB11 and ZFP131 maintain pluripotency and repress differentiation programs, we investigated their genomic binding in undifferentiated pluripotent cells. MultiGPS chromatin immunoprecipitation (ChIP)-seq analysis of HA-tagged exogenous copies identified 22,439 ZBTB11 sites and 3,635 ZFP131-binding sites (Mahony et al., 2014). The majority of ZFP131-binding sites (78%) overlap with ZBTB11 (Figure 4A). BTB domains participate in homo- and heterodimer formation (Stogios et al., 2005). To test whether these co-bound sites are ZBTB11-ZFP131 dimers, we performed co-immunoprecipitation (coIP) experiments from nuclear fraction. We could not find evidence of physical interaction (Figures S9A and S9C).
Figure 4. ZBTB11 and ZFP131 bound sites gain an H3K4me3 signal in Zbtb11Δ and Zfp131Δ knockout cells.

(A) ChIP-seq heatmap for ZBTB11 and ZFP131 binding on the genome ESC grown in +2i + LIF (n = 2). Categories were identified with ZBTB11 and ZFP131 co-binding (Z11 = Z131), differentially enriched ZBTB11 binding (Z11 > Z131), and differentially enriched ZFP131 binding (Z131 > Z11).
(B) Enriched motifs discovered via MEME-ChIP for each category of ZBTB11 and ZFP131 binding. Motif discovery was performed with centering to the peak at ZBTB11 and ZFP131 for three differential binding categories.
(C) Percentage of overlap between ZBTB11 and ZFP131 with pluripotency factors, known regulatory complex members, histone modifications, and ATAC-seq signal. ZBTB11 and ZFP131 bind at sites with active chromatin states, accessible and enriched with TrxG, Pol II S5P, NELF, and histone deacetylase members. ZBTB11 and ZFP131 do not co-bind with pluripotency TFs but bind to regions with active chromatin signatures. H3K4me1 data are obtained from GSE24165; H3K9me3 data are obtained from GSE18371; OCT4, NANOG, and SOX2 data are obtained from GSE11724; H3K4me2 data are obtained from GSE11172; and ATAC-seq data are obtained from GSE80511.
(D and E) ChIP-seq heatmap for (D) ZBTB11 and (E) ZFP131 overlap with SETD1A and H3K4me3. Categories are determined according to gain, loss, or no change in H3K4me3 signal between knockout and WT cells (n = 2 for all). ZBTB11 and ZFP131 co-occupy ESC genome with SETD1A. H3K4me3 comparison was done in −2i + LIF.
See also Figures S9 and S10.
Both ZBTB11 and ZFP131 co-bound sites (Z11 = Z131) and ZBTB11-only sites (Z11 > Z131) contain a central motif similar to that bound by ETS family TFs: CCGGAA (Figure 4B; Wilson et al., 2020). ZFP131 and ZBTB11 co-bound sites (Z11 = Z131) and ZFP131-only sites (Z131 > Z11) contain a central motif similar to that bound by bZIP TFs: TGACGTCA (Figure 4B). Using the GREAT algorithm (McLean et al., 2010), we found that a significant portion of ZBTB11 and ZFP131 sites are proximal to TSSs. 46% of ZBTB11 binding and 72% of ZFP131 binding lies within 5 kb of nearest TSSs (Figure S9D). Comparing ZBTB11 and ZFP131 binding with ESC assay for transposase-accessible chromatin using sequencing (ATAC-seq) experiments revealed that ZBTB11 and ZFP131 associate with accessible chromatin regions (Figure 4C; Velasco et al., 2017). These sites are also enriched in histone modifications associated with active transcription elements (H3K27ac) and promoter regions (H3K4me3; Figure 4C). As a whole, we noted these sites are also largely devoid of repressive histone modifications, such as H3K9me3 and H3K27me3 (Figure 4C; Bilodeau et al., 2009). Unlike ZNF114, ZNF483, and ZNF589, which are postulated to induce the strong repressive H3K9me3 histone modification, ZBTB11 and ZFP131 bind at sites containing an overall active chromatin signature (Oleksiewicz et al., 2017).
We considered several hypotheses that could explain how Zbtb11 and Zfp131 maintain pluripotency. The ZBTB family TF ZBTB3 occupies the ESC genome with core pluripotency factors to modulate their activity (Ye et al., 2018). However, low ZBTB11 and ZFP131 binding overlap (<5%) with OCT4, SOX2, and NANOG does not support the hypothesis of coregulation between ZBTB11 or ZFP131 and pluripotency factors (Figure 4C). ZBTB TFs are also known to interact with the subunits of HDAC complexes (David et al., 1998; Maeda, 2016; Masuda et al., 2016). Both ZBTB11 and ZFP131 overlap with sites occupied by CHD4 and SIN3A, known components of different HDAC complexes (Figures 4C, S10A, and S10B; Laherty et al., 1997; Xue et al., 1998). While there were small CHD4 and SIN3A decreases at ZBTB11 and ZFP131 sites in knockout cells (Figures S10C-S10F), in general, ZBTB11- or ZFP131-bound areas lost H3K27ac, which is contrary to expectation when losing HDACs (Figures S10G and S10H). Thus, these results do not support a direct role in HDAC recruitment or regulation.
ZBTB11 or ZFP131 loss increased H3K4me3 deposition and decreased NELF occupancy at their binding sites
ZBTB11 and ZFP131 binding overlap with H3K4me3 in WT ESCs, deposited by TrxG (Figure 4C). Moreover, ZBTB11 and ZFP131 binding overlap with SETD1A in WT ESCs (Figures 4C-4E; Sze et al., 2017). SETD1A is a TrxG H3K4me-depositing enzyme, expressed and required at early developmental states until inner cell mass formation in development (Bledau et al., 2014). Suggesting ZBTB11 and ZFP131 binding participates in repression, removing ZBTB11 or ZFP131 induced a significant increase of H3K4me3 at their binding sites (Figures 5A and 5B). This increase was more prominent at sites where ZBTB11 and ZFP131 binding overlaps (ZBTB11 = ZFP131; Figure 5C). Importantly, most of the regions that gained H3K4me3 in Zbtb11Δ and Zfp131Δ cells already contained SETD1A binding in the control state (Figure S9E).
Figure 5. SETD1A occupancy at ZBTB11 and ZFP131 sites.

(A–C) Metagene plots for H3K4me3 difference (A) at ZBTB11-bound sites in Zbtb11Δ versus WT (n = 2), (B) at ZFP131-bound sites in Zfp131Δ versus WT (n = 2), and (C) at ZBTB11 and ZBTB131 co-bound sites in Zbtb11Δ and Zfp131Δ versus WT. Zbtb11 and Zfp131 mutations result in an H3K4me3 gain at ZBTB11 and ZFP131 binding sites. All comparisons were done in −2i + LIF.
(D) ChIP-seq heatmap for ZBTB11 and ZFP131 overlap with RNA Pol II S5P and NELF. Categories are determined according to ZBTB11 only, ZFP131 only, and co-bound regions (n = 2 for all). ZBTB11 and ZFP131 co-occupy ESC chromatin with RNA Pol II S5P and NELF.
(E) Metagene plots for NELF signal difference in ZBTB11 only, ZFP131 only, and co-bound regions (n = 2 for all). ZBTB11 and ZFP131 sites lose NELF in Zbtb11 and Zfp131 knockout ESCs. All comparisons were done in −2i + LIF.
While H3K4me3 is generally associated with active transcription (Huang et al., 2019), it might not be instructive for it (Murray et al., 2019). Since H3K4me3 co-occupies ~90% of RNA-Pol-II-bound regions (Barski et al., 2007), we tested whether RNA Pol II binding overlaps ZBTB11 and ZFP131 in WT ESCs. To initiate transcription, RNA Pol II is loaded onto TSSs, forming a complex with the NELF, among other factors (Yamaguchi et al., 1999; Muse et al., 2007; Zeitlinger et al., 2007). Following initial elongation, RNA Pol II is phosphorylated at serine 5 (S5P), but further elongation is enabled by serine 2 phosphorylation (S2P) and with NELF removal (Egloff et al., 2012). Both ZBTB11 and ZFP131 overlap with serine 5 phosphorylated RNA Pol II (RNA Pol II S5P) and NELF-E, an essential component of NELF complex (Figures 4C and 5D). ZBTB11 and ZPF131 removal decreased NELF occupancy at these sites (Figure 5E). These results suggest that ZBTB11 or ZFP131 removal causes H3K4me3 gain and NELF displacement, leading to transcriptional upregulation of associated genes.
Loss of ZBTB11 or ZFP131 leads to upregulation of associated genes
Zbtb11Δ and Zfp131Δ cells induce the transcription of pro-differentiation genes. Since ZBTB11 or ZFP131 removal leads to gain of H3K4me3 signal and loss of NELF occupancy at their binding sites, we asked whether the associated genes mirror these changes and activate productive transcription. TSSs of ZBTB11- and ZFP131-bound genes gain H3K4me3 and lose NELF signal, marking them for increased transcription (Figures 6A and 6B). Although a fraction of these genes are bivalent, the low read count obtained from H3K27me3 at TSSs of these genes highlights that there is no global affinity or regulation of bivalency by ZBTB11 or ZFP131 (Figure 6C, please note the scale). Genes with either ZBTB11 or ZFP131 that gain H3K4me3 were upregulated in Zbtb11Δ and Zfp131Δ cells (Figures 6D and 6E). In agreement with chromatin phenotypes, the transcription of genes associated with co-bound sites (ZBTB11 = ZFP131) was strongly upregulated (Figures 6D and 6E). Genes that were bound by ZBTB11 or ZFP131 that gained H3K4me3 and mRNA upregulation in knockout lines are strongly associated with differentiation GO terms (Figures 6F and 6G). Together, these results suggest that ZBTB11 and ZFP131 bind to a subset of pro-differentiation genes that already contain SETD1A and the TrxG complex in ESCs. Upon ZBTB11 or ZFP131 removal, these sites gained H3K4me3 with the associated gene transcription, particularly at ZBTB11 and ZFP131 co-bound sites.
Figure 6. ZBTB11- and ZFP131-associated pro-differentiation genes gained H3K4me3 and lost NELF at TSSs and are transcribed in Zbtb11Δ and Zfp131Δ cells.

