

ABSTRACT
Messenger RNA (mRNA) vaccines have been studied for decades, but only recently, during the COVID-19 pandemic, has the technology garnered noteworthy attention. In contrast to traditional vaccines, mRNA vaccines elicit a more balanced immune response, triggering both humoral and cellular components of the adaptive immune system. However, some inherent hurdles associated with stability, immunogenicity, in vivo delivery, along with the novelty of the technology, have generated scepticism in the adoption of mRNA vaccines. Recent developments have pushed to bypass these issues and the approval of mRNA-based vaccines to combat COVID-19 has further highlighted the feasibility, safety, efficacy, and rapid development potential of this platform, thereby pushing it to the forefront of emerging therapeutics. This review aims to demystify mRNA vaccines, delineating the evolution of the technology which has emerged as a timely solution to COVID-19 and exploring the immense potential it offers as a prophylactic option for other cryptic diseases.
KEYWORDS: mRNA vaccine, IVT-mRNA, nucleoside modified mRNA, in vivo delivery of mRNA, vaccine efficacy, vaccine storage, COVID-19, mRNA vaccine for infectious disease
Introduction
The concept of vaccination developed in the 18th century, and since the first vaccine, the life expectancy of the human population has increased while mortality rates from infectious agents have diminished. Initially, animal viruses were used as vaccines, but the resulting virulence led to the use of microbes that had been reassorted or those that had the pathogenic components inactivated. Subsequent improvements involved attempting to utilise surface glycoproteins or proteins of the non-virulent structures as vaccines [1]. Although each of these systems generally conferred immunity against the targeted pathogens, the need for more simplified and safer methods paved the way toward using a gene-based approach to design vaccines.
The idea of genetic transfer materialised in the 1900s by a series of findings including genetic material transfer between bacterial strains, viral DNA transfer into bacteria, and the ability to rescue genetic defects via transfer of functional DNA from a foreign source [2–4]. The application of gene therapy to treat various genetic disorders over the following decades further piqued interest in the use of genetic material in vaccines.
Since the 1980s, modifications to nucleic acids have been developed towards therapy and vaccine design [5]. While DNA was the first genetic material to be engineered for this purpose, the notion of using the downstream mRNA transcript for more efficient targeting soon followed with gradual developments in mRNA technology, as highlighted in Figure 1 [6–19]. The prospect of mRNA in therapeutics was initially highlighted in 1989 with the demonstration that mRNA, within a cationic lipid, can be directly delivered and expressed in eukaryotic cells for manipulation of gene expression. Subsequently, the initial basis for mRNA as a vaccine platform was established when naked in vitro-transcribed (IVT) mRNA was directly injected into mouse skeletal muscles, and effective expression was observed in vivo [8]. The potential to use exogenous mRNA as a vaccine was further highlighted by the ability of mRNA to induce an antigen-specific immune response. This was evidenced by the presence of virus-specific cytotoxic T lymphocytes following injection of liposome-enclosed mRNA for influenza virus nucleoprotein [9]. In addition, mRNA encoding a carcinoembryonic antigen led to anti-tumoural antibody responses [20]. Overall, these findings indicated the ability of mRNA vaccines to elicit both cellular and humoral immunity in the host, making mRNA a potential vaccine candidate for infectious diseases and cancers [21].
Figure 1.
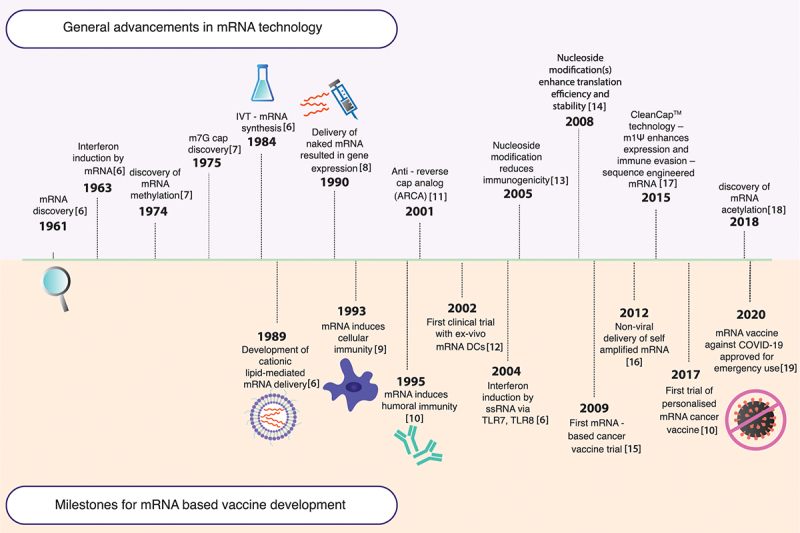
Progression of mRNA technology. The timeline illustrates the advancement of mRNA therapeutics, highlighting key milestones related to both general advancement of mRNA technology and the evolution of mRNA as a vaccine platform.
In recent times, mRNA vaccines have been widely researched for the treatment of a diverse array of diseases. Some effective mRNA vaccines have been developed for various cancers (e.g. breast and lung cancer) and one such example is BNT111 which has proven effective in clinical trials for the treatment of advanced melanoma [22]. There are also several potential mRNA vaccines for combating infectious diseases by targeting the respective causative agents (e.g. Zika virus, cytomegalovirus, influenza virus, metapneumovirus etc.). One such candidate is mRNA-1893 which is currently under clinical trial for the treatment of Zika virus infections [23]. However, the rapid emergence of mRNA vaccines has been substantially triggered by the COVID-19 pandemic. In a process that generally involves several years of testing for efficacy and safety of a prospective therapeutic, mRNA vaccines have emerged within a landmark timeframe of one year since the start of COVID-19.
In line with this renewed global interest in mRNA vaccines, this review discusses the evolution of mRNA vaccine technology, highlighting key principles, obstacles, solutions, and dissecting crucial factors that determine the acceptability of this vaccine regime. Within its scope, the review targets to resolve the apparent unfamiliarity regarding mRNA vaccines as it discusses the current approaches and future possibilities of its potential to prevent and treat obscure diseases.
The concept of mRNA vaccines
The key to mRNA vaccines gradually transitioning into one of the most versatile platforms for prophylaxis lies within their structure and mechanism of action. This transcendence can be best justified with a deeper dive into the limitations of alternative formats of vaccines.
From traditional to mRNA vaccines: the good and the bad
Traditional vaccines are typically based on the live attenuation of a pathogen to elicit an immune response in the host cells. While this is usually effective in generating a strong immune response, there is a possibility that the live-attenuated pathogen may revert to a virulent form, thereby leaving the host severely at risk of contracting an infection, especially in the case of immunocompromised individuals (e.g. HIV patients, pregnant women, individuals undergoing chemotherapy) [24]. Moreover, the production and engineering process for live-attenuated vaccines is expensive and time-consuming, historically having taken even up to a decade for development [25].
To circumvent these issues surrounding whole inactivated cells, subunit vaccines, which involve only specific parts of the pathogen, may deliver a more targeted approach. However, subunit vaccines usually require the support of adjuvants to stimulate an adequate immune response, since they lack the efficacy derived from live-attenuated vaccines [23,26]. In addition to their respective limitations, a common problem with both live-attenuated and subunit vaccines involves their inability to induce CD8-mediated cellular immunity which may render such vaccines ineffective in combating intracellular viral infections or tumours [27]. Moreover, developing these vaccines involves working with live pathogens in cell cultures which posits a health and safety complexity.
Therefore, vaccines based on plasmid DNA (pDNA) or mRNA constructs emerged to traverse many of the limitations of traditional vaccines. DNA and mRNA vaccines, being directly based on genetic constructs of the desired antigen of a target pathogen, allow for faster manufacturing, making them ideal for use in case of emerging diseases and urgent scenarios like the COVID-19 pandemic. Additionally, mRNA and DNA vectors are both intrinsically immunostimulatory, capable of modification to produce self-amplifying molecules, and can effectively induce a balanced immune response that involves both the humoral and cell-mediated components of the immune system in contrast to traditional vaccines [24,28]. DNA vaccines have been favoured generally because DNA constructs are more stable in vivo compared to mRNA and have existed longer as a vaccine technology. However, using pDNA can be detrimental due to factors such as uncontrolled long-term expression and conferring antibiotic resistance from the presence of additional foreign genes [29].
In comparison, mRNA, which is only transiently expressed, serves as a better platform since it eliminates the possibility of integration within the host genome and consequential insertional mutagenesis [23]. This transient nature of mRNA also promotes the production of higher affinity antibodies, as the mRNA-encoded antigen becomes scarcer over time. Consequently, a booster dose of the mRNA can lead to the widespread availability of these high-affinity antibodies. Another advantage that mRNA vaccines present over DNA vaccines in terms of delivery stems from the difference in the site of action of these constructs. While the DNA delivered to the cells must cross the nuclear membrane to be transcribed and its downstream mRNA transported to the cytosol to be translated, mRNA vaccines eliminate the first step of this process, simply needing to reach the cytosol to be translated [25].
Design and mechanism of action of mRNA vaccines
At the core of mRNA vaccines is an mRNA sequence designed to transiently express the antigen of interest, mimicking a key protein of the pathogen targeted for eradication. In their most basic form, mRNA vaccines contain an open reading frame (ORF) for the target antigen flanked by untranslated regions (UTRs), an N7-methyl-guanosine cap (m7G/Cap0) at the 5’-end, and a 3’-end terminal poly(A) tail. Following in vivo entry and triggering a local inflammatory response at the site of infection, the antigen of interest is generated by translation and presented as peptides on the transfected antigen-presenting cells (APCs). Consequently, a dual-pronged response, consisting of both innate and adaptive immune pathways, is stimulated comprising of humoral and cellular components, as elucidated in Figure 2 [21,30,31].
Figure 2.
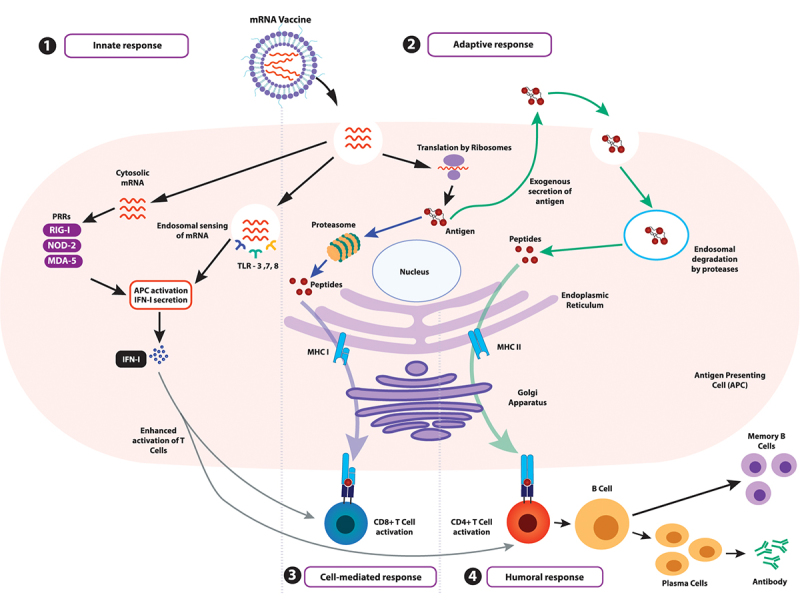
Activation of the immune system by mRNA vaccines. (1) The innate immune response is triggered via pathogen-associated molecular pattern (PAMP) recognition on non-self RNA. This recognition is mediated by pattern recognition receptors (PRRs) such as melanoma differentiation-associated 5 (MDA-5), nucleotide oligomerisation domain 2 (NOD2), and retinoic acid-inducible gene I (RIG-I) or toll-like receptors (TLRs), which ultimately results in type I interferon (IFN-I) release, allowing upregulation of proinflammatory genes, activation of the Th1 pathway and APC activation. (2) The internalised mRNA can be translated to generate the antigen, which is presented on the major histocompatibility complexes (MHC) on the APCs, initiating the adaptive response. (3) The antigen degraded by proteasomes is presented as peptides on the MHC I complex, leading to interactions with CD8+ cytotoxic T lymphocytes and triggering their maturation for a cellular response (indicated by blue arrows). (4) Additionally, antigens secreted exogenously can be internalised by other APCs, degraded into peptides within lysosomal endosomes, and presented on MHC II complexes on the cell surface (indicated by green arrows). This stimulates CD4+ T-helper cell interactions, which prime B lymphocytes on maturation for an antibody-mediated humoral response and generate memory cells specific to the target pathogen.
The innate immune response is initiated by the recognition of non-self RNA, either cytosolic or endosomal, by pathogen-associated molecular pattern (PAMP) recognition. While toll-like receptors (TLRs) sense endosomal RNA, pattern recognition receptors (PRRs) such as melanoma differentiation-associated 5 (MDA-5), nucleotide oligomerisation domain 2 (NOD2), and retinoic acid-inducible gene I (RIG-I) sense cytosolic RNA. TLR7/8 trigger the myeloid differentiation marker 88 (MyD88) pathway, successively priming B cell activation against the internalised mRNA while TLR3 leads to the production of interferon-beta (IFN-β). Furthermore, TLR7 enhances antigen presentation, promotes inflammatory cytokine secretion and enhances memory B cell survival. The cascade crescendoes with the release of type I interferon (IFN-I) which is instrumental in establishing an antiviral response by the release of proinflammatory cytokines, T-helper 1 (Th1) pathway activation, and APC activation [30].
On the other hand, the adaptive immune response requires the manifestation of the antigen translated from the mRNA. This antigen has to be presented on major histocompatibility complexes (MHCs) on APCs after degradation by intracellular proteasomes. If the antigen epitopes are presented on MHC I complex, a cytotoxic cellular response is initiated when the antigen interacts with CD8+ T lymphocytes. Conversely, exogenously secreted antigens can be endocytosed, degraded by other circulating APCs and consequently presented on MHC II complexes, fostering CD4+ T-helper lymphocyte- and B lymphocyte-mediated response. The combined interaction potentiates the production of high-affinity antibodies specific to the antigen and thereby the pathogen alongside the development of a memory B cell repertoire stowed away until the next infection by this pathogen arises [21].
mRNA vaccines can be categorised into two groups: conventional mRNA vaccines and self-amplifying mRNA (SAM) vaccines. While conventional mRNA vaccines are designed to code for only the antigen of interest, SAM contains an additional ORF encoding a viral RNA-dependent RNA polymerase, which in turn amplifies the production of the mRNA of interest, thereby significantly enhancing the potency of the vaccine at a much lower dose [32].
Synthesis pipeline of mRNA vaccines
The fast-track development of mRNA vaccines compared to other platforms is due to the ability to synthesise IVT-mRNA in a cell-free environment, as illustrated in Figure 3 [33,34]. The mRNA developed by this process mimics endogenous mRNA to the extent that it can undergo translation and regulation by the intracellular machinery to synthesise the antigen of interest. Following synthesis and purification, the mRNA transcript is introduced within a delivery vehicle to be injected intramuscularly. To deliver synthetic mRNA in vivo, an array of exploratory options exists, ranging from encapsulation within non-viral vectors (e.g. polymer or lipid nanoparticles) to utilising modified retroviral particles (e.g. foamy virus particles) to facilitate efficient transfection within the host [35].
Figure 3.
