

Abstract
The cerebellum has been increasingly implicated in autism spectrum disorder (ASD) with many ASD-linked genes impacting both cerebellar function and development. However, the precise timing and critical periods of when abnormal cerebellar neurodevelopment contributes to ASD-relevant behaviors remains poorly understood. In this study, we identify a critical period for the development of ASD-relevant behaviors in a cerebellar male mouse model of tuberous sclerosis complex (TSC), by using the mechanistic target of rapamycin (mTOR) inhibitor, rapamycin, to pharmacologically inhibit dysregulated downstream signaling. We find independent critical periods during which abnormal ASD-relevant behaviors develop for the two core ASD diagnostic criteria, social impairments and behavioral flexibility, and delineate an anatomic, physiological, and behavioral framework. These findings not only further our understanding of the genetic mechanisms underlying the timing of ASD-relevant behaviors but also have the capacity to inform potential therapies to optimize treatment interventions.
SIGNIFICANCE STATEMENT No targeted treatments currently exist for autism spectrum disorder (ASD). This complex developmental disorder has established links to genetic and circuit aberrations, yet the precise timing and coordination of these underlying mechanisms that contribute to the spectrum of physiological and behavioral abnormalities remains unclear. Cerebellar pathology is consistently seen in ASD individuals; therefore, we sought to identify the specific windows for cerebellar involvement in the development of ASD-relevant behaviors. Using pharmacologic treatment paradigms, we outline distinct critical periods of developmental vulnerability for ASD-relevant social and inflexible behaviors. From this study, we posit a refined window of time during which ASD symptoms develop that will inform therapeutic timing.
Keywords: autism, cerebellum, critical period, Purkinje cell, tuberous sclerosis
Introduction
Many developmentally acquired behaviors emerge during discrete time windows or critical periods. A critical period is defined as the time window during which behaviors or functions go through experience-dependent modification, stabilize, and become resistant to subsequent perturbation (Blows, 2003). Because of the developmental origins of autism spectrum disorder (ASD)-relevant behaviors, it has been postulated that critical periods for development of these abnormal behaviors might also exist. Critical periods for social circuity have been identified. In animal models, early social isolation results in autistic-like behaviors (Hofer, 1970, 1994; Harlow and Suomi, 1971; Neumann et al., 2005). Similarly, evidence for early developmental vulnerability of social behaviors in humans has emerged from studies of Romanian orphanages in the 1980s. Children exposed to long-term sensory deprivation developed severe developmental delays, intellectual disability, and neuropsychiatric symptoms including ASD-relevant social deficits and repetitive behaviors (Nelson et al., 2007; Mackes et al., 2020). Taken together, these data support the presence of critical periods, during which developmental disruptions have significant behavioral impacts.
Alternatively, these periods are time windows after which disruptions may not have a significant impact. If early development is a time of significant vulnerability, there may be translational opportunity to bypass critical periods of developmental vulnerability and prevent the development of abnormal behaviors if diagnosed sufficiently early. In the neurodevelopmental disorder, tuberous sclerosis complex (TSC), which is associated with significant neurologic and neuropsychiatric comorbidity, including high rates of ASD (Jeste et al., 2016), many individuals are diagnosed prenatally owing to the presence of cardiac rhabdomyomas observable on prenatal ultrasound studies (Milunsky et al., 2009). This early diagnosis is an opportunity for early therapeutic intervention (Hsieh and Thiele, 2013). TSC is characterized by aberrant mechanistic target of rapamycin (mTOR) signaling leading to unregulated translation, cell metabolism and growth (Lipton and Sahin, 2014). Drugs which target mTOR show significant benefits in both rodent models and in humans (Meikle et al., 2008b; Muncy et al., 2009; Sato et al., 2012; Tsai et al., 2012, 2018). However, chronic inhibition by drugs that target mTOR can be accompanied by severe side effects (Verhave et al., 2014). Thus, understanding the time windows when an abnormal behavior develops could help inform the duration of therapy.
The cerebellum has been increasingly implicated in the pathogenesis of ASD; cerebellar injury increases the risk of an ASD diagnosis (Limperopoulos et al., 2008), and cerebellar abnormalities are the most consistent observation in postmortem studies of individuals with ASD (Bailey et al., 1998; Whitney et al., 2008). Nearly half of all individuals with TSC are diagnosed with ASD (Jeste et al., 2016), with cerebellar abnormalities and dysfunction correlating with the presence of ASD (Asano et al., 2001; Eluvathingal et al., 2006). Disruption of cerebellar circuits (Stoodley et al., 2017; Badura et al., 2018; Kelly et al., 2020) or loss of ASD-linked genes such as Tsc1/2 (Tsai et al., 2012; Reith et al., 2013), PTEN (Cupolillo et al., 2016), or Shank2 (Peter et al., 2016) in cerebellar Purkinje cells (PCs) is sufficient to generate ASD-relevant behaviors. The developing cerebellum has been suggested to shape and refine neocortical circuitry, specifically during sensitive periods of development (Volpe, 2009; Wang et al., 2014). Expanding on these ideas, others found that altering cerebellar function in a region-specific manner during a developmental time window induces long-lasting ASD-relevant behavioral impairments (Badura et al., 2018). Taken together these studies highlight the existence of distinct time windows and underscore the contribution of the developing cerebellum in ASD.
Here, we used a PC-Tsc1 model in which loss of Tsc1 in PCs results in autism relevant-behaviors, pathology, and cerebellar dysfunction (Tsai et al., 2012). Early treatment of this model with the mTOR-specific inhibitor, rapamycin, from postnatal day (P)7 prevents the development of these phenotypes. Thus, to define the existence of critical periods of development of abnormal ASD-relevant behaviors, we investigate mutant mice in two rapamycin treatment paradigms: from P7 to P63 or P7 to P35. These studies reveal differential critical periods for the development of social versus inflexible/repetitive behaviors in addition to delineating pathologic and electrophysiological mechanisms contributing to these periods.
Materials and Methods
Experimental models and subject details
Mice
L7/Pcp2-Cre (L7Cre) mice (Barski et al., 2000; The Jackson Laboratory, IMSR_JAX004146) were crossed with loxP-flanked Tsc1 (Tsc1loxP/loxP) mice (Meikle et al., 2007; The Jackson Laboratory, IMSR_JAX005680) to generate L7cre+; Tsc1loxP/loxP mutant animals, and L7cre-; Tsc1flox/flox control animals. Only male animals were used for behavior experiments. Mice were of mixed genetic backgrounds (C57Bl/6 J, 129 SvJae, BALB/cJ). Littermate controls were used for all behavioral experiments. Age of mice at time of behavior tests stated in Tables 1, 2. All animals were group housed and maintained on a 12/12 h light/dark cycle. All experimental protocols were approved by University of Texas Southwestern Institutional Animal Care and Uses Committee.
Table 1.
Statistics for P7–P63 cohort
| Test/experiment | ||
|---|---|---|
| Figure 1, behavior characterization | ||
| Social approach | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.0001 | F(3,118) = 7.344 |
| Preference | p < 0.0001 | F(1,118) = 52.56 |
| Genotype | p = 0.2661 | F(3,118) = 1.336 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Novel animal, novel object | ||
| Control VEH n = 18 | p = 0.0022 | |
| Mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 17 | p < 0.0001 | |
| Mutant RAPA n = 14 | p < 0.0001 | |
| Social novelty | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.2465 | F(3,112) = 1.400 |
| Preference | p < 0.0001 | F(1,112) = 23.17 |
| Genotype | p = 0.1095 | F(3,112) = 2.060 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Novel animal, novel object | ||
| Control VEH n = 17 | p = 0.0199 | |
| Mutant VEH n = 12 | p > 0.9999 | |
| Control RAPA n = 17 | p = 0.0059 | |
| Mutant RAPA n = 14 | p = 0.0051 | |
| Social olfaction | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p < 0.0001 | F(42,735) = 2.641 |
| Scent | p < 0.0001 | F(14,735) = 39.46 |
| Group | p < 0.0001 | F(3,735) = 37.52 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Water | ||
| Control VEH n = 19 vs mutant VEH n = 12 | p = 0.8170 | |
| Control RAPA n = 12 vs mutant RAPA n = 10 | p > 0.9999 | |
| Control VEH n = 19 vs control RAPA n = 12 | p > 0.9999 | |
| Mutant VEH n = 12 vs mutant RAPA n = 10 | p > 0.9999 | |
| Almond | ||
| Control VEH n = 19 vs mutant VEH n = 12 | p > 0.9999 | |
| Control RAPA n = 12 vs mutant RAPA n = 10 | p > 0.9999 | |
| Control VEH n = 19 vs control RAPA n = 12 | p = 0.3913 | |
| Mutant VEH n = 12 vs mutant RAPA n = 10 | p = 0.1125 | |
| Social scent | ||
| Control VEH n = 19 vs mutant VEH n = 12 | p = 0.0149 | |
| Control RAPA n = 12 vs mutant RAPA n = 10 | p > 0.9999 | |
| Control VEH n = 19 vs control RAPA n = 12 | p = 0.0010 | |
| Mutant VEH n = 12 vs mutant RAPA n = 10 | p < 0.0001 | |
| Grooming | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.7009 | F(1,55) = 0.1491 |
| Treatment | p = 0.6617 | F(1,55) = 0.1935 |
| Group | p < 0.0001 | F(1,55) = 19.07 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 10 vs mutant VEH n = 14 | p = 0.0067 | |
