

ABSTRACT
Multiple myeloma (MM) is characterized by an accumulation of monoclonal plasma cells within the bone marrow (BM). Macrophages are an abundant component of myeloma BM microenvironment and support survival of the malignant cells and foster myeloma development and progression by suppression of the immune response. In our previous study, we found that MM patients overexpress pro-inflammatory cytokine interleukin-32 (IL-32). The present study aimed to investigate the role of IL-32 in myeloma progression and mechanisms of IL-32 on macrophages functions. We discovered that the expression of IL-32 was associated with the disease stage in myeloma patients. MM-derived exosomes containing IL-32γ promoted the expression of programmed death-ligand 1(PD-L1) by macrophages, thus promoting immune evasion. Mechanistically, myeloma-secreted IL-32γ acted via proteinase 3 (PR3) to enhance 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) dependent glycolysis and subsequent PD-L1 expression. Moreover, the PFKFB3-Janus kinase 1 (JAK1) axis might contribute to the expression of PD-L1 by macrophages. To sum up, we concluded that IL-32 was a critical mediator in myeloma progression, and targeting IL-32 signaling might be used as a potential strategy for treating myeloma.
KEYWORDS: Multiple myeloma, IL-32, macrophage, glycolysis, immune checkpoint
Introduction
Multiple myeloma (MM) is a hematological malignancy characterized by expansion of clonal plasma cells infiltration in the bone marrow (BM). Despite the dramatic advancements in therapeutics over recent years, MM remains an incurable disease, with relapsed or refractory multiple myeloma (RRMM) being the major challenge in clinical practice.1
Although multiple factors have been shown to contribute to the transition through different stages of the disease, BM microenvironment is a dominant driver that regulates tumor initiation and progression. It has been suggested that the expression of immune checkpoint molecules by macrophages leads to suppression of T-cell activation and effector functions;2 nevertheless, the exact underlying mechanisms remain unclear.
Interleukin-32 (IL-32) is a novel cytokine involved in inflammation and cancer development.3 Previous studies have found that patients with newly diagnosed multiple myeloma (NDMM) overexpressing IL-32 have an advanced International Staging System (ISS) stage, and this process has been associated with the immunosuppression function of macrophages.4–6 In addition, a growing number of studies have highlighted the crucial role of metabolic reprogramming in macrophage phenotype and function.7 Previous studies suggested that lactate secretion by endothelial cells enables macrophages to support the formation of new muscle fibers and further stimulate blood vessel formation.8 Furthermore, tumor-derived hyaluronan fragments can facilitate glycolysis, which fosters immune privilege by promoting programmed death-ligand 1(PD-L1) expression in monocytes/macrophages.9 Lauterbach et al reported that after Toll-like Receptor 4 activation, macrophages increase glycolysis and tricarboxylic acid cycle volume, which then promotes histone acetylation that is relevant for gene induction.10 Another important finding demonstrated that enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages (TAMs).11 In addition, Ferreira et al indicated that dietary glutamine supplementation might potentiate parasite clearance by developing a more effective anti-Leishmania adaptive immune response.12 However, the mechanism through which metabolic reprogramming of macrophages affects their effector functions upon IL-32 stimulation is not well understood.
The binding of PD-L1 to programmed cell death protein 1 (PD-1) on activated T cells acts as a major co-inhibitory checkpoint signaling that controls T cell-activating signals.13 The inhibition of the PD-L1/PD-1 pathway has shown a remarkable anti-tumor effect in patients with advanced cancers. However, the results of monotherapy with PD-1 inhibitors have been unsatisfactory in MM,14,15 and anti-PD-1 combined with immunomodulatory drugs have presented toxicities that are incompletely understood.16 Therefore, further research is needed for alternative combinations of drugs or different approaches to target this pathway.17 Expression of PD-L1 is frequently observed in advanced stages of MM and the presence of bone marrow stromal cells.17 Previous studies have reported that TAMs express PD-L1 and contribute to the immune-suppressive tumor microenvironment.18,19 Moreover, one study reported that PD-L1 antibody-treated macrophages exhibited greater proliferation, survival, and pro-inflammatory activation.20 These increasing body of evidence may provide a rationale for the adoption of new strategies directly targeting PD-L1 protein.
In this study, we examined the mechanisms through which IL-32γ promotes the expression of immune checkpoint molecules in macrophages. We found that myeloma-secreted IL-32γ acted via proteinase 3 (PR3) to enhance 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) dependent glycolysis and subsequent PD-L1 expression, which then resulted in the suppression of the activation and function of CD8 + T cells. We also demonstrated that the PFKFB3-Janus kinase 1 (JAK1) axis might mediate the PD-L1 expression in IL-32γ stimulated macrophages. These data suggested that IL-32γ derived from MM cells promoted the immunosuppressive function of macrophages and targeting IL-32 signaling might be used as a potential strategy for MM therapy.
Materials and methods
Cell culture
Human MM cell lines MM.1S, CAG, JJN3, H929, U266, ARP-1, and RPMI 8226 were kindly provided by Dr. Qing Yi (Center for Hematologic Malignancies, Research Institute, Houston Methodist, Houston, TX, USA). All cell lines were cultured in complete RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) in 5% CO2 at 37°C.
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors after obtaining informed consent. Human macrophages were generated from PBMCs as previously described.21 Briefly, monocytes (2 × 106 cells/well) were incubated in 6-well cell culture plates for 0.5–1 h in RPMI 1640 media. Non-adherent cells were then removed. The adherent human monocytes were differentiated to macrophages for 7 d in RPMI 1640 media containing 10% heat-inactivated FBS and 20 ng/ml macrophage colony-stimulating factor (M-CSF) (R&D Systems, MN, USA).
Patients and clinical specimens
Sixteen pairs of primary and relapsed or refractory myeloma specimens were obtained from patients who were enrolled in the First Affiliated Hospital, School of Medicine, Zhejiang University between 2015 and 2020. All patients signed the informed consent, which was approved by the Research Ethics Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University (Ethical approval number: IIT20210619A). Clinical information was retrospectively collected from the hospital archives, as shown in Table 1.
Table 1.
