

Abstract
Classically, mitochondrial respiration responds to decreased membrane potential (ΔΨ) by increasing respiration. However, we found that for succinate‐energized complex II respiration in skeletal muscle mitochondria (unencumbered by rotenone), low ΔΨ impairs respiration by a mechanism culminating in oxaloacetate (OAA) inhibition of succinate dehydrogenase (SDH). Here, we investigated whether this phenomenon extends to far different mitochondria of a tissue wherein ΔΨ is intrinsically low, i.e., interscapular brown adipose tissue (IBAT). Also, to advance our knowledge of the mechanism, we performed isotopomer studies of metabolite flux not done in our previous muscle studies. In additional novel work, we addressed possible ways ADP might affect the mechanism in IBAT mitochondria. UCP1 activity, and consequently ΔΨ, were perturbed both by GDP, a well‐recognized potent inhibitor of UCP1 and by the chemical uncoupler carbonyl cyanide m‐chlorophenyl hydrazone (FCCP). In succinate‐energized mitochondria, GDP increased ΔΨ but also increased rather than decreased (as classically predicted under low ΔΨ) O2 flux. In GDP‐treated mitochondria, FCCP reduced potential but also decreased respiration. Metabolite studies by NMR and flux analyses by LC‐MS support a mechanism, wherein ΔΨ effects on the production of reactive oxygen alters the NADH/NAD+ ratio affecting OAA accumulation and, hence, OAA inhibition of SDH. We also found that ADP‐altered complex II respiration in complex fashion probably involving decreased ΔΨ due to ATP synthesis, a GDP‐like nucleotide inhibition of UCP1, and allosteric enzyme action. In summary, complex II respiration in IBAT mitochondria is regulated by UCP1‐dependent ΔΨ altering substrate flow through OAA and OAA inhibition of SDH.
Keywords: bioenergetics, brown adipose tissue, metabolism, metabolomics, mitochondria, mitochondrial metabolism, reactive oxygen species (ROS), uncoupling protein
Abbreviations
- 2DOG
2‐deoxyglucose
- 2DOGP
2‐deoxyglucose phosphate
- DHPA
10‐acetyl‐3,7‐dihydroxyphenoxazine
- Got2
mitochondrial aspartate aminotransferase
- IBAT
interscapular brown adipose tissue
- NMR
nuclear magnetic resonance
- OAA
oxaloacetate
- ODX
oxaloacetate decarboxylase
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- SOD
superoxide dismutase
- TPP
tetraphenylphosphonium
- ΔΨ
mitochondrial membrane potential
1. INTRODUCTION
Studies dating back several decades 1 , 2 , 3 , 4 , 5 , 6 , 7 show that OAA inhibits succinate dehydrogenase (SDH). However, the effect of this phenomenon on mitochondrial function and the mechanism by which this might occur have been largely neglected. We think this is because OAA is unstable and difficult to quantify by mass spectroscopy or other means. 8 , 9 Another reason that the effect of OAA has not received attention likely involves the classical use of rotenone to block electron flow through complex I when assessing complex II respiration on succinate. But rotenone also blocks malate conversion to OAA, thus obscuring any feedback effect of OAA to inhibit complex II.
We recently developed a sensitive and highly specific NMR method to quantify OAA in mitochondrial extracts. 10 , 11 Using this methodology, we found that skeletal muscle mitochondrial respiration energized by the complex II substrate, succinate (in the absence of rotenone often added to inhibit complex I), is regulated by the accumulation of oxaloacetate (OAA), a known potent inhibitor of succinate dehydrogenase (SDH). 1 , 3 , 12 , 13 The accumulation of OAA is dependent on inner membrane potential (ΔΨ), as we showed by manipulating ΔΨ with incremental clamped concentrations of ADP or by chemical uncoupling. 10 , 11 When succinate‐energized muscle mitochondria were titrated with ADP, respiration initially increased as ATP production consumed ΔΨ. However, at a certain point, although ΔΨ continued to drop, OAA began to accumulate inhibiting succinate dehydrogenase (SDH) and, therefore, decreasing respiration. 10 , 11
The mechanism, as we proposed for muscle mitochondria, 10 is schematically depicted in Figure 1. The sequence of events involves succinate‐driven reverse electron transport to complex I which maintains NADH in the reduced state 14 , 15 impairing dinucleotide (NADH/NAD+) cycling, altering the activity of malate dehydrogenase and, thus, OAA concentrations. Reverse electron transport is known to be very sensitive to membrane potential. 16 , 17 Hence, the effect of lower ΔΨ is to reduce the reverse electron transport, allow NADH/NAD+ cycling, increase OAA, and impair complex II driven O2 flux through OAA inhibition of SDH. Consistent with this mechanism, respiration can be rapidly restored by the addition of pyruvate to clear OAA to citrate. 10 , 11 Moreover, as a part of the past work focused on an inhibitor of complex I electron flow in heart, muscle, and IBAT, 13 we observed that enhancing potential in IBAT mitochondria with GDP increased succinate‐energized respiration, although we did not address the mechanism.
FIGURE 1.
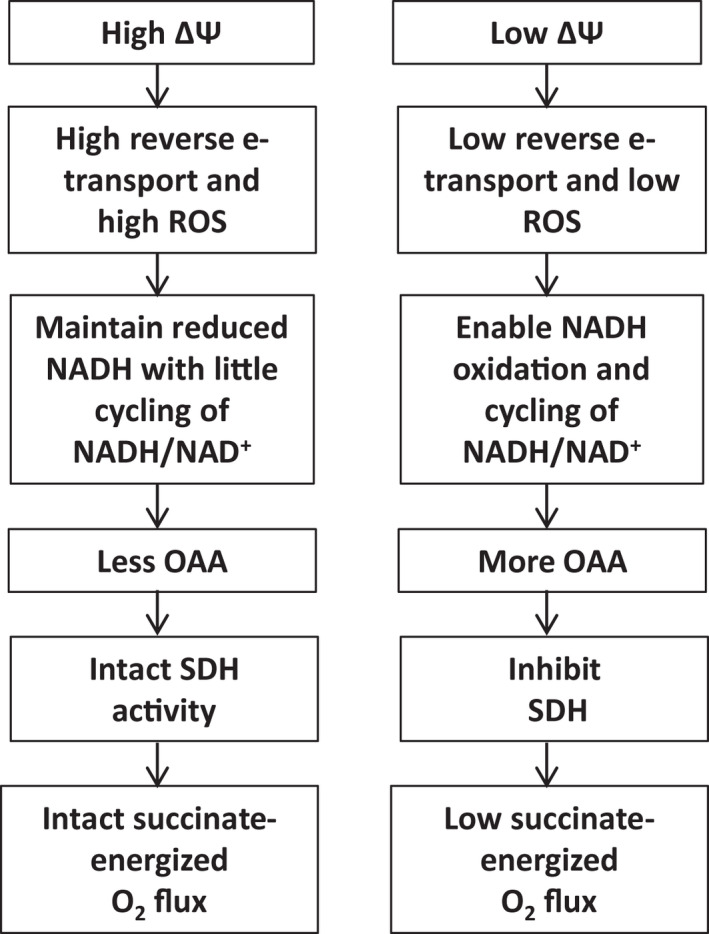
Schematic diagram depicting the mechanism by which inner membrane potential (ΔΨ) alters the accumulation of oxaloacetate (OAA) and consequent regulation of O2 flux in complex II energized skeletal muscle mitochondria
In the current work, we carried out detailed studies to address the question of whether the mechanism for skeletal muscle mitochondria depicted in Figure 1 can be extended to a tissue wherein the potential is intrinsically low i.e., IBAT. Understanding the mechanism in IBAT is important since these mitochondria are quite different from the muscle. Interscapular brown adipose tissue (IBAT) mitochondria, as opposed to muscle, are maintained at a low ΔΨ by uncoupling protein 1 (UCP1) but are, nonetheless, susceptible to changes in ΔΨ under conditions such as cold exposure or stress. To address the mechanism for the phenomenon referenced above, 13 we performed new studies of OAA and other metabolite flow, ROS, and NADH/NAD+ abundance under conditions of perturbed potential. We used 13C labeled compounds to energize IBAT mitochondria under conditions of perturbed ΔΨ enabling detailed studies of metabolite concentrations and flow by both NMR and LC‐MS. In additional new work, we questioned whether ADP (knowing that IBAT mitochondria are not particularly programmed to generate ATP) might modulate ΔΨ and downstream events. Such effects could involve ADP acting to trigger ATP synthesis, to act like GDP to induce nucleotide inhibition of UCP1, or by altering various enzyme reactions through allosteric effects.