(A–C) Metagene plots for (A) H3K4me3 signal distribution, (B) NELF signal distribution, and (C) H3K27me3 signal distribution at ZBTB11-associated genes in Zbtb11Δ versus WT (n = 2), at ZFP131 associated genes in Zfp131Δ versus WT (n = 2), and at co-occupied genes in Zbtb11Δ and Zfp131Δ versus WT. All comparisons were done in −2i + LIF. TTS, transcription termination site.
(D and E) Violin plots for Z score mRNA expression of genes that are (D) ZBTB11 bound along with NELF and H3K4me3 increase in Zbtb11 knockout (n = 1,191) and ZBTB11 and ZFP131 bound along with NELF and H3K4me3 increase in Zbtb11 knockout (n = 216) and (E) ZFP131 bound along with NELF and H3K4me3 increase in Zfp131 knockout (n = 915) and ZBTB11 and ZFP131 bound along with NELF and H3K4me3 increase in Zfp131 knockout (n = 216).
(F and G) GO enrichment analysis of significantly upregulated genes (F) with ZBTB11 binding and H3K4me3 increase (n = 531) and (G) with ZFP131 binding and H3K4me3 increase (n = 264) in +2i + LIF conditions.
The double ZBTB11-ZFP131 knockout has more profound phenotypes than the single knockouts
Single Zbtb11Δ and Zfp131Δ knockout lines differentiate and ZBTB11 and ZFP131 co-occupy a significant amount of their sites in ESC genome. Therefore, we asked whether the double-knockout phenotype would phenocopy a singleknockout phenotype or have additive effects on the transcription of pro-differentiation genes. To that end, we generated a clonal Zbtb11Δ Zfp131Δ ESC knockout line (Z11Z131Δ). Z11Z131Δ cells upregulated 1,312 genes and downregulated 770 genes compared with WT ESCs (Figure 7A). The Z11Z131Δ transcriptome was also different compared with single knockouts: 704 genes upregulated compared with Zbtb11Δ and 379 compared with Zfp131Δ and 509 genes downregulated compared with Zbtb11Δ and 181 compared with Zfp131Δ (Figures 7A and S11A). Z11Z131Δ cells further downregulated pluripotency TFs and increased upregulation of TFs associated with mesoderm, endoderm, ectoderm, and trophectoderm development when compared with single knockouts (Figure 7B). However, a fraction of genes are differentially regulated only in single knockouts and double-knockout cells (Figures S11B-S11E). Thus, the double knockout is not an additive phenotype of both knockouts and single knockouts are not epistatic to the other. Although all cells differentiate into three germ layers, each knockout line has a differentiation bias (Figure S8). The genes upregulated in the single knockout, but not present in the double, are likely to be incompatible alternative fates. As in single knockouts, binding sites of ZBTB11 and ZFP131 lose NELF in double knockout versus WT (Figure 7C). Also, upregulated genes bound by ZBTB11 and ZFP131 gain H3K4me3 in the double-knockout cells (Figure 7D). The double-knockout cell comparison reveals that Zbtb11 and Zfp131 are necessary for ESC fate and have non-redundant roles.
Figure 7. Double-knockout cells further increase pro-differentiation gene expression and H3K4me3 signal.

(A) Volcano plots of log2 fold change of transcripts expressed in Z11Z131Δ cells versus WT, Zbtb11, and Zbtb131Δ cells grown in the absence of doxycycline for 3 days in +2i + LIF media (n = 3). Significant changes are marked in orange. Double knockout of Zbtb11 and Zfp131 mutations in ESCs induce further aberrant gene expression.
(B) The log2 fold change of developmentally regulated TFs obtained from bulk RNA-seq experiment in Z11Z131Δ cells versus WT, Zbtb11Δ, and Zfp131Δ in +2i + LIF and −2 + LIF conditions (n = 3). Z11Z131Δ mutations induced higher expression of TFs associated with all three embryonic layers compared with single knockout lines.
(C) Metagene plots for NELF signal difference in ZBTB11 only, ZFP131 only, and co-bound regions in double knockout versus WT (n = 2 for all). ZBTB11 and ZFP131 sites lose NELF in Z11Z131Δ cells. All comparisons were done in −2i + LIF.
(D) Metagene plots for H3K4me3 signal distribution at ZBTB11, ZFP131, and co-occupied genes in Z11Z131Δ cells versus WT (n = 2 for all). All comparisons were done in −2i + LIF. TTS, transcription termination site.
See also Figures S11 and S12.
Zbtb11Δ, Zfp131Δ, and double-knockout cells have a strong differentiation signature, even under stringent pluripotency-promoting conditions. We tested whether the knockout ESCs have the ability to be directed to differentiate into specific cell fates. Multiple differentiations approaches require the classic embryoid body (EB) formation. Knockout cells did not form EBs that survived for more than 3 days under all conditions we tested. We believe the media and EB culture conditions do not support already differentiated cells. Then, we tested adherent protocols with WT and single- and double-knockout cells being differentiated into neuronal progenitor cells (NPCs) (Figures S12A and S12B; Ying et al., 2003). Z11Z131Δ and Zfp131Δ did not produce healthy cultures under these conditions. The Zbtb11Δ cells were able to generate SOX1-positive NPCs but very inefficiently compared with WT (Figure S12B). These results suggest that single- and double-knockout cells differentiate and do not respond like control ESC to differentiation cues. To determine whether the forced expression of Zbtb11 and/or Zfp131 block differentiation, we measured differentiation efficiency of NPCs while maintaining Zbtb11 or Zfp131 expression alone or in combination (Figure S12A). Maintaining ZBTB TF expression reduces the ability to differentiate, reinforcing their role in maintaining pluripotency and preventing differentiation (Figure S12B). These results reinforce the model where ZBTB11 and ZFP131 repress pro-differentiation genes.
DISCUSSION
Transcription factors are critical regulators of cell fate, typically playing instructive roles in cell-fate maintenance and differentiation. Decades of screens and studies have yielded a profound understanding of the functions of many TFs. However, there is still a large fraction of TFs with no described role, even in one of the most molecularly well-characterized cellular states: pluripotent stem cells. To gain insights into TF-driven gene-regulatory networks, we designed a loss-of-function CRISPR-Cas9 screen targeting all annotated TFs in the genome and deployed this unique tool to identify key TFs that regulate pluripotency. We expect this strategy to shed light on other TF-dependent regulatory processes, such as differentiation and development of diverse cell lineages.
Retention of self-renewal capacity and decrease in proliferation rate upon differentiation is a hallmark of pluripotency (Mitsui et al., 2003; Chambers et al., 2007). Therefore, by implementing the CRISPR TF screen in a competition setting, we discovered that ZBTB11 and ZFP131 are required for pluripotency. Loss of each TF led to a similar abnormal colony morphology as seen in other pluripotency TF mutants (Nichols et al., 1998; Niwa et al., 2000). Although OCT4, SOX2, and NANOG are activating TFs in a strong pluripotency positive-feedback loop (Boyer et al., 2005; Kim et al., 2008; Festuccia et al., 2012), they also bind proximally to pro-differentiation genes (Boyer et al., 2005; Jaenisch and Young, 2008). However, these TFs are not ad hoc repressors, suggesting that additional TFs with repressive functions are required. ZBTB11 and ZFP131 do not co-occupy ESC chromatin with core pluripotency TFs and are thus unlikely to form a repressive complex with them.
ZBTB TFs generally have repressive roles, with BTB domains interacting with HDACs, nuclear co-repressors (NCoR1), silencing mediator of retinoic acid and thyroid hormone receptor (NCoR2/SMRT), and Sin3A (Bardwell and Treisman, 1994; Albagli et al., 1995; Melnick et al., 2002; Ahmad et al., 2003; Ghetu et al., 2008). However, ZBTB11 and ZFP131 binding neither overlaps with NCOR nor impacts HDAC activity. Finally, although ZBTB11 and ZFP131 bind proximal to some bivalent genes, they do not overlap with PcG deposited H3K27me3 signal, suggesting that they have no global affinity for bivalent chromatin state or its regulation. Unlike other zinc finger TFs expressed in ESCs, ZBTB11 and ZFP131 do not overlap with H3K9me3 and thus are unlikely to be members of TRIM28/KRAB-ZF protein complexes (Oleksiewicz et al., 2017).
ZBTB11 and ZFP131 binding overlaps with the catalytic domain of TrxG complex and H3K4me3 signal; such loci are generally termed as transcriptionally active domains. However, associated genes have restricted expression in ESCs, and only upon loss of Zbtb11 and Zfp131 do these areas further gain H3K4me3 signal and upregulate transcription. We propose that ZBTB11 and ZFP131 prevent aberrant induction of pro-differentiation genes in pluripotent stem cells (Figure S12C). Another ZBTB TF family member, ZBTB17 (MIZ-1), was previously described to repress a set of Hox genes in pluripotent stem cells (Varlakhanova et al., 2011). It would be interesting to test whether ZBTB17 uses a similar repression mechanism as ZBTB11 and ZFP131.
The BTB domain can form homo- and heterodimers (Stogios et al., 2005). The similar phenotype of both knockouts and the binding overlap, particularly for ZFP131, suggests that they might coregulate a set of developmentally regulated genes. However, we did not find evidence for heterodimerization and non-redundancy, given the additive effects of double knockout at pro-differentiation genes. The fact that both genes were recovered from the functional screen with a similar phenotype for ZBTB11 and ZFP131 suggests that these TFs perform similar but non-redundant functions to maintain pluripotency.
Zbtb11 and Zfp131 are expressed in pluripotent stem cells and at early pluripotent developmental stages (Boroviak et al., 2018; Nowotschin et al., 2019). Although pluripotency can be maintained indefinitely by chemical signals in vitro, it is a transient in vivo state. We speculate that ZBTB11 and ZFP131 safeguard the transient state of pluripotency by preventing premature activation of differentiation during development. Broadly, the temporal gene activation in pluripotent and multipotent states is tightly regulated during vertebrate development. Since only a fraction of all TFs expressed during development are functionally characterized, we predict that TFs similar to ZBTB11 and ZFP131 have equivalent roles in maintaining poised genes in transient multipotent progenitors at later developmental stages.