Patient characteristics
| Variables | NDMM | RRMM* |
|---|---|---|
| Median age, years(range) | 56.5 (46–78) | |
| Sex (M/F) | 11/5 | |
| Durie-Salmon staging, n(%) | ||
| Stage IA | 1 (6.25%) | |
| Stage IB | - | |
| StageIIA | 1 (6.25%) | |
| StageIIB | - | |
| StageIIIA | 12 (75%) | |
| StageIIIB | 2 (12.5%) | |
| R-ISS, n(%) | ||
| Stage I | - | |
| Stage II | 14 (87.5%) | |
| Stage III | 2 (12.5%) | |
| Median Glb(g/L), (range) | 47.7 (17.3–95.3) | 25.5 (15.3–36.8) |
| ≥35 g/L | 9 (56.25%) | 1 (6.25%) |
| <35 g/L | 7 (43.75%) | 15 (93.75%) |
| Median β2-microglobulin (mg/L), (range) | 2.135 (1.041–14.4) | 2.2 (1.059–6.62) |
| ≥5.5 mg/L | 3 (18.75%) | 1 (6.25%) |
| <5.5 mg/L | 13 (81.25%) | 14 (87.5%) |
| Unknown | - | 1 (6.25%) |
| Median Hb (g/L), (range) | 107 (61–153) | 128 (77–179) |
| ≥105 g/L | 9 (56.25%) | 13 (81.25%) |
| <105 g/L | 7 (43.75%) | 3 (18.75%) |
| Median Cre (μmol/L), (range) | 78.5 (54–488) | 76 (49–578) |
| ≥176.8 μmol/L | 2 (12.5%) | 2 (12.5%) |
| <176.8 μmol/L | 14 (87.5%) | 14 (87.5%) |
| Median Ca (mmol/L), (range) | 2.295 (1.91–2.58) | 2.255 (1.91–2.42) |
| ≥2.65 mmol/L | - | - |
| <2.65 mmol/L | 16 (100%) | 16 (100%) |
| Median LDH (U/L), (range) | 194 (123–292) | 234.5 (166–477) |
| Normal | 14 (87.5%) | 8 (50%) |
| High | 1 (6.25%) | 6 (37.5%) |
| Unknown | 1 (6.25%) | 2 (12.5%) |
| M protein subtype, n (%) | ||
| IgG kappa | 3 (18.75%) | |
| IgG lambda | - | |
| IgA kappa | 2 (12.5%) | |
| IgA lambda | 2 (12.5%) | |
| IgD lambda | 2 (12.5%) | |
| Kappa light chain | 1 (6.25%) | |
| Lambda light chain | 6 (37.5%) | |
| Median Plasma cells in bone marrow (%), (range) | 28 (5–94.5) | 9.5 (0–71) |
| Median TTP, months (range) | 14.5 (6–46) |
Abbreviations: M: male, F: female, R-ISS: Revised International Staging System, Glb: globulin, Hb: hemoglobin, Cre: creatinine, Ca: calcium, LDH: lactate dehydrogenase, TTP:Time to progression.
* All patients received chemotherapy based on the bortezomib regimen.
Statistical analysis
The data are presented as the mean± standard deviation (SD) of at least three independent assays. GraphPad Prism 7.0 software (GraphPad Software, CA, USA) was used for all statistical analyses. Statistical analysis of the differences between two groups was performed using a two-tailed Student’s t-test, and multiple group comparisons were performed using a one-way or two-way ANOVA test. The significance thresholds were defined as statistically not significant (#), statistically significant (*p < .05), highly significant (**p < .01), or extremely significant (***p < .001).
Further details on the materials and methods that were used in this study can be found in the supplementary notes.
Results
IL-32 and PD-L1 expression in myeloma patients are associated with the disease stage
We performed a retrospective analysis of IL-32 expression in BM biopsies collected from patients with newly diagnosed multiple myeloma (NDMM) and relapsed or refractory multiple myeloma (RRMM), aiming to find a correlation between IL-32 expression and disease stage. The obtained results revealed that RRMM samples had a higher IL-32 staining intensity compared to NDMM (Figure 1(a,b)).
Figure 1.
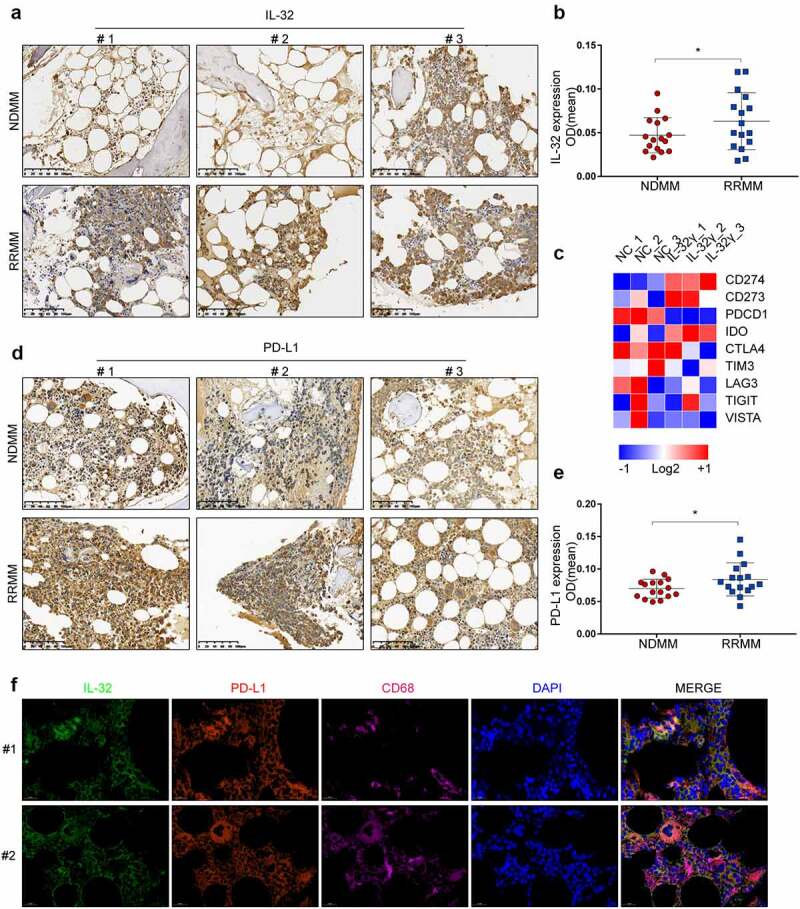
IL-32 and PD-L1 are highly expressed in myeloma patients and correlated with disease stage. (a) Immunohistochemistry (IHC) analysis of BM biopsies from patients with NDMM and RRMM showing IL-32 expression. RRMM samples showed a higher IL-32 staining intensity compared to NDMM (scale bar, 100 μm). (b) The scores were determined for quantitative staining of IL-32 in patients with NDMM and RRMM. Results are expressed as mean ± SD (n = 16). (c) Macrophages generated from PBMCs of three independent, healthy donors were treated with IL-32γ (40 ng/ml) for 24 h. RNA was extracted for RNA sequencing. The heat map on immune checkpoint-related genes was shown. (d) Representative IHC images in BM biopsies from patients with NDMM and RRMM showing PD-L1 expression (Scale bar, 100 μm). (e) The scores were determined for quantitative staining of PD-L1 in patients with NDMM and RRMM. Results are expressed as mean ± SD (n = 16). (f) Immunofluorescence analysis of IL-32 (Green), PD-L1 (Red), and CD68 (Pink) expression in tumor tissues. Scale bar, 20 μm.
Previous studies demonstrated IL-32γ as the most active isoform among the different IL-32 isoforms.6 To examine whether IL-32γ was implicated in the function of macrophages, we used our global transcriptomic analysis of macrophages treated with IL-32γ for 24 h and identified a list of immune checkpoint-related genes. Among the key immune checkpoint-related genes up-regulated in IL-32γ treated macrophages, PD-L1 exhibited the most significant increase in mRNA expression levels (Figure 1(c)).
Considering that RRMM patients have a higher IL-32 expression and knowing that IL-32γ can promote the expression of PD-L1 in macrophages, we hypothesized that PD-L1 expression is increased in RRMM patients. To test this hypothesis, we analyzed the PD-L1 expression in BM biopsies of MM patients. Consistently, the results showed that RRMM had a higher PD-L1 expression compared to NDMM (Figure 1(d,e)).
