

Abstract

Imines are multipurpose pharmacophores, simply accessible compounds, and have a broad range of usage in several areas of chemistry especially in medicine. Two novel compound imines, (E)-4-methyl-2-((o-tolylimino)methyl)phenol (1) and (E)-2-(((4-methoxybenzyl)imino)methyl)-4-methylphenol (2), were synthesized with effective product via reported protocol in the literature. Single crystal X-ray diffraction (SCXRD) was employed for structural exposition, disclosing that both compounds are orthorhombic. To optimize the newly designed imines, a B3LYP functional with a basis set 6-31G(d,p) was mainly considered. DFT results were utilized to check correlation between the data recovered from SCXRD outcomes and also to measure the energy difference. Hirshfeld surface study was done to demonstrate the intermolecular contacts along the percentage of interaction in the overall crystalline compound. Molecular operating environment program was tested against AChE and BChE enzymes to perform a modeling study of the compounds. The docking score and binding affinity of the compounds revealed that 2 showed comparatively more inhibition than 1. In silico ADMET studies exposed the physiochemical nature of these novel compounds, and it also unveiled that both compounds behaved as drug-like candidates.
1. Introduction
Imines are now well recognized by the name of Schiff bases. Amines are reacted by a condensation process with derivatives of aldehydes and ketones to give imines. Dyes and coordination polymers are synthesized using Schiff bases on a large scale.1 Imines based ligands are considered the most reliable ligands owing to the lesser efforts required for their synthesis and notable versatility. Transition metals formed various types of stable complexes with these ligands and consequently played an extensive part in the advancement of coordination chemistry.2 The condensation process of diamines with salicylaldehyde yields imine based ligands that have [N2O2] donor sets and are available for formation of stable coordination compounds.3 The biological activity of salicylaldehydes and their derivatives was studied.4,5
The process of biosynthesis of an ergosterol is inhibited by using a derivative of benzylamine that was also prescribed to cure not only tinea pedis and tinea corporis but also tinea cruris.6 Transition metals form a very stable complex assembly with salicylaldehyde that is used as an antimalarial.7 The cell toxicity of imines was determined by a LDH cell toxicity test, and antiproliferative activity was determined by means of MTT cell proliferation.8
Schiff bases are known as an imperative class of carbon-based compounds with extensive biological applications.9 Synthesis of novel therapeutic imines is now under an important consideration of pharmaceutical researchers for treatment of several illnesses. A number of studies have discussed the biotic actions of imines, together with their herbicidal, anticancer, and antimycotic activities.10
Molecules with an active site of receptors can be studied by molecular modeling which also supports the analysis of the structure–activity relationship (SAR) of compounds.11 Modeling studies also exhibit the binding energies, contact approach, and locations of interactions.12 The interactions of molecules fitted with receptor protein are helpful in examining the nonhydrophilic interaction, hydrogen bonding, and binding energy.13
Density Functional Theory (DFT) was accomplished to minimize the energy of those solids molecules having large numbers of electrons. In the past, DFT was used just for calculations of bond structure and properties of solids molecules; however, now DFT calculations are carried out for quantum chemical analysis.14 The best methodology to devise DFT is given by Kohn and Sham in which they explained a practical approach for measuring the energy properties. The Thomas Fermi approximation which considers the energy and density relationship and its effects supports much of the DFT studies.15 DFT is also helpful in order to determine the strength of the bond, electron affinities, and ionization energies.16 Molecular assembly directs the various electrochemical parameters as well as the contact of drug molecular binding sites. Hence, the foundation of in silico study is mainly the assessment of the lowest most energy value of a molecule. DFT is used for computation of relative conformational energies.17,18
A neurodegenerative syndrome often known as Alzheimer’s disease (AD) is common among old age people and develops as continuous memory loss and cognitive failure. The lower level of acetylcholine (Ach) concentration in the brain is the foremost cause of AD. A number of reported methods have been observed for improvement of cholinergic neurotransmission, including the rise of acetylcholine synthesis, presynaptic acetylcholine release, and lessening the synaptic acetylcholine’s degradation with cholinesterase inhibitors. The inhibitors of acetylocholinoesterase (AChE) and butyrylcholinoesterase (BChE) are well thought as significant targets for therapeutic plans of AD.19 In a current study new salicylaldehyde derived imines have been synthesized with imperative biological activity. The structure of their compounds has been elucidated by means of SCXRD and their computational studies have been performed by density functional theory, molecular modeling, adsorption, distribution, metabolism, excretion, and toxicity (ADMET).
2. Materials and Methods
In this work the chemicals used were ethanol, amine, aromatic aldehyde, and others reagents/solvents. They were of analytical grade and purchased from Merck (Germany).
2.1. Synthesis of (E)-4-methyl-2-((o-tolylimino)methyl)phenol imine (1) and (E)-2-(((4-methoxybenzyl)imino)methyl)-4-methylphenol imines (2)
Imines were designed by the stated method with some modifications.20 (E)-4-methyl-2-((o-tolylimino)methyl)phenol 1 and (E)-2-(((4-methoxybenzyl)imino)methyl)-4-methylphenol 2 were synthesized in ethanol (20 mL) by refluxing o-toluidine (0.02 mmol), (4-methoxyphenyl)methanamine (0.02 mmol), respectively, with 2-hydroxy-5-methylbenzaldehyde (0.02 mmol) in ethanol (20 mL). Crystal materials were attained after the mixture was stirred up to 5 h under reflux, washed with ethanol, and dried at room temperature. The synthetic route for 1 and 2 is given in Scheme 1.
Scheme 1. Synthetic Scheme of 1 and 2.