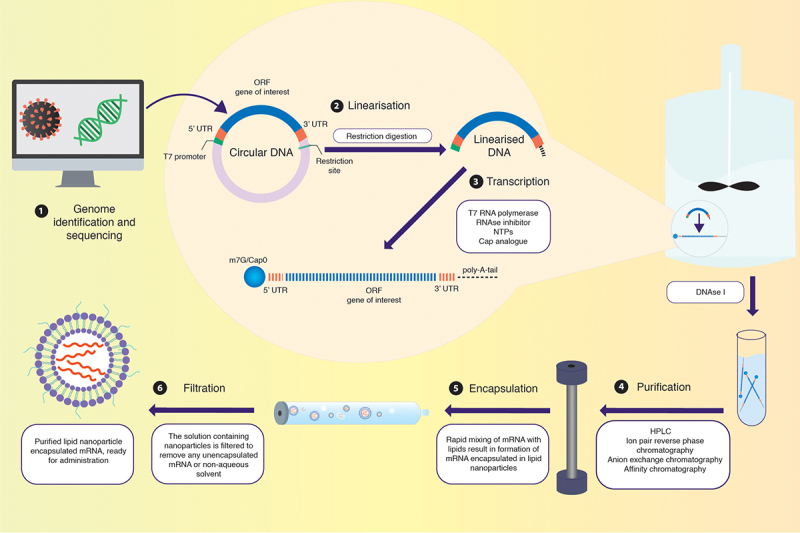
Synthesis of IVT-mRNA. (1) IVT-mRNA synthesis begins with a DNA template, usually pDNA, engineered to contain the gene of interest coding for the desired antigen identified by sequencing the genome of the target pathogen. (2) The pDNA template must include a bacteriophage promoter (T7), the ORF, a poly-deoxyribose T sequence (to code for the poly-A tail), and a restriction site which allows for restriction enzyme-mediated linearisation. (3) The linearised template undergoes in vitro transcription using a bacteriophage-derived T7 RNA polymerase. Following transcription, a mixture containing the desired mRNA, phage RNA polymerase, and nucleoside triphosphates is obtained. The 5’-cap can be introduced either enzymatically or be yielded in the transcription stage by including N7-methyl-guanosine analogue residues in excess of guanosine triphosphate (GTP) residues present. The 3’-tail can also be enzymatically added in this step, if not already incorporated. (4) The mixture is then purified using DNase I to degrade any contaminants and the template, followed by purification of the desired mRNA transcripts from a mix of abortive transcripts, longer transcripts with a 3’-overhang, oligodeoxynucleotides, and free nucleotides. While purification can be achieved by a series of precipitation and extraction steps, a chromatographic process is best suited for separating the transcripts of varied sizes. Techniques like high-performance liquid chromatography (HPLC) further refine the quality of the product, diminishing possibilities of unnecessarily activating innate immune sensors via contaminants. (5) Rapid mixing of the obtained mRNA with lipid via the utilisation of microfluidics allows for the self-assembly of mRNAs within lipid nanoparticles. (6) The obtained solution containing nanoparticles has to undergo further dialysis or filtration to remove any unencapsulated mRNA or non-aqueous solvent. Post filtration, the purified solution containing mRNA within lipid nanoparticles is ready for administration within the host.
Therapeutic evolution of mRNA vaccines
Despite the novel mechanisms and promising therapeutic avenues offered by mRNA vaccines, the adaptation of this system as a commercially mainstream vaccine platform has been hindered by several obstacles. First, being a relatively large molecule (105–106 Daltons) with a net negative charge, exogenous mRNA faces electrostatic repulsion from negatively charged carbohydrate side chains on membrane proteins, thereby leading to inefficient permeation across the membrane [23,36]. Therefore, specialised delivery systems are required for efficient in vivo delivery of mRNA. Second, native mRNA is inherently unstable with a half-life of a few hours due to its high susceptibility to degradation by nucleases, and this necessitates structural modifications to mRNA to minimise degradation [37,38]. Third, exogenous mRNA may have immunogenic properties due to its ability to act as a self-adjuvant, ultimately triggering the secretion of inflammatory cytokines and T-cell activation [36,39]. Although this immunogenic property of mRNA boosts its efficacy as a vaccine, an unchecked immune response may lead to mRNA degradation and cause adverse effects. Therefore, strategies to achieve a balanced immune response toward mRNA vaccines are required. To circumvent some of these hurdles, the past decade has witnessed substantial breakthroughs, focusing on the structure and delivery of mRNA, some of which are summarised in Figure 1.
5’ capping
An evolutionarily conserved hallmark of eukaryotic mRNAs, capping at the free 5’-end of mRNA transcripts with m7G/Cap0 plays a crucial role in cap-dependent translation initiation, RNA splicing, and intracellular transport of mRNA [40]. Similarly, IVT-mRNA requires 5’ capping for stability and robust translation efficiency.
However, in in vitro transcription, m7G/Cap0 competes with GTP for incorporation into the mRNA, resulting in inactive and/or uncapped IVT-mRNA [41,42]. Although the cap analogue m7GpppG was initially used to counteract this issue, this resulted in the formation of mRNAs with a mixture of cap analogues in the correct and reverse directions, leading to sub-standard IVT-mRNA synthesis and poor translation efficiency [41]. Since then, translation efficiency and half-life have been substantially increased by introducing an anti-reverse cap analogue (ARCA, m7,3’-OGpppG), where replacing the 3’-OH of the m7G moiety with a 3’-O-methyl group forces ARCA incorporation in the correct orientation and escape from the mRNA-decapping enzyme, Dcp2 [43].
Additional methylation at the 2’-OH of the first nucleotide generates Cap1 (m7GpppNm) [44]. Compared to Cap0, Cap1 reduces immunogenicity by eluding the RNA sensor, retinoic acid-inducible gene I (RIG-I) and RNA binding proteins, such as IFN-induced proteins with tetratricopeptide repeats (IFIT 1/5) [42]. Another co-transcriptional capping system called CleanCap exploits the capped trimer which results in a naturally occurring Cap1 structure without altering immunogenic response. CleanCap additionally may increase the efficiency of Cas9 mRNA [7].
3’ poly(A) tail modification
The 3’ poly(A) tail is another feature of eukaryotic mRNAs that has a crucial role in protecting mRNA from enzymatic degradation which conclusively increases stability and extends gene expression [30]. Together with the 5’ cap, it forms a ribonucleoprotein (RNP) complex that regulates translation.
The length of the 3’ poly(A) tail influences the stability of an IVT-mRNA. It has been reported that the appropriate length of the 3’ poly(A) tail for higher translation efficiency varies between cell types and, therefore, needs optimisation before being employed in the design of mRNA vaccines for clinical use [45].
In IVT-mRNA, a 3’ poly(A) tail can be incorporated by encoding a stretch of adenine nucleotides into the template DNA or by employing a two-step enzymatic reaction using a poly(A) polymerase during transcription [46]. However, enzymatic polyadenylation has the disadvantage of generating a 3’ poly(A) tail with an undefined length in each IVT-mRNA preparation. On the contrary, mRNA synthesis from template DNA results in a predetermined length of the 3’ poly(A) tail which is ideal for its utilisation in clinical purposes [14].
Optimising 5’ and 3’ untranslated regions
Untranslated regions (UTRs) are the noncoding sequences located upstream and downstream of the ORF in an mRNA denoted as 5’ and 3’ UTRs, respectively. Physiologically, UTRs play a critical role in mRNA stability and translation efficiency by interacting with RNA-binding proteins [30,33]. During the design of mRNA vaccines, it is imperative to optimise these UTRs for better stability and translation efficiency.
The 5’ UTR, which influences the translation of the downstream coding region, can be optimised in several ways. By ensuring that the 5’ UTR has a sequence that is not similar to the ORF sequence, undesirable disruptions to the translation of mRNA can be avoided [47]. It has been reported that inserting GCC-(A/G)-CCAUGG sequence in the 5’ UTR results in a more accurate initiation of mRNA translation [48]. A short 5’ UTR has also been suggested to enhance translation efficiency in mRNAs where highly stable secondary structures obstruct translation [49]. It has also been reported that reduced secondary structures in the 5’ UTR and the first ~30 codons of the CDS results in enhanced gene expression [45].
On the other hand, the 3’ UTR regulates mRNA stability and translation efficiency. As the 3’ UTR is notorious for its unstable regulatory elements, more stable α- and β-globin 3’ UTRs are widely used in IVT-mRNA to circumvent this problem [50,51]. It has been reported that the incorporation of two human β-globin 3’ UTRs in a head-to-tail orientation can enhance the stability of mRNAs [50]. Alternatively, AU and GU rich sequences can be used to further improve the stability of mRNAs [52,53]. Unexpectedly, increased secondary structures in the CDS (after the first ~30 codons) and 3’ UTR are associated with higher translation efficiency [45].
Base modification
Chemical modification of nucleoside base(s) in mRNA plays an important role in enhancing gene expression and significantly reducing immunogenicity. However, among 172 base modifications [54], only a few have been reported to facilitate gene expression, reduce immunogenicity, and increase stability. One example is the methylation of cytosine to 5-methylcytosine (m5C) in GC-rich regions of RNA, catalysed by NOP2/Sun RNA methyltransferase 2. The recognition of 5-methylcytosine by the mRNA export adaptor protein, Aly/REF export factor, results in enhanced gene expression [55].
Another base modification involves pseudouridylation, where pseudouridine synthase catalyses the site-specific isomerisation of uridine into pseudouridine (ψ) during post-transcriptional modification of RNA [56]. Through RNA-RNA and RNA-protein interactions [57], pseudouridine increases gene expression and reduces immunogenicity [58][59].
Another uridine base modification, 2-thiouridine (s2U), facilitates increased binding affinity through Watson-Crick base pairing with adenosine and wobble pairing with guanosine at the third codon [60], enhancing downstream translation efficiency [61]. 5-methylcytosine, pseudouridine, and 2-thiouridine when implemented in synthetic mRNA, enable the mRNA to escape detection by the innate immune system by significantly reducing TLR3/7/8 and RIG-I activation [42].
The second generation of uridine modifications includes N1-methylpseudouridine (m1ψ), which is the most frequently incorporated base modification in mRNA for therapeutic purposes. This is due to its robust ability to increase translation efficiency by promoting dephosphorylation of eukaryotic initiation factor 2 alpha (eIF2α) and to reduce immunogenicity compared to previous generations of uridine modifications [62,63]. m1ψ has been also reported to increase base pair stability resulting in complex secondary structures which enhance translation efficiency [45].
The most common modification in eukaryotic RNA, N6-methyladenosine (m6A), is important for a range of processes from RNA splicing to regulation of gene expression [64]. However, its specific role in IVT-mRNA is still obscure and may be related to increased stability [65].
More recently, N4-acetylcytidine (ac4C), a new class of RNA modification catalysed by N-acetyltransferase 10, has been reported to improve translational yield and mRNA stability [18].
Among other modifications, the site-specific role of 5-methoxyuridine (5moU) is associated with decreased immunogenicity [7]. Hence, undesirable immunogenicity can be diminished by using a range of naturally occurring nucleoside modifications and modulating expression kinetics while monitoring immunogenicity in real-time with RNA immunogenic assay [66].
Engineering open reading frames
There are 61 specialised tRNAs to decode codons required for protein synthesis. However, because of wobble base pairing, not all the tRNAs are required to successfully translate mRNAs. Harnessing this codon redundancy, ORF sequences can be engineered to increase translation efficiency by replacing the codon of a target sequence without altering the resulting amino acid composition. This can be achieved by incorporating synonymous frequent codons and/or codons with increased tRNA abundance to replace codons with rare tRNAs [67].
It has also been reported that ORFs with a higher GC content result in a 100-fold higher translation rate compared to ORFs with a low GC content [68]. An optimised ORF in IVT-mRNA further enhances translation efficiency by escaping RNA sensors (RIG-I, TLR3/7/8 and MDA5) [7]. The utilisation of sequence-engineered unmodified and pseudouridine triphosphate-modified mRNA robustly enhances translation efficiency [17,69]. Intriguingly, some proteins require a delayed translation rate to undergo proper folding and this can be readily accomplished by employing rare codons in the ORF [30].
High-temperature IVT-mRNA synthesis
One of the major problems associated with IVT-mRNA synthesis is immunogenicity caused by double-stranded RNA (dsRNA). The conventional IVT-mRNA synthesis process includes the use of phage RNA polymerases (RNAPs) which bears the burden of generating dsRNA as a by-product, predominantly through the run-off product of 3’ extension and the production of antisense RNAs [70].
Recently, utilisation of thermostable T7 RNAPs have been reported to minimise the production of 3’ extension of run-off products and are functional in vivo with reduced immunogenicity. Furthermore, combining template encoding a long poly(A) tail with thermostable T7 RNAPs prevent both kinds of by-product formation [70]. Therefore, high-temperature IVT-mRNA synthesis can help bypass post-synthesis purification, making mRNA vaccine production more economically feasible.
Circularisation of mRNA
To counteract problems associated with the short half-life of mRNA, circularisation of the linear mRNA may present as a promising solution. Circular RNAs do not have the free ends that are present in linear RNA, making them resistant to degradation by nucleases, thereby enhancing overall stability and half-life [71].
Protein-coding mRNAs of interest can be circularised via the inclusion of self-splicing introns during mRNA design in vitro. Circularisation has been shown to enhance the stability and translation efficiency of the full-length mRNA in vitro in eukaryotic cells [72]. The efficacy of circular RNA has also been demonstrated in vivo in mice, where it was efficiently translated without adversely stimulating the immune system. Furthermore, the protein expressed from circular RNA was more stable compared to that expressed by linear mRNA [73].
Modulation of mRNA expression with riboswitch
Riboswitch is a regulatory element usually located in the 5’ UTR of some mRNAs and it regulates the magnitude of gene expression. In the past, extensive efforts were invested towards exploiting this crude technology to control gene expression at the mRNA level [74–78].
Only recently, it has been reported that the expression of synthetic mRNA can be controlled both in vitro and in vivo with a kill switch by adding the US Food and Drug Administration (FDA)-approved drug, trimethoprim (TMP) [75,79]. In this mechanism, the designed SAM contains two sub-genomic promoters allowing the initiation of replication of two genes. While the first sub-genomic promoter allows encoding of the TMP-responsive fusion protein destabilising domain L7Ae (DD-L7Ae), the second promoter encodes the therapeutic or reporter protein of interest. As a result, the protein of interest is translated and DD-L7Ae is degraded in the absence of TMP. However, when added, TMP binds to DD-L7Ae which enables the TMP-DD-L7Ae complex to bind to k-turns upstream of the coding sequence of the protein of interest, enabling temporal control of gene expression [75,79]. As such, successful integration of TMP-responsive synthetic mRNA may enhance safety in case of unwanted side effects. This technology may also present an immense potential toward the next generation of therapeutics, vaccines, and diagnostics tools.
Counteracting immunogenicity with innate inhibiting proteins
SAM has also been used as a highly efficient vaccine platform for Ebola [80], HIV-1 [81], influenza [82], respiratory syncytial virus (RSV) [83], rabies [84], and COVID-19 [85]. However, unwanted immunogenicity has long been rooted within the mRNA therapy platform. With the activation of the innate immune system, protein kinase R (PKR) and 2′-5′-oligoadenylate synthetase (OAS)/ ribonuclease L (RNase L) lead to the degradation and inhibition of mRNA [86].
In addition to the strategies used to minimise immunogenicity related to synthetic mRNA (as discussed in the previous sections), it has recently been reported that innate inhibiting proteins (IIPs) can be incorporated into the mRNA to mitigate the activation of innate immune responses and to enhance gene expression [87]. When parainfluenza virus 5 (PIV-5 V) and Middle East respiratory syndrome coronavirus (MERS-CoV) ORF4a IIPs were encoded in the cis region of the mRNA transcript, protein expression was significantly increased in vitro. MERS-CoV ORF4a also enhanced gene expression in vivo in mice. Importantly, both IIPs reduced immunogenicity by downregulating the activation of interferon regulatory transcription factor 3 (IRF3) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). Because self-replication results in an exponential rise in RNA transcripts and dsRNA intermediates in the cytoplasm, offsetting the activation of the innate immune system is critical for SAM therapy [88,89].
In vivo delivery mechanisms
In vivo delivery of exogenous mRNAs is one of the major challenges in mRNA therapy. Therefore, substantial research has focused on developing more efficient delivery systems. Since a detailed discussion of each delivery platform is beyond the scope of this review, the interested reader is referred to other excellent reviews for more in-depth information on in vivo delivery of mRNA [90–92].
Among the available delivery platforms for mRNA therapeutics, lipid nanoparticles (LNPs), which are composed of a complex of synthetic or naturally occurring lipids or lipid-like molecules, are the most widely used. Generally, LNPs consist of ionisable lipids and stabilising molecules such as phospholipids, cholesterol, and polyethylene glycol (PEG). The exogenous mRNA is encapsulated within the lipid shell by electrostatic interactions between the ionisable lipid and the negatively charged mRNA [90]. LNP-encapsulation has been used for targeted delivery of mRNAs to specific cell types [93] as well as for CRISPR-Cas9-mediated genome editing in vivo in mice [94]. Furthermore, a recent study where LNPs were used to deliver mRNA in utero into mouse foetuses highlights the immense prospect of this platform to aid mRNA therapy in correcting genetic defects before birth [95].