Given these considerations, the objectives of the work reported herein were to: 1) Modulate ΔΨ in IBAT mitochondria by GDP inhibition of UCP1 and by chemical uncoupling to evaluate the ΔΨ and OAA‐dependent control of complex II respiration; 2) further delineate the mechanism through 13C isotopomer studies of metabolite flux and metabolite assessment by NMR; 3) Determine whether ΔΨ‐dependent complex II respiration is affected by the addition of ADP and whether this might involve a GDP‐like effect on UCP1, an effect on ΔΨ mediated through activation of ATP synthase, and/or another mechanism.
2. MATERIALS AND METHODS
2.1. Reagents and supplies
GDP, ADP, [U‐13C] succinate, and [U‐13C] malate were obtained from Millipore Sigma, Burlington, MA. 6‐13C‐labeled 2‐deoxyglucose was purchased from Cambridge Isotope Laboratories, Tewksbury, MA. Otherwise, reagents, kits, and supplies were as specified or purchased from the standard sources.
2.2. Animal procedures
Animals were maintained according to National Institute of Health guidelines and the protocol was approved by our Institutional Animal Care and Use Committee. Male C57BL/6J mice (Jackson Laboratories, Bar Harbor, Maine) were fed a normal rodent diet (diet 7001, Teklad, Envigo, Indianapolis, IN) until sacrifice at age 6 to 10 weeks. Mice were euthanized by isoflurane overdose and cardiac puncture.
2.3. Preparation of mitochondria
Mitochondria were prepared by differential centrifugation with further purification using a Percoll gradient as we have described in the past. 18 Mitochondrial integrity was assessed by cytochrome C release using a commercial kit (Cytochrome C Oxidase Assay Kit, Millipore‐Sigma, St. Louis), indicating a mean of 96% intact mitochondria over three assays, well within an acceptable range compared to mitochondrial preparations from several sources. 19
2.4. Respiration and membrane potential
All studies of mitochondrial respiration and inner membrane potential utilized freshly isolated and purified mitochondria on the day of the experiments. Respiration was determined using an Oxygraph‐2k high‐resolution respirometer (Oroboros Instruments, Innsbruck, Austria). In experiments where ΔΨ was measured, this was carried out simultaneously with respiration using a potential sensitive tetraphenylphosphonium (TPP+) electrode fitted into the Oxygraph incubation chamber with a volume of 2 ml. A TPP+ standard curve was performed in each run by adding tetraphenylphosphonium chloride at concentrations of 0.25, 0.5, and 0.75 μM prior to the addition of mitochondria to the chamber. Mitochondria (0.35 mg/ml for Oxygraph incubations) were incubated at 37°C in 2 ml of ionic respiratory buffer (105 mM KCl, 10 mM NaCl, 5 mM Na2HPO4, 2 mM MgCl2, 10 mM HEPES pH 7.2, 1 mM EGTA, 0.2% defatted BSA) with 10 U/ml hexokinase (Worthington Biochemical), and 10 mM 2‐deoxyglucose (2DOG).
When ADP was included in incubations, the concentration was clamped (see below) at the desired level throughout the 20‐min incubation time. Although the O2 tension in the Oxygraph drops with time, the rate of respiration is little affected until levels become very low. However, since incubations were carried out for 20 min, it was necessary to open the chamber at certain points to prevent marked deterioration in the oxygen content of the medium. Representative Oxygraph tracings are shown in supplemental Figure 1.
2.5. ADP recycling and generation of the 2‐deoxyglucose ATP energy clamp
We used a method that we previously developed to carry out bioenergetic studies of isolated mitochondria under conditions of clamped ADP and membrane potential. 18 , 20 Mitochondrial incubations were carried out in the presence of hexokinase, excess 2‐deoxyglucose (2DOG), and varying amounts of added ADP. ATP generated from ADP under these conditions drives the conversion of 2DOG to 2DOG phosphate (2DOGP) while regenerating ADP. The reaction occurs rapidly and irreversibly, thereby effectively clamping membrane potential determined by available ADP. This was in fact the case as we have demonstrated in the past for rat and mouse muscle, 10 , 20 mouse liver, 21 and mouse heart 21 mitochondria.
2.6. Use of the 2DOG ATP energy clamp to quantify ATP production
ATP production was quantified by the conversion of 2DOG to 2DOGP as we previously described. 18 , 20 Mitochondria (0.1 mg/ml) were incubated in 96‐well microplates in a total volume of 80 µl. Incubations were carried out at 37°C with shaking for 20 min in ionic respiratory buffer plus 10 units/ml hexokinase (Worthington Biochemical) and 10 mM 6‐13C‐labeled 2DOG, mimicking the conditions utilized for our Oxygraph incubations. After incubation for 20 min with orbital shaking, the contents of the microplate wells were removed to tubes on ice containing 1 µl of 160 µM oligomycin to inhibit ATP synthase. Tubes were then centrifuged for 4 min at 14,000 × g to pellet the mitochondria. Supernatants were transferred to new tubes and stored at −20°C for quantification of 13C‐labeled 2DOGP by NMR spectroscopy.
To prepare the NMR sample, 50 µl of assay supernatant was added to a 5 mm (OD) standard NMR tube (Norell, Inc.) along with 50 µl of deuterium oxide (D2O) and 380 µl of a buffer consisting of 120 mM KCl, 5 mM KH2PO4, and 2 mM MgCl2, pH 7.2. NMR spectra were collected at 37°C on a Bruker Avance II 500 MHz NMR spectrometer. Mitochondrial samples were studied by acquiring two‐dimensional (2D) 1H/13C HSQC NMR spectra using 13C‐labeled 2DOG at C6‐position ([6‐13C] 2DOG) as we previously described. 20 The amount of 2DOG and 2DOGP present in the NMR samples was quantitatively measured using the peak intensities of the assigned resonances of these compounds. NMR spectra were processed with the NMRPipe package 22 and analyzed using NMRView software. 23 ATP production rates were calculated based on the percent conversion of 2DOG to 2DOGP, the initial 2DOG concentration, incubation volume, and incubation time. This NMR method is highly specific and 40–50 times more sensitive than direct NMR detection of ATP and can be efficiently carried out in an automated fashion. 18 , 20
2.7. Mitochondrial ROS production as hydrogen peroxide
H2O2 production was determined simultaneously with ATP production as we previously described. 18 Mitochondria were incubated in microplate wells as described above. H2O2 production was assessed using the fluorescent probe 10‐acetyl‐3,7‐dihydroxyphenoxazine (DHPA or Amplex Red, ThermoFisher), a highly sensitive and stable substrate for horseradish peroxidase and a well‐established probe for isolated mitochondria. 24 Fluorescence was measured and quantification was carried out as we previously described. 25
2.8. Metabolite measurements
Metabolite measurements were performed by NMR spectroscopy as we previously described 10 , 11 , 26 on the contents of the Oxygraph chamber after mitochondrial incubation with 13C‐labeled substrates for 20 min in the same media used for measuring respiration. Immediately after mitochondrial incubations, 1.5 ml of the chamber content was placed in tubes on ice and acidified with 91 µl of 70% perchloric acid. The solutions were then thoroughly mixed, sonicated on ice for 30 s at a power setting of 4 Watts, and then stored at −80°C for up to 2 weeks. The sample tubes were then thawed on ice and centrifuged at 50,000 × g for 20 min at 4°C. Supernatants were removed from the centrifuge tube and 10 N KOH was added to bring the solution pH to 7.4, followed by centrifugation at 16,000 × g for 15 min at 4°C to remove precipitated salts. The cleared, neutralized supernatants were then stored at −80°C prior to NMR studies. For NMR sample preparations, 350 µl of the stored supernatant was added to 150 µl of 50 mM sodium phosphate, pH 7.4 in deuterium oxide for metabolite measurement. 13C and 1H NMR assignments of succinate, malate, fumarate, oxaloacetate (OAA), citrate, pyruvate, aspartate, and α‐ketoglutarate were obtained using standard compounds. OAA was found to be unstable with a half‐life about 14 h when tested at pH 7.4 and temperature at 25°C. Therefore, after mitochondrial incubation, perchloric acid extraction was carried out as quickly as possible to destroy the mitochondrial enzymes and minimize the degradation of OAA. In addition, for the determination of stability, known amounts of OAA were subjected to parallel incubation, perchloric acid extraction, neutralization, and storage.