2.2. Single Crystal Structure Analysis
The image plate diffractometer STOE IPDS II was run up to 296 K to obtain the SCXRD. The structural assemblies of compounds were elucidated by SHELXT as a direct method21 and processed completely via full-matrix least-squares process along with the WinGX program22 associated also with the SHELXL module.23 The chief parameters of anisotropic displacement were added to refine the non-hydrogen atoms. The crystallographic tools were PLATON,24 ORTEP-3,22 and MERCURY,25 utilized for the structural demonstration along the evaluation of consequences.
2.3. Density Functional Theory
A density functional theory (DFT) study was positively led by means of Gaussian 09 using B3LYP as a functional method along the 6-31G(d,p) basis set to minimize the energy of compounds.26−28 The B3LYP method mainly comprises Becke’s three-parameter (B3) exchange functional in conjunction with the Lee Yang and Parr (LYP) associated functional.29 Gauss View 5.0 software was run to calculate the energy gap measurement of the synthesized compounds and other outcomes of the DFT studies.30 To optimize compounds, the input records were obtained from the crystal assembly data of the corresponding compound in order to get good coherence using the empirical data.31
2.4. Hirshfeld Surfaces Analysis
Hirshfeld surface analysis (HS) was executed to explore and determine the involvement of the various interactions in a crystalline environment.32 Crystal Explorer 17.5 software was used to calculate two-dimensional (2D) fingerprint plots for analysis of surface contact on a Hirshfeld surface.33 The dnorm is known as normalized contact distance that depends upon de and di that are measured from a standard equation.34
2.5. Molecular Modeling Studies
Compounds 1 and 2 were docked by Molecular Operating Environment (2016.08) software.2 In molecular modeling two PDB files 1EVE and 1P0I, the first one for AChE (acetylocholinoesterase) and the second for BChE (butyrylcholinoesterase) enzymes, were chosen. The 2D molecule-receptor interactions were exhibited by means of MOE. The docking outcomes, contacts of ligand, and surface analyses were viewed by means of the Discovery Studio program.
2.6. In Silico Adsorption, Distribution, Metabolism, Excretion, and Toxicity
The evaluation profile of the ADMET is significant for newly discovered drugs and assessment of their pharmacodynamic activities. ADMET deals with promising parameters as physiochemical properties, pharmacokinetics, synthetic accessibility, drug likeness, and lipophilicity of newly synthesized compounds. At present, a number of sources are available as online and offline to check the drug-like potential of synthesized compounds.35 In the current study, in silico analysis was done using the admetSAR online available prediction tool (http://lmmd.ecust.edu.cn:8000/).
3. Results and Discussion
3.1. SCXRD Assessment of 1 and 2
Compounds were synthesized by the reported protocol with good yield and recrystallized just on the slow rate of solvent evaporation at room temperature. The reaction completion was analyzed via thin layer chromatography (TLC). The structure elucidation of 1 and 2 was done by single crystal X-ray diffraction analysis. X-ray diffraction assessment of 1 was accomplished and the outcomes are arranged in Table 1. The geometry of the crystallized compound is orthorhombic, with formula unit Z = 4, C15H15NO, and also having space group P212121. The structure of the compound is illustrated in Figure 1 that also displays the intramolecular (O1–H1···N1) H-bonding. However, intra-atomic distances in the molecular structure C15H15NO are 1.291(3) Å and 1.429(3) Å between C8–N1 and N1–C7 groups, respectively. In a unit cell, the crystal assembly of the compound is exhibited in Figure 2. In the case of 2 X-ray analysis data are also given in Table 1. Data have revealed that the compound crystallized in orthorhombic form, with formula unit Z = 4, C16H17NO2 and also has P212121 space group. The structure of the compound, shown in Figure 3, also possesses intramolecular (O1–H1···N1) H-bonding, although the intra-atomic distances in the structure of C16H17NO2 are 1.469(3) Å and 1.270(3) Å between N1–C9 and N1–C8, respectively. The crystal assembly of 2 in a unit cell is exhibited in Figure 4.
Table 1. Single Crystal X-ray Diffraction Data of Synthesized Compounds.
| crystal data | 1 | 2 |
|---|---|---|
| CCDC | 2090884 | 2090885 |
| empirical formula | C15H15NO | C16H17NO2 |
| formula weight | 225.28 | 255.30 |
| temp/K | 296(2) | 293(2) |
| crystal system | orthorhombic | orthorhombic |
| space group | P212121 | P212121 |
| a/Å | 7.6501(6) | 5.7310(2) |
| b/Å | 12.0632(10) | 12.9388(8) |
| c/Å | 13.6914(16) | 18.8260(10) |
| α/deg | 90 | 90 |
| β/deg | 90 | 90 |
| γ/deg | 90 | 90 |
| vol/Å3 | 1263.5(2) | 1395.99(12) |
| Z | 4 | 4 |
| ρcalcg/cm3 | 1.184 | 1.215 |
| μ/mm–1 | 0.074 | 0.080 |
| F(000) | 480.0 | 544.0 |
| crystal size/mm3 | 0.72 × 0.323 × 0.09 | 0.72 × 0.323 × 0.09 |
| radiation | Mo Kα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 4.5 to 51.97 | 3.82 to 56.134 |
| index ranges | –8 ≤ h ≤ 9, –14 ≤ k ≤ 14, –16 ≤ l ≤ 16 | –7 ≤ h ≤ 6, –17 ≤ k ≤ 17, –24 ≤ l ≤ 24 |
| reflections collected | 8899 | 24167 |
| independent reflections | 2485 [Rint = 0.0525, Rsigma = 0.0412] | 3369 [Rint = 0.0655, Rsigma = 0.0313] |
| data/restraints/parameters | 2485/0/158 | 3369/0/176 |
| goodness-of-fit on F2 | 0.968 | 0.947 |
| final R indexes [I ≥ 2σ(I)] | R1 = 0.0395, wR2 = 0.0779 | R1 = 0.0369, wR2 = 0.0867 |
| final R indexes [all data] | R1 = 0.0635, wR2 = 0.0849 | R1 = 0.0579, wR2 = 0.0959 |
| largest diff. peak/hole/e Å–3 | 0.10/–0.10 | 0.08/–0.12 |
| Flack parameter | 1(2) | –0.8(17) |
Figure 1.