The versatility of LNPs in mediating mRNA delivery is generally due to the ability to tailor the composition of the individual components within the complex. In this regard, modulating the type(s) of the ionisable lipids in the LNP plays a crucial role in the stability and successful intracellular delivery of the mRNA cargo. This has been most prominently demonstrated during the design of LNPs used for the delivery of COVID-19 mRNA vaccines. For example, BNT162b2 vaccine (Pfizer/BioNTech) utilises the cationic lipid ALC-0315 in its carrier LNP whereas mRNA-1273 vaccine (Moderna) utilises a different ionisable lipid, SM-102 [96]. In other examples, LNPs formulated with C1, a cationic lipid-like molecule, have been used to deliver mRNA to dendritic cells leading to efficient antigen presentation to elicit a significant T-cell response. C1 also acted as an adjuvant by inducing an inflammatory cytokine response in dendritic cells. The C1-LNP formulation was also able to effectively deliver tumour antigen-encoding mRNA in tumour models where a significant anti-tumour response was induced with no cytotoxic effects [97]. In place of cationic lipids, other lipid-like molecules can be used in a subtype of LNP known as lipid-like nanomaterials (LLNs). For example, functionalised TT derivative 5 (FTT5) has been used to deliver human factor VIII mRNA, which is a relatively long mRNA, into haemophilic mice. FTT5-encapsulated mRNA had a slower biodegradability in vivo, thereby persisting longer in the intracellular environment to allow for better mRNA expression without significant cytotoxicity [98].
The inclusion of an adjuvant to stimulate an immune response from mRNA vaccines has also been used in other LNPs. For example, LNP with the adjuvant C16-R848 (a modified TLR7/8 agonist) has been used to enhance mRNA transfection efficiency, antigen presentation, and T-cell response in tumour models, leading to both prevention and suppression of tumour growth [99].
However, in contrast to using adjuvants to boost immune responses, certain circumstances may necessitate the dampening of an excessive immune response. For example, LNPs to deliver human fibroblast growth factor 21 mRNA subcutaneously in vivo in mice have shown immunogenic effects due to the route of administration. Subsequently, the incorporation of hydrocarbon-based ester prodrugs of anti-inflammatory steroids into the LNP alleviated this excessive immune response. Additionally, this enhanced the longevity of protein expression and release into the systemic circulation, highlighting the potential of this system for use in protein replacement therapies. Furthermore, from the patients’ perspective, subcutaneous delivery of mRNA has the additional benefit of therapy self-administration, whereas intravenous administration requires trained personnel [100].
A key limitation with intravenous administration of LNP-encapsulated mRNA is the low bioavailability as a result of scavenging in the liver by the reticuloendothelial system (RES), which is involved with the clearance of toxins [101,102]. To circumvent this, the RES cells can be primed with a nanoprimer before LNP-mRNA injection. Nanoprimers are specifically designed liposomes that, due to their molecular properties, can transiently occupy RES cells, thereby preventing the uptake of LNP-mRNA by these cells. This was efficacious when introduced before LNPs encapsulating human erythropoietin mRNA in mice [103]. In addition to intravenous and subcutaneous routes of administration, aerosolised LNPs have also been shown to facilitate localised delivery of mRNAs into the pulmonary system in vivo in mice [104]. More recently, it has been demonstrated that LNP-encased mRNA may also be delivered orally. In an elegant study, LNP-encased mRNA was further contained in a self-injecting capsule known as ‘self-orienting millimetre-scale applicator (SOMA)’. SOMA capsules, while bypassing degradation by digestive enzymes, were able to inject the LNP-mRNA formulation directly into the gastric lining in vivo in mice and pigs leading to a high transfection efficiency both locally and in the systemic circulation. These findings represent a unique paradigm for mRNA vaccine delivery in circumstances where intravenous or subcutaneous delivery modes may prove difficult [105].
LNPs can be further modulated by the inclusion of branched polymers called dendrimers which contain an inner core and several layers of branched structures, at the exterior of which different functional groups can be incorporated. Drug cargoes can be encapsulated within dendrimers either via covalent or non-covalent interactions [106]. Dendrimer-based lipid nanoparticles (DLNPs) are robust in terms of tissue-targeted delivery of mRNA and monitoring of therapy. By incorporating PEGylated fluorescent dyes along with the mRNA within the DNLP, mRNA expression may be enhanced and simultaneously tumours may be visualised upon imaging [107].
Lipid-based vectors smaller than LNPs have also been employed for mRNA packaging and delivery. Nanolipoprotein particles (NLPs), which are analogous to high-density lipoprotein, are discoidal complexes consisting of a lipid bilayer with apolipoproteins. With a smaller yet adjustable diameter, NLPs are more compact compared to LNPs [108,109]. NLPs composed of cationic lipids may have the potential for enhanced in vivo delivery of large self-replicating mRNAs. For example, cationic NLP encasing of mRNA enhanced protection from RNAse degradation and boosted mRNA expression in vivo in mice. By fine-tuning the composition of the cationic lipids and including additional components, such as glycerol monooleate, the translation efficiency of the mRNA could be further improved [110].
Compact packaging of mRNA may also be achieved by bundling mRNA molecules by hybridisation with oligonucleotide crosslinkers and then packaging within PEG-coated polyplex micelles (PMs). This was shown to improve mRNA stability and expression in vivo in mice [111]. Delivery of mRNA using PEG-coated PMs has also been used for CRISPR-Cas9-mediated gene editing in vivo. By co-encapsulating Cas9 mRNA and single-guide RNA (sgRNA), which have drastically different sizes, the stability of both RNAs as well as genome-editing efficiency in mice significantly improved [112].
Another promising mRNA carrier is the mineral-coated particle (MCM), which has been shown to improve the duration of protein expression in vivo from a single dose of mRNA. The mRNA was transiently translated at high efficiency, and the overexpressed protein was then sequestered within the MCMs to enhance the longevity of the biological response. Furthermore, co-delivering anti-inflammatory proteins with the mRNA using MCMs enhanced transfection efficiency to a level comparable to that of chemically modified mRNA, making this system a viable approach when chemical modification of mRNA is undesirable [113].
A further class of polymers, known as viromers, have also exhibited potential for mRNA delivery. In contrast to LNPs, viromers have a more neutral surface charge and, therefore, do not aggregate in serum. These polymers have been used to successfully deliver therapeutic mRNAs in mouse models of inflammation [114].
A major drawback to using synthetic lipid carriers such as LNPs for mRNA delivery is the poor ability of these carriers to escape from endosomes upon entry into the cell. As a result, only a portion of the mRNA cargo is delivered to the cytosol. To counteract this issue, recently a delivery system utilising multi-tailed ionisable phospholipids (iPhos) was used to efficiently deliver mRNA for CRISPR-Cas9 gene editing with significantly enhanced endosomal escape. Furthermore, incorporating these ionisable phospholipids with other helper lipids to form hybrid LNPs (iPLNPs) led to mRNA delivery selectively to specific organs in vivo in mice [115].
Apart from using external carriers such as LNPs and other vehicles, a recent study harnessed the potential of retroviral elements within the human genome, thereby using the body's mechanisms as a delivery platform. This was shown using PEG10, a retroviral protein that binds to its mRNA and secretes it extracellularly. By flanking mRNA-encoding sequences with the 5’ and 3’ UTRs of Peg10, this system termed selective endogenous encapsidation for cellular delivery (SEND), was used as an endogenous vector to deliver mRNAs into mammalian cells [116].
On the whole, a myriad of options exists for in vivo delivery of mRNA with newer strategies emerging at a fascinating pace. In addition, key discoveries centred on the evolution of mRNA technology have generated intellectual property (Box 1). In essence, mRNA vaccines can be optimised at the level of the structural mRNA as well as at the supramolecular assembly, with a vast range of effective strategies at both levels, as indicated in Figure 4. Some of these interventions have recently been incorporated during the design and manufacture of mRNA vaccines targeting COVID-19 and are, therefore, centre-stage to the success of potential vaccines in the future.
Figure 4.
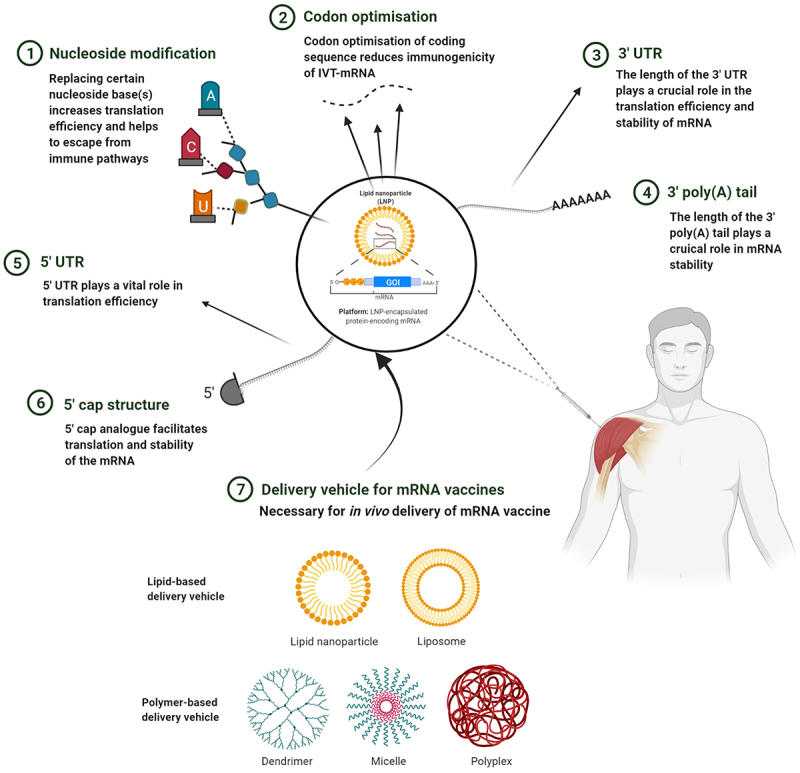
Strategy to optimise mRNA vaccines. mRNA can be modulated according to cell specificity in a number of ways: (1) By replacing nucleoside base(s) with chemically modified nucleoside(s), the translation efficiency and immunogenicity of mRNA can be altered. (2) Codon optimisation employed with GC-rich transcripts reduces immunogenicity to further increase translation efficiency and the safety profile of the mRNA. (3) By introducing stable 3’ UTR sequences, translation efficiency and stability can be modulated. (4) Manipulating the length of the 3’ poly(A) tail can improve mRNA stability. (5) Within the 5’ UTR, incorporation of GCC-(A/G)-CCAUGG sequence or avoidance of sequences similar to that of the ORF can enhance translation efficiency. (6) The 5’ cap promotes translation and further stabilises the mRNA. (7) The mRNA can be encapsulated within a variety of nanoparticle carriers for delivery into target tissues. This figure was created with BioRender.com.
Box 1-.
The patent landscape of mRNA technology
The first patent family for mRNA-based technology was identified in 1990 with a maturation phase (the time during which the number of patents submitted and granted was relatively low) up to 2010 [117]. Rapid degradation of naked mRNA and poor mRNA delivery systems were mostly associated with the maturation phase. However, with the discovery of modified nucleosides (patent granted to the University of Pennsylvania, USA) the mRNA vaccine field expanded [118]. Subsequently, the utilisation of LNPs to deliver mRNA also played a major role in the growth of the technology (patent granted to the University of British Columbia, Canada) with an increase of approximately nine-fold patent publications from 2009 to 2020 [119]. Through a chain of licence acquisitions, pharmaceuticals such as BioNTech (Mainz, Germany), Moderna (Cambridge, USA), and CureVac (Tübingen, Germany) eventually gained access to the relevant patents enabling the manufacture of COVID-19 mRNA vaccines [118]. CureVac, with more than 800 patents, has the highest number of mRNA-related patents [117]. The recent successful clinical trials of COVID-19 mRNA vaccines have further facilitated an investment surge in companies specialising in mRNA technology as well as in academia, both of which will likely lead to more patent applications in the coming years.
A new era of mRNA vaccines: COVID-19
The immense potential of mRNA vaccines is best exemplified by the advent of this therapeutic platform in light of the recent COVID-19 pandemic. Although no vaccine for COVID-19, apart from BNT162b2 (Pfizer/BioNTech) and mRNA-1273 (Moderna), has obtained full approval by the FDA [120], the World Health Organisation (WHO) has approved some vaccines for emergency use, as listed in Table 1 [121–130]. Some of these vaccines utilise conventional platforms, such as live-attenuated or inactivated viruses (e.g. CoronaVac and BBIBP-CorV). However, the possibility of reversion to a pathogenic form in the case of live-attenuated vaccines and the requirement for a multiple-dose regimen to counteract the reduced immune response in the case of inactivated virus vaccines present a challenge. Other COVID-19 vaccines are based on viral vectors, such as the adenovirus vector vaccines (e.g. ChAdOx1 and Ad26.COV2.S), but pre-existing immunity in humans may reduce the efficiency of the adenoviral vector. Therefore, mRNA vaccines have emerged as the most appealing option owing to their safety, production speed, stability, and scalability [131,132]. Currently, 25 different mRNA vaccines against SARS-CoV-2 have advanced to clinical trials [133]. Two of these, manufactured by Pfizer/ BioNTech and Moderna, were found to be approximately 95% efficacious in preventing infection.
Table 1.