| Control RAPA n = 18 vs mutant RAPA n = 17 | p = 0.0053 | |
| Control VEH n = 10 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 17 | p > 0.9999 | |
| Water Y maze | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p < 0.0001 | F(15,378) = 3.671 |
| Day | p < 0.0001 | F(5,378) = 83.08 |
| Group | p < 0.0001 | F(3,378) = 12.10 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Trial 1 | ||
| Control VEH n = 20 vs mutant VEH n = 17 | p > 0.9999 | |
| Control RAPA n = 17 vs mutant RAPA n = 13 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 17 | p > 0.9999 | |
| Mutant VEH n = 17 vs mutant RAPA n = 13 | p > 0.9999 | |
| Trial 2 | ||
| Control VEH n = 20 vs mutant VEH n = 17 | p > 0.9999 | |
| Control RAPA n = 17 vs mutant RAPA n = 13 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 17 | p > 0.9999 | |
| Mutant VEH n = 17 vs mutant RAPA n = 13 | p > 0.9999 | |
| Trial 3 | ||
| Control VEH n = 20 vs mutant VEH n = 17 | p > 0.9999 | |
| Control RAPA n = 17 vs mutant RAPA n = 13 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 17 | p > 0.9999 | |
| Mutant VEH n = 17 vs mutant RAPA n = 13 | p > 0.9999 | |
| Rev. trial 1 | ||
| Control VEH n = 20 vs mutant VEH n = 17 | p < 0.0001 | |
| Control RAPA n = 17 vs mutant RAPA n = 13 | p < 0.0001 | |
| Control VEH n = 20 vs control RAPA n = 17 | p > 0.9999 | |
| Mutant VEH n = 17 vs mutant RAPA n = 13 | p > 0.9999 | |
| Rev. trial 2 | ||
| Control VEH n = 20 vs mutant VEH n = 17 | p = 0.0186 | |
| Control RAPA n = 17 vs mutant RAPA n = 13 | p = 0.0846 | |
| Control VEH n = 20 vs control RAPA n = 17 | p > 0.9999 | |
| Mutant VEH n = 17 vs mutant RAPA n = 13 | p > 0.9999 | |
| Rev. trial 3 | ||
| Control VEH n = 20 vs mutant VEH n = 17 | p > 0.9999 | |
| Control RAPA n = 17 vs mutant RAPA n = 13 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 17 | p > 0.9999 | |
| Mutant VEH n = 17 vs mutant RAPA n = 13 | p > 0.9999 | |
| Rotarod | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p < 0.0001 | F(12,390) = 3.767 |
| Day | p < 0.0001 | F(4,390) = 16.40 |
| Group | p < 0.0001 | F(3,390) = 70.61 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Day 1 | ||
| Control VEH n = 25 vs mutant VEH n = 15 | p = 0.1112 | |
| Control RAPA n = 19 vs mutant RAPA n = 23 | p = 0.9056 | |
| Control VEH n = 25 vs control RAPA n = 19 | p > 0.9999 | |
| Mutant VEH n = 15 vs mutant RAPA n = 23 | p = 0.0071 | |
| Day 2 | ||
| Control VEH n = 25 vs mutant VEH n = 15 | p = 0.0001 | |
| Control RAPA n = 19 vs mutant RAPA n = 23 | p = 0.4920 | |
| Control VEH n = 25 vs control RAPA n = 19 | p > 0.9999 | |
| Mutant VEH n = 15 vs mutant RAPA n = 23 | p < 0.0001 | |
| Day 3 | ||
| Control VEH n = 25 vs mutant VEH n = 15 | p < 0.0001 | |
| Control RAPA n = 19 vs mutant RAPA n = 23 | p > 0.9999 | |
| Control VEH n = 25 vs control RAPA n = 19 | p = 0.1609 | |
| Mutant VEH n = 15 vs mutant RAPA n = 23 | p < 0.0001 | |
| Day 4 | ||
| Control VEH n = 25 vs mutant VEH n = 15 | p < 0.0001 | |
| Control RAPA n = 19 vs mutant RAPA n = 23 | p > 0.9999 | |
| Control VEH n = 25 vs control RAPA n = 19 | p = 0.0024 | |
| Mutant VEH n = 15 vs mutant RAPA n = 23 | p < 0.0001 | |
| Day 5 | ||
| Control VEH n = 25 vs mutant VEH n = 15 | p < 0.0001 | |
| Control RAPA n = 19 vs mutant RAPA n = 23 | p > 0.9999 | |
| Control VEH n = 25 vs control RAPA n = 19 | p < 0.0001 | |
| Mutant VEH n = 15 vs mutant RAPA n = 23 | p < 0.0001 | |
| Open field | ||
| Two-way ANOVA | ||
| Distance moved | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.9653 | F(42,989) = 0.6378 |
| Minute | p = 0.0038 | F(14,989) = 2.318 |
| Group | p < 0.0001 | F(3,989) = 10.53 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| 0:00–1:00 (60 seconds) | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p = 0.6273 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| 1:00–2:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p = 0.2082 | |
| 2:00–3:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| 3:00–4:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| 4:00–5:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| 5:00–6:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p = 0.0411 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p = 0.8482 | |
| 6:00–7:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| 7:00–8:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p = 0.4288 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| 8:00–9:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| 9:00–10:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p = 0.4031 | |
| Control VEH n = 20 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p = 0.0953 | |
| 10:00–11:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| 11:00–12:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| 12:00–13:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p > 0.9999 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| 13:00–14:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p = 0.4511 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p = 0.7478 | |
| 14:00–15:00 | ||
| Control VEH n = 20 vs mutant VEH n = 14 | p > 0.9999 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p > 0.9999 | |
| Control VEH n = 20 vs control RAPA n = 18 | p = 0.1904 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| Time in left | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.9524 | F(1,71) = 0.003591 |
| Treatment | p = 0.3550 | F(1,71) = 0.8669 |
| Genotype | p = 0.0429 | F(1,71) = 4.250 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 20 vs mutant VEH n = 14 | p = 0.3036 | |
| Control RAPA n = 18 vs mutant RAPA n = 18 | p = 0.2924 | |
| Control VEH n = 20 vs control RAPA n = 18 | p = 0.9581 | |
| Mutant VEH n = 14 vs mutant RAPA n = 18 | p > 0.9999 | |
| Elevated plus maze | ||
| Two-way ANOVA | ||
| Open arms | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.8544 | F(1,58) = 0.03400 |
| Treatment | p = 0.5133 | F(1,58) = 0.4326 |
| Genotype | p = 0.0375 | F(1,58) = 4.535 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 13 vs mutant VEH n = 11 | p = 0.4375 | |
| Control RAPA n = 17 vs mutant RAPA n = 21 | p = 0.1367 | |
| Control VEH n = 13 vs control RAPA n = 17 | p > 0.9999 | |
| Mutant VEH n = 11 vs mutant RAPA n = 21 | p > 0.9999 | |
| Figure 2, cell survival | ||
| Cell count | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p < 0.0001 | F(1,11) = 38.68 |
| Treatment | p < 0.0001 | F(1,11) = 53.47 |
| Genotype | p < 0.0001 | F(1,11) = 103.0 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 4 vs mutant VEH n = 3 | p < 0.0001 | |
| Control RAPA n = 4 vs mutant RAPA n = 3 | p = 0.0547 | |
| Control VEH n = 4 vs control RAPA n = 4 | p = 0.8911 | |
| Mutant VEH n = 3 vs mutant RAPA n = 3 | p < 0.0001 | |
| Mutant VEH n = 3 vs control RAPA n = 4 | p < 0.0001 | |
| Figure 3, physiology | ||
| PC intrinsic firing rate | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.0225 | F(1,142) = 5.320 |
| Treatment | p = 0.0026 | F(1,142) = 9.406 |
| Genotype | p = 0.0001 | F(1,142) = 15.54 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 45 vs mutant VEH n = 36 | p < 0.0001 | |
| Control RAPA n = 30 vs mutant RAPA n = 35 | p = 0.5465 | |
| Control VEH n = 45 vs control RAPA n = 30 | p > 0.9999 | |
| Mutant VEH n = 36 vs mutant RAPA n = 35 | p = 0.0005 | |
| Mutant VEH n = 36 vs control RAPA n = 30 | p < 0.0001 | |
| PC evoked responses | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p < 0.0001 | F(87,1852) = 2.112 |
| Current step | p < 0.0001 | F(29,1852) = 100.9 |
| Group | p < 0.0001 | F(3,1852) = 313.6 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 13 vs mutant VEH n = 10 | p < 0.0001 | |
| Control RAPA n = 29 vs mutant RAPA n = 14 | p < 0.0001 | |
| Control VEH n = 13 vs control RAPA n = 29 | p = 0.0255 | |
| Mutant VEH n = 10 vs mutant RAPA n = 14 | p = 0.0075 | |
| Mutant VEH n = 10 vs control RAPA n = 29 | p < 0.0001 | |
| Membrane input resistance | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.8539 | F(1,86) = 0.03412 |
| Treatment | p = 1.127 | F(1,86) = 1.254 |
| Genotype | p < 0.0001 | F(1,86) = 18.67 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 12 vs mutant VEH n = 17 | p = 0.0283 | |
| Control RAPA n = 34 vs mutant RAPA n = 27 | p = 0.0003 | |
| Control VEH n = 12 vs control RAPA n = 34 | p = 0.7549 | |
| Mutant VEH n = 17 vs mutant RAPA n = 27 | p = 0.9845 | |
| Mutant VEH n = 17 vs control RAPA n = 34 | p = 0.0004 | |
| Membrane capacitance | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.9078 | F(1,23) = 0.01373 |
| Treatment | p = 0.9270 | F(1,23) = 0.008569 |
| Genotype | p = 0.7735 | F(1,23) = 0.08483 |
| Membrane time constant | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.2842 | F(1,23) = 1.203 |
| Treatment | p = 0.5662 | F(1,23) = 0.3388 |
| Genotype | p = 0.0022 | F(1,23) = 11.86 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 6 vs mutant VEH n = 6 | p = 0.0114 | |
| Control RAPA n = 8 vs mutant RAPA n = 7 | p = 0.1840 | |
| Control VEH n = 6 vs control RAPA n = 8 | p > 0.9999 | |
| Mutant VEH n = 6 vs mutant RAPA n = 7 | p = 0.5081 | |
| Mutant VEH n = 6 vs control RAPA n = 8 | p = 0.0496 | |
| Mouse age at time of testing (all experiments) | ||
| 8-week treatment paradigm | ||
| Rotarod | 13 weeks | Rotarod |
| Open field | 14 weeks | Open field |
| Elevated plus maze | 14 weeks | Elevated plus maze |
| Social interaction (three-chamber) | 15 weeks | Social interaction (three-chamber) |
| Social olfaction | 15 weeks | Social olfaction |
| Grooming | 15 weeks | Grooming |
| Water Y maze | 16 weeks | Water Y maze |
| Immunohistochemistry | 13–16 weeks | Immunohistochemistry |
| Acute slice physiology | 13–16 weeks | Acute slice physiology |
Table 2.