A single crystal structure of 1.
Figure 2.

A close crystal assembly of 1.
Figure 3.
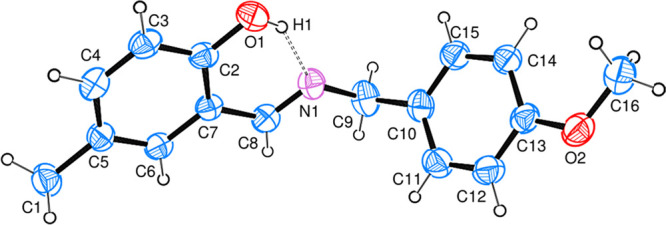
A single crystal structure of 2.
Figure 4.

A close crystal assembly of 2.
3.2. HOMO–LUMO Analysis
Gaussian 09 software was used to optimize the newly prepared imine based compounds using the B3LYP functional and with basis set of 6-31G (d,p). The energy minimized structures of 1 and 2 were illustrated in Figure 5. To compare DFT and XRD outcomes, optimized values of bonds (length and angle) were attained. It has depicted from evaluation of experimental and theoretical results that there is a strong coherence found as shown in Tables 2–5. Moreover, agreement in DFT and XRD estimations were also expressed on the basis of correlation coefficient (R2). In 1, R2 values measured both for bond length and bond angle are 0.9581 and 0.6454, respectively, also shown in Figures 6 and 7. In 2, R2 values are 0.9861 (bond length) and 0.9424 (bond angle) as presented in Figures 8 and 9. These consequences determined that the electrochemical parameters were approximately the same; therefore, the correlation coefficient is near to 1.0. An inconsequential variance in 1 was due to the different design phase of both studies. The HOMO, HOMO–1, LUMO, and LUMO+1 energy orbitals and their energy gaps between the different orbitals were also determined by DFT. The calculated energy gap in HOMO and LUMO of 1 was 0.14361, although for HOMO–1 and LUMO+1 it was 0.2296 as given in Figure 10. The energy gap for the HOMO and LUMO of 2 was 0.1592 and in the same way for HOMO–1 and LUMO+1 was 0.21745 (Figure 11). The energy gap in 1 and 2 is relatively satisfactory to stabilize the compounds.36
Figure 5.