A systematic comparison of COVID-19 vaccines
| BNT162b2 (Pfizer/BioNTech) | mRNA-1273 (Moderna) | ChAdOx1-S/ AZD1222 (Oxford/Astrazeneca) | Ad26.COV2.S (Johnson & Johnson/Janssen) | CoronaVac (Sinovac) | BBIBP-CorV (Sinopharm) | |
|---|---|---|---|---|---|---|
| Vaccine platform | Nucleoside-modified mRNA that encodes the prefusion-stabilised viral spike (S) glycoprotein of SARS-CoV-2 | Nucleoside-modified mRNA that encodes the prefusion-stabilised viral spike (S) glycoprotein of SARS-CoV-2 | Recombinant, replication-deficient chimpanzee adenovirus vector that encodes the viral spike (S) glycoprotein of SARS-CoV-2 | Replication-incompetent, adenovirus type 26 (AD26)-vectored monovalent vaccine that encodes the viral spike (S) glycoprotein of SARS-CoV-2 | Vero cell-based, aluminium hydroxide-adjuvanted, and β-propiolactone-inactivated vaccine based on the CZ02 strain | Vero cell-based, aluminium hydroxide-adjuvanted, and β-propiolactone-inactivated vaccine based on the 19nCOV-CDC-TAN-HB02 strain (HB02 strain) |
| Delivery system | mRNA encapsulated by lipid nanoparticle | mRNA encapsulated by lipid nanoparticle | Adenovirus-based delivery system; adenoviruses are non-enveloped icosahedral particles (virions) | Double-stranded DNA encapsulated by an icosahedral protein structure | ·· | ·· |
| Dosage | 30 μg of nucleoside-modified mRNA; 2 doses | 100 μg of nucleoside-modified mRNA; 2 doses | 0.5 ml of 5 × 1010 recombinant particles; 2 doses | 0.5 ml of 5 × 1010 AD26.COV2.S viral particles; 1 dose | 0.5 ml of 3 μg inactivated vaccine; 2 doses | 0.5 ml of 4 μg inactivated vaccine; 2 doses |
| Clinical trial population | Argentina, Brazil, Germany, South Africa, Turkey, and USA; 43,448 participants | USA; 30,000 participants | Brazil, South Africa, and UK; 23,745 participants | Argentina, Brazil, Chile, Colombia, Mexico, Peru, South Africa, and USA; 43,783 participants | Brazil, Chile, China, Indonesia, and Turkey: 25,000 participants | Bahrain, Egypt, Jordan, UAE; 41,301 participants |
| T-cell activation | Robust expression of T-helper 1 (Th1) and T follicular helper (Tfh) type CD4+ responses as well as a robust IFNγ+ IL-2+ CD8+ T-cell responses in participants aged ≥16 years | Robust CD4+ T-cell responses and low CD8+ T-cell responses in adults aged 18–55 years and in older adults ≥65 years | CD4+ and CD8+ T-cell responses in adults aged 18–55 years | CD4+ and CD8+ T-cell responses by day 15 and up to day 29 in the majority of adults aged 18–55 years and in older adults aged ≥65 years | CD4+ and CD8+ T-cell responses in adults aged 18–59 years and in older adults ≥60 years | CD4+ and CD8+ T-cell responses in adults aged 18–59 years and in older adults ≥60 years |
| Recommended for ages | ≥12 years | ≥18 years | ≥18 years | ≥18 years | ≥18 years | ≥18 years |
| Suggested route and site of administration | Intramuscular, deltoid muscle | Intramuscular, deltoid muscle | Intramuscular, deltoid muscle | Intramuscular, deltoid muscle | Intramuscular, deltoid muscle | Intramuscular, deltoid muscle |
| Efficacy * | 95% | 94.1% | 63.1% | 66.1% | 50.7% | 78.1% |
| Effectiveness against new variants of concern ** | 93.7% against Alpha 75% against Beta 88% against Delta |
100% against Alpha 96.4% against Beta |
74.5% against Alpha 67% against Delta |
·· | ·· | ·· |
| Cost per dose *** | Approximately USD 20 | Approximately USD 20 | Below USD 6(as low as USD 2.15) | USD 10 | ·· | USD 19–36 |
| Preparation | Diluted in 1.8 ml of 0.9% sodium chloride solution before use | Ready to use | Ready to use | Ready to use | Ready to use | Ready to use |
| Safety concerns | Known history of anaphylaxis and immediate allergic reaction (e.g. anaphylaxis, urticaria, angioedema, respiratory distress) to the first dose | Known history of anaphylaxis | Known history of anaphylaxis | Known history of anaphylaxis and capillary leak syndrome | Known history of anaphylaxis | Known history of anaphylaxis |
| Storage temperature | Undiluted: −90 to −60°C for 6 months from the time of formulation Undiluted, thawed: 2–8°C for 120 hours before dilution (up to 30°C for 2 hours) Diluted: 2–30°C for 6 hours |
Frozen, unopened: −25 to −15°C until expiration date Thawed, unopened: 2–8°C for 30 days (8–25°C for 12 hours) Thawed, punctured: 2–25°C for 6 hours after withdrawing first dose |
Frozen, unopened: 2–8°C for 6 months Opened: 2–8°C for 48 hours (up to 30°C for 6 hours) |
Frozen, unopened: −20°C for 24 months Thawed, unopened: 2–8°C for 3 months Thawed, opened: 2–8°C for 6 hours |
Unopened: 2–8°C for 12 months or until expiration date | Unfrozen, pre-filled syringe: 2–8°C for 24 months |
| Light sensitivity | Avoid direct exposure to light | Avoid direct exposure to light | Avoid direct exposure to light | Avoid direct exposure to light | Avoid direct exposure to light | Avoid direct exposure to light |
| Side effects | Very common (≥1/10): Injection site swelling, pain, headache, chills, arthralgia, myalgia, and pyrexia (higher frequency after 2nd dose Common (≥1/100 to ˂1/10): Injection site redness and nausea Uncommon (≥1/1000 to ˂1/100): Injection site itching, lymphadenopathy, insomnia, pain in extremities, and malaise Rare (≥1/10,000 to ˂ 1/1000): Bell<apos;>s palsy (acute peripheral facial paralysis), myocarditis, and pericarditis |
Very common (≥1/10): Injection site pain, headache, nausea, vomiting, fatigue, chills, fever, myalgia, arthralgia, stiffness, and lymphadenopathy Common (≥1/100 to ˂1/10): Injection site redness or swelling, rash, vomiting, and diarrhoea Uncommon (≥1/1000 to ˂1/100): Injection site itching Rare (≥1/10,000 to ˂ 1/1000): Facial swelling, Bell<apos;>s palsy (acute peripheral facial paralysis), myocarditis, and pericarditis |
Very common (≥1/10): Injection site pain, tenderness, warmth, itching, bruising, fatigue, chills, headache, nausea, vomiting, myalgia, and arthralgia Common (≥1/100 to ˂1/10): Injection site swelling, redness, and fever (≥38°C) Uncommon (≥1/1000 to ˂1/100): Lymphadenopathy, decreased appetite, dizziness, abdominal pain, hyperhidrosis (abnormal sweating), pruritus, and rash Very rare (˂1/10,000): Neuroinflammatory disorder (transverse myelitis) has been reported but a causal relationship has not been established |
Very common (≥1/10): Injection site pain, headache, nausea, fatigue and myalgia Common (≥1/100 to ˂1/10): Injection site swelling, redness, chills, arthralgia, cough, and fever (≥38°C) Uncommon (≥1/1000 to ˂1/100): Rash, muscle weakness, arm or leg pain, feeling weak and generally unwell, sneezing, sore throat, back pain, tremor, and hyperhidrosis (abnormal sweating) Rare (≥1/10,000 to ˂ 1/1000): Allergic reaction and hives |
Very common (≥1/10): Injection site pain, headache, and fatigue Common (≥1/100 to ˂1/10): Injection site swelling, itching, redness, induration, chills, myalgia, arthralgia, nasal congestion, cough, runny nose, sore throat, pruritus, loss of appetite, nausea, diarrhoea, and abdominal pain Uncommon (≥1/1000 to ˂1/100):): Injection site warmth, swelling, fever (≥37.3°C), tremor, dizziness, drowsiness, flushing, vomiting, mucocutaneous rash, and hypersensitivity Rare (≥1/10,000 to ˂ 1/1000): Ocular congestion, eyelid swelling, hot flashes, nose bleeds, diminished sense of smell, abdominal distension, constipation, and muscle spasms |
Very common (≥1/10): Injection site pain and headache Common (≥1/100 to ˂1/10): Fever, fatigue, myalgia, arthralgia, cough, dyspnoea, nausea, diarrhoea, and pruritus Uncommon (≥1/1000 to ˂1/100): Redness, swelling, induration, itching, dizziness, anorexia, oropharyngeal pain, dysphagia, runny nose, constipation, and hypersensitivity Rare (≥1/10,000 to ˂ 1/1000): Lethargy, drowsiness, insomnia, sneezing, nasopharyngitis, nasal congestion, dry throat, influenza, hypoesthesia, limb pain, palpitations, abdominal pain, rash, abnormal skin mucosa, acne, ophthalmodynia, ear discomfort, and lymphadenopathy Very rare (˂1/10,000): Chills, taste dysfunction, loss of taste, paresthesia, tremor, attention disorder, epistaxis, asthma, throat irritation, tonsillitis, physical discomfort, neck pain, jaw pain, neck lump, mouth ulcers, toothache, oesophagus disorders, gastritis, faecal discoloration, ophthalmodynia, blurred vision, eye irritation, earache, tension, hypertension, hypotension, urinary incontinence, and delayed menstruation |
* Efficacy defines the measurement of the vaccine<apos;>s ability in preventing disease in a controlled and double-blinded clinical trial
** Effectiveness of a vaccine is measured in the real world and counted 14 or more days after the second dose
*** All prices are subject to trademark agreements
·· Information not available on peer-reviewed sources
The design of the COVID-19 mRNA vaccines stems from the attributes of the entry mechanism of the virus into host cells. SARS-CoV-2, the causative agent of COVID-19, attaches to a cell-surface receptor on host cells via a surface-anchored protein called SARS-CoV-2 spike (S) glycoprotein and enters via endocytosis [134–136]. S glycoprotein is found as a trimer in mature viruses with a receptor-binding subunit (S1), a membrane fusion subunit (S2), a transmembrane anchor (TM), and an intracellular tail (IC). The S1 subunit includes a receptor-binding domain (RBD), which binds to angiotensin-converting enzyme 2 (ACE2) on the host cell [137–139]. Upon binding of RBD and ACE2, conformational changes in the S1 and S2 subunits allow virus entry into the cell [140,141]. This conformational change to allow virus attachment is facilitated by cell surface proteases. Inside the host cell, lysosomal protease cathepsins promote the release of the viral genome from the endosome [142,143]. Since the S glycoprotein is necessary for attachment and entry of the virus into the host cells, it is an ideal target for vaccines against SARS-CoV-2. Both the approved Pfizer/BioNTech and Moderna COVID-19 vaccines incorporate the S glycoprotein-encoding genetic sequence. Following intramuscular injection of these LNP-encapsulated mRNA vaccines, the mRNA is taken up by host cells and delivered to the cytosol where the mRNA sequence is translated into the S glycoprotein. Following post-translational modification by host cells, the protein is displayed at the cell surface as a membrane-bound antigen in its prefusion shape, providing an antigen target for B cells towards mounting an antibody response in the host [144].
BNT162b2 vaccine (Pfizer and BioNTech)
BNT162b2 mRNA, which encodes the full-length membrane-anchored S glycoprotein of SARS-CoV-2 [145], was co-developed by Pfizer (New York, USA) and BioNTech (Mainz, Germany). Also known as Comirnaty, it was the first vaccine against SARS-CoV-2 to have obtained full approval from the FDA [120].
In phase one trials of this vaccine, participants were administered two vaccinations with doses of either 10 μg, 30 μg, or 100 μg on the first and 22nd days [146]. The 30 ug dose was found to be the most efficacious with minimum safety concerns. The average antibody titres in participants who received the 30 μg dose surpassed that of a human convalescent serum panel obtained from patients who had recently recovered from COVID-19. There was also a high titre of SARS-CoV-2 neutralising antibodies and antigen-specific T-cell responses in the trial participants [147].
Based on this preliminary data from the phase one trials, phase two/three trials further established the efficacy of the vaccine. Whereas 162 participants who received a placebo contracted COVID-19, only eight vaccinated participants had the disease, indicating a 95% vaccine efficacy. Ultimately, BNT162b2 reached the primary efficacy endpoints, with a greater than 99.99% chance of actual vaccine efficacy greater than 30%. These findings fulfilled the pre-determined requirements, which were to create a chance greater than 98.6% of true vaccine efficacy being greater than 30%, well surpassing FDA's minimum authorisation criteria [145,148].
In terms of safety, even after the second dose, most participants only experienced mild-to-moderate local reactions which subsided within one to two days. These included pain at the injection site, fatigue, and headache but no long-term side effects. Overall, a two-dose regimen of BNT162b2 provided 95% protection against COVID-19, and for a median of two months, the safety of BNT162b2 was comparable to that of other viral vaccines [145].
mRNA-1273 vaccine (Moderna and NIAID)
The mRNA-1273 vaccine was co-developed by the National Institute of Allergy and Infectious Diseases (NIAID, the trial sponsor) and Moderna (Cambridge, USA).
In phase one trial of this vaccine, participants received mRNA-1273 in doses of either 25 μg, 100 μg, or 250 μg on the first and 29th days. Higher doses of the vaccine resulted in a higher antibody titre [149,150]. Those who received 100 μg and 250 μg of the vaccine developed antibodies specific to SARS-CoV-2 within 15 days of the first vaccination. The 100 ug dose was selected for use in further trials since 250 μg of the vaccine caused severe side effects in a few participants [149,151].
In phase three trials of mRNA-1273, 185 participants in the placebo group had onset of symptomatic COVID-19, while only 11 vaccinated participants contracted the disease. Therefore, the efficacy of mRNA-1273 was established as 94.1%. All the extreme COVID-19 cases occurred in the placebo group, implying that mRNA-1273 may have an impact on reducing the severity of the disease, ultimately preventing deaths. Additionally, there were only temporary mild-to-moderate local and systemic reactions (e.g. pain at the injection site, headache, chills, and fatigue) in most participants, thereby highlighting the safety of mRNA-1273 [152]. Overall, a double dose of mRNA-1273 provided 94.1% protection against COVID-19, with only short-term side effects.
More recently, a trial of the mRNA-1273 vaccine was conducted in transplant patients who had already received two doses of the vaccine to assess if a third dose would be safe and effective in improving the immune response in immunocompromised patients. In these individuals, a third dose of the vaccine elicited a significantly stronger immune response compared to the placebo according to analyses of both primary and secondary trial endpoints, thereby indicating the benefits of a booster dose in enhancing immunity [153].
In summary, strong clinical evidence supports the effectiveness of these COVID-19 mRNA vaccines. However, the unfamiliarity within the wider public regarding this new form of vaccination has generated some debate, most of which stems from myths that can be refuted using scientific proof (Box 2). With the growing use of these vaccines, this scepticism is starting to be addressed and it is expected that approval of more mRNA vaccines in the future can elevate this technology to a more widely accepted standard model of vaccination.
Box 2-.
Debunking COVID-19 mRNA vaccine myths
Myth: mRNA vaccines can alter the DNA
Fact: mRNA vaccines do not enter the nucleus where the DNA resides. Additionally, mRNA itself cannot be incorporated into DNA.
Myth: Clinical trials on mRNA vaccines were too fast and the vaccines are unsafe
Fact: Three decades of research and several clinical trials have generated substantial data on the safety of mRNA vaccines. In contrast to traditional vaccines, mRNA vaccine formulations can be designed in only a few weeks, making it possible to progress to clinical trials faster. Furthermore, the COVID-19 pandemic created an urgency leading to Pfizer/BioNTech conducting phase 2/3 trials simultaneously to fasten data collection.
Myth: COVID-19 mRNA vaccines affect fertility and are unsafe for pregnant women
Fact: False media reports created the confusion that the SARS-CoV-2 spike protein is the same as the spike protein of syncitin-1 which plays a crucial role in placental development. Additionally, it was falsely reported that the COVID-19 mRNA vaccines would generate an immune response against syncitin-1. The similarities between SARS-CoV-2 and syncitin-1 spike proteins are only 8.8% and 15.8% for amino acid identity and amino acid sequence respectively. Moreover, there is no cross-reactivity of SARS-CoV-2 and syncitin-1 spike proteins [154]. Thus, there is no high risk associated with COVID-19 vaccination during pregnancy and no adverse effects of the vaccine on fertility [155,156].
Myth: Vaccination is not required if one has already had COVID-19
Fact: Since vaccines work differently in individuals, it is difficult to predict the level of antibody one may acquire. Non-vaccinated individuals who recover from COVID-19 are twice as more susceptible to reinfections compared to those who recover and receive the vaccine [157].
Myth: COVID-19 mRNA vaccines contain microchips
Fact: The basic ingredients of mRNA-based vaccines are sucrose, cholesterol, and lipids among others [158].
Myth: COVID-19 mRNA vaccines can generate a magnetic field in the body
Fact: The vaccine does not contain any metal components and thus cannot create an electromagnetic field anywhere in the body [158].
Core factors in the sustainability of mRNA vaccines
Is efficacy the sole determining factor?
Based on the COVID-19 mRNA vaccine trials, both the Pfizer/BioNTech and Moderna mRNA vaccines had very similar efficacies of 95% and 94.1%, respectively. Consequently, these candidates were the first mRNA vaccines to achieve FDA ‘emergency use authorisation’ and the European Medicines Agency (EMA) ‘conditional approval’.
In any vaccine trial, efficacy plays a pivotal role in measuring the vaccine's ability to prevent the indicated disease. Although both the BNT162b2 and mRNA-1273 vaccines had similar efficacies, to better understand the term ‘efficacy’, one needs to look closer at the experimental design of the trials.
Variabilities in factors, such as time, population, vaccine regime, course-of-time between vaccinations, and exposure can affect the efficacy of any vaccine, inordinately. For example, if a clinical trial is conducted in a region where new pathogenic variants can suppress the vaccine, it is expected to have a lower efficacy rate as the vaccine was originally not generated against that particular variant of the pathogen. However, the vaccine can show better efficacy in a different region where those variants are not dominant in the population during the clinical trial. Therefore, having a lower efficacy does not necessarily indicate that a vaccine is not effective enough against the target disease.
Efficacy is based on the outcome of a controlled clinical trial where the participant pool is aimed to be representative of the population in terms of age, gender, and ethnicity, among other factors. Vaccine effectiveness, on the other hand, is a more real-world measure of a vaccine's ability to protect against a disease within a wider community where more variabilities exist that cannot be accounted for in a clinical trial.
Therefore, a vaccine's effectiveness may be different to its efficacy with the ultimate decision of a vaccine's effectiveness requiring careful consideration of the aforementioned factors that are closely associated with efficacy [159].
Storage and distribution of mRNA vaccines
A significant post-approval hindrance with the COVID-19 mRNA vaccines has been associated with the storage temperature of these vaccines, which is in the range of −20°C to −80°C for long-term storage (three to six months) [160].
Storage at such low temperatures represents a logistical roadblock to the distribution of these vaccines, with the majority of developing and under-developed countries lacking the appropriate infrastructure to support long-term acquisition. Therefore, both the Pfizer/BioNTech and Moderna mRNA vaccines have been beyond the reach of a vast majority of the world population.
However, efforts are underway to counteract this issue regarding storage. For example, CVnCoV, an mRNA vaccine manufactured by CureVac (Tübingen, Germany), used a platform that extends the stability of the mRNA for up to three months at 2–8°C [160]. Other than that, the second generation of CureVac COVID-19 vaccine and ARCoV COVID-19 vaccine (co-developed by the Academy of Military Science, Walvax Biotechnology, and Suzhou Abogen Biosciences), have been reported to be thermostable and can be stored for at least one week at room temperature [161]. Consequently, the approval of these vaccines would significantly change the global distribution of mRNA vaccines.