We then used IL-32, PD-L1, and CD68 to further perform colocalization analysis with MM tissues. As a result, immunofluorescence analysis revealed that CD68 was expressed in macrophages and co-localized with IL-32 and PD-L1 in human myeloma tissues (Figure 1(f)). These data indicated that RRMM had higher IL-32 and PD-L1 expression than NDMM, and IL-32 expression overlapped with CD68 and PD-L1 expression in MM tissues.
IL-32γ promotes PD-L1 expression in macrophages
Next, we investigated the role of IL-32γ in PD-L1 expression. Exposure of macrophages to IL-32γ resulted in a rapid increase in the expression of PD-L1 detected by flow cytometry; the expression peaked approximately 24 h after the treatment (Figure 2(a)). Additionally, IL-32γ increased the level of PD-L1 mRNA, and the highest peak was observed approximately 12 h after treatment (Figure 2(b)). Results from western blotting were consistent with flow cytometry results (Figure 2(c)).
Figure 2.
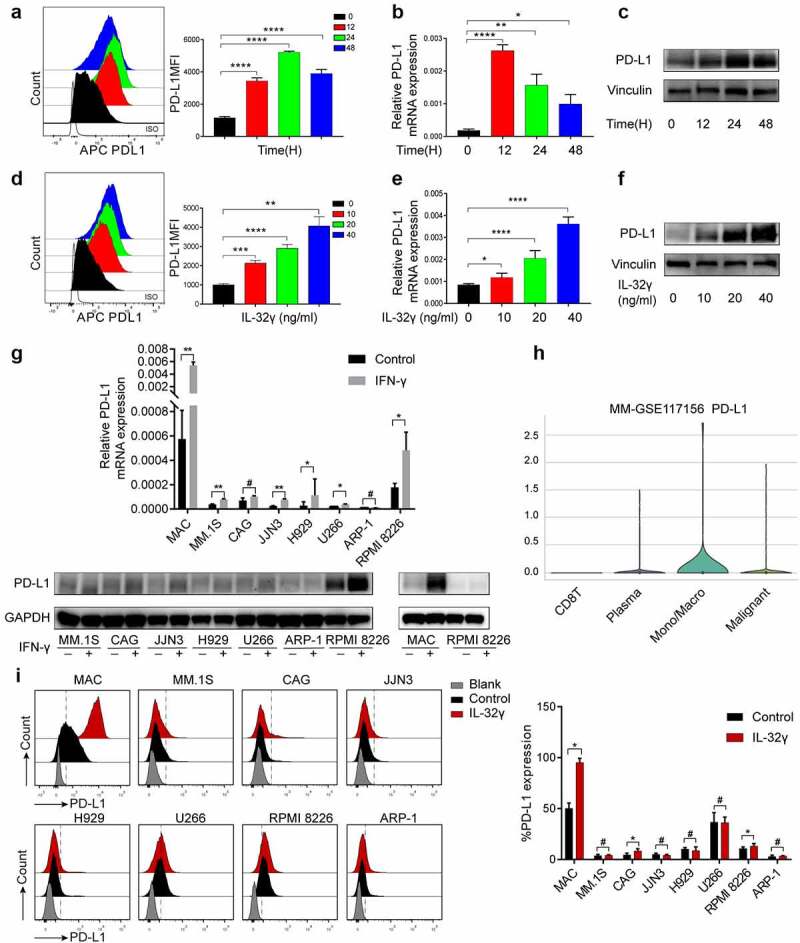
IL-32γ promotes PD-L1 expression in macrophages. (a) Flow cytometry analysis of PD-L1 level in macrophages treated with IL-32γ (40 ng/ml) at different time points. (b) qRT-PCR analysis of PD-L1 mRNA level in macrophages treated with IL-32γ (40 ng/ml) at different time points. (c) Western blot analysis of PD-L1 level in macrophages treated with IL-32γ (40 ng/ml) at different time points. (d) Flow cytometry analysis of PD-L1 level in macrophages treated with IL-32γ (24 h) for the indicated concentrations. (e) qRT-PCR analysis of PD-L1 mRNA level in macrophages treated with IL-32γ (24 h) for the indicated concentrations. (f) Western blot analysis of PD-L1 level in macrophages treated with IL-32γ (24 h) for the indicated concentrations. (g) PD-L1 expression in 7 MM cell lines and macrophages treated with IFN-γ (20 ng/ml) was measured by qRT-PCR and western blotting, respectively. (h) Distribution of the PD-L1 expression in different cell types in MM (Violin plot) by using the TISCH database. (i) Histograms representing cell surface expression of PD-L1 on macrophages (MAC), MM.1S, CAG, JJN3, H929, U266, RPMI 8226, and ARP-1 after vehicle or IL-32γ (20 ng/ml) treatment for 24 h. Data are presented as the mean ± SD of at least three independent experiments; *p < .05, **p < .01, ***p < .001.
Next, we cultured macrophages with human recombinant IL-32γ for 24 h. Flow cytometry, quantitative reverse transcription polymerase chain reaction (qRT-PCR), and western blotting showed that IL-32γ significantly induced PD-L1 expression in a dose-dependent manner (Figure 2(d-f)). Previous studies have suggested that plasma cells express PD-L1. Thus, in this study, we explored the difference in PD-L1 expression between macrophages and plasma cells. Given that PD-L1 is usually an inducible molecule, we investigated the PD-L1 expression in 7 different human multiple myeloma cell lines (HMCLs) and macrophages after interferon-γ (IFN-γ) treatment by qRT-PCR and western blotting. The results showed that macrophages expressed higher levels of PD-L1 compared to HMCLs. Moreover, IFN-γ could promote the expression of PD-L1 in most MM cell lines, especially in the RPMI 8226 cell line, and the response of macrophages to IFN-γ was stronger than that of RPMI 8226 (Figure 2(g)). Similarly, the violin plot revealed that monocytes or macrophages expressed higher levels of PD-L1 compared with malignant plasma cells by using Tumor Immune Single Cell Hub (TISCH), a comprehensive web resource enabling interactive single-cell transcriptome visualization of the tumor microenvironment (Figure 2(h)). Additionally, we investigated whether IL-32γ could induce the expression of PD-L1 in MM cells. The obtained results revealed that cell surface expression of PD-L1 was significantly increased in macrophages, with a small increase in CAG and RPMI 8226. On the other hand, there was no statistically significant change in MM.1S, JJN3, H929, U266 and ARP-1 after IL-32γ treatment (Figure 2(i)). Thus, we concluded that macrophages highly expressed PD-L1 and were more responsive to IL-32γ compared to HMCLs.
Myeloma-derived IL-32γ promotes PD-L1 expression in macrophages through PR3
To detect whether endogenous IL-32γ affects the expression of PD-L1 in macrophages, we examined IL-32γ mRNA and IL-32 protein levels in multiple myeloma cell lines compared to monocyte-derived macrophages. IL-32γ mRNA and IL-32 protein levels were abundant in H929 and JJN3 cell lines. In addition, we found almost no expression of IL-32γ in macrophages (Figure 3(a,b)).
Figure 3.