Both 13C/1H HSQC and HMQC spectra were collected at 25°C on a Bruker Avance II 800 MHz NMR spectrometer equipped with a sensitive cryoprobe for the perchloric acid‐extracted samples for quantification of metabolites of the mitochondrial incubations. All NMR spectra were processed using the NMRPipe package 22 and analyzed using NMRView. 23 Peak heights were used for quantification.
2.9. [13C] isotopomer flux analysis
LC‐MS was utilized to determine 13C‐isotopologue enrichments of mitochondrial metabolites following incubation with uniformly labeled [U‐13C]‐succinate. Reactions were carried out in the Oxygraph 2k respirometer for 20 min. Chamber contents were transferred to 2‐ml tubes and immediately snap‐frozen in liquid nitrogen. The frozen samples were lyophilized overnight and extracted in ice‐cold 2:2:1 acetonitrile:methanol:water containing D8‐valine as an internal standard. Crude extracts were centrifuged to remove insoluble material, dried using a SpeedVac vacuum concentrator, and resuspended to a 10× concentrate based on the dried extract's original volume using 1:1 acetonitrile:water for LC‐MS analysis.
Two microliters of the prepared samples were separated using a Millipore SeQuant ZIC‐pHILIC (2.1 × 150 mm, 5 µm particle size) column with a ZIC‐pHILIC guard column (20 × 2.1 mm) attached to a Thermo Vanquish Flex UHPLC. Mobile phase comprised Buffer A [20 mM (NH4)2CO3, 0.1% NH4OH] and Buffer B [acetonitrile]. The chromatographic gradient was run at a flow rate of 0.150 ml/min as follows: 0–21 min‐linear gradient from 80 to 20% Buffer B; 20–20.5 min‐linear gradient from 20 to 80% Buffer B; and 20.5–28 min‐hold at 80% Buffer B. Data was acquired using a Thermo Q Exactive MS operated in negative polarity targeted selected ion monitoring (tSIM) mode with a spray voltage set to 3.0 kV, the heated capillary held at 275°C, and the HESI probe held at 350°C. The sheath gas flow was set to 40 units, the auxiliary gas flow was set to 15 units, and the sweep gas flow was set to 1 unit. MS data resolution was set at 70,000, the AGC target at 10e6, and the maximum injection time at 200 ms. The mass isolation window was set to 12 m/z and the isolation offset to 5 allowing for the observation of −1 to +11 m/z of each metabolite targeted. The tSIM inclusion list was populated using target metabolites chemical formulas and their corresponding retention times previously determined using neat standards.
2.10. Quantification of NADH and NAD+
The redox state of mitochondrial nicotinamide adenine dinucleotide (NAD) was measured using a commercially available NAD+/NADH Assay kit #G9071 (Promega, Madison, WI). Mitochondrial incubations were carried out for 20 min in microplates as we did for quantification of ATP production (see above). Samples were processed and assayed according to the kit directions for separate measurements of NADH and NAD+. Molar quantities of NADH or NAD+ were determined in duplicate from a standard curve run in parallel on the same assay plate.
2.11. Statistics
Data were analyzed by two‐tailed, unpaired t test or two‐factor ANOVA with multiple comparisons as indicated in the figure legends using GraphPad Prism (GraphPad Software, Inc.). Significance was considered at p < 0.05.
3. RESULTS
3.1. GDP inhibition of UCP1 differentially perturbs mitochondrial respiration dependent on activation of complex I or complex II
IBAT mitochondria were incubated in the presence or absence of 1 mM GDP (Figure 2), a well‐recognized potent inhibitor of UCP1, in order to perturb (increase) ΔΨ. In the presence of the complex I substrates, pyruvate + malate, the effects of GDP in the absence of added ADP were as expected. GDP increased ΔΨ (Figure 2A) and decreased respiration (Figure 2B). In contrast to the effects of GDP in mitochondria energized at complex I, the effects of GDP in mitochondria energized at complex II (Figure 2C,D) were quite different. In mitochondria energized by the complex II substrate, succinate, GDP increased ΔΨ (Figure 2C) but also markedly increased respiration (Figure 2D) — as opposed to the decrease in Figure 2B.
FIGURE 2.
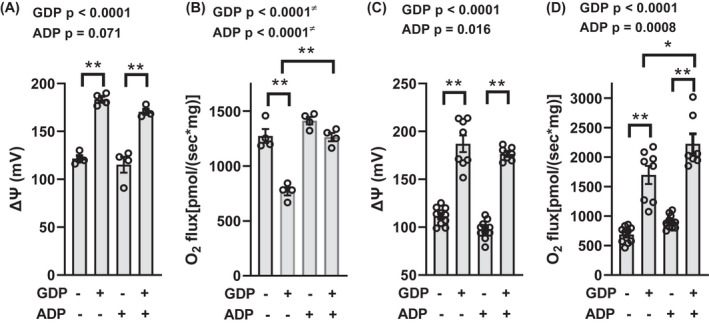
ΔΨ and O2 flux in IBAT mitochondria energized by complex I or complex II substrates. Panels (A and B) ΔΨ and O2 flux energized by 5 mM pyruvate +1 mM malate. Panels (C and D) ΔΨ and O2 flux energized by 10 mM succinate. Mitochondria were incubated for 20 min in the presence or absence of 1 mM GDP and/or 32 µM ADP as indicated below the x‐axis. n = 4 for panels A and B, n = 7–11 for panels C and D. Data were analyzed by two‐factor (GDP × ADP) ANOVA. Factor significance is shown above the panels. *p < 0.01, **p < 0.001 for multiple comparisons as indicated. ≠ Interaction is significant, so the interpretation of p values is confounded. Data represent individual values, mean, and SE
3.2. ADP effects on IBAT mitochondria energized at complex I or II
Although ΔΨ in IBAT mitochondria is directed mainly at heat production rather than ATP synthesis, we also carried out incubations in the presence or absence of added ADP clamped at a concentration of 32 µM. We reasoned that ADP might increase ΔΨ by nucleotide inhibition of UCP1 or decrease ΔΨ through ATP synthesis or perturb ΔΨ by another mechanism such as allosteric activation of TCA enzymes. We chose the concentration of 32 µM based on our past observation that O2 flux and ΔΨ were at a stable plateau at that concentration and because that is in a range we have used the past to induce ATP synthesis in isolated mitochondria. 13 , 27 In mitochondria energized by pyruvate and malate in the absence of added GDP, 32 µM ADP did not affect ΔΨ or O2 flux (Figure 2A,B). Interestingly, however, 32 µM ADP mitigated the decrease in respiration induced by GDP (Figure 2B). In mitochondria energized by succinate at complex II in the absence of GDP, ADP did not affect ΔΨ or O2 flux (Figure 2C,D). However, in the presence of GDP, 32 µM ADP mildly increased O2 flux without significantly altering ΔΨ (Figure 2C,D).
We also examined the effect of ADP on succinate‐energized mitochondria using a higher, 1 mM, concentration of ADP (same concentration as GDP used in Figure 2) thinking that there may be evidence of nucleotide inhibition of UCP1 at a high enough concentration. We found that ADP at 1 mM increased ΔΨ (Figure S2A), as opposed to the non‐significant decrease at 32 µM (Figure 2C). This effect of 1 mM ADP was associated with a large increase in respiration (Figure S2B). This is consistent with a GDP‐like effect of nucleotide inhibition of UCP1 evident only at the higher concentration of ADP.
3.3. FCCP effects on ΔΨ and O2 flux in succinate‐energized mitochondria
In mitochondria treated with GDP to increase ΔΨ, the chemical uncoupler, FCCP (1 µM), reduced ΔΨ (Figure 3A). Again, as opposed to the classical effect of reduced ΔΨ to increase respiration, FCCP reduced O2 flux in these mitochondria (Figure 3B). In mitochondria not exposed to GDP, wherein potential is intrinsically very low, 1 µM FCCP did not affect ΔΨ or O2 flux (Figure S3).
FIGURE 3.