Statistics for P7–P35 cohort
| Test/experiment | ||
|---|---|---|
| Figure 4, behavior characterization | ||
| Social approach | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.1321 | F(3,90) = 1.920 |
| Preference | p < 0.0001 | F(1,90) = 62.64 |
| Genotype | p = 0.1111 | F(3,90) = 2.060 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Novel animal, novel object | ||
| Control VEH n = 15 | p < 0.0001 | |
| Mutant VEH n = 10 | p = 0.4167 | |
| Control RAPA n = 13 | p = 0.0001 | |
| Mutant RAPA n = 14 | p < 0.0001 | |
| Social novelty | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.2467 | F(3,96) = 1.403 |
| Preference | p < 0.0001 | F(1,96) = 27.22 |
| Genotype | p = 0.5971 | F(3,96) = 0.6305 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Novel animal, novel object | ||
| Control VEH n = 17 | p = 0.0497 | |
| Mutant VEH n = 12 | p > 0.9959 | |
| Control RAPA n = 17 | p = 0.0002 | |
| Mutant RAPA n = 14 | p = 0.0380 | |
| Social olfaction | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p < 0.0001 | F(42,1050) = 4.653 |
| Scent | p < 0.0001 | F(14,1050) = 191.2 |
| Group | p < 0.0001 | F(3,1050) = 46.70 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Water | ||
| Control VEH n = 16 vs mutant VEH n = 16 | p > 0.9999 | |
| Control RAPA n = 21 vs mutant RAPA n = 21 | p > 0.9999 | |
| Control VEH n = 16 vs control RAPA n = 21 | p > 0.9999 | |
| Mutant VEH n = 16 vs mutant RAPA n = 21 | p > 0.9999 | |
| Almond | ||
| Control VEH n = 16 vs mutant VEH n = 16 | p = 0.4580 | |
| Control RAPA n = 21 vs mutant RAPA n = 21 | p > 0.9999 | |
| Control VEH n = 16 vs control RAPA n = 21 | p > 0.9999 | |
| Mutant VEH n = 16 vs mutant RAPA n = 21 | p = 0.6805 | |
| Social scent | ||
| Control VEH n = 16 vs mutant VEH n = 16 | p < 0.0001 | |
| Control RAPA n = 21 vs mutant RAPA n = 21 | p < 0.0001 | |
| Control VEH n = 16 vs control RAPA n = 21 | p > 0.9999 | |
| Mutant VEH n = 16 vs mutant RAPA n = 21 | p = 0.0003 | |
| Water Y maze | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p < 0.0001 | F(15,474) = 4.925 |
| Day | p < 0.0001 | F(15,474) = 120.6 |
| Group | p < 0.0001 | F(15,474) = 16.75 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Trial 1 | ||
| Control VEH n = 15 vs mutant VEH n = 17 | p > 0.9999 | |
| Control RAPA n = 26 vs mutant RAPA n = 25 | p > 0.9999 | |
| Control VEH n = 15 vs control RAPA n = 26 | p > 0.9999 | |
| Mutant VEH n = 17 vs mutant RAPA n = 25 | p > 0.9999 | |
| Trial 2 | ||
| Control VEH n = 15 vs mutant VEH n = 17 | p > 0.9999 | |
| Control RAPA n = 26 vs mutant RAPA n = 25 | p > 0.9999 | |
| Control VEH n = 15 vs control RAPA n = 26 | p > 0.9999 | |
| Mutant VEH n = 17 vs mutant RAPA n = 25 | p > 0.9999 | |
| Trial 3 | ||
| Control VEH n = 15 vs mutant VEH n = 17 | p > 0.9999 | |
| Control RAPA n = 26 vs mutant RAPA n = 25 | p > 0.9999 | |
| Control VEH n = 15 vs control RAPA n = 26 | p > 0.9999 | |
| Mutant VEH n = 17 vs mutant RAPA n = 25 | p > 0.9999 | |
| Rev. trial 1 | ||
| Control VEH n = 15 vs mutant VEH n = 17 | p < 0.0001 | |
| Control RAPA n = 26 vs mutant RAPA n = 25 | p = 0.0041 | |
| Control VEH n = 15 vs control RAPA n = 26 | p = 0.3740 | |
| Mutant VEH n = 17 vs mutant RAPA n = 25 | p > 0.9999 | |
| Rev. trial 2 | ||
| Control VEH n = 15 vs mutant VEH n = 17 | p < 0.0001 | |
| Control RAPA n = 26 vs mutant RAPA n = 25 | p = 0.2559 | |
| Control VEH n = 15 vs control RAPA n = 26 | p > 0.9999 | |
| Mutant VEH n = 17 vs mutant RAPA n = 25 | p = 0.0006 | |
| Rev. trial 3 | ||
| Control VEH n = 15 vs mutant VEH n = 17 | p < 0.0001 | |
| Control RAPA n = 26 vs mutant RAPA n = 25 | p > 0.9999 | |
| Control VEH n = 15 vs control RAPA n = 26 | p > 0.9999 | |
| Mutant VEH n = 17 vs mutant RAPA n = 25 | p < 0.0001 | |
| Rotarod | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.9952 | F(12,235) = 0.2504 |
| Day | p < 0.0001 | F(4,235) = 12.38 |
| Group | p < 0.0001 | F(3,235) = 14.26 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Day 1 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p = 0.6610 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.2247 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| Day 2 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p = 0.0119 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.0254 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| Day 3 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p = 0.0294 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.0626 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| Day 4 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p = 0.0748 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.1765 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| Day 5 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p = 0.0073 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.3565 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p = 0.1602 | |
| Open field | ||
| Two-way ANOVA | ||
| Distance moved | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.9845 | F(42,705) = 0.5823 |
| Minute | p = 0.5856 | F(14,705) = 0.8758 |
| Group | p < 0.0001 | F(3,705) = 9.465 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| 0:00–1:00 (60 seconds) | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p = 0.7247 | |
| Control VEH n = 12 vs control RAPA n = 13 | p > 0.9999 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 1:00–2:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p = 0.3628 | |
| Control VEH n = 12 vs control RAPA n = 13 | p > 0.9999 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p = 0.2082 | |
| 2:00–3:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p = 0.4648 | |
| Control VEH n = 12 vs control RAPA n = 13 | p > 0.9999 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 3:00–4:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p > 0.9999 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 4:00–5:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p = 0.2421 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.5267 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 5:00–6:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p > 0.9999 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 6:00-7:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p > 0.9999 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 7:00–8:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p > 0.9999 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 8:00–9:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p = 0.2996 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.2294 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 9:00–10:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.7791 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p = 0.0953 | |
| 10:00–11:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p > 0.9999 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 11:00–12:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p > 0.9999 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 12:00–13:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p > 0.9999 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 13:00–14:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p = 0.8308 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.0227 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| 14:00–15:00 | ||
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.5178 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p > 0.9999 | |
| Time in center | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.7432 | F(1,47) = 0.1086 |
| Treatment | p = 0.1829 | F(1,47) = 1.827 |
| Genotype | p = 0.4546 | F(1,47) = 0.5684 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 12 vs mutant VEH n = 10 | p > 0.9999 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p = 0.8279 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.9436 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p = 0.4841 | |
| Elevated plus maze | ||
| Two-way ANOVA | ||
| Open arms | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.1322 | F(1,47) = 2.347 |
| Treatment | p = 0.6031 | F(1,47) = 0.2741 |
| Genotype | p = 0.4877 | F(1,47) = 0.4892 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 12 vs mutant VEH n = 10 | p = 0.2909 | |
| Control RAPA n = 13 vs mutant RAPA n = 16 | p > 0.9999 | |
| Control VEH n = 12 vs control RAPA n = 13 | p = 0.3207 | |
| Mutant VEH n = 10 vs mutant RAPA n = 16 | p = 0.9617 | |
| Figure 5, cell survival | ||
| Cell count | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.0006 | F(1,10) = 24.22 |
| Treatment | p = 0.0015 | F(1,10) = 18.56 |
| Genotype | p < 0.0001 | F(1,10) = 41.95 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 4 vs mutant VEH n = 3 | p < 0.0001 | |
| Control RAPA n = 4 vs mutant RAPA n = 3 | p = 0.5947 | |
| Control VEH n = 4 vs control RAPA n = 4 | p > 0.9999 | |
| Mutant VEH n = 3 vs mutant RAPA n = 3 | p = 0.0002 | |
| Mutant VEH n = 3 vs control RAPA n = 3 | p = 0.0001 | |
| Figure 6, physiology | ||
| PC intrinsic firing rate | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.0100 | F(1,113) = 6.870 |
| Treatment | p = 0.0173 | F(1,113) = 5.832 |
| Genotype | p = 0.0110 | F(1,113) = 6.681 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 43 vs mutant VEH n = 16 | p = 0.0008 | |
| Control RAPA n = 41 vs mutant RAPA n = 17 | p > 0.9999 | |
| Control VEH n = 43 vs control RAPA n = 41 | p > 0.9999 | |
| Mutant VEH n = 16 vs mutant RAPA n = 17 | p = 0.0072 | |
| Mutant VEH n = 16 vs control RAPA n = 41 | p = 0.0042 | |
| PC evoked responses | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.2388 | F(42,1344) = 1.149 |
| Current step | p < 0.0001 | F(14,1344) = 564.4 |
| Group | p < 0.0001 | F(3,1344) = 99.38 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 39 vs mutant VEH n = 17 | p < 0.0001 | |
| Control RAPA n = 26 vs mutant RAPA n = 17 | p > 0.9999 | |
| Control VEH n = 39 vs control RAPA n = 26 | p = 0.0326 | |
| Mutant VEH n = 17 vs mutant RAPA n = 17 | p = 0.0070 | |
| Mutant VEH n = 17 vs control RAPA n = 26 | p < 0.0001 | |
| Membrane input resistance | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.4496 | F(1,71) = 0.5780 |
| Treatment | p = 0.9045 | F(1,71) = 0.01451 |
| Genotype | p = 0.0316 | F(1,71) = 4.808 |
| Bonferroni's multiple comparisons test | Adjusted p value | |
| Control VEH n = 16 vs mutant VEH n = 15 | p = 0.1140 | |
| Control RAPA n = 25 vs mutant RAPA n = 19 | p = 0.5429 | |
| Control VEH n = 16 vs control RAPA n = 25 | p > 0.9999 | |
| Mutant VEH n = 15 vs mutant RAPA n = 19 | p > 0.9999 | |
| Mutant VEH n = 15 vs control RAPA n = 25 | p = 0.5985 | |
| Membrane capacitance | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.4741 | F(1,62) = 0.5186 |
| Treatment | p = 0.2037 | F(1,62) = 1.650 |
| Genotype | p = 0.1059 | F(1,62) = 2.692 |
| Membrane time constant | ||
| Two-way ANOVA | ||
| ANOVA table | p value | F (DFn,DFd) |
| Interaction | p = 0.2520 | F(1,62) = 1.337 |
| Treatment | p = 0.2431 | F(1,62) = 1.389 |
| Genotype | p = 0.2269 | F(1,62) = 1.490 |
| Mouse age at time of testing (all experiments) | ||
| 4-week treatment paradigm | ||
| Rotarod | 9 weeks | Rotarod |
| Open field | 10 weeks | Open field |
| Elevated plus maze | 10 weeks | Elevated plus maze |
| Social interaction (three-chamber) | 11 weeks | Social interaction (three-chamber) |
| Social olfaction | 11 weeks | Social olfaction |
| Water Y maze | 12 weeks | Water Y maze |
| Immunohistochemistry | 9–12 weeks | Immunohistochemistry |
| Acute slice physiology | 9–12 weeks | Acute slice physiology |
Method details
Rapamycin treatment
Rapamycin (LC Labs, R-5000) was dissolved in 0.25% polyethylene glycol and 0.25% Tween before usage. Vehicle or rapamycin was administered intraperitoneally every Monday, Wednesday, and Friday with rapamycin dosed at 6 mg/kg per injection starting at P7. Rapamycin treatment was administered for a period of eight weeks or four weeks, treatment was then stopped and followed by a four-week washout period before behavioral testing.
Electrophysiology
Acute sagittal slices (250–300 µm thick) were prepared from the cerebellar vermis of 8- and 12-week-old mutant and control littermates from each treatment group. Slices were cut in an ice-cold artificial CSF (ACSF) solution consisting of the following: 125 mm NaCl, 26 mm NaHCO3, 1.25 mm NaH2PO4, 2.5 mm KCl, 1 mm MgCl2, 2 mm CaCl2, and 25 mm glucose (pH 7.3, osmolarity 310) equilibrated with 95% O2 and 5% CO2. Slices were initially incubated at 34°C for 25 min, and then at room temperature (21–22°C) before recording in the same ACSF.
Recordings
Visually guided (infrared DIC videomicroscopy and water-immersion 40× objective) whole-cell recordings were obtained with patch pipettes (2–4 MΩ) pulled from borosilicate capillary glass (World Precision Instruments) with a Sutter P-97 horizontal puller. Electrophysiological recordings were performed at 31–33°C. For current-clamp recordings, the internal solution contained the following: 150 mm potassium-gluconate, 3 mm KCl, 10 mm HEPES, 0.5 mm EGTA, 3 mm MgATP, 0.5 mm GTP, 5 mm phosphocreatine-tris2, and 5 mm phosphocreatine-Na2; pH was adjusted to 7.2 with NaOH. Current-clamp and extracellular recordings were performed in NBQX (5 μm; Sigma, N183), R-CPP (2.5 μm; Tocris, 0247), and picrotoxin (20 μm; Tocris, 1128) to block AMPA receptors, NMDA receptors, and GABAA receptors, respectively. Recordings were taken from PCs in vermis Lobules V–VII.