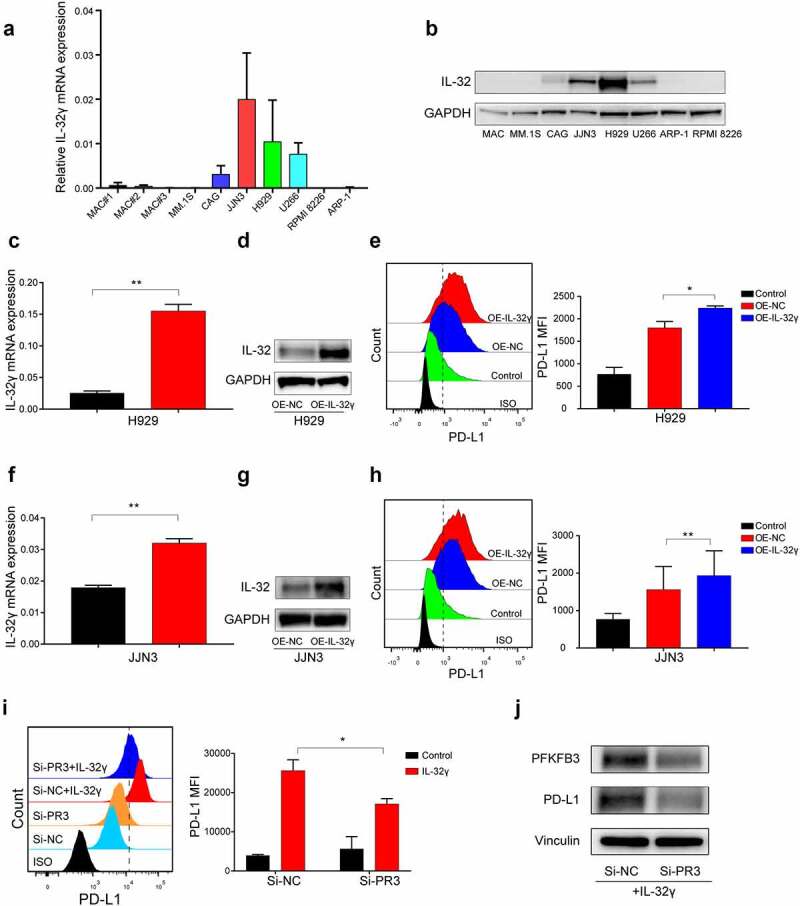
Myeloma-derived IL-32γ promotes PD-L1 expression in macrophages through PR3. (a) IL-32γ mRNA expression was quantified by qRT-PCR in monocyte-induced macrophages and MM cell lines. (b) Protein expression of IL-32 in monocyte-induced macrophages and MM cell lines was measured using western blotting. (C-D, F-G) Detection of IL-32 expression in IL-32γ-overexpressing MM cells (H929 and JJN3) by qRT-PCR (c and f) and western blotting (d and g). (e, h) Exosomes from IL-32γ-overexpressing H929 (e) and JJN3 cells (h) were incubated with macrophages, the expression of PD-L1 was examined by flow cytometry. (i) Macrophages were transfected with control siRNA or PR3 siRNA and then treated with or without IL-32γ (40 ng/ml) for 24 h; the levels of PD-L1 expression were determined by flow cytometry. (j) Western blotting was performed to measure the expression of PFKFB3 and PD-L1 when macrophages were transfected with control siRNA or PR3 siRNA and then treated with IL-32γ (40 ng/ml) for 24 h; Data are presented as the mean ± SD of at least three independent experiments; *p < .05, **p < .01, ***p < .001, #, not significant.
Next, we aimed to elucidate whether overexpression of IL-32γ in MM cell lines could promote the expression of PD-L1 by macrophages. Since previous studies have reported that IL-32 is mainly secreted by MM cells by extracellular vesicles (EVs),4 we extracted exosomes from IL-32γ-overexpressing H929 and JJN3 cells and incubated them with macrophages. We found that cell-derived exosomes from IL-32γ-overexpressing H929 and JJN3 cells could further promote the expression of PD-L1 by macrophages after 24 h of incubation (Figure 3(c-h)). These data suggested that IL-32γ in MM cells contributed to PD-L1 expression in macrophages.
PR3 is an IL-32 binding protein that may have a key role in exhibiting IL-32 bioactivity.6,22,23 To assess PR3 in IL-32γ-induced PD-L1 expression in macrophages, we downregulated PR3 expression via small interfering RNA (siRNA) (Fig. S1A), and then analyzed PD-L1 expression in macrophages in the presence or absence of IL-32γ. Notably, IL-32γ-induced PD-L1 expression was effectively abrogated in the Si-PR3 group as detected by flow cytometry (Figure 3(i)) and western blotting (Figure 3(j)). These data suggested that PR3 mediated the interaction between myeloma cells and macrophages.
IL-32γ treated macrophages mediate T cell suppression through PD-L1
To investigate whether IL-32γ treated macrophages could affect the anti-tumor immune response, we treated macrophages generated from healthy PBMCs with IL-32γ for 24 h and then co-cultured them with autologous purified, healthy CD8+ and CD4 + T cells, respectively. Flow cytometric analysis showed that CD8 + T cells co-cultured with IL-32γ treated macrophages exhibited an impaired production of Granzyme B, IFN-γ, and tumor necrosis factor-α (TNF-α) (Figure 4(a)). Moreover, the expression of the T cell activation markers CD25 and CD69 was decreased (Figure 4(b)). Similarly, IL-32γ treated macrophages suppressed IFN-γ and TNF-α production (Figure 4(c)), while there was no obvious effect on the production of interleukin-2 (IL-2) when co-cultured with CD4 + T cells (Fig. S2A). Moreover, the expression of the T cell activation markers CD25 was slightly decreased, while there was no obvious effect on CD69 expression in vitro when co-cultured with CD4 + T cells (Fig. S2B).
Figure 4.