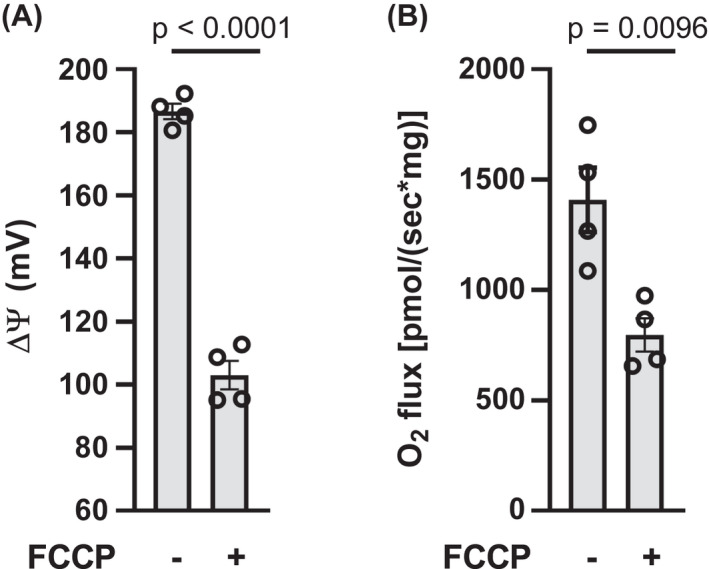
Effect of FCCP on ΔΨ (panel A) and O2 flux (panel B) in IBAT mitochondria energized by succinate. Mitochondria were energized by 10 mM succinate and incubated for 20 min in the presence of 1 mM GDP and in the presence of vehicle (−) or 1 µM FCCP (+) as indicated below the x‐axis. n = 4 for all determinations. Data analyzed by a two‐tailed, unpaired t test. Data represent individual values, mean, and SE
3.4. Effects of GDP inhibition of UCP1 on metabolite accumulation by NMR spectroscopy in complex I or complex II energized mitochondria
To assess metabolite accumulation in complex I energized mitochondria, the organelles were energized by 5 mM pyruvate +1 mM [U‐13C] malate. Under these conditions, OAA was undetectable whether GDP was present or not present (Table 1). This is consistent with the expected clearance of OAA to citrate. Aspartate was also undetectable, consistent with a lack of glutamate which would be needed for the transaminase reaction with OAA to form aspartate and α‐ketoglutarate. In the absence of ADP, GDP increased malate consistent with decreased consumption of the added substrate and decreased α‐ketoglutarate consistent with decreased respiration (Figure 2B). In the presence of 32 µM ADP, GDP did not significantly increase malate consistent with the lack of effect of ADP to decrease respiration (Figure 2B).
TABLE 1.
[13C] labeled metabolite concentrations determined by NMR spectroscopy in IBAT mitochondrial incubates
| Metabolite | Nucleotide presence (+) or absence (−) |
Nucleotide effect (p‐value) |
||||
|---|---|---|---|---|---|---|
| ADP−, GDP− | ADP−, GDP+ | ADP+, GDP− | ADP+, GDP+ | ADP | GDP | |
| Oxaloacetate (µM) | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | n/a | n/a |
| Malate (µM) | 90 ± 3 | 193 ± 24aa | 100 ± 5 | 128 ± 10b | 0.055* | <0.001* |
| Aspartate (µM) | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | n/a | n/a |
| Citrate (µM) | 277 ± 15 | 267 ± 13 | 263 ± 20 | 214 ± 13 | 0.051 | 0.082 |
| α‐ketoglutarate (µM) | 162 ± 21 | 59 ± 7a | 221 ± 23 | 203 ± 19bb | < 0.001* | 0.007* |
Mitochondria were energized by 5 mM pyruvate +1 mM [U‐13C] malate and incubated for 20 min under the same conditions used in the studies of Figure 2. Data were analyzed by 2‐factor (ADP × GDP) ANOVA. n = 4 for all values. All metabolites were determined in the same 4 individual mitochondrial preparations.
a p< 0.01, aa p < 0.001 versus ADP‐, GDP‐, b p< 0.01, bb p < 0.001 versus ADP‐, GDP+.
Interaction was significant, so interpretation is limited. Data represent mean ± SE.
To assess metabolite accumulation in complex II energized mitochondria, the organelles were energized using 10 mM [U‐13C] succinate. As shown in Table 2, OAA clearly accumulated in the absence of GDP, i.e., in the presence of uninhibited UCP1. This is consistent with low respiration (Figure 2D) due to OAA inhibition of SDH. In contrast, OAA was barely detectable in the presence of GDP wherein respiration was high (Figure 2D). GDP also decreased succinate (consumed as the added substrate) and increased the downstream metabolites malate and fumarate (Table 2) consistent with the effect of GDP to increase respiration. Aspartate was not detectable in these studies consistent with the lack of added glutamate. These studies of complex II energized mitochondrial metabolites were similar whether in the presence or absence of 32 µM ADP consistent with the data for ΔΨ and O2 flux (Figure 2C,D).
TABLE 2.
[13C] labeled metabolite concentrations determined by NMR spectroscopy in IBAT mitochondrial incubates
| Metabolite | Nucleotide presence (+) or absence (−) |
Nucleotide effect (p‐value) |
||||
|---|---|---|---|---|---|---|
| ADP−, GDP− | ADP−, GDP+ | ADP+, GDP− | ADP+, GDP+ | ADP | GDP | |
| Oxaloacetate (µM) | 22.3 ± 1.5 | 2.7 ± 0.8* | 19.3 ± 1.9 | 5.0 ± 1.2** | 0.79 | <0.001 |
| Malate (mM) | 0.70 ± 0.02 | 2.29 ± 0.05* | 0.88 ± 0.03 | 2.47 ± 0.21** | 0.12 | <0.001 |
| Fumarate (µM) | 69.0 ± 2.9 | 168 ± 5* | 79.7 ± 4.1 | 194 ± 13** | 0.030 | <0.001 |
| Succinate (mM) | 8.09 ± 0.13 | 6.58 ± 0.13* | 8.05 ± 0.14 | 6.66 ± 0.25** | 0.91 | <0.001 |
| Aspartate (µM) | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | n/a | n/a |
Mitochondria were energized by 10 mM [U‐13C] succinate and incubated for 20 min under the same conditions used in the studies of Figure 2. Data were analyzed by 2‐factor (ADP × GDP) ANOVA. n = 5 for all values. All metabolites were determined in the same 5 individual mitochondrial preparations.
Citrate, α‐ketoglutarate, glutamate, and pyruvate were also examined but results were below levels of detection. Data represent mean ± SE.
p < 0.001 vs ADP−, GDP−.
p < 0.001 vs ADP+, GDP−. Interaction was not significant for any analyses.
3.5. GDP inhibition of UCP1 perturbs ATP synthesis, reactive oxygen species (ROS) production, and NADH in mitochondria energized at complex II
Mitochondrial preparations were incubated in multiwell plates to simultaneously determine ATP production by conversion of 2‐deoxyglucose (2DOG) to 2‐deoxyglucose phosphate (2DOGP) and ROS production as Amplex Red fluorescence (Figure 4A,B, respectively). As expected, ADP was necessary for ATP production. ATP production was much greater in the presence of GDP inhibition UCP1 (Figure 4A) consistent with the effect of GDP on respiration in Figure 2D. GDP markedly increased ROS in the presence or absence of ADP (Figure 4B). However, the effect of GDP to increase ROS was markedly diminished in the presence of 32 µM ADP (note break in y‐axis). In separate incubations (Figure 4C,D), we found that GDP increased NADH and the NADH/NAD+ ratio consistent with the effect of GDP to increase ROS and maintain the NADH in the reduced state.
FIGURE 4.