Data acquisition and analysis
Electrophysiological data were acquired using a Multiclamp 700B amplifier (Molecular Devices) digitized at 20 kHz with either a National Instruments USB-6229 or PCI-MIO 16E-4 board and filtered at 2 kHz. Acquisition was controlled both with custom software written in either MATLAB or pCLAMP. Series resistance was monitored in voltage-clamp recordings with a 5-mV hyperpolarizing pulse, and only recordings that remained stable over the period of data collection were used. Glass monopolar electrodes (1–2 MΩ) filled with ACSF in conjunction with a stimulus isolation unit (WPI, A360) were used for extracellular stimulation of climbing and parallel fibers.
Immunohistochemistry
Mice were perfused and postfixed with 4% paraformaldehyde. Sections were prepared by cryostat sectioning and were stained with calbindin (Sigma, #C9848) to identify PCs. Quantification of PCs was completed by totaling Purkinje neurons from midline vermis sections from mice from each treatment group between ages 10 and 14 weeks, depending on cohort, to mirror ages at behavior testing. Cell counts were done by hand, blinded to genotype/treatment. Every PC in the slice is counted. Each slice was 20 µm thick with 10 slices (200 µm) between each.
Behavioral analysis
Behavioral testing was performed four weeks after treatment cessation; for the eight-week treatment group behavior began at 13 weeks, and for the four-week treatment group behavior began at nine weeks (Tables 1, 2). Tests were performed in the following order: rotarod, week 13/9; open field and elevated plus, week 14/10; three-chambered social interaction, olfaction (Almond, McCormick: UPC:052100070643), and grooming, week 15/11; water Y maze, week 16/12. Differences in number for behavioral testing cohorts resulted from variation between genotypes generated from crosses. In ordering and grouping these tests, every attempt has been made to refer to previous literature regarding behavioral test order in mice (McIlwain et al., 2001; Võikar et al., 2004; McFarlane et al., 2008). All studies were done blinded to genotype.
Social interaction
Animals were tested for social interaction in the three-chambered apparatus (Nationwide Plastics) as previously described (Yang et al., 2011). Animals were individually housed for 30 min before placed in the middle chamber of the three-chambered apparatus for 10 min. Next, in the habituation phase, the animals explored the entire apparatus for 10 min. A novel animal (male, age matched, C57BL/6j) and novel object were then inserted into opposite chambers in the apparatus and animals were tested for 10 min in this social approach model. Then, a novel animal was inserted into the chamber in place of the novel object and social novelty was evaluated for an additional 10 min. Time spent in each chamber and the number of crossings between chambers were recorded in an automated manner (Noldus Ethovision software version 12.5). Time spent interacting with the novel animal and novel object was scored with a stopwatch by an examiner who was blinded to experimental condition and genotype. Animals were tested between 10 and 14 weeks of age. Light at the center of the three-chambered apparatus was 30 lux for all experiments.
Open field
Open field testing was performed as described for a 15-min period (Silverman et al., 2010). Movement and time spent in the center quadrants were recorded by video camera and automated analysis were performed using Noldus Ethovison software version 12.5. Light at the center of the open field was 30 lux.
Elevated plus maze
Elevated plus maze testing was performed as previously described for a 5-min period (Silverman et al., 2010). Distance traveled and time in open arms were recorded by video camera and automated analysis were performed using Noldus Ethovision software version 12.5. Light in the open arms was 20 lux.
Grooming
Animals were removed from home cages and placed individually into new cages contained bedding only. Animals were allowed to habituate to the new cage for 10 min. Animals were then observed for 10 min, and time spent grooming was scored by an examiner blinded to experimental condition and genotype (McFarlane et al., 2008).
Water Y maze
Reversal learning was testing using the water Y maze as previously described (Roullet and Crawley, 2011). Animals were briefly habituated to the apparatus. For the first three trial sessions, mice were given 15 trials to locate a submerged platform placed in one of the maze arms. After the third trial session, the platform was moved to the other arm of the Y maze. Mice were then tested for three additional sessions with 15 trials per session (reversal trials 1–3). Animals underwent two trial sessions per day, and the number of correct trials were recorded.
Olfaction
Olfaction was tested using the habituation/dishabituation experimental model as previously described and performed (Yang and Crawley, 2009). Animals were briefly habituated to the testing environment for 30 min, then sequentially presented with cotton swabs dipped in water, almond extract or banana extract diluted at 1:200 (McCormick). Mice were exposed to each olfactory stimulus for three 2-min trials. Time spent sniffing each olfactory stimulus was recorded.
Accelerating rotarod
Animals were tested using the accelerating rotarod as previously described over five consecutive days (Buitrago et al., 2004). Latency to fall was recorded.
Statistical analysis
Statistics data are reported as a mean ± SEM, and statistical analysis was conducted with GraphPad Prism software using two-way ANOVA with Bonferroni's multiple comparison tests for post hoc analysis. Significance was defined as p < 0.05. Number of animals and statistical results used for all studies is included in Tables 1, 2. ROUT methodology in GraphPad Prism was used to determine the presence of outliers. Behavioral testing results can be found in Table 1 (eight-week Rapa treatment cohort) and Table 2 (four-week Rapa treatment cohort).
Results
Social and inflexible behaviors have distinct critical periods of development
Rapamycin is an mTOR-specific inhibitor. When administered to PC-Tsc1 mutant mice starting at P7 this treatment prevents the development of social impairments and behavioral inflexibility (Tsai et al., 2012). To identify a critical period for development of ASD-relevant symptoms using the PC-Tsc1 mutant mice, we modeled our paradigm on these prior studies. Mice received an intraperitoneal (IP) injection of 6 mg/kg rapamycin 3×/wk, starting at P7. This paradigm has been demonstrated in numerous studies to result in robust mTOR inhibition (Meikle et al., 2007, 2008a; Di Nardo et al., 2009; Tsai et al., 2012, 2018). Treatment began at P7 and lasted for a total of eight weeks completing at P63. Four weeks after treatment was stopped, we evaluated behavioral, physiological, and anatomic phenotypes (Fig. 1A; all statistical data for this eight-week cohort can be found in Table 1). This model tests whether normal mTOR signaling during this eight-week time window, from P7 to P63, can prevent the development of abnormal ASD-relevant behaviors even with the return of abnormal mTOR signaling on cessation of rapamycin treatment. Put another way, this model investigates whether disruption of mTOR signaling after this eight-week period is sufficient to disrupt normal behaviors. If subsequent behaviors are abnormal, this result would indicate a persistently open critical period during which animals continue to be vulnerable to the development of these abnormal behaviors.
Figure 1.
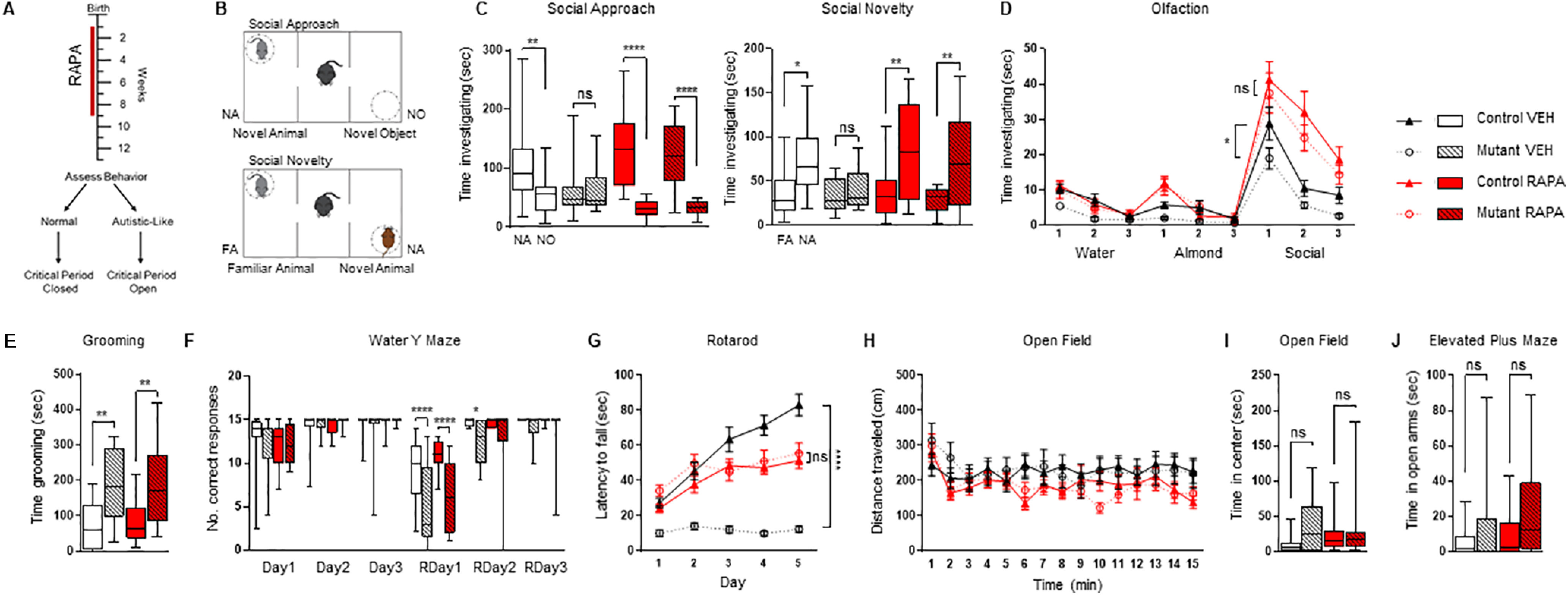
Treatment with rapamycin for eight weeks is sufficient to prevent the onset of impaired social behaviors. A, Diagram of treatment and behavior paradigm. Red line denotes period of rapamycin treatment from postnatal day 7- postnatal day 63. Behavioral assessment then begins four weeks after cessation of rapamycin treatment. Critical period denotes period to develop abnormal behaviors. B, Diagram of three-chamber social behavior test. C, D, Eight weeks of rapamycin prevents the development of a social deficit in (C) three-chamber social approach, social novelty and (D) social olfactory testing. E, F, Eight weeks of treatment do not prevent (E) repetitive grooming or (F) deficits in reversal learning behavior on a water Y maze. G, In rotarod testing, eight-week rapamycin-treated groups show improved motor learning compared with vehicle treated mutants. H, Open field testing revealed no significant impairment in locomotor function between any group. I, J, No anxiety-like behaviors were observed in (I) time spent in open field center nor (J) time spend in elevated plus maze open arms following eight weeks of rapamycin treatment. Two-way ANOVA, Bonferroni post hoc analysis. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. FA, familiar animal; NA, novel animal; NO, novel object; ns, not significant. RAPA, rapamycin; VEH, vehicle; RDay, reversal day. Data are reported as mean ± SEM.
As social impairment is the defining behavioral feature in ASD, we first examined social behavior (Fig. 1B–D). In the previously characterized PC-Tsc1 mouse, social deficits are observed by eight weeks of age (Tsai et al., 2012). This result was reproduced in the vehicle-treated mutants in all social testing performed: we observed impairments in social approach and social novelty assessments using the three-chamber assay and in social olfactory testing in vehicle-treated mutant mice (Fig. 1C,D). In contrast, mutants treated with rapamycin for eight weeks, from P7 to P63, continued to possess a social preference for social stimuli even four weeks after treatment was stopped in all social behavioral paradigms (Fig. 1C,D) at levels comparable to littermate controls. Taken together, these data identify a critical period for the development of cerebellar-regulated social behaviors that appears closed by nine weeks of age, as, even weeks after treatment has been terminated, abnormal social behaviors do not develop.