Recently, BioNTech has further introduced a low-cost mobile production unit that is equipped with 12 containers called BioNTainers that can produce 50 million doses of mRNA vaccines annually. BioNTech is expected to deliver these modular production units to Africa to combat COVID-19 [162]. Additionally, CureVac is collaborating with Tesla (Texas, USA) to develop ‘RNA printers’ which will be automated RNA vaccine production units [163].
As an alternative to relying on vaccines from multinational pharmaceuticals, WHO has recently established an mRNA vaccine technology hub to manufacture mRNA vaccines in low- and middle-income countries [164]. A prototype COVID-19 vaccine has already been designed independently by the hub and the technology along with the training for bulk production of the vaccine will be transferred to a growing list of countries including Bangladesh, Indonesia, Pakistan, Serbia, Vietnam, Egypt, Kenya, Nigeria, Senegal, Tunisia, Argentina, and Brazil [164,165]. Successful implementation of this strategy will not only help combat COVID-19 but also potentially help in the fight against other diseases.
Since mRNA vaccines are relatively new to commercialisation, better strategies to stabilise these vaccines both at the manufacturers’ and the distributors’ ends are a matter of sustained research efforts and time, as would be expected of any novel and emerging technology.
How long does protective immunity last?
Upon vaccination, the desired antigen is expressed by the targeted cell or taken up by APCs to lymph nodes which initiates the interaction between B cell, APCs, and follicular helper T cells. This interaction supports the formation of the germinal centre. The germinal centre allows the formation and maturation of B cells and immunoglobin class switching to promote the production of high-affinity neutralising antibodies against the targeted antigen and plays a crucial role in prolonged immunity against a disease [166].
However, it is difficult to precisely predict the duration of acquired immunity against COVID-19. The functioning of the immune system inherently varies between individuals even when they are vaccinated with the same vaccine or naturally exposed to the same virus. It has been observed that individuals who recovered from COVID-19 with mild-to-severe symptoms still had antibodies five to seven months post infection [167]. Even though the neutralising antibodies diminish with time, clinical trials of mRNA-1273 vaccine reported high levels of antibody in participants even after six months of their second dose [168]. BNT162b2 vaccine also documented similar antibody levels after six months [169]. It was also shown that even though antibody titres waned with time, B and T memory cells were present even after eight months, indicating the possibility of protection against COVID-19 for a longer period of time [170].
It is also known, based on data from past epidemics and pandemics, that protective immunity may last decades. For example, it was reported that individuals who contracted the 1918 H1N1 influenza virus still retained immunity against the antigen 90 years later [171]. Additionally, individuals who recovered from the more recent 2003 SARS-CoV-1 infection still harboured some CD4+ and CD8+ T cells against the antigen 17 years later [172].
Therefore, even though the immunological analysis of mRNA-based COVID-19 vaccine data is very promising, it is too early to be able to determine the longevity of protective immunity. As such, both time and further comprehensive studies are necessary in this regard.
Prospective mRNA vaccines across therapeutic avenues
With the approval of two mRNA vaccines against COVID-19, widespread acceptance of the technology has begun to come forth. By leveraging rapid development, safety, and high potency over traditional vaccine and therapeutic platforms, mRNA technologies present an immense potential to fight promptly against both infectious and non-infectious diseases.
Cancer
Cancer mRNA vaccines are being extensively studied since they present a promising alternative to conventional chemotherapy. Tumour-associated antigens which are expressed selectively in cancerous cells could be harnessed to design cancer mRNA vaccines [32]. Some key developments in mRNA vaccines for cancers utilise the power of mapping cancer-related mutations in the genome. An example is the ‘mutanome’ approach which is the overall detection and mapping of somatic mutations in tumours of individual patients using next-generation sequencing (NGS) technology [16]. Based on the identified mutations, personalised neo-epitope mRNA cancer vaccines may be designed. Neo-epitopes are small peptides that are derived from tumour-specific somatic mutations that are exposed on cancer cell surfaces and can be recognised by T cells [16,173]. Using this technology, cancer mRNA vaccines may be tailored to individual patients and this technique was successfully employed on melanoma patients [16,174]. Mutanome-based mRNA vaccines for triple-negative breast cancer are currently undergoing clinical trials [175].
Generally, clinical trials merge mRNA vaccines with cytokines or checkpoint modulators to boost immunity. This approach is currently being used in clinical trials to treat malignant and metastatic tumours including melanoma [176–178], glioblastoma [179], non-small cell lung cancer [177,180,181], colorectal cancer [177,180,182], and pancreatic cancer [180,183].
Tumour-associated antigens (TAAs) that are selectively expressed in cancer cells can also be targeted by mRNA vaccines. For example, a mixture of different mRNAs targeting TAAs for melanoma has been used in clinical trials for the treatment of metastatic melanoma. BNT111 (also known as Lipo-MERIT) is a well-known example of an mRNA vaccine in this category. BNT111 targets the TAAs – New York oesophagal squamous cell carcinoma 1 (NY-ESO-1), tyrosinase, melanoma-associated antigen 3 (MAGE-A3), and transmembrane phosphatase with tensin homology (TPTE). Phase one/two trials are currently being carried out to assess the safety, tolerability, and efficacy of this vaccine in patients suffering from advanced melanoma [184,185].
AIDS
Since its discovery in the 1980s, acquired immunodeficiency syndrome (AIDS), a chronic and life-threatening disease caused by human immunodeficiency virus (HIV) infection, has yet to find a truly effective and manageable solution. Defeating HIV is a major focus of research on mRNA vaccines. As such, there are several mRNA vaccines in clinical trials for the treatment of AIDS [32].
One strategy is to use dendritic cell delivery systems which are generally used in cancer treatment. It has been shown that dendritic cell mRNA vaccines activate CD4+ and CD8+ T-cell immune responses although there was little observed benefit during the clinical trials [32]. On the other hand, the combination of mRNA vaccine therapy and immunotherapy may achieve more promising results. This approach is currently being tested in the HIVACAR project. In this phase one/two clinical trial an mRNA vaccine that is tailored to individual patients is combined with a CD4 blocking antibody and a latency-reversing agent (LRA) which reactivates latent HIV-infected cells [186]. If successful, this approach may significantly reduce the costs associated with HIV treatment while improving patient care.
HTI-TriMix is yet another new mRNA vaccine candidate against HIV-1. Activation adjuvant TriMix and mRNA that codes for 16 conservative fragments of HIV structural proteins (Gag, Pol, Vif, and Nef) have been combined to design this vaccine. Upon intranodal injection in mice, potent antigen-specific cytotoxic T-cell responses were elicited [187]. Based on phase one and two clinical trials, this vaccine was found to be safe but the HTI protein expression was affected due to an unexpected start codon found upstream of the sequence that codes for the HTI recombinant antigen [188]. Therefore, further studies are required to overcome this problem. Additionally, there are still difficulties in the treatment of AIDS due to the poor understanding of HIV and the pathophysiology of the disease. However, if proper antigen targeting is employed, mRNA vaccines can play a crucial role in preventing HIV infection.
Influenza
Influenza viruses have a high rate of mutation which has made it difficult to eradicate influenza with traditional vaccines. Since mRNA vaccines can be rapidly and specifically developed against a target variant, they are suitably poised to tackle the high mutation rate of influenza viruses. Phase one clinical trials of two non-replicating mRNA vaccines against influenza viruses (H10N8 and H7N9) have been carried out and both vaccines were found to be safe and effective in eliciting an immune response [189]. Another vaccine platform known as RNActive, which has self-adjuvant properties, also showed promising results in pre-clinical trials [190,191].
Rabies
Individuals who are more likely to be exposed to the rabies virus are recommended to receive pre-exposure treatment [192]. CV7201 is a prophylactic vaccine for rabies virus glycoprotein (RABV-G) which, in phase one clinical trials, was found to be safe and efficacious. However, there were problems associated with unstable administration-dependent immune responses [193]. More recently, LNP-encased RABV-G mRNA was found to be better at eliciting both humoral and cell-mediated immune responses in preclinical trials with subsequent trials being currently pursued [194]. Another candidate, CV202 was also found to be safe and tolerable in patients in addition to meeting the WHO's requirements in terms of the neutralising antibody titres [195].
Zika virus infection
The lack of a safe and effective vaccine is hampering efforts to tackle Zika virus (ZIKV) infections. However, one potential vaccine has been suggested to be a nucleoside-modified mRNA that encodes viral premembrane and envelope glycoproteins. In the presence of the modified nucleoside, N1-methylpseudouridine, an mRNA encoding the prM-E region of the ZIKV H/PF/2013 was designed. Upon a single intradermal injection of the LNP-encased mRNA, significant neutralising antibody titres, as well as ZIKV-specific cellular responses, were mounted in immunocompetent mice and rhesus macaques, thereby providing full protection against the viral challenge [196].
Additionally, the vaccines mRNA-1325 and mRNA-1893 have been shown to protect against ZIKV-induced congenital disease in both immunocompetent and immunocompromised mice during pregnancy [197,198]. For both these candidates, phase one clinical studies have recently been completed, thereby paving the way for potential vaccinations in humans [199,200].
Respiratory syncytial virus infection
Respiratory syncytial virus (RSV) is a leading cause of respiratory tract infection among infants and elderly individuals and it is responsible for approximately 74,000 deaths per year [201,202]. In 1968, a formalin-inactivated RSV vaccine candidate generated vaccine-associated enhanced diseases (VAED) which resulted in severe pneumonia or bronchiolitis in 80% of the vaccinated children with two fatalities [203]. To this day no working vaccine against RSV has been developed.
Current vaccine efforts utilise the viral surface protein F which aids the fusion of the virus with the host cell membrane. Even though a few such vaccine candidates failed due to insufficient neutralising antibodies [204], a novel insight into the protein F structure has revealed that the use of perfusion conformation enhances neutralising antibodies [205–208]. In preclinical studies, mRNA vaccines encoding either the stabilised perfusion conformation or the native protein F were delivered without any VAED indication in mice and rats [83,209].
Moderna is currently developing three RSV vaccines which include mRNA-1172 and mRNA-1777 for adults and mRNA-1345 for children. Phase one clinical trial of mRNA-1777 exhibited humoral immunity with neutralising antibodies and CD4+ T cell-mediated responses against protein F without any severe side effects [210]. In mRNA-1345, further codon optimisation was used to extend translation efficiency and immunogenicity to levels that were similar to that of mRNA-1777. Interim data of phase one clinical trial shows that a 100 μg dose of mRNA-1345 vaccine, post one month, provides eightfold higher neutralising antibodies than mRNA-1777 [34]. These data highlight the potential of mRNA vaccines towards helping eradicate RSV infections where other vaccine regimes have failed to achieve significant success.
Malaria
Even though mRNA vaccines are primarily targeted against a wide range of viruses, efforts are also underway in exploiting this platform to fight other pathogens. Plasmodium, a genus of unicellular eukaryotic parasites, is the primary cause of malaria which leads to 400,000 deaths globally [211]. The lack of a target surface protein as well as the complicated life cycle of Plasmodium has so far hindered the development of an effective vaccine against malaria. However, recent studies on understanding immune responses against Plasmodium have revealed new antigen targets.
Macrophage migrating inhibitory factor (PMIF), a cytokine secreted by Plasmodium, has been identified to avert T cells from generating long-term memory responses [212]. This led to the design of a self-amplifying mRNA vaccine encoding PMIF which is complexed with squalene-based cationic nanoemulsion. Upon successful administration of two 15 μg-doses of PMIF mRNA, anti-Plasmodium antibodies were generated with memory T cell responses [212]. Subsequently, it was identified that Plasmodium falciparum glutamic acid-rich protein (PfGARP), which binds to the surface of infected erythrocytes, causes programmed cell death. This further led to the formulation of another nucleoside-modified mRNA vaccine encoding PfGARP. Upon LNP-mediated delivery of three 50 μg doses of the vaccine, parasitaemia was reduced in Aotus monkeys [213].
Lyme disease
Lyme disease is a growing concern in North America and Europe caused by the black-legged tick, Ixodes scapularis which acts as a medium for the transmission of a spectrum of pathogens including Borrelia burgdorferi, the causative agent of Lyme disease. In some animals, resistance to tick infections can be achieved by repeated exposure to ticks but this has not been demonstrated for humans, thereby necessitating a working vaccine [214].
Recently, a nucleoside-modified mRNA vaccine encoding 19 salivary proteins of I. scapularis (19ISP) was designed and injected into guinea pigs where it led to potent antibody responses. Subsequent exposure to uninfected I. scapularis nymphs in the vaccinated animals led to poor feeding and detachment of the ticks with 80% detachment in vaccinated animals compared to 20% in unvaccinated animals [215].
Multiple sclerosis
Multiple sclerosis (MS) is one of the leading causes of neurological disorders in young adults, likely disabling the brain and spinal cord with 2.2 million people affected globally [216]. Recently, a nucleoside-modified mRNA vaccine was tested in a mouse model of MS. In this disease model, the myelin oligodendrocyte glycoprotein epitope, MOG35-55, induces MS pathology. Therefore, LNP-encased N1-methylpseudouridine-containingmRNA encoding MOG35-55 was successfully administered to mice. Consequently, there was a suppression of effector T cell populations and activation of regulatory T cell populations which protected the mice from developing MS [217].
Transthyretin amyloidosis
Transthyretin amyloidosis, also known as ATTR amyloidosis, is a potentially fatal illness marked by the accumulation of misfolded transthyretin (TTR) protein fibrils in internal organs [218]. NTLA-2001 is an in vivo gene-editing therapeutic approach that is intended to cure ATTR amyloidosis by lowering serum TTR levels. It is based on the clustered regularly interspaced short palindromic repeats and Cas9 endonuclease (CRISPR-Cas9) system and consists of LNP-encased Cas9 mRNA and a single guide RNA targeting TTR. TTR knockdown was demonstrated to be persistent after a single dosage in preclinical trials with minimal adverse events, thereby highlighting the efficacy of this strategy in ATTR therapy [219].
Allergies
mRNA vaccines pave the way for a safer preventative immunisation against allergic conditions. mRNA vaccines encoding the allergen ensure higher purity of the immunising antigen compared to more traditionally used allergen extracts which may initiate further allergic responses [220].
mRNA vaccines are efficacious as preventative vaccines against type I allergy in mice due to activation of a Th1 cell response [221]. The robustness of this Th1 response has also been demonstrated in mice that were injected with mRNAs encoding a wide variety of allergens. Following immunisation with a particular mRNA, the mice were challenged with the respective allergen. Ultimately, it was observed that inflammatory signatures such as eosinophil count, IL-4 and IL-5 levels were reduced whereas anti-inflammatory phenotypes such as induction of IFN-γ producing cells were enhanced [222].
It has further been demonstrated that mRNA vaccines may be able to elicit long-term memory responses in immunised mice with subsequent re-exposure to the allergen inducing potent anti-inflammatory responses [223]. This also highlights the potential of using mRNA vaccines to target allergies without the requirement of booster vaccinations.
Overall, mRNA vaccines possess the ability to bring forth the next generation of futuristic medicine with genome editing and gene therapy for the aforementioned diseases among others. As such, significant financial investments have been made in the field of mRNA vaccine technology (Box 3). As of today, there are several mRNA vaccines and therapeutic candidates for different diseases and medical conditions in clinical trials, as indicated in Table 2. Despite this, it is unlikely that any changes would occur in the already approved and cost-effective vaccine platforms for polio, hepatitis B, chickenpox, and diphtheria, among others.