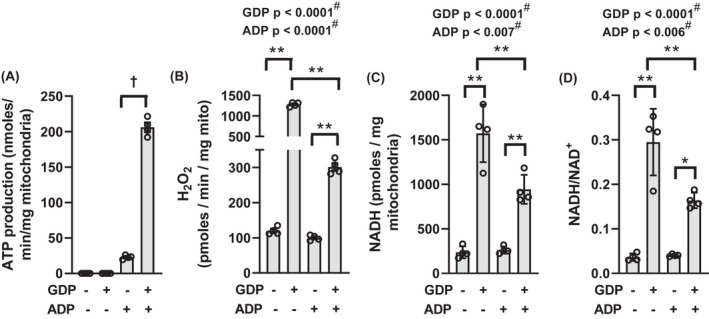
Parameters measured in mitochondria energized by 10 mM succinate and incubated for 20 min in multi‐well plates in the presence or absence of 1 mM GDP and in the presence or absence of 32 µM ADP as indicated below the x‐axis. (A) ATP production, n = 4. (B) H2O2 production, n = 4. (C and D) NADH content and the ratio of NADH to NAD+, n = 4. † p < 0.001 in panel A by unpaired, two‐tailed t test. *p < 0.01, **p < 0.001 in panels (B‐D) for multiple comparisons as indicated. Data were analyzed by two‐factor (GDP × ADP) ANOVA. Factor significance is shown above the panels. # Interaction is significant, so interpretation of p values is confounded. Data represent individual values, mean, and SE
3.6. GDP inhibition of UCP1 perturbs metabolite flux
The metabolites measured by NMR (Tables 1 and 2) determine the accumulation of molar amounts of labeled compounds generated from [13C] malate (Table 1) or [13C] succinate (Table 2). To provide further insight, we carried out studies of [13C] isotopomer flux by LC‐MS in mitochondria energized by [U‐13C] succinate. This technique quantified compounds between conditions only in relative terms but is much more sensitive and examines metabolite flux by assessing differentially 13C‐labeled metabolites (isotopologue distribution).
As shown in Figure 5, M+4 succinate (the added energy substrate), was reduced by GDP consistent with increased respiration and SDH activity while M+4 malate and M+4 fumarate were enhanced consistent with direct flow from succinate. Although OAA cannot be measured in this way due to instability, 8 , 9 conversion to aspartate by the mitochondrial transaminase, Got2, is considered a surrogate for OAA. 28 To generate aspartate, some amount of glutamate must be present for reaction with OAA to form aspartate and α‐ketoglutarate. Although aspartate was not detected by less sensitive NMR, we did detect aspartate in these LC‐MS studies (Figure 5) indicating that some glutamate remained or could be generated from other endogenous metabolites carried over in our mitochondrial preparations after isolation. The detected aspartate was largely present as the M+4 isotopologue indicating conversion from M+4 OAA, which would have derived from M+4 malate. Consistent with the ΔΨ‐dependent events in Figure 1, M+4 aspartate (and by inference, M+4 OAA) was less detectable in the presence of GDP.
FIGURE 5.
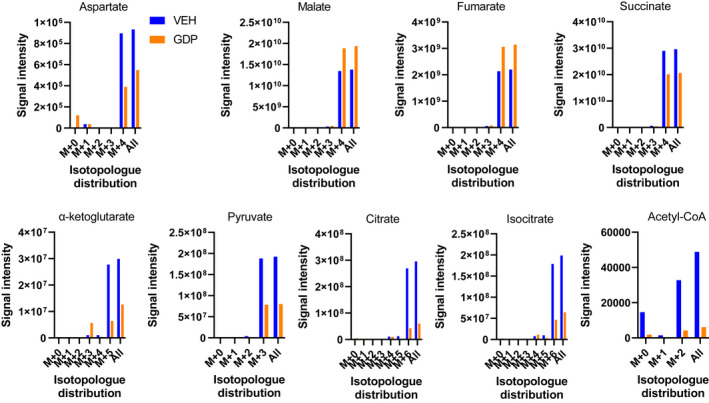
[13C] isotopomer flux analyses. Data depict the effect of 1 mM GDP versus vehicle on relative signal intensities and isotopologue distribution for several TCA metabolites. IBAT mitochondria were energized by 10 mM [U‐13C] succinate in the presence of 32 µM ADP and incubated in the Oxygraph respirometer for 20 min in the presence or absence of 1 mM GDP. Incubates were subject to LC‐MS to determine signal strength (y‐axis) and isotopologue distribution (x‐axis)
Also, M+3 pyruvate, M+2 acetyl‐CoA, and M+6 citrate were detected, less so in the presence of GDP, consistent with conversion of M+4 OAA to pyruvate which could occur in a non‐enzymatic fashion or catalyzed by oxaloacetate decarboxylase. M+3 pyruvate could then be metabolized to M+2 acetyl‐CoA leading to the formation of M+6 citrate from M+4 OAA. We also detected M+6 isocitrate and M+5 α‐ketoglutarate consistent with formation downstream from citrate, these compounds being present in lesser amounts in the presence of GDP.
3.7. GDP inhibition perturbs respiration, ΔΨ, and metabolite accumulation in mitochondria energized by succinate plus glutamate
To further assess metabolite flow related to OAA, we examined the effect of GDP in mitochondria energized with 10 mM [U‐13C] succinate but with a low (0.5 mM) concentration of [U‐13C]‐labeled glutamate to provide substrate for the Got2 catalyzed transaminase reaction converting OAA to aspartate and α‐ketoglutarate. As in the absence of glutamate, GDP increased ΔΨ and O2 flux (Figure 6). NMR studies (Table 3) demonstrated that GDP markedly decreased OAA and aspartate consistent with mitigation of OAA inhibition of SDH. Moreover, GDP decreased succinate (utilized added substrate) and increased malate and fumarate consistent with increased respiration. Note that, although the p value is not significant, less glutamate appears to be utilized. At first thought, this may seem inconsistent with increased respiration but is explainable based on less OAA accumulation and the capacity of malate (that is not converted to OAA) to exit mitochondria.
FIGURE 6.
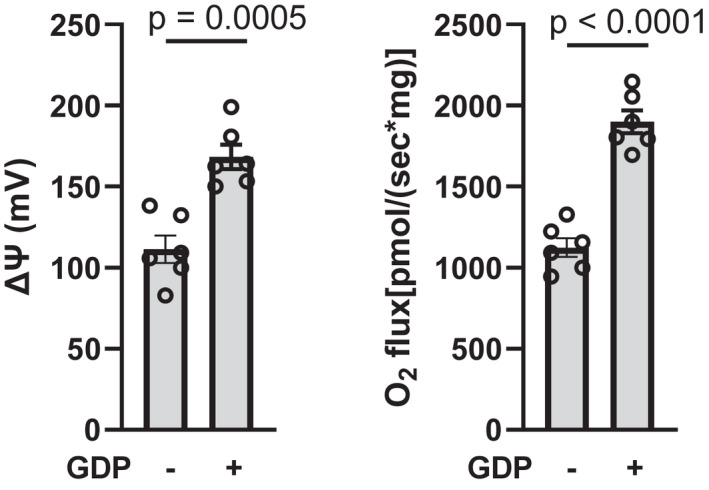
IBAT ΔΨ and O2 flux in mitochondria energized by 10 mM succinate +0.5 mM glutamate. Mitochondria were incubated for 20 min in the presence (+) or absence of 1 mM GDP as indicated below the x‐axis. n = 6 for all determinations. Data analyzed by a two‐tailed, unpaired t test. Data represent individual values, mean, and SE
TABLE 3.
[13C] labeled metabolite concentrations determined by NMR spectroscopy in IBAT mitochondrial incubates
| Metabolite | GDP (0 mM) | GDP (1 mM) | p‐value |
|---|---|---|---|
| Oxaloacetate (µM) | 17.3 ± 1.36 | 0.45 ± 0.45 | <0.0001 |
| Malate (mM) | 1.06 ± 0.034 | 2.35 ± 0.11 | <0.0001 |
| Fumarate (µM) | 95.9 ± 4.9 | 185.3 ± 6.6 | <0.0001 |
| Succinate (mM) | 8.53 ± 0.12 | 7.61 ± 0.18 | 0.0016 |
| Aspartate (µM) | 19.4 ± 2.4 | 9.0 ± 1.0 | 0.0024 |
| Glutamate (µM) | 409 ± 5 | 428 ± 8 | 0.0881 |
Mitochondria were energized by 10 mM [U‐13C] succinate plus 0.5 mM [U‐13C] glutamate and incubated for 20 min with or without GDP in the presence of 32 μM ADP under the same conditions use in the studies of Figures 6 and 7. Data were analyzed by unpaired, 2‐tailed t test, n = 6 for all values. Data represent mean ± SE.
3.8. Metabolite flux and isotopologue distribution in succinate‐energized mitochondria in the presence of glutamate
We further examined metabolite flux in mitochondria energized by 10 mM [U‐13C] succinate +unlabeled 0.5 mM glutamate (to generate aspartate from OAA) in LC‐MS isotopomer studies (Figure 7). The reason we used unlabeled glutamate in these studies, as opposed to the 13C‐labeled glutamate used in our NMR studies, is that our 2D NMR method employed a stable isotope resolved metabolomics (SIRM) approach that traces only 13C‐labeled compounds, while LC‐MS detects all isotopologues.