DFT energy minimized structures of the synthesized 1 and 2.
Table 2. Comparison of Bond Lengths for 1.
| length/Å |
length/Å |
||||||
|---|---|---|---|---|---|---|---|
| atom | atom | XRD | DFT | atom | atom | XRD | DFT |
| O1 | C14 | 1.369(3) | 1.36269 | C7 | C2 | 1.406(3) | 1.41684 |
| N1 | C7 | 1.429(3) | 1.41726 | C7 | C6 | 1.390(3) | 1.40752 |
| N1 | C8 | 1.291(3) | 1.30560 | C14 | C13 | 1.389(3) | 1.40376 |
| C9 | C10 | 1.401(3) | 1.41379 | C2 | C3 | 1.391(4) | 1.40183 |
| C9 | C14 | 1.405(3) | 1.42294 | C2 | C1 | 1.515(4) | 1.51024 |
| C9 | C8 | 1.456(3) | 1.44639 | C13 | C12 | 1.384(4) | 1.39024 |
| C10 | C11 | 1.393(3) | 1.39265 | C6 | C5 | 1.386(4) | 1.39629 |
| C11 | C12 | 1.400(4) | 1.41536 | C3 | C4 | 1.373(4) | 1.39968 |
| C11 | C15 | 1.516(3) | 1.51293 | C5 | C4 | 1.376(4) | 1.39857 |
Table 5. Comparison of Bond Angles for 2.
| angle/deg |
angle/deg |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| atom | atom | atom | XRD | DFT | atom | atom | atom | XRD | DFT |
| C13 | O2 | C16 | 117.40(17) | 118.77 | C10 | C15 | C14 | 122.14(19) | 121.38 |
| C8 | N1 | C9 | 119.75(19) | 120.34 | C3 | C4 | C5 | 121.7(2) | 121.80 |
| C6 | C7 | C2 | 118.67(17) | 119.01 | C15 | C14 | C13 | 119.2(2) | 119.33 |
| C6 | C7 | C8 | 120.39(16) | 120.59 | O2 | C13 | C14 | 124.2(2) | 124.39 |
| C2 | C7 | C8 | 120.94(18) | 120.39 | O2 | C13 | C12 | 116.67(19) | 115.52 |
| C6 | C5 | C4 | 117.12(19) | 117.66 | C12 | C13 | C14 | 119.1(2) | 120.08 |
| C6 | C5 | C1 | 121.39(19) | 121.63 | C15 | C10 | C11 | 117.6(2) | 118.31 |
| C4 | C5 | C1 | 121.5(2) | 120.70 | C15 | C10 | C9 | 120.9(2) | 120.93 |
| C5 | C6 | C7 | 122.47(18) | 122.01 | C11 | C10 | C9 | 121.4(2) | 120.72 |
| O1 | C2 | C7 | 121.33(18) | 121.46 | C4 | C3 | C2 | 120.77(19) | 120.12 |
| O1 | C2 | C3 | 119.45(17) | 119.16 | C11 | C12 | C13 | 120.7(2) | 119.78 |
| C3 | C2 | C7 | 119.23(19) | 119.36 | C12 | C11 | C10 | 121.3(2) | 121.09 |
| N1 | C8 | C7 | 121.94(18) | 122.005 | N1 | C9 | C10 | 109.66(17) | 111.58 |
Figure 6.

Correlation determination for bond lengths of 1.
Figure 7.

Correlation determination for bond angles of 1.
Figure 8.

Correlation determination for bond lengths of 2.
Figure 9.

Correlation determination for bond angles of 2.
Figure 10.

Energy Gap Measurement (HOMO–LUMO and HOMO–1–LUMO+1) of 1.
Figure 11.

Energy Gap Measurement (HOMO - LUMO and HOMO–1 - LUMO+1) of 2.
Table 3. Comparison of Bond Angles for 1.
| angle/deg |
angle/deg |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| atom | atom | atom | XRD | DFT | atom | atom | atom | XRD | DFT |
| C8 | N1 | C7 | 119.3(2) | 122.80 | O1 | C14 | C13 | 119.0(2) | 119.18 |
| C10 | C9 | C14 | 118.7(2) | 118.86 | C13 | C14 | C9 | 119.7(2) | 119.49 |
| C10 | C9 | C8 | 119.2(2) | 120.43 | N1 | C8 | C9 | 122.6(2) | 121.93 |
| C14 | C9 | C8 | 122.1(2) | 120.70 | C7 | C2 | C1 | 120.6(2) | 120.90 |
| C11 | C10 | C9 | 122.4(2) | 122.03 | C3 | C2 | C7 | 117.5(2) | 118.16 |
| C10 | C11 | C12 | 117.0(2) | 117.70 | C3 | C2 | C1 | 121.9(2) | 120.92 |
| C10 | C11 | C15 | 121.2(3) | 121.66 | C12 | C13 | C14 | 120.2(2) | 120.07 |
| C12 | C11 | C15 | 121.8(2) | 120.63 | C13 | C12 | C11 | 121.9(2) | 121.83 |
| C2 | C7 | N1 | 118.2(2) | 117.86 | C5 | C6 | C7 | 120.3(2) | 120.57 |
| C6 | C7 | N1 | 121.5(2) | 121.98 | C4 | C3 | C2 | 122.4(3) | 121.66 |
| C6 | C7 | C2 | 120.2(2) | 120.11 | C4 | C5 | C6 | 120.1(3) | 119.73 |
| O1 | C14 | C9 | 121.3(2) | 121.31 | C3 | C4 | C5 | 119.6(3) | 119.71 |
Table 4. Comparison of Bond Lengths for 2.
| length/Å |
length/Å |
||||||
|---|---|---|---|---|---|---|---|
| atom | atom | XRD | DFT | atom | atom | XRD | DFT |
| O1 | C2 | 1.349(2) | 1.36418 | C5 | C1 | 1.508(3) | 1.51307 |
| O2 | C13 | 1.369(3) | 1.39087 | C2 | C3 | 1.384(3) | 1.40416 |
| O2 | C16 | 1.427(3) | 1.45136 | C15 | C14 | 1.384(3) | 1.40049 |
| N1 | C8 | 1.270(3) | 1.29816 | C15 | C10 | 1.380(3) | 1.40006 |
| N1 | C9 | 1.469(3) | 1.47542 | C4 | C3 | 1.373(3) | 1.39055 |
| C7 | C6 | 1.397(3) | 1.41150 | C14 | C13 | 1.391(3) | 1.40059 |
| C7 | C2 | 1.406(3) | 1.42516 | C13 | C12 | 1.386(3) | 1.40358 |
| C7 | C8 | 1.459(3) | 1.45195 | C10 | C11 | 1.386(3) | 1.40704 |
| C5 | C6 | 1.385(3) | 1.39386 | C10 | C9 | 1.510(3) | 1.51644 |
| C5 | C4 | 1.399(3) | 1.41432 | C12 | C11 | 1.370(4) | 1.39202 |
3.3. Hirshfeld Surface Analysis
In crystalline molecules, Hirshfeld surface (HS) analysis is done to check the intermolecular contacts and also explain the surface features of the molecules. Crystal Explorer is run by loading up CIF files as input files for Hirshfeld analysis. The HS is diagrammed by means of distinctive colors (blue, white, and red), and is primarily governed by radii distances.37 Molecular surfaces are transparent to show the imagining in a similar alignment, wherever this was determined. The dnorm surface is valuable representing adjacent interactions and its values start from the negative side to the positive end. The more negative values denote a closer interaction as compared to standard rvdW (van der Waals radii) and vice versa for positive values, although, a reference surface resolution was considered to exhibit a HS along the 3D dnorm with the set range (−0.25 to 1.3 Å). The red spots represent nearer contacts (dnorm value: negative), white spots seemed due to an alike variation around the zero value, and blue colors displayed extended contacts (dnorm value, positive). The HS illustration of 1 and 2 is given in Figure 12 in dnorm fashions.
Figure 12.