Limitations of the study
In this work, we present a strategy to catalog and understand the role of relevant TFs for individual cell fates. The approach aims to identify the required TFs out of all TFs expressed in each cell fate and then proceed to functional characterization. In this case, we focused on pluripotent stem cells. We want to highlight some of the limitations of this study: first, although we followed up on the functional characterization of three ZBTB TFs, the screen identified other candidate pluripotency-required TFs that remain unexplored. Following studies by the community or us will shed light on their functions. Second, it is evident ZBTB11 and ZFP131 maintain pluripotency; however, we failed to place them within the known PGRN. We suspect that future PGRN modeling and epistasis experiments with known TFs will place these and other repressive TFs within the PGRN. Third, although the experiments clearly show ZBTB11 and ZFP131 repress pro-differentiation genes, the exact mechanisms of how ZBTB11 and ZFP131 pause RNA Pol II is not known. We believe co-immuno-precipitation and mass spectrometry experiments to identify ZBTB11 and ZFP131 binding partners will be a good entry point to solve this issue. Finally, this work lacks the fine temporal resolution to tackle some of the mechanistic questions presented in this section. Inducible and rapid degradable ZBTB11 and ZFP131 protein versions will allow for the identification of direct versus indirect transcriptional targets that maintain pluripotency and prevent differentiation.
STARMETHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Esteban O. Mazzoni (eom204@nyu.edu).
Materials availability
All materials and reagents will be made available upon installment of a material transfer agreement (MTA).
Data and code availability
All sequencing data (ChIP-seq, RNA-seq, scRNA-seq, GeCKO-seq) has been deposited at the GEO database under accession number GSE160966. We performed re-analysis of data sourced from GEO database entries GEO: GSE24165 (H3K4me1 ChIP-seq) (Creyghton et al., 2010), GEO: GSE18371 (H3K9me3 ChIPseq) (Bilodeau et al., 2009), GEO: GSE11724 (OCT4, NANOG, SOX2 ChIPseq) (Marson et al., 2008), GEO: GSE11172 (H3K4me2 ChIPseq) (Meissner et al., 2008), and GEO: GSE80511 (ATAC-seq) (Velasco et al., 2017).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
A17 E14Tg2a (Iacovino et al., 2011) mouse male embryonic stem cell line was used for the initial CRISPR TF screen experiments. Zbtb11 and Zfp131 were amplified from cDNA of ESCs using Zbtb11-fwd (5′ ATGTCAAGCGAGGAGAGCTACC), Zbtb11-rvs (5′ CTCTGCCTCTGGCATATGTGC), Zfp131-fwd (5′ ATGGAGGCTGAAGAGACGATGG) and Zfp131-rvs (5′ TTCTAAAACCGGCAGAG ATGTCC) primers. The cDNA of Zbtb10 was amplified from Origene (MR214195L4) Zbtb10 mouse tagged ORF clone using Zbtb10-fwd (5′ ATGTCGTTCAGTGAGATGAACCG) and Zbtb10-rvs primer (5′ TCCAGAGACATAAACGCCTCC). C-terminus of Zbtb10, Zbtb11, and Zfp131 cDNAs were fused to 1× hemagglutinin (HA) tag and cloned into p2lox plasmid using In-fusion (Clontech) cloning. To avoid sgRNAs from both landing on exogenous alleles and Cas9 from cutting at the PAM site, the sgRNA docking site on the BTB domain sequence of Zbtb10, Zbtb11 and Zfp131 was altered without changing the amino acid sequence (Figure S6C). Resulting CRISPR-proof alleles in p2lox-Zbtb10-HA, p2lox-Zbtb11-HA, p2lox-Zfp131-HA were nucleofected into mouse ESCs grown in 1 μg ml−1 doxycycline (Sigma, D9891) to induce iCre expression for 16h. Successfully inserted transgenes were selected using G418 selection (400 μg ml−1, Cellgro) and characterized for expression of tagged transgenic proteins with HA staining (anti-HA, ab9110).
To knock out endogenous alleles of Zbtb10, Zbtb11, and Zfp131, inducible Zbtb10-HA, Zbtb11-HA and Zfp131-HA lines were nucleofected with CMV-GFP and pTF vector (Figure S1D). Two pTF vectors with sgRNAs targeting 100 bp apart from each other with Zbtb10 sgRNAs (5′ TCTTTGTGATGTCAGCATTG and 5′ AGAAACGGCTGCCTGCAACC), Zbtb11 sgRNAs (5′ AGCGCACAAGTCTGTCCTCT and 5′ AGGAGCAGTTTCTAGTCACG), and Zfp131 sgRNAs (5′ TGTATGTGAACTCAATTAAG and 5′ AAGAAGAAGCCAATGATGTG). GFP expressing clones were sorted with a fluorescent activated cell sorter (FACS, BD FACSAria II) 48 h after nucleofection and cells were plated at a single-cell density. Clones were picked after 9 days and knockout alleles were verified by genotyping PCR. Similar strategy was implemented to generate Zbtb11 Zfp131 double knockout line, with first inserting V5 epitope tagged Zfp131 using piggyback cassette (De Santis et al., 2018) into inducible Zbtb11-HA Zbtb11Δ line. Successfully inserted transgenes were selected using blasticidin (100 ug ml−1, Thermofisher) and characterized for expression of tagged transgenic proteins with V5 staining (anti V5, R960-25). To knockout Zfp131, same sgRNAs and same strategy described above to generate single knockout line were used.
H2B-GFP line was generated by nucleofecting the H2B-GFP plasmid (Addgene #11680) into the A17 E14Tg2a line. Randomly inserted H2B-GFP expressing cells were selected clonally and verified by live nuclear GFP expression under Nikon Perfect Focus Eclipse Ti live-cell fluorescence microscope and with FACS Aria.
The Oct4:tdTomato Sox2:Gfp line was generated by inserting an IRES-dtTomato cassette (Swanzey and Stadtfeld, 2016) into the 3′UTR of the endogenous Oct4 locus to KH2-OKSM ESCs (Stadtfeld et al., 2010) similarly as previously described (Lengner et al., 2007). This was followed by heterozygous insertion of an EGFP cassette in the position of the start codon of the endogenous Sox2 locus, using a previously described targeting vector (Ellis et al., 2004).
Mouse ESCs were cultured as in (An et al., 2019), on 0.1% gelatin (Milipore) coated dishes at 37°C, 8% CO2 in ESC medium (Advanced DMEM/F12: Neurobasal (1:1) Medium (Gibco), supplemented with 2.5% mESC-grade fetal bovine serum (Corning), 1× N2 (Gibco), 1× B27 (Gibco), 2 mM L-glutamine (Gibco), 0.1 mM β-mercaptoethanol (Gibco)) supplemented with LIF (leukemia inhibitory factor, 1,000 U ml−1 (Millipore)) and 2-inhibitor cocktail (3 mM CHIR (BioVision) and 1 mM PD0325901 (Sigma)) unless stated otherwise.
NYGCe001-A human female embryonic stem cells (derived from HUES66) were used for the human TF CRISPR screen (Lu and Sanjana, 2019). NYGCe001-A cells were maintained using the Enhanced Culture Platform (ECP), as described previously by Cowan and colleagues (Schinzel et al., 2011). Briefly, hESCs were cultured in Essential 8 media (Thermo Fisher) supplemented with 100 μg/mL Normocin (InvivoGen) and cultured in standard tissue culture dishes coated with Geltrex (Thermo Fisher) at 37°C in 5% CO2. Accutase (STEMCELL) was used for passaging the cells. 10 μM Rho kinase inhibitor (ROCKi) Y-27632 (MilliporeSigma) was added to the culture medium at each passage. ROCKi was removed at the subsequent media change (typically 24 h later).
METHOD DETAILS
Library cloning
Synthesized oligonucleotides (Twist Bioscience) were dissolved in Buffer EB (Qiagen). 16 ng/uL single-stranded pooled oligos were amplified with NEBNext High-Fidelity 2X PCR Master Mix (NEB) with the following PCR protocol: Denaturation 98°C for 30s, 8 cycles of 98°C for 10s, 63°C for 10s, 72°C for 15s, final elongation 72°C for 3 min 40 μg pTF vector containing sgRNA-E + F scaffold with U6 promoter and Cas9-P2A-Zeocin with EFS-NS promoter is digested with Esp3l (Thermo Scientific) in FastDigest Buffer (10X, Thermo Scientific) and 1 mM DTT in 100 uL (Chen et al., 2013). The digested vector is also dephosphorylated in FastAP Thermosensitive Alkaline Phosphatase (1U/μL, Thermo Scientific) in FastDigest Buffer (10X) in 200 μL volume. Purified pooled oligos were cloned into digested pTF vector using 2X Gibson Assembly Master Mix with 10 times molar ratio of pooled oligos to digested vector and transformed to Endura Electro Competent Cells (Lucigen) and plated on LB + Amp plates (Figure S1D). 565 E.coli colonies per guide ratio (>500× coverage) was used to process the 4 Maxi-Prep (Qiagen) reactions. After bacterial transformation, the plasmid library was sequenced to verify uniform sgRNA coverage and minimal bias (Figure S1E).
Lentiviral packaging
12.5 million 293T (ATCC CRL-3216) cells were plated on 4 T225 flasks and grown in DMEM with high glucose, pyruvate (Thermo Scientific) with 10% FBS (Corning), and 2 mM L-glutamine (Gibco). When the cells were 90% confluent, 12.5 mL Opti-MEM with 630 μL Lipofectamine 3000 (Thermo Scientific) was mixed with 12.5 mL of Opti-MEM containing 61.2 μg of pMD2.G, 93.6 μg of psPAX2 and 122.4 μg of CRISPR TF library and 540 μL of P3000 enhancer (Thermo Scientific). After the mixed solution was incubated for 15 min, 6.5 mL of transfection mix is transferred per T225. 5 h later, media containing the transfection mix was replaced with fresh DMEM with 10% FBS. 48h later, the supernatant containing lentivirus was collected from 4 T225 flasks and spun down at 1000 × g for 5 min at 4°C to remove debris. The pooled virus-containing supernatant was filtered through a 0.45 μM PES membrane (VWR). The filtered supernatant was then spun in an ultracentrifuge for 2 h at 20,000 g at 4°C. 30 mL of the supernatant obtained per T225 was concentrated in 300 μL PBS1X containing 10% BSA (Sigma). Aliquots were frozen at −80°C. To prepare individual sgRNA viruses, the above-mentioned protocol for T225 was scaled down to p100 and PEI was used to replace Lipofectamine 3000. 1:2.54 DNA to PEI ratio was used for p100.