FIGURE 7.
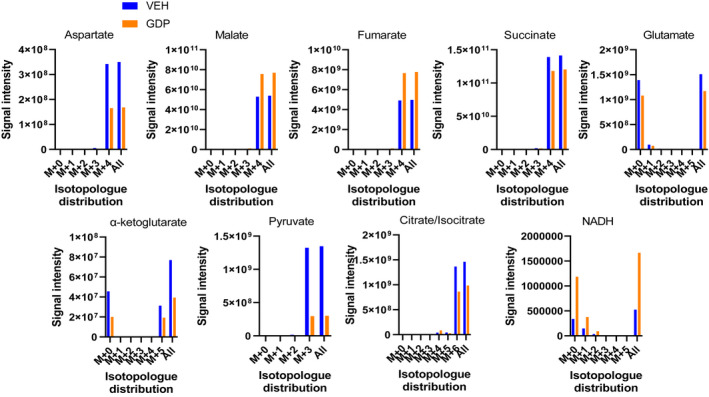
[13C] isotopomer flux analyses. Data depict the effect of 1 mM GDP versus vehicle on relative signal intensities and isotopologue distribution for several TCA metabolites. IBAT mitochondria were energized by 10 mM [U‐13C] succinate plus unlabeled 0.5 mM glutamate in the presence of 32 µM ADP and incubated in the Oxygraph respirometer for 20 min in the presence or absence of 1 mM GDP. Incubates were subject to LC‐MS to determine signal strength (y‐axis) and isotopologue distribution (x‐axis)
GDP reduced aspartate consistent with its effect to reduce OAA. As expected, in the presence of glutamate, the signal intensity for aspartate was about 300‐fold greater than that seen in Figure 5. Again, as in Figure 5, aspartate was almost entirely present as the M+4 isomer indicating direct conversion from M+4 OAA derived from uniformly labeled succinate metabolism to M+4 malate. The results otherwise resembled what we observed for incubations done on 10 mM succinate without glutamate (Figure 5). As in Figure 5, pyruvate was present as the M+3 isotopologue consistent with formation from OAA. Of note, α‐ketoglutarate was now present as both the M+0 and M+5 isotopologues likely reflecting conversion from the added unlabeled glutamate as well as from TCA flux through citrate. The decrease in citrate/isocitrate and in α‐ketoglutarate in the presence of GDP, despite increased respiration, is explainable by less OAA accumulation and malate exit, as mentioned in the above result section. We were also able to measure NADH in these experiments. GDP increased the amount of this dinucleotide, consistent with the data in Figure 4C.
4. DISCUSSION
Major new findings are as follows. First, we provide evidence that the mechanism proposed in Figure 1 involving ΔΨ‐dependent OAA accumulation can be extended to mitochondria wherein ΔΨ is intrinsically low due to chronic uncoupling. IBAT mitochondria are programmed far differently than muscle mitochondria, as the former consume ΔΨ mainly for heat rather than ATP synthesis. Second, our metabolite and isotopomer analyses (not previously done for skeletal muscle mitochondria) show that perturbation of ΔΨ alters downstream OAA flow to aspartate and α‐ketoglutarate by transamination and to citrate by citrate synthase. Moreover, our isotopomer flux experiments now provide direct evidence for malate conversion to OAA and lack of any significant OAA generation by endogenous pyruvate or aspartate that might have been present in our isolated mitochondria, i.e., changes which would have led to pyruvate, acetyl CoA, and citrate as isotopologues other than M+4. This is important since, as discussed below, only low concentrations of OAA are needed to inhibit SDH. Third, we show that ADP added to IBAT mitochondria, as opposed to GDP, has more subtle and diverse effects on complex II‐energized respiration acting in multiple ways. The rationale underlying the above contentions is discussed in the following text.
In succinate‐energized IBAT mitochondria, we observed, as expected, that ΔΨ is low but strongly increased by GDP inhibition of UCP1 (Figure 2C). However, this increase in ΔΨ is associated with increased rather than decreased respiration (Figure 2D), opposite the classical concept that higher ΔΨ results in lower respiration due to downstream electrical resistance. 29 The initiating event in this paradoxical increase in O2 flux is very likely reverse electron transport to complex I. This phenomenon is known to increase with ΔΨ 16 , 17 and is known to maintain NADH in the reduced state. 14 , 15 Indeed, we observed greater NADH and an increased NADH/NAD+ ratio with higher ΔΨ (Figures 4C,D and 7). Although we could not directly measure to reverse electron transport, the phenomenon is strongly associated with complex I superoxide, 14 , 25 which we determined as H2O2 production. Past studies in our laboratory and others 25 , 30 show that H2O2, as we measured it, largely detects H2O2 generated at complex I from superoxide catalyzed by superoxide dismutase (SOD); hence, a good surrogate marker for reverse electron transport.
Decreased NADH cycling resulting from reverse electron transport would impair the malate dehydrogenase reaction leading to reduced OAA production. Of note, the malate dehydrogenase reaction is far to the left by equilibrium dynamics. 31 This is in line with the relative amounts of malate and OAA in Tables 2 and 3. However, only small amounts of OAA (i.e., only a small shift to the right for the malate dehydrogenase reaction) are needed to impair succinate dehydrogenase as shown by Stepanova, et al 12 and as we confirmed for IBAT and muscle mitochondria. 13 In this regard, the amounts of OAA measured here by NMR (Tables 2 and 3) are within that range. To summarize, dinucleotide cycling is free to occur at low ΔΨ (intact UCP1) enabling OAA formation and inhibition of SDH. However, cycling is impaired at high ΔΨ (UCP1 inhibited) leading to NADH accumulation, inhibition of malate dehydrogenase, less OAA, less SDH inhibition, and greater respiration. Moreover, we have observed 13 that pyruvate rapidly restores succinate‐energized respiration in IBAT mitochondria consistent with its action to clear OAA to pyruvate. Taken together, the above findings provide strong evidence that low ΔΨ (due to UCP1) and consequent OAA accumulation impairs complex II respiration in IBAT mitochondria implying that the mechanism listed in Figure 1 for skeletal muscle mitochondria can be extended to IBAT.
We also modulated ΔΨ in succinate‐energized mitochondria using FCCP, in this case decreasing rather than increasing potential. Consistent with the events depicted in Figure 1, when FCCP was added to GDP‐treated mitochondria, FCCP reduced potential but also reduced, rather than increased, respiration (Figure 3). When FCCP was added in the absence of GDP, we saw no significant change in respiration and potential. However, ΔΨ was already low due to uninhibited UCP1.
A significant advance in this work, as opposed to our past work, 10 , 13 is our discovery of how these ΔΨ driven events mechanistically impact metabolite flux. Metabolite flow based on our current results are compatible with the sequence of events proposed in Figure 1. These results are summarized in schematic form in Figure 8. GDP (high ΔΨ) decreased succinate while malate and fumarate increased (Tables 2 and 3, Figures 5 and 7) consistent with the action of GDP to increase respiration (Figures 2D and 6). As hypothesized, GDP decreased OAA measured both directly by NMR (Tables 2 and 3) or indirectly as aspartate by LC‐MS (Figures 5 and 7). Moreover, OAA and aspartate, whether generated in the presence or absence of GDP, must have been derived from malate dehydrogenase since malate and aspartate were present almost totally as M+4 isotopologues. Thus, we show that only minimal, if any, OAA was derived by endogenous unlabeled pyruvate or aspartate that might have been present in our isolated mitochondria as this would have generated unlabeled or differently labeled pyruvate, acetyl CoA, and citrate isotopologues. This is important because, as discussed above, only low amounts of OAA are needed to impair SDH.
FIGURE 8.