Hirshfeld surfaces mapped for dnorm.
To study the contacts between atoms, 2D fingerprint plots of 1 and 2 are plotted in Figures 13 and 14. It was revealed from all interactions that H···H contacts are mostly considered in the prepared compounds. H···H interactions were seen as 56.2% and 54.4% of all interactions in 1 and 2, respectively. After hydrogen interactions, C···H/C···H contacts were nearly 32.3% in 1 and 30.1% in 2. The H···O/H··· O contacts were about 5.6% for 1, whereas they were 13.0% for 2. In 1 other interactions are 1.8% for N···H/N···H and 1.1% for C···C/C···C. Compound 2 also has 1.7% for N···H/N···H and 0.4% for C···N/C···N. These interactions played a major role in stabilizing the crystal assembly of both compounds.
Figure 13.

2D fingerprint plots of interactions along their corresponding percentages for 1.
Figure 14.

2D fingerprint plots of interactions along their corresponding percentages for 2.
3.4. Molecular Modeling Studies
To check the enzyme inhibition potential of synthesized imines, molecular modeling was accomplished. The binding affinity and docking score values exposed that 2 executed effective inhibition as compared to 1. For inhibition of AChE and BChE, the docking score values for 1 were −14.5496 and −16.5711 and also binding affinity values −13.0807 and −15.0943, respectively. Compound 2 exhibited contacts on AChE with docking score −19.2255 and binding affinity −14.2516, although the BChE −19.2986 docking score and −15.9512 binding affinity values are also listed in Table 6. The true docked posture of 1 and 2 with AChE and BChE are shown in Figures 15, 16, 17, and 18. Compound 1 exhibited interaction (Figure 15) with the binding position of AChE as π–π interactions with Tyr334, Phe330, and Trp279.
Table 6. Docking Outcomes of Synthesized 1 and 2.
| AChE |
BChE | |||
|---|---|---|---|---|
| enzymes compounds | docking score | binding affinity (kcal/mol) | docking score | binding affinity (kcal/mol) |
| 1 | –14.5496 | –13.0807 | –16.5711 | –15.0943 |
| 2 | –19.2255 | –14.2516 | –19.2986 | –15.9512 |
Figure 15.

Docking view of 1 with AChE.
Figure 16.

Docking view of 2 with AChE.
Figure 17.

Docking view of 1 with BChE.
Figure 18.