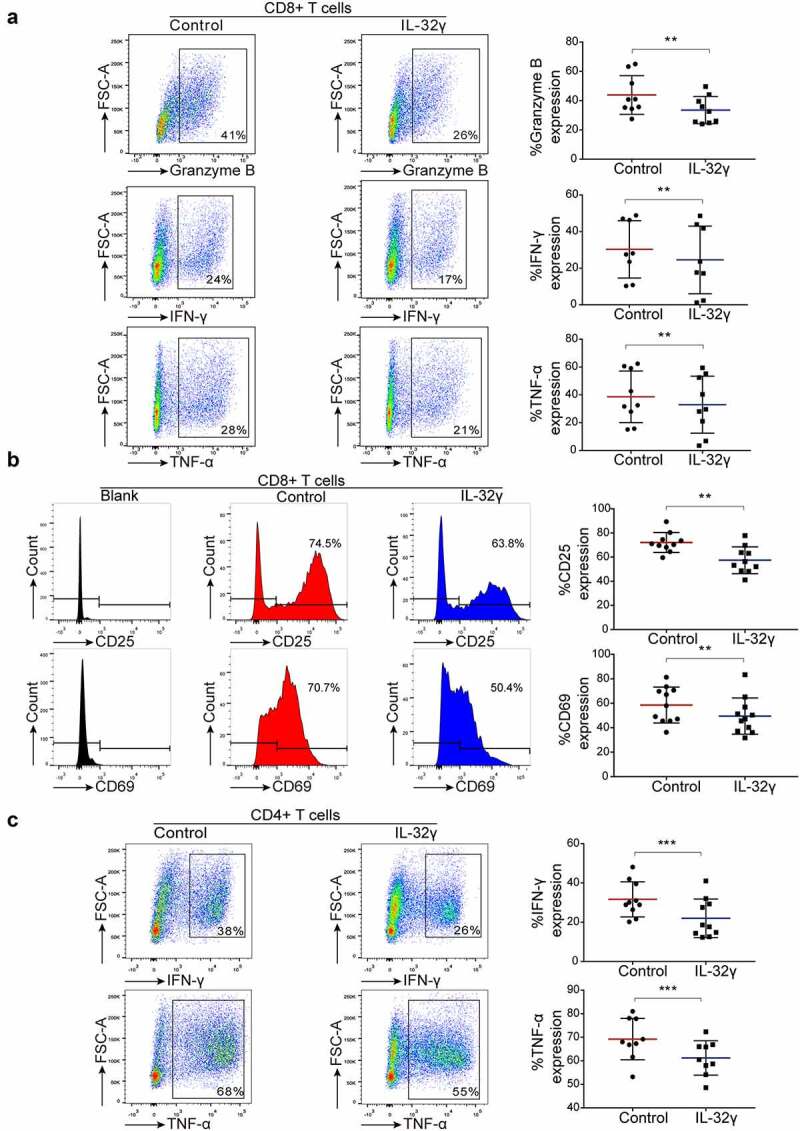
IL-32γ treated macrophages inhibit T cell function and suppress T cell activation. (a) Macrophages were exposed to medium (Control) or IL-32γ (40 ng/ml) for 24 h and then co-cultured with healthy purified CD8 + T cells (macrophages: 35,000/well; T cells:35,0000/well). The levels of Granzyme B, IFN-γ, and TNF-α production were determined by flow cytometry after 48 h. (b) The expression of the activation markers CD25 and CD69 was measured by flow cytometry after 48 h. (c) Macrophages were exposed to medium (Control) or IL-32γ (40 ng/ml) for 24 h and then co-cultured with healthy purified CD4 + T cells (macrophages: 35,000/well; T cells: 35,0000/well). The levels of IFN-γ and TNF-α were determined by flow cytometry after 48 h. Data are presented as the mean ± SD of at least three independent experiments; *p < .05, **p < .01, ***p < .001.
To further confirm the role of PD-L1 in IL-32γ treated macrophages mediated T cell suppression, we treated macrophages generated from healthy PBMCs with IL-32γ for 24 h and then co-cultured them with autologous healthy purified CD8+ and CD4 + T cells in the presence of anti-PD-L1 antibody. The inhibition of PD-L1 using anti-PD-L1 antibody effectively restored the production of Granzyme B, IFN-γ, and TNF-α (Figure 5(a)), and the expression of the activation markers CD25 and CD69 (Figure 5(b)) in autologous healthy purified CD8 + T cells. Consistently, the inhibition of PD-L1 slightly restored the production of IFN-γ and TNF-α in autologous healthy purified CD4 + T cells; yet, no statistical significance was observed (Figure 5(c)). In summary, the above results indicated that IL-32γ-induced PD-L1 expression in macrophages could induce CD8 + T cells suppression.
Figure 5.
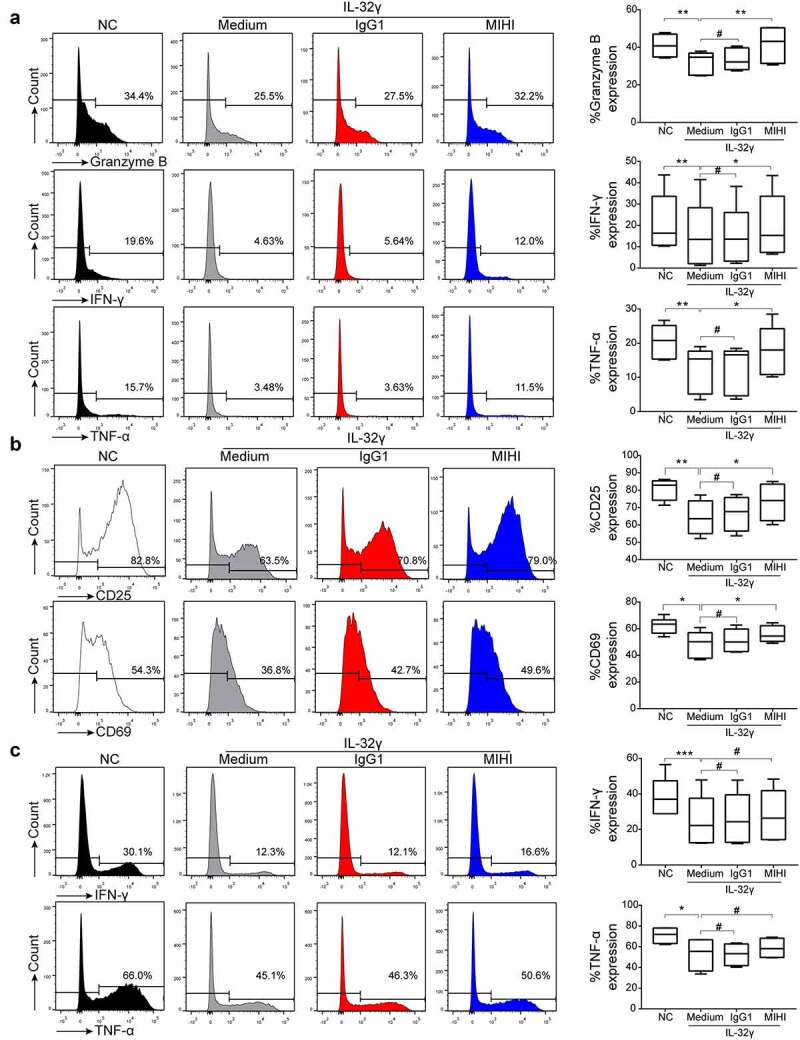
PD-L1 blockade reverses IL-32γ treated macrophages mediated CD8 + T cell suppression. (a) Macrophages or IL-32γ (40 ng/ml)-treated macrophages were incubated with autologous healthy purified CD8 + T cells (macrophages: 35,000/well; T cells:35,0000/well) in the presence or absence of 2.5 µg/ml anti-PD-L1 or control antibody as described in materials and methods. The levels of Granzyme B, IFN-γ, and TNF-α production were determined by flow cytometry after 48 h. (b) The expression of the activation markers CD25 and CD69 was measured by flow cytometry after 48 h. (c) Macrophages were exposed to medium or IL-32γ (40 ng/ml) for 24 h and then co-cultured with autologous healthy purified CD4 + T cells (macrophages: 35,000/well; T cells:35,0000/well) in the presence or absence of 2.5 µg/ml anti-PD-L1 or control antibody. The levels of IFN-γ and TNF-α were determined by flow cytometry after 48 h. Data are presented as the mean ± SD of at least three independent experiments; *p < .05, **p < .01, ***p < .001, #, not significant.