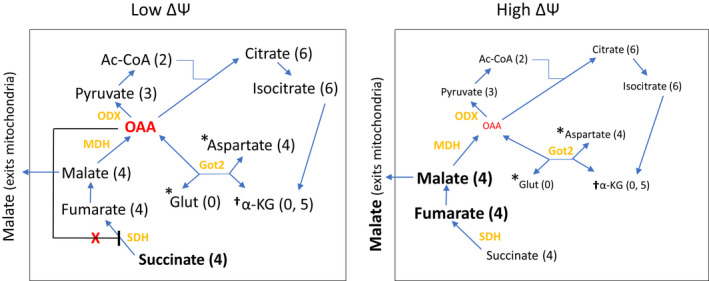
Schematic diagram depicting differences in metabolite flow at low or high ΔΨ and dominant isotopologue distribution. The diagram is based on the NMR and LC‐MS results in Tables 1, 2, and 3 and Figures 5 and 7. Differences in amounts of metabolites in the presence versus absence of GDP are reflected in the size and boldness of characters. Parentheses represent the number of labeled carbons in the dominant isotopic form(s) for each metabolite (see Figures 5 and 7). *presence dependent on added glutamate. † amount dependent on both added glutamate and conversion from isocitrate. Ac‐CoA, acetyl‐CoA; Glut, glutamate, Got2, mitochondrial aspartate aminotransferase; MDH, malate dehydrogenase; ODX, oxaloacetate decarboxylase; SDH, succinate dehydrogenase; α‐KG, α‐ketoglutarate
The formation of M+3 pyruvate and M+2 acetyl‐CoA (Figures 5 and 7) was somewhat of a surprise, but consistent with the mechanism in Figure 1. These isotopologues could arise either by non‐enzymatic conversion of OAA 26 or catalyzed by oxaloacetate decarboxylase 26 , 32 , 33 (ODX, also known as FAHD1) which has been described in human cells. 33 With GDP administration and reduction in OAA, we observed decreased M+3 pyruvate (Figures 5 and 7) and M+2 acetyl‐CoA (Figure 5) and therefore less M+6 citrate, M+6 isocitrate, and M+5 α‐ketoglutarate (Figures 5 and 7). When studies were done in the presence of glutamate (Figure 7), unlabeled α‐ketoglutarate was also present and decreased by GDP. This is consistent with the production of α‐ketoglutarate both from the unlabeled glutamate (M+0) and by TCA flow from isocitrate (M+5). We point out that the decrease in acetyl‐CoA, pyruvate, citrate, isocitrate, and α‐ketoglutarate seen with GDP is not inconsistent with the effect of GDP to increase respiration. This can be explained since there is less OAA to generate TCA flow to citrate and by malate exit from mitochondria (Figure 8), which can be substantial as we have shown in the past. 11
We also questioned how ADP, as opposed to GDP, might affect succinate‐energized O2 flux in IBAT mitochondria and whether this might involve a GDP‐like effect on UCP1, an effect mediated through ATP production, or other action. As shown in Figure 4A, for succinate‐energized IBAT mitochondria, ADP does lead to ATP formation more so in the presence of GDP. Although ATP production alone should decrease ΔΨ, ADP at 32 µM had only non‐significant effects on ΔΨ (Figure 2C). Moreover, ADP at 1 mM actually increased ΔΨ (Figure S2). This suggests that ADP, at the higher concentration, had a GDP‐like nucleotide effect to inhibit UCP1 activity. For complex I energized IBAT mitochondria, 32 µM ADP had little, if any, effect on ΔΨ (Figure 2A), but increased respiration, non‐significantly in the absence of GDP and significantly in the presence of GDP (Figure 2B). We think this might be due to the allosteric effect of ADP to activate downstream dehydrogenase reactions, isocitrate dehydrogenase in particular. 34 This is supported by the metabolic data showing that GDP did not significantly decrease α‐ketoglutarate in the presence of ADP (Table 1). Moreover, the very near significant (p = 0.051) decrease in citrate in the presence of GDP (Table 1) is consistent with downstream consumption by isocitrate dehydrogenase to form α‐ketoglutarate. So, overall, we conclude that ADP impacts ΔΨ and respiration in IBAT mitochondria but does this in a complex and multifactorial manner.
The most notable effect of 32 µM ADP in complex II energized IBAT mitochondria was on ROS production. ADP markedly reduced ROS in the presence of high ΔΨ induced by GDP (Figure 4B). As discussed above, ROS, as measured here largely, detects H2O2 generated at complex I from superoxide by SOD while superoxide, under these conditions, is generated by reverse electron transport. 14 , 25 Hence, this effect of ADP is consistent with the known high sensitivity of reverse electron transport to inner membrane potential.
In the current work, we used GDP only as a tool to modulate ΔΨ through UCP1. Intracellular amounts of GDP thought to be in the low micromolar range, 35 , 36 may not be high enough to modulate UCP1 in vivo. However, the relevant issue is the role of UCP1, as opposed to the tool used to modify it. And, there are certainly physiologic situations where UCP1 expression varies as evidenced by differences in expression in humans dependent on fat mass, 37 , 38 , 39 , 40 differences in genetic strains of rodents, 41 differences related to temperature and adrenergic input, 42 , 43 or perturbation of diet. 44
Further study will be needed to better understand the physiologic or pathophysiologic significance of oxaloacetate inhibition of SDH in whole‐cell metabolism. We can, however, offer some speculation. OAA inhibition of SDH may represent a type of metabolic brake, perhaps important in modulating reverse electron transport from SDH. This may be of particular importance during ischemia/reperfusion. 45 Moreover, alterations in SDH activity are involved in neurologic, oncologic, and cardiovascular function, 46 , 47 , 48 so OAA might prove an important control mechanism. With respect to possible in vivo treatment, we speculate that mitochondrial matrix OAA concentrations might be modulated by production or metabolism as could occur through manipulation of OAA clearance by the mitochondrial transaminase (Got2), by OAA metabolism to or from pyruvate by ODX or pyruvate carboxylase, or by production from malate dehydrogenase.
In summary, we show that respiration in complex II energized IBAT mitochondria is regulated by OAA in a manner dependent on inner membrane potential and consequent downstream events. Moreover, we delineate the consequent changes in metabolite flow. Since ΔΨ is largely dependent on UCP1, the activity and expression of this protein likely contribute to the control of OAA effects on complex II function. ADP also impacts complex II function in IBAT mitochondria but in a more subtle and multifactorial fashion.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
AUTHOR CONTRIBUTIONS
W.I. Sivitz, L. Yu, and B. D. Fink designed the research; B.D. Fink, W.I. Sivitz, L. Yu, A.J. Rauckhorst, and E.B. Taylor analyzed data; B.D. Fink, L. Yu, and A.J. Rauckhorst performed research; L. Yu and E.B. Taylor contributed analytic tools; B.D. Fink, L. Yu, and W.I. Sivitz wrote the paper; A.J. Rauckhorst, and E.B. Taylor reviewed the paper.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
These studies were supported by the NIH award, 1 R01 DK123043‐01A1, and by Merit Review Award 2 I01 BX000285‐06 from the U.S. Department of Veterans Affairs, Biomedical Laboratory Research and Development Service resources, and the use of facilities at the Department of Veterans Affairs Health Care System, Iowa City, IA 52246 and by the Iowa Fraternal Order of the Eagles. The authors thank the University of Iowa Carver College of Medicine Nuclear Magnetic Resonance Facility and the University of Iowa Metabolomics Core Facility.