Mouse TF CRISPR screen
3 × 106 ESC cells were distributed to 5 12-well dishes (Thermo Scientific) with 5 × 105 mESC cells per well. 4 h after cells were plated, the media was replaced with 8 μL of 100× concentrated library lentivirus mixed in 1 mL ESC medium with 1× polybrene (EMD Millipore). 16 h later, the virus solution was removed, cells were washed 1 time with PBS1X, and cells were split 1:2 to 10 12-well plates. 50 ng/μL Zeocin (Invivogen) was added to each well 24h later and transduced cells were maintained with Zeocin selection. To maintain 50–70% confluency throughout the screen, cells were transferred to 6-well plates (Thermo Fisher) on day 2 and selected for 3 additional days for a total of 5 days of selection. The first time point was obtained on day 5 post-selection. The second and third time-points were taken on day 8 and day 12 while growing in ESC medium containing 50 ng/μL Zeocin.
We performed library readout as described previously (Chen et al., 2015). Briefly, in a 15 mL conical tube, we added 6 mL of NK Lysis Buffer (50 mM Tris (Boston BioProducts), 50 mM EDTA (Ambion), 1% SDS (Invitrogen), pH 8), and 30 μL of 20 mg/mL Proteinase K (Qiagen) to 9 million cells (>500 coverage) and incubated at 55°C overnight. 30 μL of 10 mg/mL RNAse A (Qiagen, diluted in NK Lysis Buffer to 10 mg/mL and then stored at 4°C) was added to the lysed sample, which was then inverted 25 times and incubated at 37°C for 30 min. Samples were cooled on ice before the addition of 2 mL of pre-chilled 7.5 M ammonium acetate (Sigma) to precipitate proteins. After adding ammonium acetate, the samples were vortexed at high speed for 20 s and then centrifuged at ≥ 4,000 × g for 10 min. After the spin, a tight pellet was visible in each tube and the supernatant was carefully decanted into a new 15 mL conical tube. 6 mL 100% isopropanol was added to the tube, inverted 50 times, and centrifuged at ≥4,000 × g for 10 min. Genomic DNA was visible as a small white pellet in each tube. The supernatant was discarded, 6 mL of freshly prepared ice-cold 70% ethanol was added, the tube was inverted 10 times, and then centrifuged at ≥4,000 × g for 1 min. The supernatant was discarded by pouring; the tube was briefly spun, and the remaining ethanol was removed using a P200 pipette. After air-drying for 10–30 min, the DNA changed appearance from a milky white pellet to slightly translucent. At this stage, 500 μL of 1× TE buffer (Sigma) was added, the tube was incubated at 65°C for 1 h and then overnight at room temperature to fully resuspend the DNA. The next day, the gDNA samples were vortexed briefly. The gDNA concentration was measured using a Nanodrop (Thermo Scientific).
5 PCR1 reactions were used for 52 μg of DNA for >500× coverage. Per PCR tube, 10.4 μg of genomic DNA was used in 100 μL volume with 0.4 μL Taq-B Polymerase (Enzymatics), 10X Taq buffer (Enzymatics), 10 mM dNTPs (NEB), 10 μM Fwd (5′ GAGGGCCTATTTCCCATGATTC), and Rvs (5′ GTTGCGAAAAAGAACGTTCACGG) using the following PCR protocol: Denaturation 94°C for 30s, 25 cycles of 94°C for 10s, 55°C for 30s, 68°C for 45s, final elongation 68°C for 2 min. For the addition of Illumina barcodes, 5 staggered forward primers and 1 reverse primer with specific barcodes containing Illumina adaptors were mixed to obtain 10 μM primer mix. 2 PCR2 reactions were used per time point with 5 μL of PCR1 product amplified with 25 μL Q5 High Fidelity 2X Master Mix (NEB), 5 μL of 10 μM primer mix in 50 μL volume using the following PCR protocol: Denaturation 98°C for 30s, 10 cycles of 98°C for 10s, 65°C for 30s, 72°C for 45s, final elongation 72°C for 5 min. All samples were pooled in equimolar ratio and PCR purified pooled sample with QIAquick PCR Purification Kit (Qiagen). The purified sample was then run on a 2% E-Gel and the correct size band was extracted for PCR2 (250–270 bp) and purified with QIAquick Gel Extraction Kit (Qiagen). The pooled library was then quantified with a Low-Range Quantitative ladder on 2% E-Gel. Libraries were then sequenced on Illumina MiSeq v3 for 150 cycles single end at the genomics core facility at NYU.
Sequences flanking the 20 nucleotide sgRNA sequence were trimmed using cutadapt. The remaining 20 nucleotide sgRNA sequence reads were aligned to a library index generated by 17,820 sgRNA sequences using bowtie. Each sgRNA ratio was calculated by normalizing to the total reads captured per time point to be used for downstream analysis.
Human TF CRISPR screen
For the Human TF CRISPR-Cas9 library, we designed sgRNAs to target 1891 known human TFs using the GUIDES web tool (http://guides.sanjanalab.org) with 10 sgRNAs per TF and also included 1000 non-targeting (negative control) sgRNAs (Ravasi et al., 2010; Meier et al., 2017). The library was cloned and packaged using the same methods described above (Library cloning and Lentiviral packaging), except that no BSA was added to concentrated lentivirus.
Briefly, 80 × 106 NYGCe001-A cells were transduced with human TF CRISPR-Cas9 library lentivirus. 2 days after transduction, 4 μg/μL Zeocin (Invivogen) were added to culture media, and then cells were maintained in selection media for 2 days. After selection, cells were cultured for 24 days, then harvested for DNA extraction and library amplification (see DNA extraction and library amplification described in the Mouse TF CRISPR screen section). Cells were passaged whenever they reached approximately 75% confluence. After each passage, at least 10 × 106 cells were maintained in culture to ensure ~500-fold coverage of the total number of sgRNAs in the Human TF CRISPR library.
To identify depleted guide RNAs, we compared sgRNA representation from Day 24 post-selection to the plasmid library. To identify significantly depleted guides, we assessed how many sgRNAs for each TF were depleted below the 5th percentile of non-targeting guide RNAs and also performed robust rank aggregation (RRA) analysis.
Competition experiment with qPCR
U6-sgRNA-E + F scaffold and EFS-NS-Cas9-P2A-ZeocinR-WPRE digested from pTF vector with AflII and HindIII and cloned into the piggyBac backbone (De Santis et al., 2018). sgRNAs targeting Klf5 (5′ TGGCGAATTAACTGGCAGAG), Nanog (5′ TGTCCTTGAGTGCACACAGC), Oct4 (5′ CCGCCCGCATACGAGTTCTG), Zbtb10 (5′ AGAAACGGCTGCCTGCAACC), Zbtb11 (5′ AGCGCACAAGTCTGTCCTCT), Zfp131 (5′ GTTCTTTAAAGTGTCCAAAG) and non-targeting sgRNA (5′ GCCGCAACGTTAGATGTATA) were cloned into piggyBac plasmids. Equimolar ratio (0.5 μg) of plasmids were pooled and co-nucleofected (Lonza) with 0.5 μg pBac transposase to mESCs (De Santis et al., 2018). DNA was collected with >10,000 cells per sgRNA coverage d1, d3, d6, d9, and d13 post-transduction. PCR1 reaction described above was used to amplify the sgRNA construct from the genome. qPCR primers were designed to overlap sgRNA and scaffold (5′ CGGTGCCACTTTTTCAAGTTG). 10 μL Maxima SYBR Green brilliant PCR amplification kit (Thermo Scientific), 5 uL forward and reverse primer mix (2 nM), 5 μL of PCR1 reaction (2 ng) were combined for the qPCR reaction using CFX 96 Touch Biorad qPCR thermocycler (Biorad). Δct was calculated by subtracting the mean Cq of each sgRNA to a non-targeting control. ΔΔct was calculated by subtracting the Δct of each time point from the d1 original abundance. The average depletion rate was drawn with error bars (n = 3).
Oct4::TdTomato Sox2::Gfp experiments
5× 105 Oct4:tdTomato Sox2:Gfp cells were plated at a single-cell density to 6-well plates and after attachment to the plate, transduced with 36 sgRNAs (3 positive controls (Klf5, Nanog, Oct4), 2 non-targeting negative controls and 7 candidate TFs). Transduced cells were selected on 50 ng/μL zeocin for 5 days post-transduction. ESCs were cultured in +2i + LIF media conditions for the initial 4 days and media conditions switched to −2i-LIF for 24h before proceeded to FACS.
Immunocytochemistry
Embryonic stem cells were fixed with 4% paraformaldehyde in 1X PBS for 10 min, washed 3 times with 1X PBS, treated with 0.02% Triton X-(Thermo Fisher) for 10 min and then with blocking buffer for 30 min (10% FBS, 0.05% NaAz in 1X PBS), stained with primary antibody overnight at 4°C, washed 3 times with 1X PBS, stained with secondary antibody at room temperature for 1h, washed 3 times with 1X PBS and mounted with Fluoroshield with DAPI (Sigma). Images were acquired with a Nikon Perfect Focus Eclipse Ti live-cell fluorescence microscope. We used antibodies to V5 (R960-25, Thermo Fisher Scientific; 1:5000), OCT3/4 (Sc-5279, Santa Cruz; 2 mg/mL), GFP (ab13970, Abcam; 1:5000), HA (ab9110, Abcam; 1:5000), Alexa 488 (A-11029), Alexa 568 (A-11036) secondary antibodies were used (Thermo Fisher, 1:2000).
Competition experiments with H2B-GFP
106 H2B-GFP cells were co-plated with 106 E14Tg2a wt, iZbtb10-HA Zbtb10Δ, iZbtb11-HA Zbtb11Δ and iZfp131-HA Zfp131Δ cells on 6-well plates and grown in +2i + LIF conditions without doxycycline. Cells were fixed d1 and d5 after plated and stained for GFP to calculate GFP/DAPI ratio. Due to plating error, d1 values were normalized to 1:1 and d5 values were calculated according to the normalization. Competition rates were calculated with error bars (n = 3).
RNA-seq
Cells were collected after grown for 3d in +2i or −2i conditions in the absence of doxycycline. TRIzol LS Reagent (LifeTechnologies) was used to extract RNA and RNAeasy mini kit (Qiagen) used to purify RNA, as in (Bulajić et al., 2020). Agilent High Sensitivity RNA Screentape (Agilent) was used to determine RNA integrity. 500 ng RNA from cells that was spiked-in with ERCC Exfold Spike-in mixes (Thermo Fisher, 4456739) was used for the generation of RNA-seq libraries. TruSeq Stranded mRNA Library Preparation kit (Illumina, 20020594) was used to prepare RNA-seq libraries. High Sensitivity DNA ScreenTape (Agilent, 5067-5584) was used to verify library sizes. A KAPA library amplification kit was used on Roche Lightcycler 480 to quantify the library prior to sequencing. Libraries were sequenced on Illumina NextSeq 500 using V2.5 chemistry (75 cycles, single-end 75bp) at the Genomics Core Facility at NYU.