We then examined repetitive and inflexible behavior, both of which are present in ASD. We first assessed for stereotyped grooming behavior, and, consistent with previous data (Tsai et al., 2012), vehicle treated mutants demonstrated repetitive grooming (Fig. 1E). To test for the existence of a critical period for the onset of abnormal grooming behavior we treated mutant mice and their littermate controls as described above (P7–P63) and then measured behavior four weeks after treatment was completed. Unlike with social testing, the treatment paradigm did not rescue repetitive grooming in the mutants (Fig. 1E). Next, we used a water Y maze to evaluate behavioral flexibility. Consistent with previous data (Tsai et al., 2012, 2018) the vehicle-treated group demonstrated impaired behavioral flexibility (Fig. 1F). Rapamycin treatment for eight weeks did not improve repetitive or flexible behavior deficits observed in PC-Tsc1 mutants; thus, we cannot define a critical period for the development of these behaviors between P7 and P63.
We then examined motor behaviors and observed a persistent improvement in motor learning on the accelerating rotarod in rapamycin-treated mutants as compared with vehicle-treated mutant littermates (Fig. 1G), pointing to the closure of a critical period for the development of these motor learning deficits. Also, to confirm that any ASD-relevant behavioral changes were not a result from changes in locomotor behaviors, open field tests were conducted with no significant results observed between treatment groups (Fig. 1H). We used the elevated plus maze and time spent in the center of the open field box to evaluate anxiety behaviors. Although a slight trend toward heightened anxiety in the mutants was observed, the results did not reach significance (Fig. 1I,J).
PC survival and excitability remains intact after eight weeks of treatment
PC-Tsc1 mutant mice present with PC death starting at P42, decreased spontaneous PC activity, and reduced intrinsic PC excitability as a result of Tsc1 loss (Tsai et al., 2012, 2018). Moreover, persistent rapamycin treatment initiated at P7 prevents the development of these pathologic and electrophysiological phenotypes (Tsai et al., 2012). To assess whether these phenotypes might contribute to the sustained benefits observed from eight weeks of rapamycin treatment, we first examined PC survival. Four weeks after cessation of treatment we stained cerebellar sections for PCs with anti-calbindin antibody. We found significantly increased PC numbers in the rapamycin-treated mutant cohort compared with vehicle-treated mutant groups (Fig. 2A,B).
Figure 2.

Treatment with rapamycin for eight weeks is sufficient to prevent reduced PC survival. A, Midline vermis sagittal sections are stained with anti-calbindin antibody and show that PC loss is prevented after eight weeks of rapamycin treatment, even after treatment cessation for four weeks. B, Quantification of PC numbers. Two-way ANOVA, Bonferroni's post hoc testing. *p < 0.05, **p < 0.01, ***p < 0.001 ****, and p < 0.0001. ns, not significant; VEH, vehicle; RAPA, rapamycin. Scale bar: 100 µm. Data are reported as mean ± SEM.
We then investigated whether electrophysiological changes might also be observed in accordance with behavioral phenotypes. We performed extracellular recordings in acute slice preparations to examine spontaneous PC firing. Consistent with our previous studies in untreated and vehicle treated mutant animals (Tsai et al., 2012, 2018), we observed reduced spontaneous firing and decreased PC excitability in vehicle-treated mutant mice (Fig. 3A,B). However, in rapamycin-treated mutants we observed persistent normalization of both tonic PC firing rates and PC excitability at levels comparable to control littermates even four weeks after cessation of treatment (Fig. 3A,B). Additionally, we examined PC membrane properties and found decreased membrane input resistance and membrane time constants (Fig. 3C–E).
Figure 3.
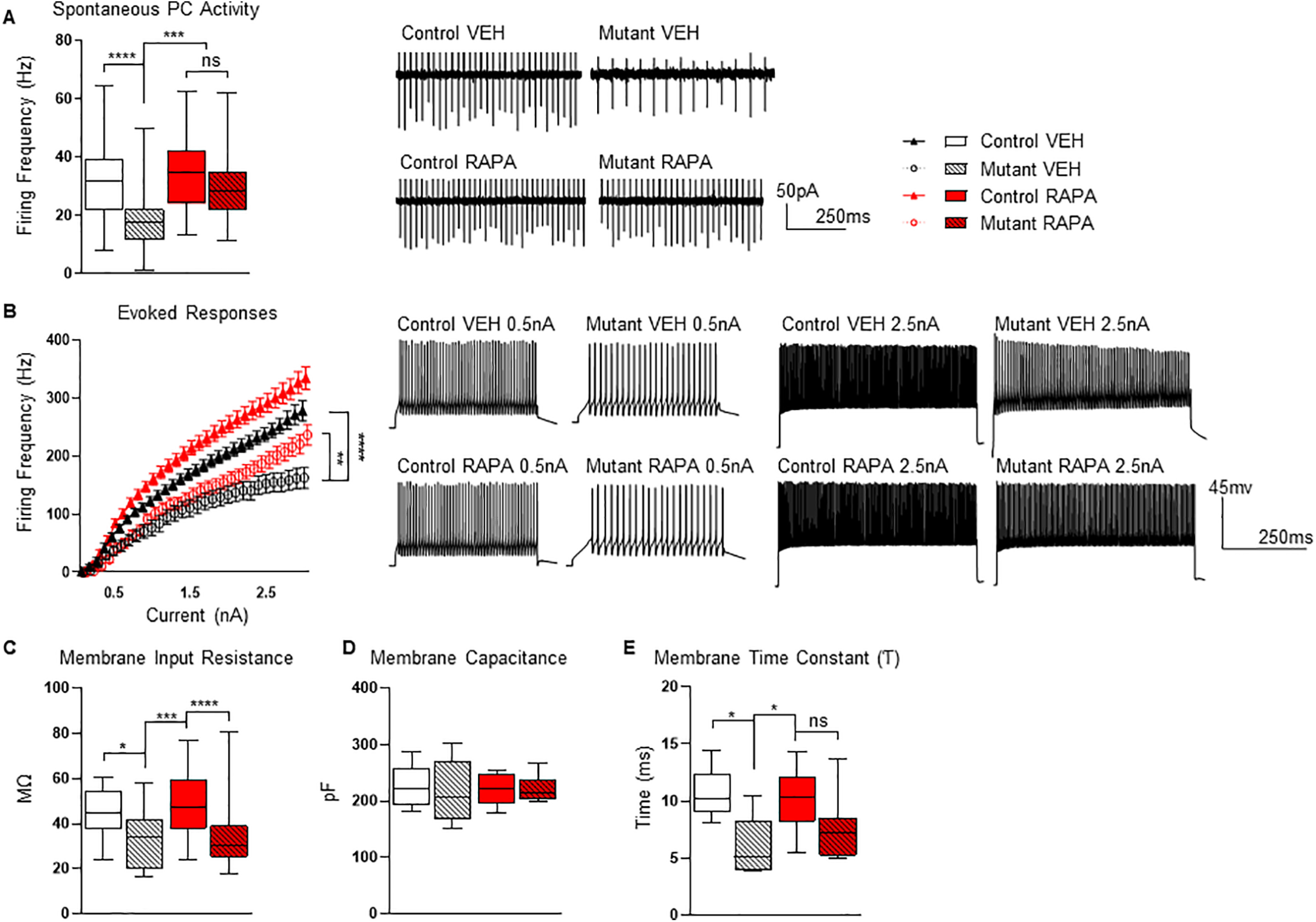
Impact of eight-week rapamycin treatment on PC function. A, B, Rapamycin treatment paradigm results in improved PC (A) intrinsic firing rates and (B) excitability in mutant mice. C–E, Cell properties during recordings show (C) decreased membrane input resistance in mutant mice, (D) membrane capacitance remained unaffected, and (E) a decreased membrane time constant in the mutants. Recordings were performed at 13 weeks of age (4 weeks after treatment was stopped at 9 weeks of age). Two-way ANOVA, Bonferroni's post hoc testing. *p < 0.05, **p < 0.01, ***p < 0.001 ****, and p < 0.0001. ns, not significant; VEH, vehicle; RAPA, rapamycin. Scale bar: 100 µm. Data are reported as mean ± SEM.
Social behaviors remain intact after four-week treatment window
Our data above demonstrate that rapamycin treatment from P7 to P63 results in persistently intact social behaviors (Fig. 1). These results suggest that the critical period during which social circuits are vulnerable is closed by nine weeks of age. Furthermore, these data indicate that development has stabilized and is no longer susceptible to dysregulated mTOR signaling. We next evaluated whether we could refine this critical window further and whether a shortened treatment duration would be sufficient to prevent the future development of social impairments. A defined critical period could be crucial for the refinement of treatment strategies, as a shorter critical period could reduce the duration of treatment therapy and thereby minimize potential therapeutic side effects. Therefore, we asked whether the critical period for social behaviors in a PC-Tsc1 mouse would be closed by five weeks of age. To assess this question, rapamycin treatment was initiated at P7 and continued for only four weeks until P35. Treatment was then stopped, and behavioral assessments commenced following an additional four weeks without treatment (Fig. 4A; all statistical data for four-week cohort can be found in Table 2).
Figure 4.