Fink BD, Rauckhorst AJ, Taylor EB, Yu L, Sivitz WI. Membrane potential‐dependent regulation of mitochondrial complex II by oxaloacetate in interscapular brown adipose tissue. FASEB BioAdvances. 2022;4:197–210. doi: 10.1096/fba.2021-00137
REFERENCES
- 1. Azzone GF, Ernster L, Klingenberg M. Energetic aspects of the mitochondrial oxidation of succinate. Nature. 1960;188:552‐555. [DOI] [PubMed] [Google Scholar]
- 2. Chance B, Hagihara B. Activation and inhibition of succinate oxidation following adenosine diphosphate supplements to pigeon heart mitochondria. J Biol Chem. 1962;237:3540‐3545. [PubMed] [Google Scholar]
- 3. Chance B, Hollunger G. Energy‐linked reduction of mitochondrial pyridine nucleotide. Nature. 1960;185:666‐672. [DOI] [PubMed] [Google Scholar]
- 4. Panov AV, Kubalik N, Zinchenko N, et al. Metabolic and functional differences between brain and spinal cord mitochondria underlie different predisposition to pathology. Am J Physiol Regul Integr Comp Physiol. 2011;300:R844‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Panov AV, Vavilin VA, Lyakhovich VV, Brooks BR, Bonkovsky HL. Effect of bovine serum albumin on mitochondrial respiration in the brain and liver of mice and rats. Bull Exp Biol Med. 2010;149:187‐190. [DOI] [PubMed] [Google Scholar]
- 6. Schollmeyer P, Klingenberg M. Oxaloacetate and adenosinetriphosphate levels during inhibition and activation of succinate oxidation. Biochem Biophys Res Comm. 1961;4:43‐47. [DOI] [PubMed] [Google Scholar]
- 7. Wojtczak AB, Wojtczak L. The effect of oxaloacetate on the oxidation of succinate in liver mitochondria. Biochim Biophys Acta. 1964;89:560‐563. [DOI] [PubMed] [Google Scholar]
- 8. Al Kadhi O, Melchini A, Mithen R, Saha S. Development of a LC‐MS/MS method for the simultaneous detection of tricarboxylic acid cycle intermediates in a range of biological matrices. J Anal Methods Chem. 2017;2017:5391832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zimmermann M, Sauer U, Zamboni N. Quantification and mass isotopomer profiling of alpha‐keto acids in central carbon metabolism. Anal Chem. 2014;86:3232‐3237. [DOI] [PubMed] [Google Scholar]
- 10. Fink BD, Bai F, Yu L, et al. Oxaloacetic acid mediates ADP‐dependent inhibition of mitochondrial complex II‐driven respiration. J Biol Chem. 2018;293:19932‐19941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bai F, Fink BD, Yu L, Sivitz WI. Voltage‐dependent regulation of complex II energized mitochondrial oxygen flux. PLoS One. 2016;11:e0154982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stepanova A, Shurubor Y, Valsecchi F, Manfredi G, Galkin A. Differential susceptibility of mitochondrial complex II to inhibition by oxaloacetate in brain and heart. Biochim Biophys Acta. 2016;1857:1561‐1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fink BD, Yu L, Sivitz WI. Modulation of complex II‐energized respiration in muscle, heart, and brown adipose mitochondria by oxaloacetate and complex I electron flow. Faseb J. 2019;33:11696‐11705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lambert AJ, Brand MD. Inhibitors of the quinone‐binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). J Biol Chem. 2004;279:39414‐39420. [DOI] [PubMed] [Google Scholar]
- 15. Scialo F, Fernandez‐Ayala DJ, Sanz A. Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease. Front Physiol. 2017;8:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brand MD, Buckingham JA, Esteves TC, et al. Mitochondrial superoxide and aging: uncoupling‐protein activity and superoxide production. Biochem Soc Symp. 2004;71:203‐213. [DOI] [PubMed] [Google Scholar]
- 17. Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochem Biophys Acta. 1998;1363:100‐124. [DOI] [PubMed] [Google Scholar]
- 18. Yu L, Fink BD, Sivitz WI. Simultaneous quantification of mitochondrial ATP and ROS production. Methods Mol Biol. 2015;1264:149‐159. [DOI] [PubMed] [Google Scholar]
- 19. Wojtczak L, Zaluska H, Wroniszewska A, Wojtczak AB. Assay for the intactness of the outer membrane in isolated mitochondria. Acta Biochim Pol. 1972;19:227‐234. [PubMed] [Google Scholar]
- 20. Yu L, Fink BD, Herlein JA, Sivitz WI. Mitochondrial function in diabetes: novel methodology and new insight. Diabetes. 2013;62:1833‐1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu L, Fink BD, Herlein JA, Oltman CL, Lamping KG, Sivitz WI. Dietary fat, fatty acid saturation and mitochondrial bioenergetics. J Bioenerg Biomembr. 2014;46(1):33‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277‐293. [DOI] [PubMed] [Google Scholar]
- 23. Johnson BA, Blevins RA. NMR View: a computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603‐614. [DOI] [PubMed] [Google Scholar]
- 24. Rhee SG, Chang TS, Jeong W, Kang D. Methods for detection and measurement of hydrogen peroxide inside and outside of cells. Mol Cells. 2010;29:539‐549. [DOI] [PubMed] [Google Scholar]
- 25. O'Malley Y, Fink BD, Ross NC, Prisinzano TE, Sivitz WI. Reactive oxygen and targeted antioxidant administration in endothelial cell mitochondria. J Biol Chem. 2006;281:39766‐39775. [DOI] [PubMed] [Google Scholar]
- 26. Yu L, Sivitz WI. Oxaloacetate mediates mitochondrial metabolism and function. Curr Metabolomics Syst Biol. 2020;7:11‐23. [Google Scholar]
- 27. Herlein JA, Fink BD, O'Malley Y, Sivitz WI. Superoxide and respiratory coupling in mitochondria of insulin‐deficient diabetic rats. Endocrinology. 2009;150:46‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Buescher JM, Antoniewicz MR, Boros LG, et al. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol. 2015;34:189‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;437:297‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lambert AJ, Boysen HM, Buckingham JA, et al. Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell. 2007;6:607‐618. [DOI] [PubMed] [Google Scholar]
- 31. McEvily AJ, Mullinax TR, Dulin DR, Harrison JH. Regulation of mitochondrial malate dehydrogenase: kinetic modulation independent of subunit interaction. Arch Biochem Biophys. 1985;238:229‐236. [DOI] [PubMed] [Google Scholar]
- 32. Etemad S, Petit M, Weiss AKH, Schrattenholz A, Baraldo G, Jansen‐Dürr P. Oxaloacetate decarboxylase FAHD1 ‐ a new regulator of mitochondrial function and senescence. Mech Ageing Dev. 2019;177:22‐29. [DOI] [PubMed] [Google Scholar]
- 33. Petit M, Koziel R, Etemad S, Pircher H, Jansen‐Dürr P. Depletion of oxaloacetate decarboxylase FAHD1 inhibits mitochondrial electron transport and induces cellular senescence in human endothelial cells. Exp Gerontol. 2017;92:7‐12. [DOI] [PubMed] [Google Scholar]
- 34. Ma T, Zou F, Pusch S, Xu Y, von Deimling A, Zha X. Inhibitors of mutant isocitrate dehydrogenases 1 and 2 (mIDH1/2): an update and perspective. J Med Chem. 2018;61:8981‐9003. [DOI] [PubMed] [Google Scholar]
- 35. Dudzinska W, Lubkowska A, Dolegowska B, Safranow K, Jakubowska K. Adenine, guanine and pyridine nucleotides in blood during physical exercise and restitution in healthy subjects. Eur J Appl Physiol. 2010;110:1155‐1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1‐22. [DOI] [PubMed] [Google Scholar]
- 37. Cypess AM, Lehman S, Williams G, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saito M, Okamatsu‐Ogura Y, Matsushita M, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58(7):1526‐1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, et al. Cold‐activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500‐1508. [DOI] [PubMed] [Google Scholar]
- 40. Virtanen KA, Lidell ME, Orava J, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518‐1525. [DOI] [PubMed] [Google Scholar]
- 41. Fink BD, Herlein JA, Almind K, Cinti S, Kahn CR, Sivitz WI. The mitochondrial proton leak in obesity‐resistant and obesity‐prone mice. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1773‐1780. [DOI] [PubMed] [Google Scholar]
- 42. Ricquier D, Fleury C, Larose M, et al. Contributions of studies on uncoupling proteins to research on metabolic diseases. J Intern Med. 1999;245:637‐642. [DOI] [PubMed] [Google Scholar]
- 43. Almind K, Manieri M, Sivitz WI, Cinti S, Kahn CR. Ectopic brown adipose tissue in muscle provides a mechanism for differences in risk of metabolic syndrome in mice. Proc Natl Acad Sci USA. 2007;104:2366‐2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Paulo E, Zhang Y, Masand R, et al. Brown adipocyte ATF4 activation improves thermoregulation and systemic metabolism. Cell Rep. 2021;36:109742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chouchani ET, Pell VR, Gaude E, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jespersen NR, Hjortbak MV, Lassen TR, et al. Cardioprotective effect of succinate dehydrogenase inhibition in rat hearts and human myocardium with and without diabetes mellitus. Sci Rep. 2020;10:10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rasheed M, Tarjan G. Succinate dehydrogenase complex: an updated review. Arch Pathol Lab Med. 2018;142:1564‐1570. [DOI] [PubMed] [Google Scholar]
- 48. Jodeiri Farshbaf M, Kiani‐Esfahani A. Succinate dehydrogenase: prospect for neurodegenerative diseases. Mitochondrion. 2018;42:77‐83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material