ChIPseq
Wildtype and knockout cells were collected after being grown for 3d in +2i or −2i conditions in the presence or absence of 3 ng/uL doxycycline. 1 mM DSG (ProteoChem) was used for crosslinking at RT for 15min. 1% Formaldehyde (vol/vol) was added for an additional 15min until quenched with glycine (Sigma). Cells were separated into aliquots of 25 × 106 and centrifuged to freeze pellets at −80°C. Thawed cells were lysed in 5 mL of 50 mM HEPES-KOH pH 7.5 (Gibco), 140 mM NaCl (Thermo Fisher), 1 mM EDTA pH 8.0, 10% glycerol (vol/vol) (Thermo Fisher), 0.5% Igepal (vol/vol) (Sigma), 0.25% Triton X-100 (vol/vol) with 1×protease inhibitors (Roche) for 10min at 4°C. Cells were centrifuged for 5min at 1100 rpm and resuspended in 5 mL of 10 mM Tris-HCl pH 8.0 (Boston BioProducts), 200 mM NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0 (Boston BioProducts) with 1×protease inhibitors, and incubated for an additional 10 min at 4°C on a rotating platform. Cells were centrifuged for 5min at 1100 rpm and resuspended in 2 mL of Sonication Buffer (50 mM Hepes pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8.0, 1 mM EGTA pH 8.0, 1% Triton X-100 (vol/vol), 0.1% sodium deoxycholate (wt/vol), 0.1% SDS (vol/vol) with 1×protease inhibitors). Sonication was performed in two Bioruptor tubes per sample with 0.45 g sonication beads. Bioruptor Pico (Diagenode) was used for 18 cycles of 30 sec on and 30 sec off to sonicate to an average size of approximately 200 bp. To immunoprecipitate, Dynabeads protein-G (Thermo Fisher) and HA (Abcam ab9110), H3K4me3 (ActiveMotif 39159), H3K27me3 (ActiveMotif 39155), H3K27ac (Abcam ab4729), SIN3A (Abcam ab3479), CHD4 (Abcam ab70469), SETD1A (Abcam ab70378), NELF (Sigma HPA007187) and POL2S5P (Abcam ab5131) antibodies were incubated with sonicated chromatin for 16h at 4°C on a rotating platform. After the immunoprecipitation, the sample was washed with sonication buffer, high salt sonication buffer (500 mM NaCl, LiCl wash buffer (20 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 250 mM LiCl (Sigma), 0.5% Igepal (vol/vol), 0.5% sodium deoxycholate (wt/vol)), and TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8). The sample was eluted in elution buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA pH 8.0, 1% SDS (vol/vol)) by incubating for 45min at 65°C. Crosslinks were reversed by incubating the sample for 16h at 65°C. RNA was removed by 0.2 mg/mL RNAse A (Sigma) in 200 μL of TE for 2h at 37°C. To remove protein, 0.2 mg/mL Proteinase K (Invitrogen) and CaCl2 (Sigma) were added and the sample incubated at 55°C for 30min. DNA was first purified with phenol:chloroform:isoamyl alcohol (25:24:1; vol/vol) (Invitrogen) and then by performing salt-ethanol precipitation. DNA pellets were resuspended in 50 uL H2O. Illumina DNA sequencing libraries were prepared with half of the ChIP sample and a 1:100 dilution of the input sample in H2O. Library preparation was performed with end repair, A-tailing, and ligating multiplexed adapters (Illumina-compatible Bioo Scientific). Agencourt AmpureXP beads (Beckman Coulter) were used to remove unligated adapters. Libraries were then amplified by PCR with TruSeq primers (Sigma) and Phusion polymerase (New England Biolabs). Libraries with sizes ranging between 250-550bp were gel purified (Qiagen). KAPA library amplification kit was used on Roche Lightcycler 480 to quantify the library prior to sequencing. Libraries were sequenced on Illumina NextSeq 500 using V2.5 chemistry (75 cycles, single-end 75bp) at the Genomics Core Facility at NYU.
scRNAseq
Zbtb11Δ and Zfp131Δ cells were collected after being grown for 3d in −2i conditions and mixed with H2B-GFP expressing wt cells in a 9:1 ratio to have 1000 cells/ul. CellTrics 30 μM (Cat #04–004-2326) was used to remove debris and clumps. 10X Genomics Chromium Single Cell 3’ library kit was used to generate the single-cell library as in (Aydin et al., 2019), with a cell recovery rate of 10.000 cells (120262 Chromium™ i7 Multiplex Kit, 120236 Chromium™ Single Cell 3′ Chip Kit v2, 120237 Chromium™ Single Cell 3′ Library & Gel Bead Kit v2). Agilent High Sensitivity DNA D1000 Screentape (5067– 5585) was used to detect fragment length distribution of the library. KAPA library amplification kit was used on Roche Lightcycler 480 to quantify the library prior to sequencing. Libraries were sequenced on Illumina NovaSeq 6000 using SP chemistry (100 cycles, 26 × 98 bp) at the Genomics Core Facility at NYU.
Co-IP experiments
Co-IP was done using Active Motif Nuclear Complex Co-IP Kit (54001) with IP-low buffer and no supplement. Protein G beads (Dynabeads, ThermoFisher, 10004D) were used for IP. All steps followed the standard protocol. IP samples were separated in 6% SDS-PAGE, the proteins were transferred to a PVDF membrane for 15h at 15V in TRIS-Glycine buffer. For the detection of the proteins of interest, membranes were blocked in a non-mammalian blocking buffer in TTBS (LiCOR Biosciences, #927-60001) for 1h at room temperature. The membranes were incubated with primary antibodies in blocking buffer overnight at 4°C, HA mouse monoclonal antibody (1:1000, Thermo Fisher Scientific, #MA1-12429), V5 mouse monoclonal antibody (1:1000, Thermo Fisher Scientific, R960-25). Then, the membranes were incubated with mouse secondary antibody (LiCOR Biosciences, #925-32210) in TBS for 1h in room temperature. The membrane images were captured in Odyssey Clx (LiCOR Biosciences).
NPC differentiation
NPC differentiation experiment was performed as in (Ying et al., 2003). Briefly, dissociated 10,000 single cells were plated on 0.1% gelatin coated coverslips in 24-well plate in +2i + LIF medium. Medium was changed 4h after plating to N2B27 differentiation medium. Media change was also performed after 48h and 72h. 96h after plating, SOX1 was expressed in most of the wt cells. Cells were subjected to above described immunocytochemistry protocol and stained with SOX1 antibody (R&D Systems, AF3369-SP, 1:100). Images were acquired with a Nikon Perfect Focus Eclipse Ti live-cell fluorescence microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Candidate TF identification with false discovery rate
In the False Discovery Rate method, for each time point, normalized counts of each sgRNA were calculated. Log2 scores were then calculated and ranked by taking the log2 of the final count divided by the initial count. We set the thresholds for calling depleted and enriched targeting sgRNAs setting an empirical <0.05 false discovery rate (FDR). Any targeting sgRNA that was underrepresented less than 50th most underrepresented non-targeting sgRNA was determined to be a depleted sgRNA with FDR <0.05 (marked by orange in Figures 1C and S2C). Conversely, any sgRNA that was overrepresented more than the 50th most overrepresented non-targeting sgRNA was determined to be an enriched sgRNA with FDR<0.05 (marked by blue in Figures 1C and S2C). Depleting and enriching sgRNAs were then appointed to genes to determine how many sgRNAs were depleted or enriched per each gene.
Candidate TF identification with linear model
After normalization, a mixed linear model (Gelman et al., 2010) was used to evaluate guide enrichment or depletion. The model has the following formula:
| (Equation 1) |
Where yij is the log counts of guide i at the j-th time point; β0 is the overall intercept; β1 is the overall slope for time; xj is the j-th time point; b0i is the random intercept for guide i and εj is the random error.
A random intercept model was chosen to account for the variability in the guides’ initial observed counts. Hypothesis testing was done using the likelihood ratio test via ANOVA (Kaufmann and Schering, 2014). The model was compared to a reduced version that does not account for time. p value adjustment was performed using the Holm (Holm, 1979) method. An adjusted p value < 0.05 was used to identify candidates.
To take advantage of the time course data captured throughout our experiment, the linear model was modified to classify each of the hits as early, late, or a stably depleting gene. In this new model, the time parameter was encoded using the following factors:
| (Equation 2) |
| timeearly | timelate | Classification |
|---|---|---|
| True | True/False | Early |
| False | False | Stable |
| False | True | Late |
Hypothesis testing was performed as described above. Depending on the significance of each time factor, our genes were classified as follows:
“Early depleting” genes include core cellular machinery and the core PGRN TFs Oct4 and Nanog (Figures S4B-S4D). “Stable and late depleting” genes include cellular stress and DNA repair factors along with accessory TFs in the PGRN, such as Tfcp2l1 (“stable depleting”), Stat3 (“late depleting”), and Nr0b1 (“late depleting”) (Figures S4B and S4E-S4G). Accessory TFs do not rapidly deplete due to the time required for their mutations to exert an effect on the levels of core TFs. The screen depth and time series were sensitive enough to identify sgRNAs targeting partially redundant genes like Klf2 and Klf5 as “late depleting” TFs (Figures S4H and S4I).
RNAseq data processing
RNA-seq fastq files were aligned to the mouse genome (version mm10) using Hisat2 (version 2.1.0) (Kim et al., 2015). FeatureCounts R package was used to assign reads to genomic features (Liao et al., 2014). The DESeq2 package was used for differential gene expression analysis and data visualization (volcano plots, heatmaps, PCA plots) (Love et al., 2014). A q-value cutoff of <0.01 was used for significantly misregulated genes. PANTHER (version 14) (http://geneontology.org) was used to perform Gene Ontology term enrichment analysis (Mi et al., 2019).
scRNAseq data processing
Fastq files were generated by using 10X Genomics CellRanger (version 4.0.0) with default settings. New fasta and annotation files were created using mkref in mm10 for the detection of the H2B:GFP sequence (Addgene #11680). CellRanger count function was used to assign reads to genomic features. 7510 cells were estimated for Zbtb11Δ and H2B-GFP population whereas 6982 cells were estimated for Zfp131Δ and H2B-GFP population. Seurat R package (version 3.0) was used for differential gene expression analysis and data visualization (UMI plots, violin plots, gene-specific UMI plots) (Stuart et al., 2019).