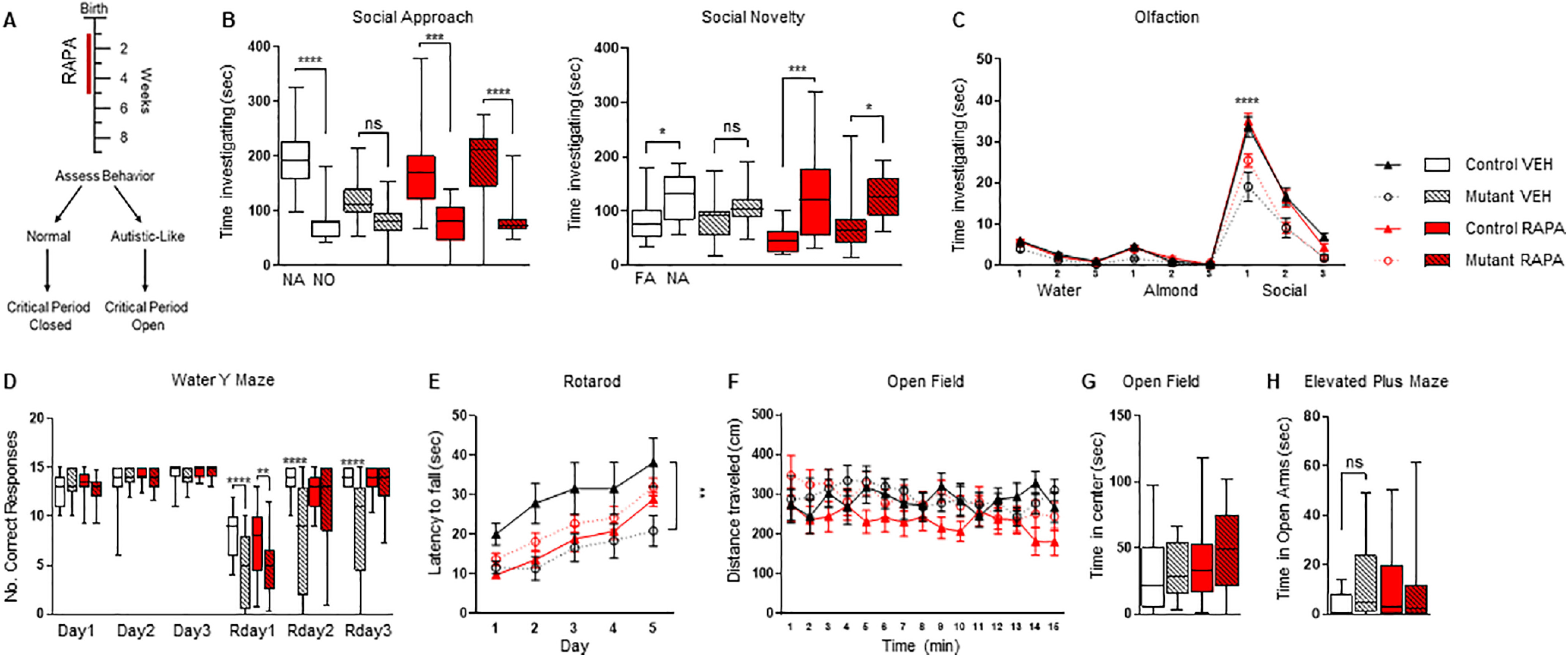
Treatment with rapamycin for four weeks is sufficient to prevent the onset of social impairments. A, Four-week rapamycin treatment paradigm (denoted by red line). Behavioral assessment then begins four weeks after cessation of rapamycin treatment. Critical period denotes period to develop abnormal behaviors. B, C, Treatment with rapamycin for four weeks prevents the onset of social deficits in (B) three-chamber social approach, social novelty, and (C) social olfactory testing. D, No changes in behavioral flexibility were observed between genotypes in the water Y maze; four weeks of rapamycin treatment had no effect on performance on Rday1. E, Motor learning in the accelerating rotarod. F, Open field testing did not reveal any differences between locomotor function between any group. G, H, No significant differences were found in (G) center time in open field or (H) time in elevated plus maze open arms between any group. Two-way ANOVA, Bonferroni's post hoc testing. *p < 0.05, **p < 0.01, ***p < 0.001 ****, and p < 0.0001. ns, not significant; VEH, vehicle; RAPA, rapamycin. Data are reported as mean ± SEM.
As previously observed in our eight-week cohort, vehicle-treated mutants displayed social impairments with no social preference noted during social approach and novelty testing in the three-chambered apparatus (Fig. 4B). In contrast, rapamycin-treated mutant mice retained their social preference, even in this shortened treatment paradigm of four weeks on treatment and subsequent assessment after four weeks off treatment (Fig. 4B). These results indicate that a shortened rapamycin treatment period is sufficient to prevent the onset of social deficits and maintain the absence of those social deficits. We then examined social olfactory behavior and similarly found that mutant mice treated with rapamycin for four weeks retained social preferences even after treatment cessation (Fig. 4C). Taken together, these data point to the critical period of developmental vulnerability for disruption of social behaviors being closed by five weeks of age.
We then evaluated inflexible behaviors and found that four weeks of rapamycin treatment from P7 to P35 did not rescue performance on reversal learning paradigms in the water Y maze in mutant mice (Fig. 4D), similar to data from the eight-week treatment cohort. In contrast, during rotarod testing, we did see an improvement in motor learning behavior in the four-week rapamycin-treated group even after treatment cessation (Fig. 4E). Open field testing did not reveal any deficits in overall locomotor function (Fig. 4F). Anxiety-like behaviors were tested with elevated plus maze and open field studies with no significant results observed between treatment groups (Fig. 4G,H).
PC survival and intrinsic excitability remain intact despite only four weeks of rapamycin treatment
We then evaluated whether pathology and electrophysiological findings would similarly support the behavioral observations in this cohort. We first examined whether this shortened treatment period would be sufficient to protect PC survival. Vehicle-treated mutants show significantly reduced PC numbers; however, rapamycin treatment from P7 to P35, resulted in continued PC survival even 4+ weeks after cessation of treatment (Fig. 5A,B).
Figure 5.

Impact of four-week rapamycin treatment on PC survival. Treatment with rapamycin for four weeks is sufficient to prevent reduced PC function. A, Midline vermis sagittal sections are stained with anti-calbindin antibody. Studies show PC loss is prevented by four-week treatment paradigm. B, PC numbers quantified. Two-way ANOVA, Bonferroni's post hoc testing. *p < 0.05, **p < 0.01, ***p < 0.001 ****, and p < 0.0001. ns, not significant; VEH, vehicle; RAPA, rapamycin. Scale bar: 100 µm. Data are reported as mean ± SEM.
We then examined spontaneous PC firing in extracellular recordings from acute slice preparations. Similar to the eight-week (P7–P63) treatment cohort, PC spontaneous firing rates and intrinsic excitability were significantly reduced in vehicle treated mutants (Fig. 6A,B). Rapamycin treatment from P7 to P35 in mutant mice, however, resulted in persistent normalization of both tonic firing frequency and intrinsic excitability (Fig. 6A,B). Cell membrane properties were evaluated with no significant differences between groups (Fig. 6C–E).
Figure 6.
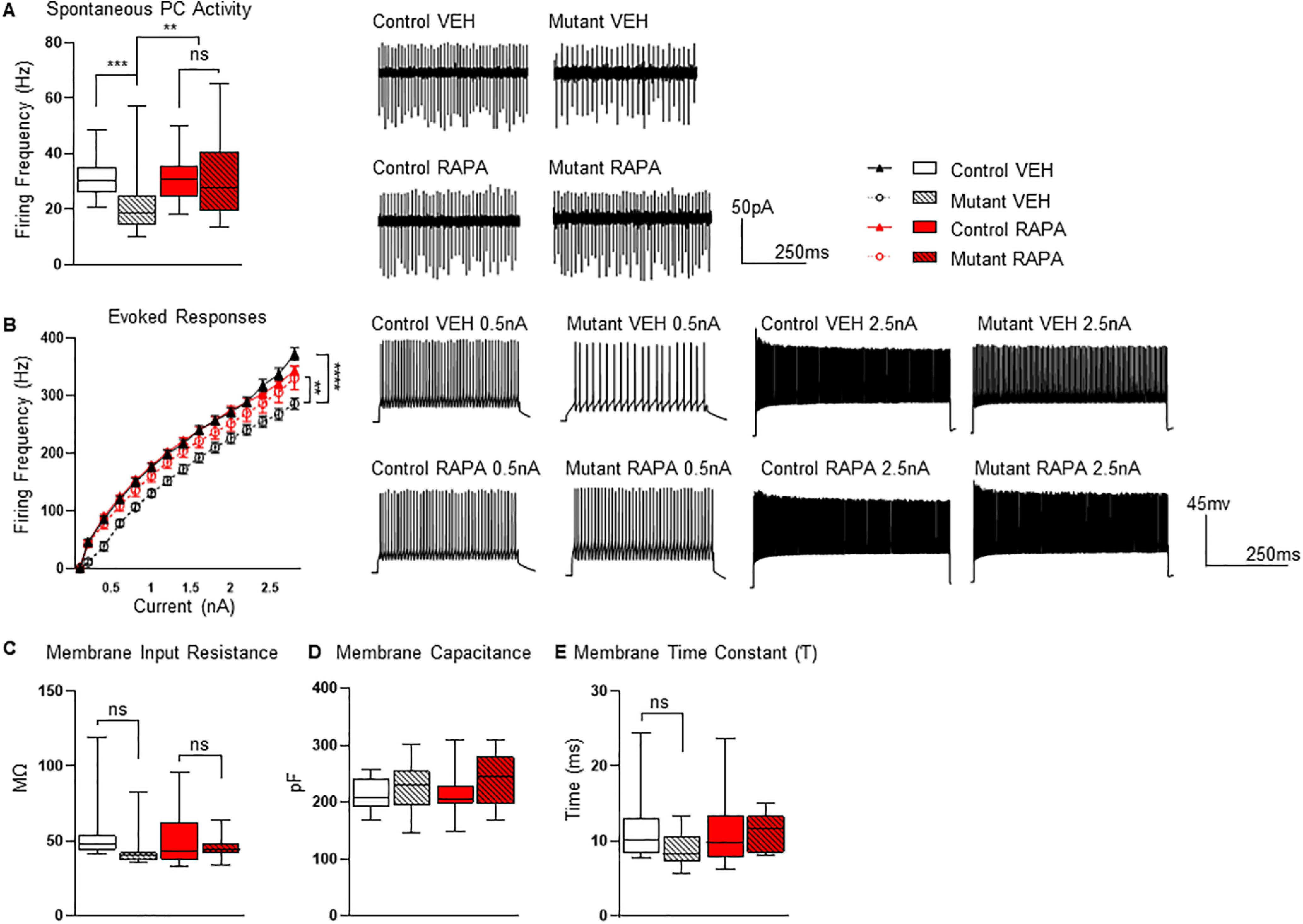
Impact of four-week rapamycin treatment on PC function. A, B, Four weeks of rapamycin treatment prevented deficits in (A) PC intrinsic firing rates and (B) excitability. C–E, Cell properties during recordings revealed no significant differences in (C) membrane input resistance, (D) membrane capacitance, or (E) membrane time constant. Recordings were performed at nine weeks of age (4 weeks after treatment was stopped at 5 weeks of age). Two-way ANOVA, Bonferroni's post hoc testing. *p < 0.05, **p < 0.01, ***p < 0.001 ****, and p < 0.0001. ns, not significant; VEH, vehicle; RAPA, rapamycin. Scale bar: 100 µm. Data are reported as mean ± SEM.
Discussion
The developing brain can adapt and change with the surrounding environmental and sensory stimuli that an organism experiences (Kolb et al., 2013). This development occurs during distinct time windows termed critical periods that are a crucial part of the development and formation of neural circuitry necessary for complex behaviors. These periods close once structural consolidation of circuit connectivity is achieved, thereby reducing if not preventing further occurrence of plasticity in the mature animal. Although these time windows offer enhanced plasticity and thus enhanced learning, the brain is also vulnerable to disruptions during these windows of neural circuit formation. Disruptions during early development results in numerous neurodevelopmental disorders including disorders marked by social impairment such as ASD (LeBlanc and Fagiolini, 2011). Once closure of these periods occurs, individuals may be less susceptible to genetic or other disruption.
Multiple studies have examined sensitive periods, time windows for treatment, of ASD-relevant behaviors in neurodevelopmental disorders and models of ASD (Silva-Santos et al., 2015; Mei et al., 2016; Ure et al., 2016), including for cerebellar associated ASD-relevant behaviors (Tsai et al., 2018). Unlike sensitive periods, critical periods are not windows when treatment can be effective but are the times when disruptions may result in developmental disability. Beyond these time windows of developmental vulnerability, disruption may result in less severe or no consequences, consistent with the lack of emergence of ASD during adulthood even with sensory deprivation during later childhood/adulthood (Nelson et al., 2007). However, insults during a critical period of early childhood will lead to abnormal behavior and neurodevelopmental disability (Sonuga-Barke et al., 2017). Thus, closure of these developmental periods affords a translational, clinically relevant opportunity; if one can bypass these periods of developmental vulnerability, an individual may avoid the most detrimental consequences of environmental or genetic insults.