Enhanced glycolysis mediates IL-32γ-induced PD-L1 expression in macrophages
Next, we investigated the underlying mechanism through which IL-32γ induces PD-L1 expression. Given that metabolic reprogramming of macrophages has a predominant role in regulating their phenotype and function,7,24 we employed inhibitors to interfere with different metabolic pathways. Measurements of the expression of PD-L1 were captured to test the relevance of metabolic reconfiguration during IL-32γ induced PD-L1 expression in macrophages. Our results revealed that the inhibition of glycolysis by 2-deoxy-D-glucose (2DG) significantly reduced PD-L1 expression after IL-32γ stimulation in macrophages (Figure 6(a,b)). Unlike 2-DG, oxidative phosphorylation inhibitor oligomycin (Oli), fatty acid synthesis inhibitor C75, fatty acid oxidation inhibitor etomoxir (ETO), and glutaminase inhibitor CB839 did not decrease the expression of PD-L1 (Fig. S3A-S3D).
Figure 6.
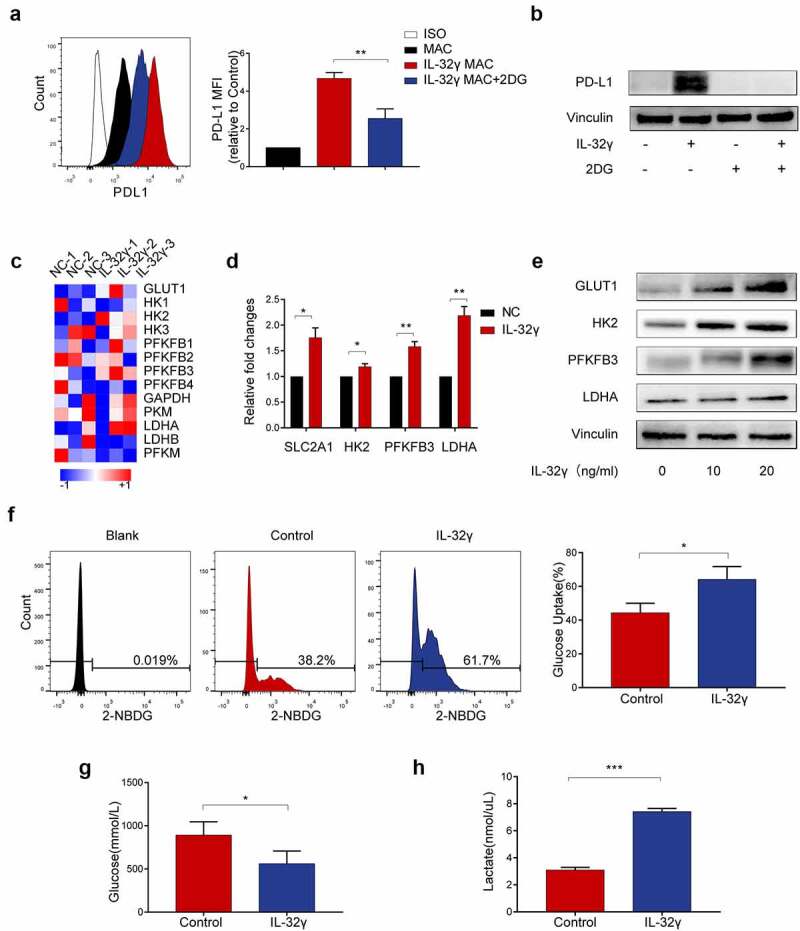
Enhanced glycolysis mediates IL-32γ-induced PD-L1 expression in macrophages. (a, b) Flow cytometry and western blot analysis of PD-L1 expression in macrophages pretreated with 2DG (5 mM) for 2 h before stimulation with IL-32γ (40 ng/ml) for 24 h. (c) Microarray analysis of RNA transcripts of IL-32γ (40 ng/ml) treated macrophages and control, with heatmap display of key glycolytic genes up-regulated in IL-32γ treated macrophages. (d) Macrophages were left untreated or treated with IL-32γ (40 ng/ml) for 24 h, the levels of glycolysis-related gene expression were determined by qRT-PCR. (e) Western blot analysis of glycolysis-related gene expression in macrophages treated with IL-32γ (0, 10, 20 ng/ml) for 24 h. (f, g) Glucose uptake rate (f) and glucose content (g) in macrophages treated with IL-32γ (20 ng/ml) for 24 h were determined with Glucose Uptake Assay Kit and Glucose Assay Kit, respectively. (h) The levels of lactate production were determined with a lactate assay kit. Data are presented as the mean ± SD of at least three independent experiments; *p < .05, **p < .01, ***p < .001, #, not significant.
To determine the levels of glycolysis in IL-32γ treated macrophages, we performed an RNA microarray to profile gene expression in IL-32γ treated macrophages. Our results showed that the IL-32γ treated subset had higher expression levels of glucose transporter 1(GLUT1), Hexokinase-2 (HK2), PFKFB3 and lactate dehydrogenase A (LDHA) (Figure 6(c)). We then confirmed the results via qRT-PCR, which showed that IL-32γ treated macrophages had significantly increased expression levels of key glycolytic enzymes (GLUT1[or SLC2A1], HK2, PFKFB3 and LDHA compared to untreated control (Figure 6(d)). Similarly, we observed increased protein levels of GLUT1, HK2, PFKFB3 and LDHA in macrophages stimulated with IL-32γ (Figure 6(e)). Consistently, increased glucose uptake (Figure 6(f)) and glucose consumption (Figure 6(g)) and upregulation of lactate production (Figure 6(h)) were observed in macrophages treated with IL-32γ compared with control. These data indicated that IL-32γ treated macrophages exhibited enhanced glycolysis, which in turn mediated IL-32γ induced PD-L1 expression.
PFKFB3 mediates glycolysis-induced PD-L1 expression in IL-32γ treated macrophages
Among the key glycolytic enzymes up-regulated in IL-32γ-exposed macrophages, GLUT1, PFKFB3 and LDHA exhibited a significant increase in mRNA expression levels. To explore which glycolytic enzymes participate in IL-32γ-induced PD-L1 expression, macrophages were treated with the GLUT1 inhibitor BAY876, LDHA inhibitor AZ33, and PFKFB3 inhibitor PFK15 before IL-32γ stimulation (Fig. S4A-S4B and Figure 7(a,b)). Among these inhibitors, we found that PFK15 inhibited IL-32γ-induced elevation in PD-L1 expression, as measured by western blotting (Figure 7(a)) and flow cytometry (Figure 7(b)). Then, we investigated the expression of PFKFB3 in macrophages stimulated by IL-32γ. PFKFB3 mRNA level was markedly increased with the rising doses of IL-32γ for 24 h, as was PFKFB3 protein (Figure 7(c,d)). Differently, exposure of macrophages to IL-32γ resulted in a rapid increase in the expression of PFKFB3 mRNA and protein, both of which peaked at approximately 12 h after treatment (Figure 7(e,f)). Additionally, immunofluorescence results confirmed that upon IL-32γ treatment, expression of PFKFB3 was significantly increased compared to untreated control (Figure 7(g)).
Figure 7.