ChIP-seq data processing and differential analysis
Fastq files were aligned to the mouse genome (mm10) using Bowtie (v1.0.1) (Langmead et al., 2009) with options “-q –best–strata -m 1 –chunkmbs 1024”. MultiGPS (v.0.74) was used to call peaks after alignment on all transcription factors (Mahony et al., 2014). A q-value cutoff of <0.01 was used to identify significant binding events. Histone modification and ATAC-seq peaks were called using the DomainFinder module in SeqCode (https://github.com/seqcode/seqcode-core/blob/master/src/org/seqcode/projects/seed/DomainFinder.java). Contiguous 50-bp genomic bins with significantly higher read enrichment compared with normalized input were identified (binomial test, p < 0.05). Differential binding and modification analyses were performed using DESeq2 (v1.28.1) with an adjusted p value cutoff of <0.05 and all default options. To create the count matrix for DESeq2, peaks were first called independently in the wildtype and knockout ChIP-seq datasets. Overlapping peak regions were merged between the wt and KO. Peak regions found exclusively in either file were also included. The final count matrix was created using Bedtools coverage with the “-counts” option separating each replicate into its count column. Before running DESeq2 on the matrix, counts were normalized by the length of the corresponding region for histone modifications (domains have differing lengths).
Motif finding
For motif finding, the top 1000 peaks were selected from each zinc finger ChIP-seq dataset based on MultiGPS q-value. Following this, bedtools getFasta was used to retrieve the sequences underlying these binding events. Sequences were fed through Repeat-Masker (v4.0.7) on default parameters (Smit et al., 1996). Finally, meme-chip from the MEME suite (v4.11.3) (Bailey et al., 2015) was performed on the masked sequences using the options “-meme-nmotifs 5 -meme-mod zoops -meme-minw 6 -meme-maxw 20”.
Data visualization for heatmaps and profile plots
Heatmaps and profile plots were generated using Deeptools (v3.1.3) (Ramírez et al., 2016). Bigwigs were created directly from the BAM files using RPKM normalization within Deeptools bamCoverage (−binSize 50 −normalizeUsing RPKM). Other normalizations were also tested. Using RPKM normalization kept the noise the same visually between replicates and conditions when plotting 5000 random regions in the genome (for within-study datasets). Replicates were concatenated together before creating the bigwig file as suggested by the Deeptools manual. Matrices for the heatmaps and profile plots were created using Deeptools computeMatrix with the options “reference-point −referencePoint center −a 1000 −b 1000”. Heatmaps themselves were generated using Deeptools plotHeatmap with all default options outside of changing sample colors. Max values were chosen by comparing 5000 random noise regions in each sample for within-study data and inspecting these visually. The datasets retrieved from other studies had significantly more noise than from our own. Profile plots were created using Deeptools plotProfile with the option “−perGroup”.
Overlap analysis for ChIPseq data
Overlap percentages were calculated using bedtools intersect with the “−u” option. The first file or “−a” was always the zinc finger TF. In other words, the percentages represent the number of zinc finger TF peaks (A) that intersect the other transcription factor or histone modification regions (B) in question. For Figure 4A, ZBTB11 was used as “−a.” Observed/expected values in Figure 4B were calculated using a python script. In brief, this script created bed files that had the same number of regions and lengths of the regions within the second bed file (B) and intersected these with the zinc finger bed files (A). The script discounted blacklist regions to increase accuracy. This was performed 30 times to get the average number of times one would expect file A to overlap with file B. The mean was used as expected in Figure 5 and the actual value from our data was used as the observed.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-V5 | Thermo Fisher Scientific | Cat#R960-25; RRID:AB_2556564 |
| Mouse monoclonal anti-Oct3/4 | Santa Cruz | Cat#sc-5279; RRID:AB_628051 |
| Chicken polyclonal anti-GFP | Abcam | Cat#ab13970; RRID:AB_300798 |
| Rabbit polyclonal anti-HA | Abcam | Cat#ab9110; RRID:AB_307019 |
| Rabbit polyclonal anti-H3K4me3 | Active Motif | Cat#39159; RRID:AB_2615077 |
| Rabbit polyclonal anti-H3K27me3 | Active Motif | Cat#39155; RRID:AB_2561020 |
| Rabbit polyclonal anti-H3K27ac | Abcam | Cat#ab4729; RRID:AB_2118291 |
| Rabbit polyclonal anti-V5 | Abcam | Cat#ab15828; RRID:AB_443253 |
| Rabbit polyclonal anti-mSIN3A | Abcam | Cat#ab3479; RRID:AB_303839 |
| Mouse monoclonal anti-CHD4 | Abcam | Cat#ab70469; RRID:AB_2229454 |
| Rabbit polyclonal anti-SETD1A | Abcam | Cat#ab70378; RRID:AB_1951955 |
| Rabbit polyclonal anti-NELF | Sigma | Cat#HPA007187; RRID:AB_1856161 |
| Rabbit polyclonal anti-POLS5P | Abcam | Cat#ab5131; RRID:AB_449369 |
| Goat polyclonal anti-SOX1 | R&D Systems | Cat#AF3369; RRID:AB_2239879 |
| Goat polyclonal anti-mouse | LI-COR Biosciences | Cat#925-32210; RRID:AB_2687825 |
| Goat polyclonal anti-mouse, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11029; RRID:AB_2534088 |
| Goat polyclonal anti-rabbit, Alexa Fluor 568 | Thermo Fisher Scientific | Cat#A-11036; RRID:AB_10563566 |
| Bacterial and virus strains | ||
| Endura Electro Competent Cells | Lucigen | Cat#60242 |
| 10-beta Competent E.Coli | NEB | Cat#C3019 |
| Chemicals, peptides, and recombinant proteins | ||
| CHIR 99021 | BioVision | Cat#1677-5; CAS:252917-06-9 |
| PD0325901 | Sigma | Cat#PZ0162; CAS:391210-10-9 |
| LIF | Millipore | Cat#ESG1107 |
| DSG | ProteoChem | Cat#c1104; CAS:79642-50-5 |
| Dynabeads protein-G | Thermo Fisher Scientific | Cat#10004D |
| Doxycycline | Sigma | Cat#D9891 |
| Lipofectamine 3000 Transfection Reagent | Thermo Scientific | Cat#100022052 |
| G418 | Sigma | Cat#1720; CAS: 108321-42-2 |
| Polybrene | EMD Millipore | Cat#TR1003G; CAS: 28728-55-4 |
| Zeocin | Invivogen | Cat#ant-zn-05; CAS: 11006-33-0 |
| PEI | Fisher Scientific | Cat# NC1014320; CAS: 9002-98-6 |
| Agencourt AmpureXP beads | Beckman Coulter | Cat#A63880 |
| Critical commercial assays | ||
| TruSeq Stranded mRNA Library Preparation kit | Illumina | Cat#20020594 |
| 10X Genomics Chromium Single Cell 3’ library kit | 10× Genomics | Cat#PN-120267 |
| Deposited data | ||
| Raw and analyzed sequencing data | This paper | GEO: GSE160966 |
| H3K4me1 ChIPseq data | (Creyghton et al., 2010) | GEO: GSE24165 |
| H3K9me3 ChIPseq data | (Bilodeau et al., 2009) | GEO: GSE18371 |
| OCT4, NANOG, and SOX2 ChIPseq data | (Marson et al., 2008) | GEO: GSE11724 |
| H3K4me2 ChIPseq data | (Meissner et al., 2008) | GEO: GSE11172 |
| ATAC-seq data | (Velasco et al., 2017) | GEO: GSE80511 |
| Experimental models: Cell lines | ||
| Mouse: A17 E14Tg2a | (Iacovino et al., 2011) | Wildtype |
| Mouse: iZbtb10-HA mESC line | This paper | iZbtb10-HA |
| Mouse: iZbtb11-HA mESC line | This paper | iZbtb11-HA |
| Mouse: iZfp131-HA mESC line | This paper | iZfp131-HA |
| Mouse: iZbtb10-HA Zbtb10Δ mESC line | This paper | iZbtb10-HA Zbtb10Δ |
| Mouse: iZbtb11-HA Zbtb11Δ mESC line | This paper | iZbtb11-HA Zbtb11Δ |
| Mouse: iZfp131-HA Zfp131Δ mESC line | This paper | iZfp131-HA Zfp131Δ |
| Mouse: iZbtb11-HA iZfp131-V5 Zbtb11Δ mESC line | This paper | iZbtb11-HA iZfp131-V5 Zbtb11Δ |
| Mouse: iZbtb11-HA iZfp131-V5 Zbtb11Δ Zfp131Δ mESC line | This paper | iZbtb11-HA iZfp131-V5 Zbtb11Δ Zfp131Δ |
| Mouse: Oct4::tdTomato Sox2::Gfp mESC line | This paper | Oct4::tdTomato Sox2::Gfp |
| Mouse: H2B-GFP mESC line | This paper | H2B-GFP |
| Human: HEK 293T cells | ATCC | Cat# CRL-3216, RRID:CVCL_0063 |
| Human: NYGCe001-A hESC line | (Lu and Sanjana, 2019) | RRID:CVCL_YC46 |
| Oligonucleotides | ||
| See Table S3 for all oligonucleotides | This paper | N/A |
| Recombinant DNA | ||
| See Table S3 for all plasmids | This paper | N/A |
| Software and algorithms | ||
| Hisat2 (version 2.1.0) | (Kim et al., 2015) | http://daehwankimlab.github.io/hisat2/ |
| featureCounts (version 1.22.2) | (Liao et al., 2014) | https://www.rdocumentation.org/packages/Rsubread/versions/1.22.2/topics/featureCounts |
| DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| PANTHER (version 16) | (Mi et al., 2019) | http://geneontology.org |
| 10X Genomics CellRanger (version 4.0.0) | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Seurat R package (version 3.0) | (Stuart et al., 2019) | https://satijalab.org/seurat/ |
| MultiGPS (version 0.74) | (Mahony et al., 2014) | https://mahonylab.org/software/multigps/ |
| Bowtie (version 1.0.1) | (Langmead et al., 2009) | http://bowtie-bio.sourceforge.net/index.shtml |
| RepeatMasker (version 4.0.7) | (Smit et al., 1996) | https://www.repeatmasker.org/ |
| MEME suite (version 4.11.3) | (Bailey et al., 2015) | https://meme-suite.org/meme/doc/download.html |
| Deeptools (version 3.1.3) | (Ramírez et al., 2016) | https://deeptools.readthedocs.io/en/develop/ |
Highlights.