Docking view of 2 with BChE.
π–Alkyl contacts with His440, Tyr334, Phe330, and Tyr121 and conventional interaction by Asp72 were also seen with 1. Similarly, Trp279, Tyr121, Tyr334, Asp72, Phe330, and His440 are amino acid positioned on the binding furrow, 2 showed various interactions (Figure 16) with these residues. π–π stacked interaction with Tyr 121, Tyr334, and Phe330 through π–alkyl interactions were shown by Trp279, Phe330, and His 440. The newly synthesized compounds also showed intermediate types of interaction with the 1POI receptor site. Compound 1 displayed π–π connections with Trp82 and Phe329. Compound 1 also had π–alkyl contacts with Leu286 and Phe329 and conventional contact with Gly116. (Figure 17). The contacts of BChE (Figure 18) were also presented with Trp82 and Leu286 by means of π–alkyl and π–π interactions with Leu286 but conventional contacts with Gly116.
3.5. In Silico ADMET Evaluation
Currently, various parameters associated to drug properties of formulated compounds have been determined by in silico ADMET studies using the Swiss online available AdmetSAR tool. Most of the parameters discussed are lipophilicity, water-solubility, drug likeness, pharmacokinetics, and medicinal chemistry. These distinctive characteristics explain that either the formulated compounds exhibit drug-likeness or not. According to the Lipinski rule, the values of log Po/w (iLOGP) for both synthesized 1 and 2 were 2.41 and 2.83, respectively. Similarly, the solubility log S (ESOL) values were −3.90 and −3.60, respectively for 1 and 2. These prepared compounds also showed Lipinski zero violation. The synthetic accessibility of compounds was in good range and 2 gave a higher value of 2.59 for it. The gastrointestinal absorption rate is also high in both compounds. Compounds 1 and 2 showed inhibition of cytochrome as CYP2C19 and CYP1A2, while 2 also inhibited CYP2D6 and CYP3A4. The physicochemical parameters of the compounds are presented in Tables 7–12.
Table 7. Physicochemical Parameters of the Synthesized Compoundsa.
| code | formula | MW | NHA | NAHA | F Csp3 | NRB | NHBA | NHBD | MR | TPSA (Å2) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | C15H15NO | 225.29 | 17 | 12 | 0.13 | 2 | 2 | 1 | 72.09 | 32.59 |
| 2 | C16H17NO2 | 255.31 | 19 | 12 | 0.19 | 4 | 3 | 1 | 77.87 | 41.82 |
Notation: number of heavy atoms (NHA), number of aromatic heavy atoms (NAHA), F (fraction), number of rotatable bonds (NRB), number of H-bond acceptors (NHBA), number of H-bond donors (NHBD), molar refractivity (MR), topological polar surface area (TPSA).
Table 12. Medicinal Chemistry of Synthesized Compounds.
| code | PAINS | Brenk | lead likeness | synthetic accessibility |
|---|---|---|---|---|
| 1 | 0 alert | 1 alert | no | 2.46 |
| 2 | 0 alert | 1 alert | yes | 2.59 |
Table 8. Lipophilicity of Synthesized Compounds.
| code | log Po/w (iLOGP) | log Po/w (XLOGP3) | log Po/w (WLOGP) | log Po/w (MLOGP) | log Po/w (SILICOS-IT) | consensus log Po/w |
|---|---|---|---|---|---|---|
| 1 | 2.41 | 3.60 | 3.76 | 3.13 | 4.36 | 3.45 |
| 2 | 2.83 | 3.14 | 3.18 | 2.49 | 4.26 | 3.18 |
Table 9. Water Solubility of Synthesized Compounds.
| code | log S (ESOL) | solubility (mg/mL) | class | log S (Ali) | solubility (mg/mL) | class | log S (SILICOS-IT) | solubility (mg/mL) | class |
|---|---|---|---|---|---|---|---|---|---|
| 1 | –3.90 | 2.87 × 10–02 | soluble | –3.97 | 2.41 × 10–02 | soluble | –5.21 | 1.38 × 10–03 | moderately soluble |
| 2 | –3.60 | 6.35 × 10–02 | soluble | –3.69 | 5.24 × 10–02 | soluble | –5.36 | 1.12 × 10–03 | moderately soluble |
Table 10. Pharmacokinetics of Synthesized Compounds.
| code | GI absorption | BBB permeant | P-gp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | log Kp cm/s |
|---|---|---|---|---|---|---|---|---|---|
| 1 | high | yes | no | yes | yes | no | no | no | –5.12 |
| 2 | high | yes | no | yes | yes | no | yes | yes | –5.63 |
Table 11. Drug Likeness of Synthesized Compounds.
| Code | Lipinski | Ghose | Veber | Egan | Muegge | Bio. score |
|---|---|---|---|---|---|---|
| 1 | yes; 0 violation | no | yes | yes | yes | 0.55 |
| 2 | yes; 0 violation | yes | yes | yes | yes | 0.55 |
4. Conclusions