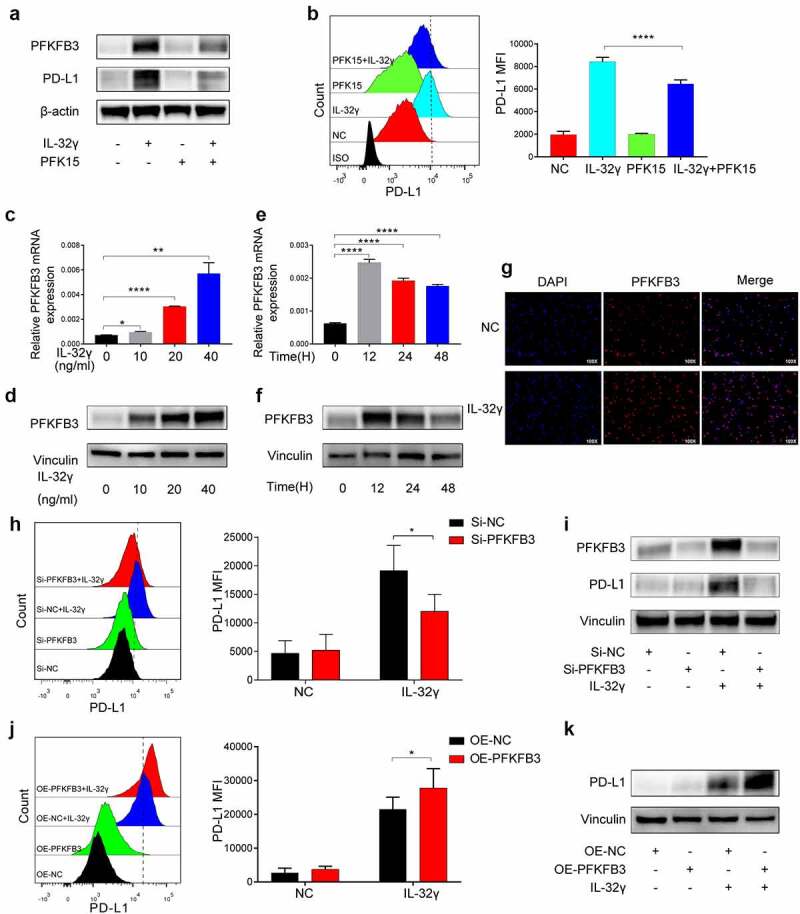
PFKFB3 mediates glycolysis-induced PD-L1 expression in IL-32γ treated macrophages. (a) Western blot analysis of PD-L1 expression by macrophages pretreated with PFK15 (10 uM) for 2 h before stimulation with IL-32γ (40 ng/ml) for 24 h. (b) Flow cytometry analysis of PD-L1 expression by macrophages pretreated with PFK15(10 uM) for 24 h before stimulation with IL-32γ (40 ng/ml) for another 24 h. (c, d) Macrophages were treated with IL-32γ at indicated concentrations for 24 h. The expression of PFKFB3 was analyzed by qRT-PCR (c) and western blotting (d), respectively. (e, f) Macrophages were treated with IL-32γ (40 ng/ml) at the indicated time. The expression of PFKFB3 was analyzed by qRT-PCR (e) and western blotting (f), respectively. (g) Immunofluorescence staining was performed to investigate PFKFB3 expression in macrophages treated with IL-32γ (40 ng/ml) for 24 h. (h, i) Macrophages were transfected with control siRNA (Si-NC) or PFKFB3 siRNA (Si-PFKFB3) and then treated with or without IL-32γ (40 ng/ml) for 24 h, the levels of PD-L1 expression were determined by flow cytometry (h), and the levels of PFKFB3 and PD-L1 expression were determined by western blotting (i). (j, k) Macrophages were transfected with empty vector (OE-NC) or PFKFB3 overexpressing vector (OE-PFKFB3) and then treated with or without IL-32γ (40 ng/ml) for 24 h; the levels of PD-L1 expression were determined by flow cytometry (j) and western blotting (k), respectively. Data are presented as the mean ± SD of at least three independent experiments; *p < .05, **p < .01, ***p < .001, #, not significant.
To further verify the role of PFKFB3, we depleted PFKFB3 using siRNA in macrophages (Fig. S4C). The results showed that knockdown of PFKFB3 markedly abrogated the induction of PD-L1 expression in IL-32γ-exposed macrophages as detected by flow cytometry and western blotting (Figure 7(h,i)). Moreover, we transduced macrophages with adeno-associated virus expressing GFP alone or wild-type PFKFB3 sequence (Fig. S4D). Consistently, overexpression of PFKFB3 promoted IL-32γ induced PD-L1 expression (Figure 7(j,k)). In order to assess PR3 in IL-32γ-induced PFKFB3 dependent PD-L1 expression in macrophages, we analyzed PFKFB3 expression in macrophages transfected with control siRNA (Si-NC) or PR3 siRNA (Si-PR3) in the presence of IL-32γ. Notably, IL-32γ-induced PFKFB3 was effectively abrogated in the Si-PR3 group as detected by western blotting (Figure 3(j)). Together, these data indicated that PFKFB3 could mediate the effects of IL-32γ-induced glycolysis-dependent PD-L1 expression in macrophages.
PFKFB3-JAK1 axis induces PD-L1 expression in IL-32γ treated macrophages
We have previously shown that IL-32γ could activate the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) and nuclear factor kB (NF-kB) signaling pathways in macrophages.6 To understand the role of the JAK family in modulating IL-32γ induced PD-L1 expression, we first detected the expressions of JAK family members after IL-32γ stimulation by using western blotting. The results showed that IL-32γ treatment promoted the levels of Phospho-JAK1 (p-JAK1) and Phospho-JAK3 (p-JAK3) (Figure 8(a)). The data of Phospho-JAK2 (p-JAK2) and Phospho-Tyrosine kinase 2 (p-TYK2) are not shown because the expression level is too low. Next, we studied IL-32γ stimulated PD-L1 expression after JAK1 siRNA or JAK3 siRNA transfection. We found that after JAK1 knockdown, the effect of IL-32γ on the expression of PD-L1 in macrophages was attenuated (Figure 8(b-c), S5A), while JAK3 knockdown had a limited role in this process (Figure 5(d) and S5B). These results indicated that JAK1 had a dominant role in the IL-32γ-induced expression of PD-L1 by macrophages. To investigate the effects of an NF-kB signaling pathway in IL-32γ induced PD-L1 expression, we inhibited this pathway by pretreating macrophages with NF-kB inhibitor (QNZ). Our data showed that the NF-kB inhibitor does not affect the IL-32γ-triggered PD-L1 upregulation in macrophages (Figure 8(e)).
Figure 8.