A CRISPR screen identifies ZBTB11 and ZFP131 as required TFs for pluripotency
ZBTB11 and ZFP131 bind proximally to pro-differentiation genes
Zbtb11 or Zfp131 mutant ESCs express genes associated with three germ layers
As a result, Zbtb11 or Zfp131 mutant cells lose pluripotency and differentiate
ACKNOWLEDGMENTS
This work was supported by the NICHD (R01HD079682) and NINDS R01NS100897 to E.O.M. N.E.S. is supported by New York University and New York Genome Center startup funds, National Institutes of Health (NIH)/National Human Genome Research Institute (grant nos. R00HG008171 and DP2HG010099), NIH/National Cancer Institute (grant no. R01CA218668), Defense Advanced Research Projects Agency (grant no. D18AP00053), the Sidney Kimmel Foundation, the Melanoma Research Alliance, the Cancer Research Institute, and the Brain and Behavior Foundation. A.M. is supported by the CBIOS training grant from NIGMS (T32GM102057). The Mahony lab is supported by NIGMS (R01GM125722) to S.M. L.S.L.-Z. is supported by the MDC-NYU exchange program. M.S. is supported by grants from Simons Foundation and the Tri-Institutional Stem Cell Initiative by the Starr Foundation (Tri-SCI). The authors would like to thank the Mazzoni lab members, Claude Desplan, Danny Reinberg, and Ana Pombo, for feedback and constructive comments; the NYU Genomics Core facility for technical support and data processing; Disi An for establishing the H2B::GFP reporter line; Silvia Velasco for generating input control for ChIP-seq in ESCs; Roland Schwarz for helpful input on the linear models; Albert Tan for quantifying competition experiments; and BioRender.com for creation of graphical abstract and Figure 7C.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110524.
DECLARATION OF INTERESTS
G.G. is a current employee of Patch Biosciences. S.E.V. is a current employee of Retro Biosciences. N.E.S. is an adviser to Vertex and Qiagen.
REFERENCES
- Ahmad KF, Melnick A, Lax S, Bouchard D, Liu J, Kiang CL, Mayer S, Takahashi S, Licht JD, and Privé GG (2003). Mechanism of SMRT corepressor recruitment by the BCL6 BTB domain. Mol. Cell 12, 1551–1564. 10.1016/S1097-2765(03)00454-4. [DOI] [PubMed] [Google Scholar]
- Albagli O, Dhordain P, Deweindt C, Lecocq G, and Leprince D (1995). The BTB/POZ domain: a new protein-protein interaction motif common to DNA- and actin-binding proteins. Cell Growth Differ. 6, 1193–1198. [PubMed] [Google Scholar]
- An D, Fujiki R, Iannitelli DE, Smerdon JW, Maity S, Rose MF, Gelber A, Wanaselja EK, Yagudayeva I, Lee JY, et al. (2019). Stem cell-derived cranial and spinal motor neurons reveal proteostatic differences between ALS resistant and sensitive motor neurons. Elife 8, e44423. 10.7554/eLife.44423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin B, Kakumanu A, Rossillo M, Moreno-Estellés M, Garipler G, Ringstad N, Flames N, Mahony S, and Mazzoni EO (2019). Proneural factors Ascl1 and Neurog2 contribute to neuronal subtype identities by establishing distinct chromatin landscapes. Nat. Neurosci 22, 897–908. 10.1038/s41593-019-0399-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuara V, Perry P, Sauer S, Spivakov M, Jørgensen HF, John RM, Gouti M, Casanova M, Warnes G, et al. (2006). Chromatin signatures of pluripotent cell lines. Nat. Cell Biol 8, 532–538. 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME suite. Nucleic Acids Res. 43, W39–W49. 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell VJ, and Treisman R (1994). The POZ domain: a conserved protein-protein interaction motif. Genes Dev. 8, 1664–1677. 10.1101/gad.8.14.1664. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, and Zhao K (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. (2006). A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 125, 315–326. 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bilodeau S, Kagey MH, Frampton GM, Rahl PB, and Young RA (2009). SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 23, 2484–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bledau AS, Schmidt K, Neumann K, Hill U, Ciotta G, Gupta A, Torres DC, Fu J, Kranz A, Stewart AF, et al. (2014). The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development 141, 1022–1035. [DOI] [PubMed] [Google Scholar]
- Boroviak T, Stirparo GG, Dietmann S, Hernando-Herraez I, Mohammed H, Reik W, Smith A, Sasaki E, Nichols J, and Bertone P (2018). Single cell transcriptome analysis of human, marmoset and mouse embryos reveals common and divergent features of preimplantation development. Development 145, dev167833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. (2005). Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–956. 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulajić M, Srivastava D, Dasen JS, Wichterle H, Mahony S, and Mazzoni EO (2020). Differential abilities to engage inaccessible chromatin diversify vertebrate HOX binding patterns. Development 147, dev.194761. 10.1242/dev.194761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012).The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2, 401–404. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, Vrana J, Jones K, Grotewold L, and Smith A (2007). Nanog safeguards pluripotency and mediates germline development. Nature 450, 1230–1234. 10.1038/nature06403. [DOI] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, et al. (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491. 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, Scott DA, Song J, Pan JQ, Weissleder R, et al. (2015). Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160, 1246–1260. 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Markoulaki S, Levine SS, Hanna J, Lodato MA, Sha K, Young RA, Jaenisch R, and Boyer LA (2008). H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell 135, 649–661. 10.1016/j.cell.2008.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Nat. Acad. Sci 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Alland L, Hong S-H, Wong C-W, DePinho RA, and Dejean A (1998). Histone deacetylase associated with mSin3A mediates repression by the acute promyelocytic leukemia-associated PLZF protein. Oncogene 16, 2549–2556. 10.1038/sj.onc.1202043. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovey OM, Foster CT, and Cowley SM (2010). Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc. Nat. Acad. Sci 107, 8242–8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni S, Duttagupta R, Cheng J, Dehghani H, Hoeppner DJ, Dash C, Bazett-Jones DP, Le Grice S, McKay RDG, Buetow KH, et al. (2008). Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2, 437–447. 10.1016/j.stem.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff S, Dienstbier M, and Murphy S (2012). Updating the RNA polymerase CTD code: adding gene-specific layers. Trends Genet. 28, 333–341. 10.1016/j.tig.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Ellis P, Fagan BM, Magness ST, Hutton S, Taranova O, Hayashi S, McMahon A, Rao M, and Pevny L (2004). SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev. Neurosci 26, 148–165. 10.1159/000082134. [DOI] [PubMed] [Google Scholar]
- Evans MJ, and Kaufman MH (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156. 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- Ferrai C, Torlai Triglia E, Risner-Janiczek JR, Rito T, Rackham OJ, Santiago I, Kukalev A, Nicodemi M, Akalin A, Li M, et al. (2017). RNA polymerase II primes Polycomb-repressed developmental genes throughout terminal neuronal differentiation. Mol. Syst. Biol 13, 946. 10.15252/msb.20177754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festuccia N, Osorno R, Halbritter F, Karwacki-Neisius V, Navarro P, Colby D, Wong F, Yates A, Tomlinson SR, and Chambers I (2012). Esrrb is a direct Nanog target gene that can substitute for Nanog function in pluripotent cells. Cell Stem Cell 11, 477–490. 10.1016/j.stem.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman A, and Hill J (2010). Multilevel linear models: the basics. In Data Analysis Using Regression and Multilevel/Hierarchical Models, pp. 251–278. 10.1017/cbo9780511790942.016. [DOI] [Google Scholar]
- Ghetu AF, Corcoran CM, Cerchietti L, Bardwell VJ, Melnick A, and Privé GG (2008). Structure of a BCOR corepressor peptide in complex with the BCL6 BTB domain dimer. Mol. Cell 29, 384–391. 10.1016/j.molcel.2007.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S (1979). Board of the Foundation of the Scandinavian Journal of Statistics, 6 (Wiley; ), pp. 65–70. http://www.jstor.org/stable/4615733. [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol 31, 827–832. 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Gao X, Li W, Jiang S, Li R, Hong H, Zhao C, Zhou P, Chen H, Bo X, et al. (2019). Stable H3K4me3 is associated with transcription initiation during early embryo development. Bioinformatics 35, 3931–3936. 10.1093/bioinformatics/btz173. [DOI] [PubMed] [Google Scholar]
- Iacovino M, Bosnakovski D, Fey H, Rux D, Bajwa G, Mahen E, Mitanoska A, Xu Z, and Kyba M (2011). Inducible cassette exchange: a rapid and efficient system enabling conditional gene expression in embryonic stem and primary cells. Stem Cell. 29, 1580–1588. 10.1002/stem.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihry RJ, Salick MR, Ho DJ, Sondey M, Kommineni S, Paula S, Raymond J, Henry B, Frias E, Wang Q, Worringer KA, Ye C, Russ C, Reece-Hoyes JS, Altshuler RC, Randhawa R, Yang Z, McAllister G, Hoffman GR, Dolmetsch R, and Kaykas A (2019). Genome-scale CRISPR screens identify human pluripotency-specific genes. Cell Rep. 27, 616–630.e6. 10.1016/j.celrep.2019.03.043. [DOI] [PubMed] [Google Scholar]
- Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, Schafer X, Lun Y, and Lemischka IR (2006). Dissecting self-renewal in stem cells with RNA interference. Nature 442, 533–538. 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, and Young R (2008). Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 132, 567–582. 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamaladdin S, Kelly RDW, O’Regan L, Dovey OM, Hodson GE, Millard CJ, Portolano N, Fry AM, Schwabe JWR, and Cowley SM (2014). Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. Proc. Natl. Acad. Sci. U S A 111, 9840–9845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantzal E, Schulz H, Hummel O, Hubner N, Hatzopoulos A, and Kretsovali A (2008). Histone deacetylase inhibition accelerates the early events of stem cell differentiation: transcriptomic and epigenetic analysis. Genome Biol. 9, R65. 10.1186/gb-2008-9-4-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann J, and Schering A (2014).Analysis of Variance ANOVA (Wiley Stats-Ref: Statistics Reference Online). 10.1002/9781118445112.stat06938. [DOI] [Google Scholar]