Table 2.
mRNA-based vaccines and therapeutics in clinical trials
| Antigen encoded by mRNA | Route of administration | Target condition | Phase | Status | Clinical trial number | |
|---|---|---|---|---|---|---|
| Infectious disease vaccines | ||||||
| mRNA-1273 | Full-length, prefusion-stabilised spike (S) protein of SARS-CoV-2 | Intramuscular | COVID-19 | Phase 4 | Recruiting | NCT04780659 |
| BNT162 (Comirnaty) | Full-length, membrane-anchored spike (S) protein of SARS-CoV-2 | Intramuscular | COVID-19 | Phase 4 | Recruiting | NCT04760132 |
| CVnCoV | Full-length, prefusion-stabilised spike (S) protein of SARS-CoV-2 | Intramuscular | COVID-19 | Phase 3 | Active, not recruiting | NCT04674189 |
| ARCT-021 | Spike (S) protein of SARS-CoV-2 | Intramuscular | COVID-19 | Phase 2 | Active, not recruiting | NCT04668339 |
| ARCoV | RBD domain of SARS-CoV-2 spike (S) protein | Intramuscular | COVID-19 | Phase 2 | Completed | ChiCTR2100041855 |
| ChulaCov19 | Undisclosed | Intramuscular | COVID-19 | Phase 1 | Not yet recruiting | NCT04566276 |
| PTX-COVID19-B | Full-length, membrane-anchored spike (S) protein of SARS-CoV-2 | Intramuscular | COVID-19 | Phase 1 | Active, not recruiting | NCT04765436 |
| CoV2 SAM (LNP) | Spike (S) protein of SARS-CoV-2 | Intramuscular | COVID-19 | Phase 1 | Active, not recruiting | NCT04758962 |
| mRNA-1273.351 | Full-length, prefusion-stabilised spike (S) protein of SARS-CoV-2 B.1.351 variant | Intramuscular | COVID-19 | Phase 1 | Active, not recruiting | NCT04785144 |
| MRT5500 | Full-length, prefusion-stabilised spike (S) protein of SARS-CoV-2 | Intramuscular | COVID-19 | Phase 1/2 | Recruiting | NCT04798027 |
| CV7202 | Rabies virus glycoprotein, RABV-G | Intramuscular | Rabies | Phase 1 | Active, not recruiting | NCT03713086 |
| mRNA-1647 | Subunits of the CMV pentamer complex and the glycoprotein B (gB) protein | Intramuscular | Cytomegalovirus infection | Phase 1 | Completed | NCT03382405 |
| mRNA-1944 | A fully human IgG antibody originally isolated from B cells of a patient with a prior history of potent immunity against Chikungunya infection | Intravenous | Chikungunya | Phase 1 | Completed | NCT03829384 |
| Cancer vaccines | ||||||
| CV8102 | A noncoding single-stranded RNA complexed with a cationic peptide that functions as a strong immunomodulator based on TLR7/8 and RIG-1 activation | Intratumoural | Cutaneous melanoma, adenoidcystic carcinoma, squamous cell cancer of skin, head, and neck | Phase 1 | Recruiting | NCT03291002 |
| BI1361849 (CV9202) | Six non-small cell lung cancer (NSCLC)-associated antigens (NY-ESO-1, MAGE-C1, MAGE-C2, survivin, 5T4, and MUC-1) | Intradermal | Non-small cell lung cancer | Phase 1/2 | Active, not recruiting | NCT03164772 |
| BNT111 (FixVac) | Liposomal RNA (RNA-LPX) vaccine, which targets four non-mutated, tumour-associated antigens that are prevalent in melanoma | Intravenous | Advanced melanoma (adjuvant and metastatic) | Phase 1 | Active, not recruiting | NCT02410733 |
| MVT-5873 (BNT321) | A fully human IgG1 monoclonal antibody targeting sialyl Lewis A (sLea), an epitope on CA19-9 that is expressed in pancreatic and other gastrointestinal cancers that plays a role in tumor adhesion and metastasis formation, and is a marker of an aggressive cancer phenotype | Intravenous | Pancreatic cancer | Phase 1 | Recruiting | NCT02672917 |
| BNT122 (Autogene cevumeran) | Targets locally advanced or metastatic solid tumors | Intravenous | Metastatic melanoma and other solid tumors | Phase 2 | Recruiting | NCT03289962 |
| mRNA-4157 | Personalised cancer vaccine (PCV) with a combination of validated defined neoantigens, predicted neoepitopes, and mutations in driver genes | Intramuscular | Solid tumours | Phase 1 | Recruiting | NCT03313778 |
| mRNA-5671/V941 | Four common mutations of an oncogene known as KRAS | Intramuscular | Non-small cell lung cancer, pancreatic neoplasms, and colorectal neoplasms | Phase 1 | Recruiting | NCT03948763 |
| Curative and protein supplementation therapeutics | ||||||
| AZD8601 | Vascular endothelial growth factor-A (VEGF-A) | Epicardial | Myocardial ischemia | Phase 2 | Active, not recruiting | NCT03370887 |
| MRT5005 | Cystic fibrosis transmembrane conductance regulator (CFTR) | Inhalation | Cystic fibrosis | Phase 1/2 | Recruiting | NCT03375047 |
| LUNAR-OTC (ARCT-810) | Ornithine transcarbamylase (OTC) | Intravenous | Ornithine transcarbamylase deficiency | Phase 1b | Recruiting | NCT04442347 |
| mRNA-3927 | Propionyl-CoA carboxylase | Intravenous | Propionic acidemia | Phase 1/2 | Recruiting | NCT04159103 |
| NTLA-2001 | Cas9 | Intravenous | Transthyretin amyloidosis | Phase 1 | Recruiting | NCT04601051 |
Box 3-.
Outlook on the economic impact of mRNA vaccines
Borne out of the COVID-19 pandemic, appreciation of mRNA vaccines has initiated substantial investments in the technology. There has been a sharp growth of combined market capitalisation for five publicly listed mRNA companies in the USA from USD 15 billion in 2019 to USD 300 billion in 2021, indicating the economic impact of mRNA vaccines [224].
This boost is further backed by the government and non-government funding agencies in North America and Europe which constituted cumulative global funding in 2020 of over USD 9 billion for mRNA therapeutics [225]. In 2021, two new RNA start-ups, Laronde (Cambridge, USA) and Orna Therapeutics (Cambridge, USA) secured USD 440 million and USD 80 million, respectively [226]. Early investments of this scale reflect the triumph of mRNA vaccines against COVID-19 with the market value solely based on COVID-19 vaccines estimated at USD 50 billion in 2021 [224].
Multinational pharmaceutical companies are also investing in the research and development of mRNA vaccines. Sanofi S.A. (Paris, France) recently acquired a clinical-stage mRNA therapeutic company, Translate Bio (Massachusetts, USA) with USD 3.2 billion [227]. Additionally, Sanofi S.A. plans to annually invest USD 400 million in its one-of-a-kind ‘mRNA Center of Excellence’ [228]. Furthermore, Novartis (Basel, Switzerland) has signed a contract with BioNTech for the bulk production of BioNTech's COVID-19 vaccine in 2022 [229]. GlaxoSmithKline (Brentford, UK) has also initiated a €150 million collaboration with CureVac to develop the second generation of COVID-19 vaccines to address the emerging SARS-CoV-2 variants [230].
All of these collaborations indicate the interest of global healthcare companies in mRNA-based vaccines and therapeutics which may significantly play a role in expanding its market. However, there is a possibility of a decline in the mRNA vaccine market from 2023 to 2025 due to decreased demand for COVID-19 vaccines as a result of the waning pandemic. Despite this, the affirming nature of COVID-19 vaccines could facilitate efforts to address the lag that currently exists in clinical trials for other prospective mRNA vaccines. As such, it may be expected that from 2028 onwards new mRNA vaccine products targeting a myriad of diseases will come forth [224].
By 2035, the projected market value is set to reach USD 23 billion with prophylactic vaccines dominating by generating 50% of the revenue while therapeutic vaccines and other therapeutics may account for 30% and 20% of the profits [224].
Conclusion
From their early indication as a therapeutic tool in the 1990s to exhibiting their flair as a pioneering vaccination programme in 2020 amidst one of the greatest pandemics in recent times, mRNA vaccines have traversed difficult roadblocks to deliver their promise. In line with the success of COVID-19 mRNA vaccines, initiatives are being undertaken by the government, corporations and academia to further utilise this exemplary vaccine regime for other diseases. As such, the future of vaccines, gene therapy, and genome editing are likely to be hinging on mRNA technologies. With the foundation set by the approved COVID-19 mRNA vaccines, faster approval of future vaccines will lead to a further demystification of the technology. However, the bench-to-bedside translation of mRNA vaccines will necessitate adequate funding, international collaborations between policymakers, industry, and academia as well as a consensus within the public health community. Although some of these requirements often tend to be unmet in a pandemic-free world, their overall importance has received prominence during the challenging times of COVID-19. With a growing number of mRNA vaccine candidates entering clinical trials and production pipelines, the cloud of scepticism surrounding this technology is starting to clear. Like every monumental therapeutic platform, mRNA vaccines have their limitations. However, massive strides have been made in adapting them for better efficacy, safety, delivery, and tolerance with efforts on this front remaining a continued research priority. With well-equipped manufacturing capabilities for mRNA-based vaccine production worldwide, thorough guidelines can be expected for the production and evaluation of this new age of therapeutics in the years to come.
Acknowledgments
The authors would like to thank their respective PhD research funding bodies. S.B. is supported by the University Hospital Giessen and Marburg (15/2020MR to Dr. Moritz Lindner). N.T. is supported by a Wellcome Trust 4-year PhD Studentship (102171/Z/13/Z).
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Author contributions
N.Z.R., S.B., and N.T. are shared first authors and have contributed equally to the conceptualisation and writing of the article. N.T. has also been involved in the drafting and editing of the article. Additionally, S.B. has designed Figure 4 with BioRender. L.M.O. is the second author and has illustrated Figures 1, 2, and 3 and written the section ‘The concept of mRNA vaccines’. M.M.M. is the third author and has written the ‘Introduction’ and assisted N.Z.R., S.B., and N.T. in the design of tables.
Data availability
No data was generated and/or analysed for the review article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Plotkin S. History of vaccination. Proc Natl Acad Sci. USA. 2014;111(34):12283–12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Griffith F. The significance of pneumococcal types. J Hyg (Lond.). 1928;27(2):113–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Szybalska EH, Szybalski W. Genetics of human cell line. IV. DNA-mediated heritable transformation of a biochemical trait. Proc Natl Acad Sci. USA. 1962;48(12):2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zinder ND, Lederberg J. Genetic exchange in Salmonella. J Bacteriol. 1952;64(5):679–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Maruggi G, Zhang C, Li J, et al. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol Ther. 2019;27(4):757–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sahin U, Karikó K, Ö T. mRNA-based therapeutics - developing a new class of drugs. Nat Rev Drug Discov. 2014;13(10):759–780. [DOI] [PubMed] [Google Scholar]
- [7].Vaidyanathan S, Azizian KT, Haque A, et al. Uridine depletion and chemical modification increase Cas9 mRNA activity and reduce immunogenicity without HPLC purification. Mol Ther Nucleic Acids. 2018;12:530–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wolff JA, Malone RW, Williams P, et al. Direct gene transfer into mouse muscle in vivo. Science. 1990;247(4949 Pt1):1465–1468. [DOI] [PubMed] [Google Scholar]
- [9].Martinon F, Krishnan S, Lenzen G, et al. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol. 1993;23(7):1719–1722. [DOI] [PubMed] [Google Scholar]
- [10].Esprit A, de Mey W, Bahadur Shahi R, et al. Neo-antigen mRNA vaccines. Vaccines (Basel). 2020;8(4):776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Stepinski J, Waddell C, Stolarski R, et al. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3’-O-methyl)GpppG and 7-methyl (3’-deoxy)GpppG. RNA. 2001;7(10):1486–1495. [PMC free article] [PubMed] [Google Scholar]
- [12].Benteyn D, Heirman C, Bonehill A, et al. mRNA-based dendritic cell vaccines. Expert Rev Vaccines. 2015;14(2):161–176. [DOI] [PubMed] [Google Scholar]
- [13].Karikó K, Buckstein M, Ni H, et al. Suppression of RNA recognition by toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23(2):165–175. [DOI] [PubMed] [Google Scholar]
- [14].Kim SC, Sekhon SS, Shin W-R, et al. Modifications of mRNA vaccine structural elements for improving mRNA stability and translation efficiency. Mol Cell Toxicol. 2022;18(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Weide B, Pascolo S, Scheel B, et al. Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. J Immunother. 2009;32(5):498–507. [DOI] [PubMed] [Google Scholar]
- [16].Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. [DOI] [PubMed] [Google Scholar]
- [17].Thess A, Grund S, Mui BL, et al. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol Ther. 2015;23(9):1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Arango D, Sturgill D, Alhusaini N, et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell. 2018;175(7):1872–1886.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lamb YN. BNT162b2 mRNA COVID-19 vaccine: first approval. Drugs. 2021;81(4):495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Conry RM, LoBuglio AF, Wright M, et al. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995;55(7):1397–1400. [PubMed] [Google Scholar]
- [21].Verbeke R, Lentacker I, De Smedt S, et al. Three decades of messenger RNA vaccine development. Nano Today. 2019;28:100766. [Google Scholar]
- [22].Sahin U, Oehm P, Derhovanessian E, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585(7823):107–112. [DOI] [PubMed] [Google Scholar]
- [23].Wadhwa A, Aljabbari A, Lokras A, et al. Opportunities and challenges in the delivery of mRNA-based vaccines. Pharmaceutics. 2020;12(2):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Reichmuth AM, Oberli MA, Jaklenec A, et al. mRNA vaccine delivery using lipid nanoparticles. Ther Deliv. 2016;7(5):319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu MA. A comparison of plasmid DNA and mRNA as vaccine technologies. Vaccines (Basel). 2019;7(2):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Iavarone C, O’hagan DT, Yu D, et al. Mechanism of action of mRNA-based vaccines. Expert Rev Vaccines. 2017;16(9):871–881. [DOI] [PubMed] [Google Scholar]
- [27].Pollard C, De Koker S, Saelens X, et al. Challenges and advances towards the rational design of mRNA vaccines. Trends Mol Med. 2013;19(12):705–713. [DOI] [PubMed] [Google Scholar]
- [28].Tan L, Sun X. Recent advances in mRNA vaccine delivery. Nano Res. 2018;11(10):5338–5354. [Google Scholar]
- [29].Zhong Z, Mc Cafferty S, Combes F, et al. mRNA therapeutics deliver a hopeful message. Nano Today. 2018;23:16–39. [Google Scholar]
- [30].Linares-Fernández S, Lacroix C, Exposito J-Y, et al. Tailoring mRNA vaccine to balance innate/adaptive immune response. Trends Mol Med. 2020;26(3):311–323. [DOI] [PubMed] [Google Scholar]
- [31].Lee K, Kim M, Seo Y, et al. Development of mRNA vaccines and their prophylactic and therapeutic applications. Nano Res. 2018;11(10):5173–5192. [Google Scholar]
- [32].Pardi N, Hogan MJ, Porter FW, et al. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov. 2018;17(4):261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schlake T, Thess A, Fotin-Mleczek M, et al. Developing mRNA-vaccine technologies. RNA Biol. 2012;9(11):1319–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chaudhary N, Weissman D, Whitehead KA. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat Rev Drug Discov. 2021;20(11):817–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schott JW, Morgan M, Galla M, et al. Viral and synthetic RNA vector technologies and applications. Mol Ther. 2016;24(9):1513–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kowalski PS, Rudra A, Miao L, et al. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther. 2019;27(4):710–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136(4):763–776. [DOI] [PubMed] [Google Scholar]
- [38].Sharova LV, Sharov AA, Nedorezov T, et al. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res. 2009;16(1):45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].De Beuckelaer A, Grooten J, De Koker S. Type I interferons modulate CD8(+) T cell immunity to mRNA vaccines. Trends Mol Med. 2017;23(3):216–226. [DOI] [PubMed] [Google Scholar]
- [40].Ramanathan A, Robb GB, Chan S-H. mRNA capping: biological functions and applications. Nucleic Acids Res. 2016;44(16):7511–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pasquinelli AE, Dahlberg JE, Lund E. Reverse 5’ caps in RNAs made in vitro by phage RNA polymerases. RNA. 1995;1(9):957–967. [PMC free article] [PubMed] [Google Scholar]
- [42].Sahu I, Haque A, Weidensee B, et al. Recent developments in mRNA-based protein supplementation therapy to target lung diseases. Mol Ther. 2019;27(4):803–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mockey M, Gonçalves C, Dupuy FP, et al. mRNA transfection of dendritic cells: synergistic effect of ARCA mRNA capping with poly(A) chains in cis and in trans for a high protein expression level. Biochem Biophys Res Commun. 2006;340(4):1062–1068. [DOI] [PubMed] [Google Scholar]