Delineating the timing of these critical periods of developmental opportunity and potential vulnerability affords critical translational utility. Pharmacologic agents that target signaling dysfunction in many neurodevelopmental disorders are being developed; however, many of these signaling pathways are fundamental pathways and these therapeutic opportunities also come with risk of potential side effects. For instance, epilepsy therapies have significant side effects on cognition and metabolic function (Mintzer, 2010; Beltramini et al., 2015). Similarly, the drug rapamycin has proven efficacious for treatment of many facets of dysfunction associated with TSC; however, its use comes at significant cost including immunosuppression among others (Palavra et al., 2017). Thus, identifying the critical periods after which treatment could ultimately be stopped, thereby bypassing neurodevelopmental disruption but limiting side effects from treatment as well, would be a critical intervention for treatment optimization.
In this study, we delineate critical periods for the cerebellar contribution to ASD-relevant behaviors. We find that treatment with rapamycin to five weeks of age is sufficient to facilitate the development and maintenance of normal social behaviors and that social behaviors remain comparable to control littermates even after cessation of treatment for a month or greater. In the mouse, five weeks of age is a postweaning time frame in which the animal is approaching sexual maturity, and thus is consistent with late childhood/early adolescence (Dutta and Sengupta, 2016). Thus, similar to what has been observed in some developmental epilepsies which often change behaviors around puberty, critical periods of vulnerability for cerebellar-regulated social behaviors appear to close by this time point. Taken together, these results add to the growing literature (Volpe, 2009; Tsai et al., 2012, 2018; Wang et al., 2014; Stoodley et al., 2017; Badura et al., 2018; Kelly et al., 2020), to outline a timetable and clear cerebellar contribution to developing ASD-relevant behaviors (Table 3). Differences observed between studies could be attributed to multiple differences in methodology, targeted cell types between models, regional versus global cerebellar involvement, in addition to the tested circuit versus genetic contributions across development examined in these studies.
Table 3.
Results comparisons
| Cell type targeted | Social: three-chamber, olfaction | Locomotor: Rotarod, open field, gait | Reversal learning: water Y/T maze | Repetitive: grooming | PC viability: survival/function | |
|---|---|---|---|---|---|---|
| Rescue experiments | ||||||
| Tsai et al. (2012); | ||||||
| Rapamycin: P7–indefinitely |
PC | No impairment | No impairment | No impairment | X | No impairment |
| Tsai et al. (2018); | ||||||
| Rapamycin: P42–indefinitely |
PC | No impairment | No impairment | Impairment | Impairment | No impairment |
| Rapamycin: P70–indefinitely |
PC | Impairment | No impairment | Impairment | Impairment | Impairment |
| Loss of function experiments | ||||||
| Stoodley et al. (2017); | ||||||
| P56–acute chemogenetic inhibition | PC | Impairment* | No impairment* | Impairment* | Impairment* | No impairment |
| Badura et al. (2018); | ||||||
| P30–P56 chemogenetic inhibition | Molecular layer interneuron | Impairment* | No impairment* | Impairment* | Impairment* | No impairment |
| P57–acute chemogenetic inhibition | Molecular layer interneuron | Impairment* | No impairment* | Impairment* | No impairment* | No impairment |
| Kelly et al. (2020); | ||||||
| P56–acute chemogenetic inhibition | PC | Impairment* | No impairment* | Impairment* | Impairment* | No impairment |
| This study | ||||||
| Rapamycin: P7–P63 | PC | No impairment | No impairment | Impairment | Impairment | No Impairment |
| Rapamycin: P7–P35 | PC | No impairment | No impairment | Impairment | X | No Impairment |
| * for selected regions tested; X not determined | ||||||
Mechanistically, we observe the continued survival of PCs in rapamycin-treated mutants with persistent normalization of spontaneous firing rates and intrinsic excitability. Maintenance of these cellular properties is likely critical to prevent the development of ASD behaviors, as PC loss is the most consistently identified pathology in ASD, and cerebellar dysfunction is consistently noted in ASD (Bauman and Kemper, 1985; Ryu et al., 1999; D'Mello and Stoodley, 2015). Importantly, although cell survival is necessary, preservation of intrinsic cell function is equally critical. Even in the absence of PC death, intrinsic excitability and firing rate deficits in PCs are associated with abnormal behaviors (De Zeeuw et al., 2011; Stoodley et al., 2017; Kelly et al., 2020). The importance of PC function is further underscored by the fact that the inhibitory PC efferents are the sole output to downstream cerebellar nuclear neurons, which are major sources of excitatory input to the rest of the brain (Baumel et al., 2009). Thus, reductions in PC function and subsequent loss of inhibition onto cerebellar nuclear output result in increased excitatory input to the cortex (Stoodley et al., 2017; Kelly et al., 2020) and significant excitatory-inhibitory imbalance that has been documented in ASD (Antoine et al., 2019).
In this study, critical periods also do not appear to be monolithic constructs, with critical period closure depending on the specific behavior. We were unable to define a critical period for behavioral inflexibility, which is a hallmark of ASD and is robustly observed in this ASD model (Tsai et al., 2012, 2018; Stoodley et al., 2017; Kelly et al., 2020) even with treatment into adulthood and persistent cell survival and normalization of PC firing rates and intrinsic excitability. A possible explanation for this discrepancy is that these repetitive behaviors and behavioral flexibility continue to be plastic through the adult years and/or that a critical period for the contribution of mTOR signaling to these behaviors ends beyond the time window tested in this study. This hypothesis is consistent with data showing that repetitive and inflexible behaviors emerge from brain insults in adulthood (Wood and Worthington, 2017). This occurrence differs from the social impairments in ASD which emerge during early development (Lord et al., 2018). Additionally, the difference in responses between these autism-relevant social and repetitive/inflexible behaviors observed under this treatment regimen also likely relate to these behaviors being governed by divergent circuitry (Kelly et al., 2020) and may therefore have discrete critical periods. These observations suggest that normal development during multiple critical periods is essential for neuro-typical development and may be a contributing factor to the etiology of the disorder.
Few preclinical studies have examined the existence of critical periods in animal models of neurodevelopmental disorders. A recent study in a rodent model of Shank3 dysfunction points to continued vulnerability for the development of both social and repetitive behavior at least through P30 in the mouse (Mossa et al., 2021), in contrast to the distinct critical periods between these two behaviors we observed in this study. In our study, treatment until P35 with a four-week washout, results in persistently normalized social behaviors. The differences between the two studies are numerous, from duration of treatment, genetic model, to the cerebellar focus of this study, all distinctions that could promote differences between the studies. Another important factor to consider is the discrepancy in severity across genetic neurodevelopmental disorders, which may be a causal factor in the differing phenotypes across studies of critical periods. Further evaluation of these factors in addition to comprehensive study of additional models will be critical to evaluate whether findings are specific to certain factors or more generalizable across studies.
These studies clearly raise the possibility for translational application. With the advent of genetic testing and continued biomarker development, the prospect of early diagnosis of neurodevelopmental disorders such as ASD will continue to increase, thereby raising the possibility that treatment initiated early could provide therapeutic benefit. Thus, knowing the parameters of critical periods when therapy could be halted at a precise time point to prevent the development of abnormal behavior instead of continuous treatment to manage abnormal behaviors which are fully developed and no longer plastic, would be critically important in minimizing side effects from unnecessarily prolonged therapies. For TSC in particular, this strategy has greater potential. TSC is characterized by hamartomatous lesion development involving regions beyond the brain. Cardiac rhabdomyomas develop in utero and are detected during routine prenatal ultrasound screening (Milunsky et al., 2009). The presence of these lesions is a major indicator prompting testing for TSC, which often results in diagnosis before birth. Thus, this early diagnosis might provide the opportunity for neurodevelopmental disorders such as TSC to be treated early with potential opportunity to stop treatment once the critical period has been bypassed. From a social impairment point of view, findings from this study may point to preadolescence for that timing.
Thus, we provide a framework for the timing of a critical period for the development of ASD-relevant behaviors, while providing cellular and physiological mechanisms that contribute to these behaviors. Delineation of this timing has the capacity to inform clinical therapy for treatment of neurodevelopmental disorders in general and TSC in specific.
Footnotes
Acknowledgements: We thank the University of Texas Southwestern Medical Center Whole Brain Microscopy Core Facility, RRID:SCR_017949, for assistance with microscopy and imaging. We also thank the University of Texas Southwestern Medical Center Behavioral Phenotyping Core. J.M.G. was supported by the National Heart, Lung, and Blood Institute Grant 1T32HL139438-01A1. P.T.T. was supported by the National Institute of Neurologic Disorders and Stroke of the National Institutes of Health (NIH) Grant K08 NS083733 and National Institute of Mental Health of the NIH Grant R01 MH116882.
The authors declare no competing financial interests.