To treat the existing and new diseases, a number of research efforts were carried out to synthesize novel therapeutical agents. Nitrogen-based compounds have attracted the attention of pharmaceutical chemists toward the designing of innovative chemical agents against several diseases. In the present study, two active imines were synthesized starting from different amines using a very easy route and also inexpensive resources. Structural confirmation of both compounds was carried out by single-crystal X-ray diffraction analysis (SCXRD). DFT results showed that a close agreement exists between values of practical and computational studies. To study several contacts of the crystalline compounds, Hirshfeld surface analysis (HS) was also done, illustrating that the H···H bond contacts were the foremost contributors in interactions of imines. However, to check the in silico inhibitory action of both compounds, a molecular modeling study was executed against enzymes. 2D and 3D molecule–enzyme contacts and their docking positions also proposed that compounds are moderate inhibitors against esterase enzymes. ADMET studies of compounds elucidated the pharmacokinetic parameters of compounds. Both compounds gave good values against all parameters although 2 exhibits higher synthetic accessibility than 1. The computational and practical investigations recommended that these imines might be utilized in the therapeutic field based on their in vivo and supplementary studies.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00102.
The authors declare no competing financial interest.
Supplementary Material
References
- Abuamer K. M.; Maihub A. A.; El-Ajaily M. M.; Etorki A. M.; Abou-Krisha M. M.; Almagani M. A. The role of aromatic Schiff bases in the dyes techniques. Int. J. Org. Chem. 2014, 4, 7–15. 10.4236/ijoc.2014.41002. [DOI] [Google Scholar]
- Raza M. A.; Necmi D.; Doǧan O. E.; Tuǧgan A.; Sumrra S. H. Synthesis of two new Schiff bases; crystal structure, Hirshfeld surface analysis, density functional theory and molecular docking. J. Mol. Struct. 2021, 1226, 129330. 10.1016/j.molstruc.2020.129330. [DOI] [Google Scholar]
- McCleverty J. A.Comprehensive coordination chemistry II, 2nd ed.; Elsevier: Amsterdam, 2003; pp 809–847. [Google Scholar]
- Jahnke K.; Podschun B.; Schnackerz K. D.; Kautz J.; Cook P. F. Acid-base catalytic mechanism of dihydropyrimidinase from pH studies. Biochem. 1993, 32, 5160–5166. 10.1021/bi00070a027. [DOI] [PubMed] [Google Scholar]
- Fillebeen C.; Pantopoulos K. Iron inhibits replication of infectious hepatitis C virus in permissive Huh7. 5.1 cells. J. Hepatol. 2010, 53, 995–999. 10.1016/j.jhep.2010.04.044. [DOI] [PubMed] [Google Scholar]
- Zhang A. Y.; Camp W. L.; Elewski B. E. Advances in topical and systemic antifungals. Dermatol. Clin. 2007, 25, 165–183. 10.1016/j.det.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Scovill J. P.; Klayman D. L.; Franchino C. F. 2-Acetylpyridine thiosemicarbazones. 4. Complexes with transition metals as antimalarial and antileukemic agents. J. Med. Chem. 1982, 25, 1261–1264. 10.1021/jm00352a036. [DOI] [PubMed] [Google Scholar]
- Taş N. A.; Şenocak A.; Aydın A. Preparation and cytotoxicity evaluation of some amino acid methyl ester Schiff bases. J. Turk. Chem. Soc. 2018, 5, 585–606. [Google Scholar]
- Aliaksandr Dzeikala; Anna Sykula Schiff bases as important class of pharmacological agents. J. Pharm. Pharmacol. 2018, 6, 989–1009. 10.17265/2328-2150/2018.12.002. [DOI] [Google Scholar]
- Kostova I.; Saso L. Advances in research of Schiff-base metal complexes as potent antioxidants. Curr. Med. Chem. 2013, 20, 4609–4632. 10.2174/09298673113209990149. [DOI] [PubMed] [Google Scholar]
- Badavath V. N.; Kumar A.; Samanta P. K.; Maji S.; Das A.; Blum G.; Jha A.; Sen A. Determination of potential inhibitors based on isatin derivatives against SARS-CoV-2 main protease (mpro): A molecular docking, molecular dynamics and structure-activity relationship studies. J. Biomol. Struct. Dyn. 2020, 1–19. 10.1080/07391102.2020.1845800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.; Sun H.; Wang J.; Wang Z.; Liu H.; Zhang J. Z.; Hou T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- Azam S. S.; Abbasi S. W.. Molecular docking studies for the identification of novel melatoninergic inhibitors for acetylserotonin-O-methyltransferase using different docking routines. Theor. Biol. Med. Model. 2013, 10.1186/1742-4682-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y.; Song L.; Lin Y. Block-localized wavefunction (BLW) method at the density functional theory (DFT) level. J. Phys. Chem. A 2007, 111, 8291–8301. 10.1021/jp0724065. [DOI] [PubMed] [Google Scholar]
- Yu H. S.; Li S. L.; Truhlar D. G. Perspective: Kohn-Sham density functional theory descending a staircase. J. Chem. Phys. 2016, 145, 130901. 10.1063/1.4963168. [DOI] [PubMed] [Google Scholar]
- Sajjad S.; Hashmi M. A.; Mahmood T.; Ayub K. Density functional theory study of structural, electronic and CO adsorption properties of anionic Scn-(n= 2–13) clusters. Comput. Theor. Chem. 2019, 1163, 112511. 10.1016/j.comptc.2019.112511. [DOI] [Google Scholar]
- Li P.; Bu Y.; Ai H. Conformational study of glycine amide using density functional theory. J. Phys. Chem. A 2003, 107, 6419–6428. 10.1021/jp034886f. [DOI] [Google Scholar]
- Lambie B.; Ramaekers R.; Maes G. Conformational behavior of serine: an experimental matrix-isolation FT-IR and theoretical DFT (B3LYP)/6-31++ G** study. J. Phys. Chem. A 2004, 108, 10426–10433. 10.1021/jp047192v. [DOI] [Google Scholar]