- [44].Furuichi Y, Shatkin AJ. Viral and cellular mRNA capping: past and prospects. Adv Virus Res. 2000;55:135–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mauger DM, Cabral BJ, Presnyak V, et al. mRNA structure regulates protein expression through changes in functional half-life. Proc Natl Acad Sci USA. 2019;116(48): 24075–24083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Quabius ES, Krupp G. Synthetic mRNAs for manipulating cellular phenotypes: an overview. N Biotechnol. 2015;32(1):229–235. [DOI] [PubMed] [Google Scholar]
- [47].Gray NK, Wickens M. Control of translation initiation in animals. Annu Rev Cell Dev Biol. 1998;14:399–458. [DOI] [PubMed] [Google Scholar]
- [48].Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol. 1987;196(4):947–950. [DOI] [PubMed] [Google Scholar]
- [49].Pelletier J, Sonenberg N. Insertion mutagenesis to increase secondary structure within the 5’ noncoding region of a eukaryotic mRNA reduces translational efficiency. Cell. 1985;40(3):515–526. [DOI] [PubMed] [Google Scholar]
- [50].Holtkamp S, Kreiter S, Selmi A, et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108(13):4009–4017. [DOI] [PubMed] [Google Scholar]
- [51].Karikó K, Kuo A, Barnathan ES. Overexpression of urokinase receptor in mammalian cells following administration of the in vitro transcribed encoding mRNA. Gene Ther. 1999;6(6):1092–1100. [DOI] [PubMed] [Google Scholar]
- [52].Ferizi M, Leonhardt C, Meggle C, et al. Stability analysis of chemically modified mRNA using micropattern-based single-cell arrays. Lab Chip. 2015;15(17):3561–3571. [DOI] [PubMed] [Google Scholar]
- [53].von Niessen Ag O, Poleganov MA, Rechner C, et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3’ UTRs identified by cellular library screening. Mol Ther. 2019;27(4):824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Boccaletto P, Machnicka MA, Purta E, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yang X, Yang Y, Sun BF, et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017;27(5):606–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ge J, Yu YT. RNA pseudouridylation: new insights into an old modification. Trends Biochem Sci. 2013;38(4):210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Carlile TM, Martinez NM, Schaening C, et al. mRNA structure determines modification by pseudouridine synthase 1. Nat Chem Biol. 2019;15(10):966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Haque A Ashiqul, Weinmann P, Biswas S, Handgretinger R, Mezger M, Kormann M S and Antony J S. (2020). RNA ImmunoGenic Assay: Simple method for detecting immunogenicity of in vitro transcribed mRNA. Adv Cell Gene Ther, 3(2), 10.1002/acg2.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Karikó K, Muramatsu H, Welsh FA, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. 2008;16(11):1833–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Duechler M, Leszczyńska G, Sochacka E, et al. Nucleoside modifications in the regulation of gene expression: focus on tRNA. Cell Mol Life Sci. 2016;73(16):3075–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rodriguez-Hernandez A, Spears JL, Gaston KW, et al. Structural and mechanistic basis for enhanced translational efficiency by 2-thiouridine at the tRNA anticodon wobble position. J Mol Biol. 2013;425(20):3888–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Andries O, Mc Cafferty S, De Smedt SC, et al. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J Control Release. 2015;217:337–344. [DOI] [PubMed] [Google Scholar]
- [63].Svitkin YV, Cheng YM, Chakraborty T, et al. N1-methyl-pseudouridine in mRNA enhances translation through eIF2α-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res. 2017;45(10):6023–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhang C, Fu J, Zhou Y. A review in research progress concerning m6A methylation and immunoregulation. Front Immunol. 2019;10:922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149(7):1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Haque AA, Weinmann P, Biswas S, et al. RNA immunoGenic assay: a method to detect immunogenicity of in vitro transcribed mRNA in human whole blood. Biol Protoc. 2020;10(24):e3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22(7):346–353. [DOI] [PubMed] [Google Scholar]
- [68].Kudla G, Lipinski L, Caffin F, et al. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 2006;4(6):e180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Karikó K, Muramatsu H, Keller JM, et al. Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol Ther. 2012;20(5):948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wu MZ, Asahara H, Tzertzinis G, et al. Synthesis of low immunogenicity RNA with high-temperature in vitro transcription. RNA. 2020;26(3):345–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Enuka Y, Lauriola M, Feldman ME, et al. Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res. 2016;44(3):1370–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wesselhoeft RA, Kowalski PS, Anderson DG. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat Commun. 2018;9(1):2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wesselhoeft RA, Kowalski PS, Parker-Hale FC, et al. RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol Cell. 2019;74(3):508–520.e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wroblewska L, Kitada T, Endo K, et al. Mammalian synthetic circuits with RNA binding proteins for RNA-only delivery. Nat Biotechnol. 2015;33(8):839–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wagner TE, Becraft JR, Bodner K, et al. Small-molecule-based regulation of RNA-delivered circuits in mammalian cells. Nat Chem Biol. 2018;14(11):1043–1050. [DOI] [PubMed] [Google Scholar]
- [76].Bell CL, Yu D, Smolke CD, et al. Control of alphavirus-based gene expression using engineered riboswitches. Virology. 2015;483:302–311. [DOI] [PubMed] [Google Scholar]
- [77].Weigand JE, Suess B. Tetracycline aptamer-controlled regulation of pre-mRNA splicing in yeast. Nucleic Acids Res. 2007;35(12):4179–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wachsmuth M, Findeiß S, Weissheimer N, et al. De novo design of a synthetic riboswitch that regulates transcription termination. Nucleic Acids Res. 2013;41(4):2541–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mc Cafferty S, De Temmerman J, Kitada T, et al. In vivo validation of a reversible small molecule-based switch for synthetic self-amplifying mRNA regulation. Mol Ther. 2021;29(3):1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Chahal JS, Khan OF, Cooper CL, et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc Natl Acad Sci. USA. 2016;113(29): E4133–E4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Brito LA, Chan M, Shaw CA, et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol Ther. 2014;22(12):2118–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Blakney AK, Zhu Y, McKay PF, et al. Big is beautiful: enhanced saRNA delivery and immunogenicity by a higher molecular weight, bioreducible, cationic Polymer. ACS Nano. 2020;14(5):5711–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Geall AJ, Verma A, Otten GR, et al. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci. USA. 2012;109(36): 14604–14609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].GlaxoSmithKline . A study to evaluate the safety and immunogenicity of GlaxoSmithKline (GSK) biologicals’ experimental rabies vaccine in healthy adults [Internet]. 2019. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT04062669
- [85].Arcturus Therapeutics I. A trial evaluating the safety and effects of an RNA vaccine ARCT-021 in healthy adults [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT04668339
- [86].Liang SL, Quirk D, Zhou A. RNase L: its biological roles and regulation. IUBMB Life. 2006;58(9):508–514. [DOI] [PubMed] [Google Scholar]
- [87].Blakney AK, McKay PF, Bouton CR, et al. Innate inhibiting proteins enhance expression and immunogenicity of self-amplifying RNA. Mol Ther. 2021;29(3):1174–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kim DY, Atasheva S, McAuley AJ, et al. Enhancement of protein expression by alphavirus replicons by designing self-replicating subgenomic RNAs. Proc Natl Acad Sci. USA. 2014;111(29): 10708–10713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Pepini T, Pulichino AM, Carsillo T, et al. Induction of an IFN-mediated antiviral response by a self-amplifying RNA vaccine: implications for vaccine design. J Immunol. 2017;198(10):4012–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Miao L, Zhang Y, Huang L. mRNA vaccine for cancer immunotherapy. Mol Cancer. 2021;20(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Xu S, Yang K, Li R, et al. mRNA vaccine era - mechanisms, drug platform and clinical prospection. Int J Mol Sci. 2020;21(18):6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zeng C, Zhang C, Walker PG, et al. Formulation and delivery technologies for mRNA vaccines. Curr Top Microbiol Immunol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kim M, Jeong M, Hur S, et al. Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Sci Adv. 2021;7(9):eabf4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Qiu M, Glass Z, Chen J, et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc Natl Acad Sci. USA. 2021;118(10): e2020401118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Riley RS, Kashyap MV, Billingsley MM, et al. Ionizable lipid nanoparticles for in utero mRNA delivery. Sci Adv. 2021;7(3):eaba1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Schoenmaker L, Witzigmann D, Kulkarni JA, et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int J Pharm. 2021;601:120586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Zhang H, You X, Wang X, et al. Delivery of mRNA vaccine with a lipid-like material potentiates antitumor efficacy through toll-like receptor 4 signaling. Proc Natl Acad Sci. USA. 2021;118(6): e2005191118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Zhang X, Zhao W, Nguyen GN, et al. Functionalized lipid-like nanoparticles for in vivo mRNA delivery and base editing. Sci Adv. 2020;6(34):eabc2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Islam MA, Rice J, Reesor E, et al. Adjuvant-pulsed mRNA vaccine nanoparticle for immunoprophylactic and therapeutic tumor suppression in mice. Biomaterials. 2021;266:120431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Davies N, Hovdal D, Edmunds N, et al. Functionalized lipid nanoparticles for subcutaneous administration of mRNA to achieve systemic exposures of a therapeutic protein. Mol Ther Nucleic Acids. 2021;24:369–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Tavares AJ, Poon W, Zhang YN, et al. Effect of removing Kupffer cells on nanoparticle tumor delivery. Proc Natl Acad Sci. USA. 2017;114(51):E10871–E10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Tsoi KM, MacParland SA, Ma XZ, et al. Mechanism of hard-nanomaterial clearance by the liver. Nat Mater. 2016;15(11):1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Saunders NRM, Paolini MS, Fenton OS, et al. A nanoprimer to improve the systemic delivery of siRNA and mRNA. Nano Lett. 2020;20(6):4264–4269. [DOI] [PubMed] [Google Scholar]
- [104].Zhang H, Leal J, Soto MR, et al. Aerosolizable lipid nanoparticles for pulmonary delivery of mRNA through design of experiments. Pharmaceutics. 2020;12(11):1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Abramson A, Kirtane AR, Shi Y, et al. Oral mRNA delivery using capsule-mediated gastrointestinal tissue injections. Matter. 2022;5(3):975–987. [Google Scholar]
- [106].Carvalho MR, Reis RL, Oliveira JM. Dendrimer nanoparticles for colorectal cancer applications. J Mater Chem B. 2020;8(6):1128–1138. [DOI] [PubMed] [Google Scholar]
- [107].Xiong H, Liu S, Wei T, et al. Theranostic dendrimer-based lipid nanoparticles containing PEGylated BODIPY dyes for tumor imaging and systemic mRNA delivery in vivo. J Control Release. 2020;325:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Fischer NO, Rasley A, Corzett M, et al. Colocalized delivery of adjuvant and antigen using nanolipoprotein particles enhances the immune response to recombinant antigens. J Am Chem Soc. 2013;135(6):2044–2047. [DOI] [PubMed] [Google Scholar]
- [109].Fischer NO, Weilhammer DR, Dunkle A, et al. Evaluation of nanolipoprotein particles (NLPs) as an in vivo delivery platform. PloS One. 2014;9(3):e93342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].He W, Evans AC, Rasley A, et al. Cationic HDL mimetics enhance in vivo delivery of self-replicating mRNA. Nanomedicine. 2020;24:102154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Koji K, Yoshinaga N, Mochida Y, et al. Bundling of mRNA strands inside polyion complexes improves mRNA delivery efficiency in vitro and in vivo. Biomaterials. 2020;261:120332. [DOI] [PubMed] [Google Scholar]
- [112].Abbasi S, Uchida S, Toh K, et al. Co-encapsulation of Cas9 mRNA and guide RNA in polyplex micelles enables genome editing in mouse brain. J Control Release. 2021;332:260–268. [DOI] [PubMed] [Google Scholar]
- [113].Khalil AS, Yu X, Umhoefer JM, et al. Single-dose mRNA therapy via biomaterial-mediated sequestration of overexpressed proteins. Sci Adv. 2020;6(27):eaba2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Jansig E, Geissler S, Rieckmann V, et al. Viromers as carriers for mRNA-mediated expression of therapeutic molecules under inflammatory conditions. Sci Rep. 2020;10(1):15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Liu S, Cheng Q, Wei T, et al. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR–Cas gene editing. Nat Mater. 2021;20(5):701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Segel M, Lash B, Song J, et al. Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science. 2021;373(6557):882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Martin C, Lowery D. mRNA vaccines: intellectual property landscape. Nat Rev Drug Discov. 2020;19(9):578. [DOI] [PubMed] [Google Scholar]
- [118].Gaviria M, Kilic B. A network analysis of COVID-19 mRNA vaccine patents. Nat Biotechnol. 2021;39(5):546–548. [DOI] [PubMed] [Google Scholar]
- [119].KnowMade RNA vaccine patent landscape analysis January 2021. [Internet]. [cited 2022 Mar 6]. Available from: https://www.knowmade.com/wp-content/uploads/2021/01/RNA-Vaccine-Patent-landscape-2021-FLYER.pdf
- [120].US Food and Drug Administration . Comirnaty and Pfizer-BioNTech COVID-19 vaccine [Internet]. 2022. [cited 2022 Mar 6]. Available from: https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/comirnaty-and-pfizer-biontech-covid-19-vaccine
- [121].Abu-Raddad LJ, Chemaitelly H, Butt AA. Effectiveness of the BNT162b2 covid-19 vaccine against the B.1.1.7 and B.1.351 variants. N Eng J Med. 2021;385(2):187–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Baraniuk C. What do we know about China’s covid-19 vaccines? BMJ. 2021;373:n912. [DOI] [PubMed] [Google Scholar]
- [123].Chemaitelly H, Yassine HM, Benslimane FM, et al. mRNA-1273 COVID-19 vaccine effectiveness against the B.1.1.7 and B.1.351 variants and severe COVID-19 disease in Qatar. Nat Med. 2021;27(9):1614–1621. [DOI] [PubMed] [Google Scholar]
- [124].Lopez Bernal J, Andrews N, Gower C, et al. Effectiveness of covid-19 vaccines against the B.1.617.2 (delta) variant. N Eng J Med. 2021;385(7):585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].World Health Organisation . Background document on mRNA vaccine BNT162b2 (Pfizer-BioNTech) against COVID-19 [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://www.who.int/publications/i/item/background-document-on-mrna-vaccine-bnt162b2-(pfizer-biontech)-against-covid-19
- [126].World Health Organisation . Background document on the mRNA-1273 vaccine (Moderna) against COVID-19 [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://www.who.int/publications/i/item/background-document-on-the-mrna-1273-vaccine-(moderna)-against-covid-19
- [127].World Health Organisation . Background document on the AZD1222 vaccine against COVID-19 developed by Oxford University and AstraZeneca [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://www.who.int/publications/i/item/background-document-on-the-azd1222-vaccine-against-covid-19-developed-by-oxford-university-and-astrazeneca
- [128].World Health Organisation . Background document on the Janssen Ad26.COV2.S (COVID-19) vaccine: background document to the WHO Interim recommendations for use of Ad26.COV2.S (COVID-19) vaccine. [Internet]. 2021. [cited 2022 Mar 6]. Available from. https://www.who.int/publications/i/item/WHO-2019-nCoV-vaccines-SAGE-recommendation-Ad26.COV2.S-background-2021.1.
- [129].World Health Organisation . Background document on the inactivated COVID-19 vaccine BIBP developed by China National Biotec Group (CNBG). Sinopharm, 7 May 2021 [Internet]. 2021 [cited 2022 Mar 6]. Available from: https://www.who.int/publications/i/item/WHO-2019-nCoV-vaccines-SAGE_recommendation-BIBP-background-2021.1
- [130].World Health Organisation . Background document on the inactivated vaccine Sinovac-CoronaVac against COVID-19. [Internet]. 2021. [cited 2022 Mar 6]. Available from. https://www.who.int/publications/i/item/WHO-2019-nCoV-vaccines-SAGE_recommendation-Sinovac-CoronaVac-background-2021.1.