- Kawamura T, Ono K, Morimoto T, Wada H, Hirai M, Hidaka K, Morisaki T, Heike T, Nakahata T, Kita T, et al. (2005). Acetylation of GATA-4 is involved in the differentiation of embryonic stem cells into cardiac myocytes. Journal of Biological Chemistry 280, 19682–19688. 10.1074/jbc.M412428200. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Chu J, Shen X, Wang J, and Orkin SH (2008). An extended transcriptional network for pluripotency of embryonic stem cells. Cell 132, 1049–1061. 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, and Eisenman RN (1997). Histone deacetylases associated with the mSin3 corepressor mediate Mad transcriptional repression. Cell 89, 349–356. 10.1016/S0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengner CJ, Camargo FD, Hochedlinger K, Welstead GG, Zaidi S, Gokhale S, Scholer HR, Tomilin A, and Jaenisch R (2007). Oct4 expression is not required for mouse somatic stem cell self-renewal. Cell Stem Cell 1, 403–415. 10.1016/j.stem.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, and Izpisua Belmonte JC (2018). Deconstructing the pluripotency gene regulatory network. Nat. Cell Biol 20, 382–392. 10.1038/s41556-018-0067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, and Sanjana NE (2019). Generation of a knock-in MAP2-tdTomato reporter human embryonic stem cell line with inducible expression of NEUROG2/1 (NYGCe001-A). Stem Cell Res. 41, 101643. 10.1016/j.scr.2019.101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T (2016). Regulation of hematopoietic development by ZBTB transcription factors. Int. J. Hematol 104, 310–323. 10.1007/s12185-016-2035-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony S, Edwards MD, Mazzoni EO, Sherwood RI, Kakumanu A, Morrison CA, Wichterle H, and Gifford DK (2014). An integrated model of multiple-condition ChIP-seq data reveals predeterminants of Cdx2 binding. PLoS Comput. Biol 10, e1003501. 10.1371/journal.pcbi.1003501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, et al. (2008). Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 134, 521–533. 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Wang X, Maeda M, Canver MC, Sher F, Funnell APW, Fisher C, Suciu M, Martyn GE, Norton LJ, et al. (2016). Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 351, 285–289. 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JA, Zhang F, and Sanjana NE (2017). GUIDES: sgRNA design for loss-of-function screens. Nat. Methods 14, 831–832. 10.1038/nmeth.4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. (2008). Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770. 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick A, Carlile G, Ahmad KF, Kiang CL, Corcoran C, Bardwell V, Prive GG, and Licht JD (2002). Critical Residues within the BTB Domain of PLZF and Bcl-6 Modulate Interaction with Corepressors. Mol. Cell Biol 22, 1804–1818. 10.1128/mcb.22.6.1804-1818.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, et al. (2017). Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545, 229–233. 10.1038/nature22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426. 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560. 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, and Yamanaka S (2003). The homeoprotein nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 113, 631–642. 10.1016/S0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- Murray SC, Lorenz P, Howe FS, Wouters M, Brown T, Xi S, Fischl H, Khushaim W, Rayappu JR, Angel A, et al. (2019). H3K4me3 is neither instructive for, nor informed by, transcription. Preprint at bioRxiv. 10.1101/709014. [DOI] [Google Scholar]
- Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, and Adelman K (2007). RNA polymerase is poised for activation across the genome. Nat. Genet 39, 1507–1511. 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Schöler H, and Smith A (1998). Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 95, 379–391. 10.1016/S0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- Niwa H, Miyazaki J.-i., and Smith AG (2000). Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet 24, 372–376. 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- Nowotschin S, Setty M, Kuo Y-Y, Liu V, Garg V, Sharma R, Simon CS, Saiz N, Gardner R, Boutet SC, et al. (2019). The emergent landscape of the mouse gut endoderm at single-cell resolution. Nature 569, 361–367. 10.1038/s41586-019-1127-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiewicz U, Gładych M, Raman AT, Heyn H, Mereu E, Chlebanowska P, Andrzejewska A, Sozańska B, Samant N, Fąk K, et al. (2017). TRIM28 and interacting KRAB-ZNFs control self-renewal of human pluripotent stem cells through epigenetic repression of pro-differentiation genes. Stem Cell Rep. 9, 2065–2080. 10.1016/j.stemcr.2017.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, et al. (2017). Identification of essential genes for cancer immunotherapy. Nature 548, 537–542. 10.1038/nature23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüing B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, Tan K, Akalin A, Schmeier S, Kanamori-Katayama M, Bertin N, et al. (2010). An Atlas of Combinatorial Transcriptional Regulation in Mouse and Man. Cell 140, 744–752. 10.1016/j.cell.2010.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santis R, Garone MG, Pagani F, de Turris V, Di Angelantonio S, and Rosa A (2018). Direct conversion of human pluripotent stem cells into cranial motor neurons using a piggyBac vector. Stem Cell Research 29, 189–196. 10.1016/j.scr.2018.04.012. [DOI] [PubMed] [Google Scholar]
- Schinzel RT, Ahfeldt T, Lau FH, Lee YK, Cowley A, Shen T, Peters D, Lum DH, and Cowan CA (2011). Efficient culturing and genetic manipulation of human pluripotent stem cells. PLoS One 6, e27495. 10.1371/journal.pone.0027495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE (1948). A Mathematical Theory of Communication. Bell Syst. Tech. J 27, 379–423. 10.1002/j.1538-7305.1948.tb01338.x. [DOI] [Google Scholar]
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, and Vakoc CR (2015). Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol 33, 661–667. 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohat S, and Shifman S (2019). Genes essential for embryonic stem cells are associated with neurodevelopmental disorders. Genome Res. 29, 1910–1918. 10.1101/gr.250019.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R, and Green P (1996). RepeatMasker Open-3.0, Repeat-Masker Open-3.0. [Google Scholar]
- Stadtfeld M, Maherali N, Borkent M, and Hochedlinger K (2010). A reprogrammable mouse strain from gene-targeted embryonic stem cells. Nat. Methods 7, 53–55. 10.1038/nmeth.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock JK, Giadrossi S, Casanova M, Brookes E, Vidal M, Koseki H, Brockdorff N, Fisher AG, and Pombo A (2007). Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat. Cell Biol 9, 1428–1435. 10.1038/ncb1663. [DOI] [PubMed] [Google Scholar]
- Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, and Privé GG (2005). Sequence and structural analysis of BTB domain proteins. Genome Biol. 6, R82. 10.1186/gb-2005-6-10-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surface LE, Thornton SR, and Boyer LA (2010). Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell 7, 288–298. 10.1016/j.stem.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Swanzey E, and Stadtfeld M (2016). A reporter model to visualize imprinting stability at the Dlk1 locus during mouse development and in pluripotent cells. Development 143, 4161–4166. 10.1242/dev.138255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CC, Cao K, Collings CK, Marshall SA, Rendleman EJ, Ozark PA, Chen FX, Morgan MA, Wang L, and Shilatifard A (2017). Histone H3K4 methylation-dependent and -independent functions of set1A/COMPASS in embryonic stem cell self-renewal and differentiation. Genes Dev. 31, 1732–1737. 10.1101/gad.303768.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varlakhanova N, Cotterman R, Bradnam K, Korf I, and Knoepfler PS (2011). Myc and Miz-1 have coordinate genomic functions including targeting Hox genes in human embryonic stem cells. Epigenetics & Chromatin 4, 20. 10.1186/1756-8935-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw NL, and Schier AF (2012). Bivalent histone modifications in early embryogenesis. Current Opinion in Cell Biology 24, 374–386. 10.1016/j.ceb.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco S, Ibrahim MM, Kakumanu A, Garipler G, Aydin B, Al-Sayegh MA, Hirsekorn A, Abdul-Rahman F, Satija R, Ohler U, et al. (2017). A multi-step transcriptional and chromatin state cascade underlies motor neuron programming from embryonic stem cells. Cell Stem Cell 20, 205–217. 10.1016/j.stem.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BC, Boehme L, Annibali A, Hodgkinson A, Carroll TS, Oakey RJ, and Seitan VC (2020). Intellectual disability-associated factor Zbtb11 cooperates with NRF-2/GABP to control mitochondrial function. Nat. Commun 11, 5469. 10.1038/s41467-020-19205-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Wong J, Moreno GT, Young MK, Côté J, and Wang W (1998). NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell 2, 851–861. 10.1016/S1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, Hasegawa J, and Handa H (1999). NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 97, 41–51. 10.1016/S0092-8674(00)80713-8. [DOI] [PubMed] [Google Scholar]
- Ye B, Liu B, Yang L, Zhu X, Zhang D, Wu W, Zhu P, Wang Y, Wang S, Xia P, et al. (2018). LncKdm2b controls self-renewal of embryonic stem cells via activating expression of transcription factor Zbtb3. EMBO J. 37, e97174. 10.15252/embj.201797174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz A, Peretz M, Aharony A, Sagi I, and Benvenisty N (2018). Defining essential genes for human pluripotent stem cells by CRISPR-Cas9 screening in haploid cells. Nat. Cell Biol 20, 610–619. 10.1038/s41556-018-0088-1. [DOI] [PubMed] [Google Scholar]
- Ying Q-L, Stavridis M, Griffiths D, Li M, and Smith A (2003). Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat. Biotechnol 21, 183–186. 10.1038/nbt780. [DOI] [PubMed] [Google Scholar]
- Ying Q-L, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, and Smith A (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–523. 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J, Stark A, Kellis M, Hong J-W, Nechaev S, Adelman K, Levine M, and Young RA (2007). RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat. Genet 39, 1512–1516. 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data (ChIP-seq, RNA-seq, scRNA-seq, GeCKO-seq) has been deposited at the GEO database under accession number GSE160966. We performed re-analysis of data sourced from GEO database entries GEO: GSE24165 (H3K4me1 ChIP-seq) (Creyghton et al., 2010), GEO: GSE18371 (H3K9me3 ChIPseq) (Bilodeau et al., 2009), GEO: GSE11724 (OCT4, NANOG, SOX2 ChIPseq) (Marson et al., 2008), GEO: GSE11172 (H3K4me2 ChIPseq) (Meissner et al., 2008), and GEO: GSE80511 (ATAC-seq) (Velasco et al., 2017).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.