- Raza M. A.; Fatima K.; Saqib Z.; Maurin J. K.; Budzianowski A. Designing of diamino based esterases inhibitors; synthesis, characterization, density functional theory and molecular modeling. J. Mol. Struct. 2019, 1195, 712–722. 10.1016/j.molstruc.2019.06.021. [DOI] [Google Scholar]
- Sumrra S. H.; Sahrish I.; Raza M. A.; Ahmad Z.; Zafar M. N.; Chohan Z. H.; Khalid M.; Ahmed S. Efficient synthesis, characterization, and in vitro bactericidal studies of unsymmetrically substituted triazole-derived Schiff base ligand and its transition metal complexes. Monatsh. Chem. Mon. 2020, 151, 549–557. 10.1007/s00706-020-02571-z. [DOI] [Google Scholar]
- Sheldrick G. M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia L. J. WinGX and ORTEP for Windows: an update. J. Appl. Crystallogr. 2012, 45, 849–854. 10.1107/S0021889812029111. [DOI] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek A. L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae C. F.; Edgington P. R.; McCabe P.; Pidcock E.; Shields G. P.; Taylor R.; Towler M.; van de Streek J. Mercury: visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. 10.1107/S002188980600731X. [DOI] [Google Scholar]
- Arshad M. N.; Asiri A. M.; Alamry K. A.; Mahmood T.; Gilani M. A.; Ayub K.; Birinji A. S. Synthesis, crystal structure, spectroscopic and density functional theory (DFT) study of N-[3-anthracen-9-yl-1-(4-bromo-phenyl)-allylidene]-N-benzenesulfonohydrazine. Spectrochim. Acta A: Mol. Biomol. Spectrosc. 2015, 142, 364–374. 10.1016/j.saa.2015.01.101. [DOI] [PubMed] [Google Scholar]
- Milenković D.; Avdović E.; Dimić D.; Sudha S.; Ramarajan D.; Milanović Ž.; Trifunović S.; Marković Z. S. Vibrational and Hirshfeld surface analyses, quantum chemical calculations, and molecular docking studies of coumarin derivative 3-(1-m-toluidinoethylidene)-chromane-2, 4-dione and its corresponding palladium (II) complex. J. Mol. Struct. 2020, 1209, 127935. 10.1016/j.molstruc.2020.127935. [DOI] [Google Scholar]
- Milenkovic D.; Dimitric Markovic J. M; Dimic D.; Jeremic S.; Amic D.; Stanojevic Pirkovic M.; Markovic Z. S Structural characterization of kaempferol: A spectroscopic and computational study. Maced. J. Chem. Chem. Eng. 2019, 38, 49–62. 10.20450/mjcce.2019.1333. [DOI] [Google Scholar]
- Wang H.; Wang X.; Wang H.; Wang L.; Liu A. DFT study of new bipyrazole derivatives and their potential activity as corrosion inhibitors. J. Mol. Model. 2006, 13, 147–153. 10.1007/s00894-006-0135-x. [DOI] [PubMed] [Google Scholar]
- Mohapatra R. K.; Sarangi A. K.; Azam M.; El-ajaily M. M.; Kudrat-E-Zahan M.; Patjoshi S. B.; Dash D. C. Synthesis, structural investigations, DFT, molecular docking and antifungal studies of transition metal complexes with benzothiazole based Schiff base ligands. J. Mol. Struct. 2019, 1179, 65–75. 10.1016/j.molstruc.2018.10.070. [DOI] [Google Scholar]
- Jawaria R.; Hussain M.; Khalid M.; Khan M. U.; Tahir M. N.; Naseer M. M.; Braga A. A. C.; Shafiq Z. Synthesis, crystal structure analysis, spectral characterization and nonlinear optical exploration of potent thiosemicarbazones based compounds: A DFT refine experimental study. Inorg. Chim. Acta 2019, 486, 162–171. 10.1016/j.ica.2018.10.035. [DOI] [Google Scholar]
- Falek W.; Benali-Cherif R.; Golea L.; Samai S.; Benali-Cherif N.; Bendeif E.-E.; Daoud I. A structural comparative study of charge transfer compounds: Synthesis, crystal structure, IR, Raman-spectroscopy, DFT computation and hirshfeld surface analysis. J. Mol. Struct. 2019, 1192, 132–144. 10.1016/j.molstruc.2019.04.084. [DOI] [Google Scholar]
- Wolff S.; Grimwood D.; McKinnon J.; Turner M.; Jayatilaka D.; Spackman M.. Crystal Explorer package, ver. 3.1; University of Western Australia: Perth, Australia, 2013. [Google Scholar]
- Shyamapada S.; Christoph M.; Mitra S. Synthesis, crystal structure, and Hirshfeld surface analysis of a new mixed ligand copper (II) complex. Acta Chim. Slov. 2016, 63, 129–137. 10.17344/acsi.2015.2024. [DOI] [PubMed] [Google Scholar]
- Raza M. A.; Fatima K. Molecular modeling approach for designing of amino-derived anti-Alzheimer agents: A computational study. J. Phys. Org. Chem. 2020, 4076. 10.1002/poc.4076. [DOI] [Google Scholar]
- Ding L. P.; Zhang F. H.; Zhu Y. S.; Lu C.; Kuang X. Y.; Lv J.; Shao P. Understanding the structural transformation, stability of medium-sized neutral and charged silicon clusters. Sci. Rep 2015, 15951. 10.1038/srep15951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hökelek T.; Özkaya S.; Necefoǧlu H. Crystal Structure and Hirshfeld Surface Analysis of AquaBis (Nicotinamide-Κn 1) Bis (2, 4, 6-TriMethylBenzoato-Κ2 O, O′) Cadmium (II). Acta Crystallogr. E 2018, 74, 246–251. 10.1107/S2056989018001494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.