- [131].Rauch S, Jasny E, Schmidt KE, et al. New vaccine technologies to combat outbreak situations. Front Immunol. 2018;9:1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Zhao J, Zhao S, Ou J, et al. COVID-19: coronavirus vaccine development updates. Front Immunol. 2020;11:602256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].World Health Organisation . COVID-19 vaccine tracker and landscape [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines
- [134].Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3(1):237–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Perlman S, Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol. 2009;7(6):439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Schoof M, Faust B, Saunders RA, et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive spike. Science. 2020;370(6523):1473–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Li F. Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J Virol. 2015;89(4):1954–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Li F, Li W, Farzan M, et al. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. [DOI] [PubMed] [Google Scholar]
- [139].Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Gui M, Song W, Zhou H, et al. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017;27(1):119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Yuan Y, Cao D, Zhang Y, et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun. 2017;8:15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Belouzard S, Millet JK, Licitra BN, et al. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. 2012;4(6):1011–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Heald-Sargent T, Gallagher TR, Set F. The coronavirus spike protein and acquisition of fusion competence. Viruses. 2012;4(4):557–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Smith TRF, Patel A, Ramos S, et al. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat Commun. 2020;11(1):2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. N Eng J Med. 2020;383(27):2603–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Walsh EE, Frenck RW, Falsey AR, et al. Safety and immunogenicity of two RNA-based covid-19 vaccine candidates. N Eng J Med. 2020;383(25):2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Sahin U, Muik A, Vogler I, et al. BNT162b2 vaccine induces neutralizing antibodies and poly-specific T cells in humans. Nature. 2021;595(7868):572–577. [DOI] [PubMed] [Google Scholar]
- [148].US Food and Drug Administration . Emergency use authorization for vaccines to prevent COVID-19 [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://www.fda.gov/media/142749/download
- [149].Anderson EJ, Rouphael NG, Widge AT, et al. Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N Eng J Med. 2020;383(25):2427–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Widge AT, Rouphael NG, Jackson LA, et al. Durability of responses after SARS-CoV-2 mRNA-1273 vaccination. N Eng J Med. 2020;384(1):80–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Jackson LA, Anderson EJ, Rouphael NG, et al. An mRNA vaccine against SARS-CoV-2 - preliminary report. N Eng J Med. 2020;383(20):1920–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Baden LR, El Sahly HM, Essink B, et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Eng J Med. 2020;384(5):403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Hall VG, Ferreira VH, Ku T, et al. Randomized trial of a third dose of mRNA-1273 vaccine in transplant recipients. N Eng J Med. 2021;385:1244–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Prasad M, Lin JL, Gu Y, et al. No crossreactivity of anti-SARS-CoV-2 spike protein antibodies with syncytin-1. Cell Mol Immunol. 2021;18(11):2566–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Shimabukuro TT, Kim SY, Myers TR, et al. Preliminary findings of mRNA Covid-19 vaccine safety in pregnant persons. N Engl J Med. 2021;384(24):2273–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Chen F, Zhu S, Dai Z, et al. Effects of COVID-19 and mRNA vaccines on human fertility. Hum Reprod. 2021;37(1):5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Cavanaugh AM, Spicer KB, Thoroughman D, et al. Reduced risk of reinfection with SARS-CoV-2 after COVID-19 vaccination - kentucky, may-june 2021. MMWR Morb Mortal Wkly Rep. 2021;70(32):1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Centers for disease control and prevention. interim clinical considerations for use of COVID-19 vaccines currently approved or authorized in the United States [Internet]. 2022. [cited 2022 Mar 6]. Available from: https://www.cdc.gov/vaccines/covid-19/clinical-considerations/covid-19-vaccines-us.html#appendix-d
- [159].Olliaro P, Torreele E, Vaillant M. COVID-19 vaccine efficacy and effectiveness-the elephant (not) in the room. Lancet Microbe. 2021;2(7):e279–e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Crommelin DJA, Anchordoquy TJ, Volkin DB, et al. Addressing the cold reality of mRNA vaccine stability. J Pharm Sci. 2021;110(3):997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Zhang NN, Li XF, Deng YQ, et al. A thermostable mRNA vaccine against COVID-19. Cell. 2020;182(2):1271–1283.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].BioNTech . BioNTech introduces first modular mRNA manufacturing facility to promote scalable vaccine production in Africa [Internet]. 2022. [cited 2022 Mar 6]. Available from: https://investors.biontech.de/news-releases/news-release-details/biontech-introduces-first-modular-mrna-manufacturing-facility
- [163].Yi HG, Kim H, Kwon J, et al. Application of 3D bioprinting in the prevention and the therapy for human diseases. Signal Transduct Target Ther. 2021;6(1):177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].World Health Organisation . The mRNA vaccine technology transfer hub [Internet]. 2022. [cited 2022 Mar 6]. Available from: https://www.who.int/initiatives/the-mrna-vaccine-technology-transfer-hub?fbclid=IwAR03_HNtbSnCZEGSlqIkhulRMTTp1O4dbTSLMyXx8R6IwEs6maGCWAmuy50
- [165].World Health Organisation . Moving forward on goal to boost local pharmaceutical production, WHO establishes global biomanufacturing training hub in Republic Of Korea [Internet]. 2022. [cited 2022 Mar 6]. Available from: https://www.who.int/news/item/23-02-2022-moving-forward-on-goal-to-boost-local-pharmaceutical-production-who-establishes-global-biomanufacturing-training-hub-in-republic-of-korea
- [166].Singh A. Eliciting B cell immunity against infectious diseases using nanovaccines. Nat Nanotechnol. 2021;16(1):16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Ripperger TJ, Uhrlaub JL, Watanabe M, et al. Orthogonal SARS-CoV-2 serological assays enable surveillance of low-prevalence communities and reveal durable humoral immunity. Immunity. 2020;53(5):925–933.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Doria-Rose N, Suthar MS, Makowski M, et al. Antibody persistence through 6 months after the second dose of mRNA-1273 vaccine for Covid-19. N Engl J Med. 2021;384(23):2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Tartof SY, Slezak JM, Fischer H, et al. Effectiveness of mRNA BNT162b2 COVID-19 vaccine up to 6 months in a large integrated health system in the USA: a retrospective cohort study. Lancet. 2021;398(10309):1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Dan JM, Mateus J, Kato Y, et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science. 2021;371(6529):eabf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Yu X, Tsibane T, McGraw PA, et al. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature. 2008;455(7212):532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Le Bert N, Tan AT, Kunasegaran K, et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584(7821):457–462. [DOI] [PubMed] [Google Scholar]
- [173].Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. [DOI] [PubMed] [Google Scholar]
- [174].Coulie PG, Van den Eynde BJ, van der Bruggen P, et al. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135–146. [DOI] [PubMed] [Google Scholar]
- [175].Heesen L, Frenzel K, Bolte S, et al. Abstract CT221: mutanome engineered RNA immuno-therapy (MERIT) for patients with triple negative breast cancer (TNBC). Cancer Res. 2019;79(13):CT221. [Google Scholar]
- [176].I M, Sharp M, Corp D. An efficacy study of adjuvant treatment with the personalized cancer vaccine mRNA-4157 and pembrolizumab in participants with high-risk melanoma (keynote-942) [Internet]. 2019. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT03897881
- [177].Genentech I, B SE. A study of Autogene cevumeran (RO7198457) as a single agent and in combination with atezolizumab in participants with locally advanced or metastatic tumors [Internet]. 2017. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT03289962
- [178].Genentech I, B SE. A study to evaluate the efficacy and safety of RO7198457 in combination with pembrolizumab versus pembrolizumab alone in participants with previously untreated advanced melanoma [Internet]. 2019. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT03815058
- [179].Florida U, Ppn-o C, University of California SF, CureSearch . A study of RNA-lipid particle (RNA-LP) vaccines for newly diagnosed pediatric high-grade gliomas (pHGG) and adult glioblastoma (GBM) [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT04573140
- [180].Sharp M, Corp D. A study of mRNA-5671/V941 as monotherapy and in combination with pembrolizumab (V941-001) [Internet]. 2019. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT03948763
- [181].Roche H-L. A study of the efficacy and safety of RO7198457 in combination with atezolizumab versus atezolizumab alone following adjuvant platinum-doublet chemotherapy in participants who are ctDNA positive after surgical resection of stage II-III non-small cell lung cancer [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT04267237
- [182].SE B. A phase II clinical trial comparing the efficacy of RO7198457 versus watchful waiting in patients with ctDNA-positive, resected stage II (high risk) and stage III colorectal cancer [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT04486378
- [183].Center MSKC, Genentech I. Study of personalized tumor vaccines (PCVs) and a PD-L1 blocker in patients with pancreatic cancer that can be treated with surgery [Internet]. 2019. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT04161755
- [184].SE B. Evaluation of the safety and tolerability of i.v. administration of a cancer vaccine in patients with advanced melanoma [Internet]. 2015. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT02410733
- [185].SE B, Pharmaceuticals R. Trial with BNT111 and cemiplimab in combination or as single agents in patients with anti-PD-1-refractory/relapsed, unresectable stage III or IV melanoma [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT04526899
- [186].Cinca DG, Sunyer I-IdIBAPi, Coordinator FG-I, Barcelona HCo . Evaluating a combination of immune-based therapies to achieve a remission of HIV infection [Internet]. 2020. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT03619278
- [187].Guardo AC, Joe PT, Miralles L, et al. Preclinical evaluation of an mRNA HIV vaccine combining rationally selected antigenic sequences and adjuvant signals (HTI-TriMix). AIDS. 2017;31(3):321–332. [DOI] [PubMed] [Google Scholar]
- [188].Jong W, Leal L, Buyze J, et al. Therapeutic vaccine in chronically HIV-1-infected patients: a randomized, double-blind, placebo-controlled phase IIa trial with HTI-TriMix. Vaccines (Basel). 2019;7(4):209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Feldman RA, Fuhr R, Smolenov I, et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine. 2019;37(25):3326–3334. [DOI] [PubMed] [Google Scholar]
- [190].Kallen K-J, Heidenreich R, Schnee M, et al. A novel, disruptive vaccination technology: self-adjuvanted RNActive(®) vaccines. Hum Vaccin Immunother. 2013;9(10):2263–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Petsch B, Schnee M, Vogel AB, et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat Biotechnol. 2012;30(12):1210–1216. [DOI] [PubMed] [Google Scholar]
- [192].Wongsaroj P, Udomchaisakul P, Tepsumethanon S, et al. Rabies neutralizing antibody after 2 intradermal doses on days 0 and 21 for pre-exposure prophylaxis. Vaccine. 2013;31(13):1748–1751. [DOI] [PubMed] [Google Scholar]
- [193].Alberer M, Gnad-Vogt U, Hong HS, et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet. 2017;390(10101):1511–1520. [DOI] [PubMed] [Google Scholar]
- [194].Lutz J, Lazzaro S, Habbeddine M, et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines. 2017;2:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Aldrich C, Leroux-Roels I, Huang KB, et al. Proof-of-concept of a low-dose unmodified mRNA-based rabies vaccine formulated with lipid nanoparticles in human volunteers: a phase 1 trial. Vaccine. 2021;39(8):1310–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Pardi N, Hogan MJ, Pelc RS, et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature. 2017;543(7644):248–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Jagger BW, Dowd KA, Chen RE, et al. Protective efficacy of nucleic acid vaccines against transmission of Zika virus during pregnancy in mice. J Infect Dis. 2019;220(10):1577–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Richner JM, Jagger BW, Shan C, et al. Vaccine mediated protection against Zika virus-induced congenital disease. Cell. 2017;170(2):273–283.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].I M, Research BA, Authority D. Safety, tolerability, and immunogenicity of mRNA-1325 in healthy adult subjects [Internet]. 2016. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT03014089
- [200].I M, Research BA, Authority D. Safety, tolerability, and immunogenicity of Zika vaccine mRNA-1893 in healthy flavivirus seropositive and seronegative adults [Internet]. 2019. [cited 2022 Mar 6]. Available from: https://ClinicalTrials.gov/show/NCT04064905
- [201].Falsey AR, Hennessey PA, Formica MA, et al. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med. 2005;352(17):1749–1759. [DOI] [PubMed] [Google Scholar]
- [202].Shi T, McAllister DA, O’Brien KL, et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet. 2017;390(10098):946–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Kim HW, Canchola JG, Brandt CD, et al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol. 1969;89(4):422–434. [DOI] [PubMed] [Google Scholar]
- [204].Mazur NI, Higgins D, Nunes MC, et al. The respiratory syncytial virus vaccine landscape: lessons from the graveyard and promising candidates. Lancet Infect Dis. 2018;18(10):e295–e311. [DOI] [PubMed] [Google Scholar]
- [205].Crank MC, Ruckwardt TJ, Chen M, et al. A proof of concept for structure-based vaccine design targeting RSV in humans. Science. 2019;365(6452):505–509. [DOI] [PubMed] [Google Scholar]
- [206].McLellan JS, Chen M, Leung S, et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science. 2013;340(6136):1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].McLellan JS, Chen M, Joyce MG, et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science. 2013;342(6158):592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Krarup A, Truan D, Furmanova-Hollenstein P, et al. A highly stable prefusion RSV F vaccine derived from structural analysis of the fusion mechanism. Nat Commun. 2015;6:8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Espeseth AS, Cejas PJ, Citron MP, et al. Modified mRNA/lipid nanoparticle-based vaccines expressing respiratory syncytial virus F protein variants are immunogenic and protective in rodent models of RSV infection. NPJ Vaccines. 2020;5(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Aliprantis AO, Shaw CA, Griffin P, et al. A phase 1, randomized, placebo-controlled study to evaluate the safety and immunogenicity of an mRNA-based RSV prefusion F protein vaccine in healthy younger and older adults. Hum Vaccin Immunother. 2021;17(5):1248–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Chaudhary N, Weissman D, Whitehead KA. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat Rev Drug Discov. 2021;20(11):817–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Baeza Garcia A, Siu E, Sun T, et al. Neutralization of the Plasmodium-encoded MIF ortholog confers protective immunity against malaria infection. Nat Commun. 2018;9(1):2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Raj DK, Das Mohapatra A, Jnawali A, et al. Anti-PfGARP activates programmed cell death of parasites and reduces severe malaria. Nature. 2020;582(7810):104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Kurokawa C, Narasimhan S, Vidyarthi A, et al. Repeat tick exposure elicits distinct immune responses in Guinea pigs and mice. Ticks Tick Borne Dis. 2020;11(6):101529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Sajid A, Matias J, Arora G, et al. mRNA vaccination induces tick resistance and prevents transmission of the lyme disease agent. Sci Transl Med. 2021;13(620):eabj9827. [DOI] [PubMed] [Google Scholar]
- [216].Wallin MT, Culpepper WJ, Nichols E, et al. Global, regional, and national burden of multiple sclerosis 1990-2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019;18(3):269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Krienke C, Kolb L, Diken E, et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science. 2021;371(6525):145–153. [DOI] [PubMed] [Google Scholar]
- [218].Gertz MA, Benson MD, Dyck PJ, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451–2466. [DOI] [PubMed] [Google Scholar]
- [219].Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385(6):493–502. [DOI] [PubMed] [Google Scholar]
- [220].Weiss R, Scheiblhofer S, Thalhamer J. Allergens are not pathogens: why immunization against allergy differs from vaccination against infectious diseases. Hum Vaccin Immunother. 2014;10(3):703–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Weiss R, Scheiblhofer S, Roesler E, et al. Prophylactic mRNA vaccination against allergy. Curr Opin Allergy Clin Immunol. 2010;10(6):567–574. [DOI] [PubMed] [Google Scholar]
- [222].Roesler E, Weiss R, Weinberger EE, et al. Immunize and disappear-safety-optimized mRNA vaccination with a panel of 29 allergens. J Allergy Clin Immunol. 2009;124(5):1070–1077.e1-11. [DOI] [PubMed] [Google Scholar]
- [223].Hattinger E, Scheiblhofer S, Roesler E, et al. Prophylactic mRNA vaccination against allergy confers long-term memory responses and persistent protection in mice. J Immunol Res. 2015;2015:797421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Xie W, Chen B, Wong J. Publisher correction: evolution of the market for mRNA technology. Nat Rev Drug Discov. 2021;20(11):880. [DOI] [PubMed] [Google Scholar]
- [225].Mikulic M. Global funding on mRNA therapeutics and vaccines from 2015 to 2020 [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://www.statista.com/statistics/1274559/funding-in-mrna-therapeutics-and-vaccines-worldwide/#statisticContainer
- [226].Cross R. This was mRNA’s breakout year, and scientists are just getting started. Chemical & Engineering News [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://cen.acs.org/pharmaceuticals/Another-year-pharma/99/i44#This-was-mRNAs-breakout-year-and-scientists-are-just-getting-started
- [227].Sanofi. Sanofi to acquire translate bio; advances deployment of mRNA technology across vaccines and therapeutics development. Internet]. 2021. [cited 2022 Mar 6]. Available from. https://www.sanofi.com/en/media-room/press-releases/2021/2021-08-03-07-00-00-2273307.
- [228].Sanofi. Sanofi launches dedicated vaccines mRNA center of excellence. Internet]. 2021. [cited 2022 Mar 6]. Available from. https://www.sanofi.com/en/media-room/press-releases/2021/2021-06-29-10-00-40-2254458#.
- [229].Novartis . Novartis signs initial agreement with CureVac to manufacture COVID-19 vaccine candidate [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://www.novartis.com/news/media-releases/novartis-signs-initial-agreement-curevac-manufacture-covid-19-vaccine-candidate
- [230].GlaxoSmithKline . GSK and CureVac to develop next generation mRNA COVID-19 vaccines [Internet]. 2021. [cited 2022 Mar 6]. Available from: https://www.gsk.com/en-gb/media/press-releases/gsk-and-curevac-to-develop-next-generation-mrna-covid-19-vaccines/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was generated and/or analysed for the review article.