References
- Antoine MW, Langberg T, Schnepel P, Feldman DE (2019) Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron 101:648–661.e4. 10.1016/j.neuron.2018.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano E, Chugani DC, Muzik O, Behen M, Janisse J, Rothermel R, Mangner TJ, Chakraborty PK, Chugani HT (2001) Autism in tuberous sclerosis complex is related to both cortical and subcortical dysfunction. Neurology 57:1269–1277. 10.1212/WNL.57.7.1269 [DOI] [PubMed] [Google Scholar]
- Badura A, Verpeut JL, Metzger JW, Pereira TD, Pisano TJ, Deverett B, Bakshinskaya DE, Wang SSH (2018) Normal cognitive and social development require posterior cerebellar activity. Elife 7:e36401. 10.7554/eLife.36401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, Rutter M, Lantos P (1998) A clinicopathological study of autism. Brain 121:889–905. 10.1093/brain/121.5.889 [DOI] [PubMed] [Google Scholar]
- Barski JJ, Dethleffsen K, Meyer M (2000) Cre recombinase expression in cerebellar Purkinje cells. Genesis 28:93–98. 10.1002/1526-968X(200011/12)28:3/4<93::AID-GENE10>3.0.CO;2-W [DOI] [PubMed] [Google Scholar]
- Bauman M, Kemper TL (1985) Histoanatomic observations of the brain in early infantile autism. Neurology 35:866–874. 10.1212/wnl.35.6.866 [DOI] [PubMed] [Google Scholar]
- Baumel Y, Jacobson GA, Cohen D (2009) Implications of functional anatomy on information processing in the deep cerebellar nuclei. Front Cell Neurosci 3:14. 10.3389/neuro.03.014.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramini GC, Cendes F, Yasuda CL (2015) The effects of antiepileptic drugs on cognitive functional magnetic resonance imaging. Quant Imaging Med Surg 5:238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blows WT (2003) Child brain development. Nurs Times 99:28–31. [PubMed] [Google Scholar]
- Buitrago MM, Schulz JB, Dichgans J, Luft AR (2004) Short and long-term motor skill learning in an accelerated rotarod training paradigm. Neurobiol Learn Mem 81:211–216. 10.1016/j.nlm.2004.01.001 [DOI] [PubMed] [Google Scholar]
- Cupolillo D, Hoxha E, Faralli A, De Luca A, Rossi F, Tempia F, Carulli D (2016) Autistic-like traits and cerebellar dysfunction in Purkinje Cell PTEN knock-out mice. Neuropsychopharmacology 41:1457–1466. 10.1038/npp.2015.339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zeeuw CI, Hoebeek FE, Bosman LW, Schonewille M, Witter L, Koekkoek SK (2011) Spatiotemporal firing patterns in the cerebellum. Nat Rev Neurosci 12:327–344. 10.1038/nrn3011 [DOI] [PubMed] [Google Scholar]
- Di Nardo A, Kramvis I, Cho N, Sadowski A, Meikle L, Kwiatkowski DJ, Sahin M (2009) Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J Neurosci 29:5926–5937. 10.1523/JNEUROSCI.0778-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello AM, Stoodley CJ (2015) Cerebro-cerebellar circuits in autism spectrum disorder. Front Neurosci 9:408. 10.3389/fnins.2015.00408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S, Sengupta P (2016) Men and mice: relating their ages. Life Sci 152:244–248. 10.1016/j.lfs.2015.10.025 [DOI] [PubMed] [Google Scholar]
- Eluvathingal TJ, Behen ME, Chugani HT, Janisse J, Bernardi B, Chakraborty P, Juhasz C, Muzik O, Chugani DC (2006) Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. J Child Neurol 21:846–851. 10.1177/08830738060210100301 [DOI] [PubMed] [Google Scholar]
- Harlow HF, Suomi SJ (1971) Social recovery by isolation-reared monkeys. Proc Natl Acad Sci USA 68:1534–1538. 10.1073/pnas.68.7.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer MA (1970) Physiological responses of infant rats to separation from their mothers. Science 168:871–873. 10.1126/science.168.3933.871 [DOI] [PubMed] [Google Scholar]
- Hofer MA (1994) Hidden regulators in attachment, separation, and loss. Monogr Soc Res Child Dev 59:192–207. 10.1111/j.1540-5834.1994.tb01285.x [DOI] [PubMed] [Google Scholar]
- Hsieh DT, Thiele EA (2013) Vigabatrin-related magnetic resonance imaging abnormalities in an infant with tuberous sclerosis complex and infantile spasms. J Pediatr 162:215. 10.1016/j.jpeds.2012.07.061 [DOI] [PubMed] [Google Scholar]
- Jeste SS, Varcin KJ, Hellemann GS, Gulsrud AC, Bhatt R, Kasari C, Wu JY, Sahin M, Nelson CA 3rd (2016) Symptom profiles of autism spectrum disorder in tuberous sclerosis complex. Neurology 87:766–772. 10.1212/WNL.0000000000003002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E, Meng F, Fujita H, Morgado F, Kazemi Y, Rice LC, Ren C, Escamilla CO, Gibson JM, Sajadi S, Pendry RJ, Tan T, Ellegood J, Basson MA, Blakely RD, Dindot SV, Golzio C, Hahn MK, Katsanis N, Robins DM, et al. (2020) Regulation of autism-relevant behaviors by cerebellar–prefrontal cortical circuits. Nat Neurosci 23:1102–1110. 10.1038/s41593-020-0665-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb B, Mychasiuk R, Muhammad A, Gibb R (2013) Brain plasticity in the developing brain. Prog Brain Res 20:35–64. [DOI] [PubMed] [Google Scholar]
- LeBlanc JJ, Fagiolini M (2011) Autism: a “critical period” disorder? Neural Plast 2011:921680. 10.1155/2011/921680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limperopoulos C, Bassan H, Sullivan NR, Soul JS, Robertson RL Jr, Moore M, Ringer SA, Volpe JJ, du Plessis AJ (2008) Positive screening for autism in ex-preterm infants: prevalence and risk factors. Pediatrics 121:758–765. 10.1542/peds.2007-2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton JO, Sahin M (2014) The neurology of mTOR. Neuron 84:275–291. 10.1016/j.neuron.2014.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Elsabbagh M, Baird G, Veenstra-Vanderweele J (2018) Autism spectrum disorder. Lancet 392:508–520. 10.1016/S0140-6736(18)31129-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackes NK, Golm D, Sarkar S, Kumsta R, Rutter M, Fairchild G, Mehta MA, Sonuga-Barke EJS; ERA Young Adult Follow-up team (2020) Early childhood deprivation is associated with alterations in adult brain structure despite subsequent environmental enrichment. Proc Natl Acad Sci USA 117:641–649. 10.1073/pnas.1911264116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane HG, Kusek GK, Yang M, Phoenix JL, Bolivar VJ, Crawley JN (2008) Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes Brain Behav 7:152–163. 10.1111/j.1601-183X.2007.00330.x [DOI] [PubMed] [Google Scholar]
- McIlwain KL, Merriweather MY, Yuva-Paylor LA, Paylor R (2001) The use of behavioral test batteries: effects of training history. Physiol Behav 73:705–717. 10.1016/s0031-9384(01)00528-5 [DOI] [PubMed] [Google Scholar]
- Mei Y, Monteiro P, Zhou Y, Kim J-A, Gao X, Fu Z, Feng G (2016) Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530:481–484. 10.1038/nature16971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ (2007) A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci 27:5546–5558. 10.1523/JNEUROSCI.5540-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ (2008a) Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci 28:5422–5432. 10.1523/JNEUROSCI.0955-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ (2008b) Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci 28:5422–5432. 10.1523/JNEUROSCI.0955-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milunsky A, Ito M, Maher TA, Flynn M, Milunsky JM (2009) Prenatal molecular diagnosis of tuberous sclerosis complex. Am J Obstet Gynecol 200:321.e1–6. [DOI] [PubMed] [Google Scholar]
- Mintzer S (2010) Metabolic consequences of antiepileptic drugs. Curr Opin Neurol 23:164–169. 10.1097/WCO.0b013e32833735e7 [DOI] [PubMed] [Google Scholar]
- Mossa A, Pagano J, Ponzoni L, Tozzi A, Vezzoli E, Sciaccaluga M, Costa C, Beretta S, Francolini M, Sala M, Calabresi P, Boeckers TM, Sala C, Verpelli C (2021) Developmental impaired Akt signaling in the Shank1 and Shank3 double knock-out mice. Mol Psychiatry 26:1928–1944. 10.1038/s41380-020-00979-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muncy J, Butler IJ, Koenig MK (2009) Rapamycin reduces seizure frequency in tuberous sclerosis complex. J Child Neurol 24:477–477. 10.1177/0883073808324535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CA 3rd, Zeanah CH, Fox NA, Marshall PJ, Smyke AT, Guthrie D (2007) Cognitive recovery in socially deprived young children: the Bucharest Early Intervention Project. Science 318:1937–1940. 10.1126/science.1143921 [DOI] [PubMed] [Google Scholar]
- Neumann ID, Wigger A, Krömer S, Frank E, Landgraf R, Bosch OJ (2005) Differential effects of periodic maternal separation on adult stress coping in a rat model of extremes in trait anxiety. Neuroscience 132:867–877. 10.1016/j.neuroscience.2005.01.034 [DOI] [PubMed] [Google Scholar]
- Palavra F, Robalo C, Reis F (2017) Recent advances and challenges of mTOR inhibitors use in the treatment of patients with tuberous sclerosis complex. Oxid Med Cell Longev 2017:9820181. 10.1155/2017/9820181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter S, Ten Brinke MM, Stedehouder J, Reinelt CM, Wu B, Zhou H, Zhou K, Boele HJ, Kushner SA, Lee MG, Schmeisser MJ, Boeckers TM, Schonewille M, Hoebeek FE, De Zeeuw CI (2016) Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat Commun 7:12627. 10.1038/ncomms12627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith RM, McKenna J, Wu H, Hashmi SS, Cho S-H, Dash PK, Gambello MJ (2013) Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol Dis 51:93–103. 10.1016/j.nbd.2012.10.014 [DOI] [PubMed] [Google Scholar]
- Roullet FI, Crawley JN (2011) Mouse models of autism: testing hypotheses about molecular mechanisms. Curr Top Behav Neurosci 7:187–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu YH, Lee JD, Yoon PH, Kim DI, Lee HB, Shin YJ (1999) Perfusion impairments in infantile autism on technetium-99m ethyl cysteinate dimer brain single-photon emission tomography: comparison with findings on magnetic resonance imaging. Eur J Nucl Med 26:253–259. 10.1007/s002590050385 [DOI] [PubMed] [Google Scholar]
- Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, Mizuguchi M (2012) Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun 3:1992. 10.1038/ncomms2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Santos S, van Woerden GM, Bruinsma CF, Mientjes E, Jolfaei MA, Distel B, Kushner SA, Elgersma Y (2015) Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin Invest 125:2069–2076. 10.1172/JCI80554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN (2010) Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 11:490–502. 10.1038/nrn2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonuga-Barke EJS, Kennedy M, Kumsta R, Knights N, Golm D, Rutter M, Maughan B, Schlotz W, Kreppner J (2017) Child-to-adult neurodevelopmental and mental health trajectories after early life deprivation: the young adult follow-up of the longitudinal English and Romanian Adoptees study. Lancet 389:1539–1548. 10.1016/S0140-6736(17)30045-4 [DOI] [PubMed] [Google Scholar]
- Stoodley CJ, D'Mello AM, Ellegood J, Jakkamsetti V, Liu P, Nebel MB, Gibson JM, Kelly E, Meng F, Cano CA, Pascual JM, Mostofsky SH, Lerch JP, Tsai PT (2017) Altered cerebellar connectivity in autism and cerebellar-mediated rescue of autism-related behaviors in mice. Nat Neurosci 20:1744–1751. 10.1038/s41593-017-0004-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, Sahin M (2012) Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488:647–651. 10.1038/nature11310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Rudolph S, Guo C, Ellegood J, Gibson JM, Schaeffer SM, Mogavero J, Lerch JP, Regehr W, Sahin M (2018) Sensitive periods for cerebellar-mediated autistic-like behaviors. Cell Rep 25:357–367.e4. 10.1016/j.celrep.2018.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ure K, Lu H, Wang W, Ito-Ishida A, Wu Z, He LJ, Sztainberg Y, Chen W, Tang J, Zoghbi HY (2016) Restoration of Mecp2 expression in GABAergic neurons is sufficient to rescue multiple disease features in a mouse model of Rett syndrome. Elife 5:e14198. 10.7554/eLife.14198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhave J, Boucher A, Dandavino R, Collette S, Senécal L, Hebert MJ, Girardin C, Cardinal H (2014) The incidence, management, and evolution of rapamycin-related side effects in kidney transplant recipients. Clin Transplant 28:616–622. 10.1111/ctr.12361 [DOI] [PubMed] [Google Scholar]
- Võikar V, Vasar E, Rauvala H (2004) Behavioral alterations induced by repeated testing in C57BL/6J and 129S2/Sv mice: implications for phenotyping screens. Genes Brain Behav 3:27–38. 10.1046/j.1601-183x.2003.0044.x [DOI] [PubMed] [Google Scholar]
- Volpe JJ (2009) Cerebellum of the premature infant: rapidly developing, vulnerable, clinically important. J Child Neurol 24:1085–1104. 10.1177/0883073809338067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SSH, Kloth AD, Badura A (2014) The cerebellum, sensitive periods, and autism. Neuron 83:518–532. 10.1016/j.neuron.2014.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney ER, Kemper TL, Bauman ML, Rosene DL, Blatt GJ (2008) Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum 7:406–416. 10.1007/s12311-008-0043-y [DOI] [PubMed] [Google Scholar]
- Wood RL, Worthington A (2017) Neurobehavioral abnormalities associated with executive dysfunction after traumatic brain injury. Front Behav Neurosci 11:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Crawley JN (2009) Simple behavioral assessment of mouse olfaction. Curr Protoc Neurosci Chapter 8:Unit 8.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Silverman JL, Crawley JN (2011) Automated three-chambered social approach task for mice. Curr Protoc Neurosci Chapter 8:Unit–Uni8.26. [DOI] [PMC free article] [PubMed] [Google Scholar]