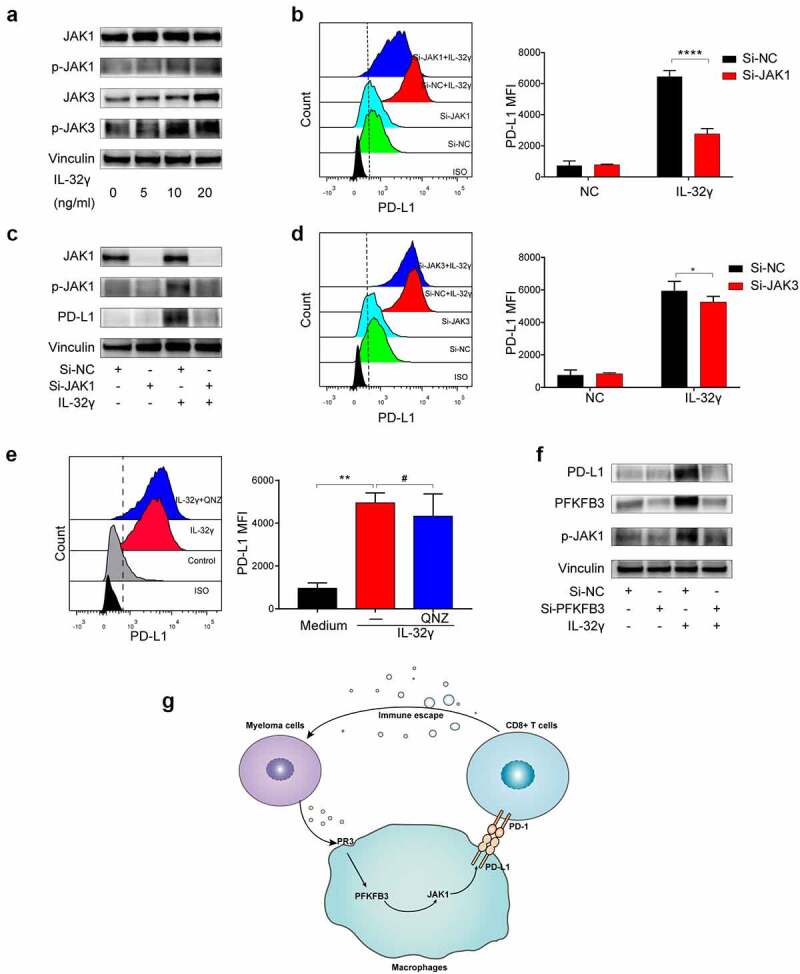
PFKFB3-JAK1 axis induces PD-L1 expression in IL-32γ treated macrophages. (a) Macrophages were treated with IL-32γ (24 h) for the indicated concentrations. Cell extracts were analyzed by western blotting using indicated antibodies. (b) Macrophages were transfected with control siRNA (Si-NC) or JAK1 siRNA (Si-JAK1) and then treated with or without IL-32γ (20 ng/ml) for 24 h. The PD-L1 expression was subsequently examined by flow cytometry. (c) The JAK1, p-JAK1, and PD-L1 expressions were analyzed by western blotting. (d) Macrophages were transfected with control siRNA (Si-NC) or JAK3 siRNA (Si-JAK3) and then treated with IL-32γ (20 ng/ml) for 24 h. The PD-L1 expression was examined by flow cytometry. (e) Macrophages were pretreated with QNZ (20uM) or not for 2 h, after which IL-32γ (20 ng/ml) was added into cell culture for 24 h, and PD-L1 expression was subsequently examined by flow cytometry. (f) Macrophages were transfected with control siRNA (Si-NC) or PFKFB3 siRNA (Si-PFKFB3) and then treated with or without IL-32γ (20 ng/ml) for 24 h. Cell extracts were analyzed by western blotting using indicated antibodies. (g) Schematic illustration of the molecular mechanism of IL-32γ induced PFKFB3 dependent PD-L1 expression in macrophages. Data are presented as the mean ± SD of at least three independent experiments; *p < .05, **p < .01, ***p < .001.
To further explore the mechanisms regulating PD-L1 induction by PFKFB3 activation, we set out to determine whether JAK1 was involved in PFKFB3-induced PD-L1 expression in macrophages. We analyzed the protein expression of the p-JAK1 by downregulating PFKFB3 expression via siRNA before IL-32γ stimulation. We observed diminished phosphorylation of JAK1 upon PFKFB3 knockdown in macrophages treated with IL-32γ (Figure 8(f)). These results suggested that the JAK1 signaling pathway might be involved in regulating the downstream functions of PFKFB3.
Discussion
IL-32 is a novel cytokine involved in inflammation and cancer development,3 which has nine alternative spliced isoforms such as IL-32α, IL-32β, IL-32γ, IL-32δ, IL-32ε, IL-32ζ, IL-32η, IL-32θ, and IL-32 small (sm).3 Previous studies demonstrated that higher IL-32 expression was positively correlated with ISS stage and levels of serum β2-microglobulin in NDMM.6 Herein, we showed that RRMM samples had a higher IL-32 staining intensity compared to NDMM. These data indicated that IL-32 was a poor prognostic factor for patients with MM.
TAMs, which are abundantly infiltrated in most tumors, have an important role in tumor progression by providing a barrier against anti-tumor immunity via the expression of inhibitory molecules or immune checkpoint ligands.25,26 Here, we found that IL-32γ could promote the expression of PD-L1 in macrophages. Consistently, RRMM samples had a higher PD-L1 expression compared to NDMM. It has been reported that macrophages expressing PD-L1 could inhibit T cell function.9,27,28 Similarly, our current results suggested that IL-32γ-induced PD-L1 expression in macrophages could induce CD8 + T cells suppression, which provides evidence that IL-32γ induces a suppressive immunological environment in MM.
Metabolic studies have revealed that specific metabolic pathways in macrophages are tightly associated with their phenotype and function. The metabolic profile of TAMs is indeed very dynamic in response to the nutritional needs of malignant cells and TME perturbations.29 Herein, we showed that IL-32γ treated macrophages exhibited enhanced glycolysis, which mediated IL-32γ-induced PD-L1 expression. Our data revealed important mechanisms through which glycolysis metabolism favored an immunosuppressive phenotype or function of macrophages upon stimulation by cytokine IL-32γ. PFKFB3, a gene that encodes 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 enzyme in humans, is known to have the highest ratio (>700) of kinase/phosphatase activity and thus favors the rise in fructose-2,6-bisphosphate, thereby sustaining a high rate of glycolysis.30,31 Importantly, our results showed that PFKFB3 has an important role in glycolysis-induced PD-L1 expression by macrophages stimulated with IL-32γ. The current study unveiled a novel mechanism, suggesting that downregulation of PFKFB3 could inhibit the expression of PD-L1 by macrophages after IL-32γ stimulation, thus identifying potential targets for future immune-based anti-myeloma therapies.
Exosomes have a key role in establishing communication networks among cells, which specifically expand the cells away from the plasma membrane.32 Previous studies have shown that IL-32 is secreted from MM cells in the form of EVs.4 In our study, exosomes derived from IL-32γ overexpression MM cell lines increased the expression levels of PD-L1 in macrophages. These results suggested that MM-derived exosomes containing IL-32γ could promote the expression of the inhibitory molecule PD-L1 by the macrophages. It is now clear that PR3 is an IL-32 binding protein that can extend the biological activity of IL-32.6,22 Herein, we observed that PR3 was essential for IL-32γ induced PFKFB3 dependent PD-L1 expression in macrophages. Our study revealed a new reciprocal interaction between cancer cells and their microenvironment, which could regulate numerous processes related to its malignant transformation during MM development.
Our previous study demonstrated that IL-32γ significantly activates the JAK-STAT and NF-kB signaling pathways in macrophages.6 In the present study, we showed that the expression of PD-L1 in macrophages stimulation with IL-32γ was significantly downregulated in the JAK1 blockade groups. All these observations, together with previous finding33 suggest that JAK1 signaling promotes the expression of the PD-L1. It has been reported that NF-kB activation is crucial for regulating PD-L1 expression.9 Yet, our current results suggested that such mechanisms might not be involved in the regulation of PD-L1 expression in macrophages by IL-32γ.
By investigating the mechanisms of PFKFB3 function in IL-32γ induced PD-L1 expression in macrophages, we found that JAK1 signaling might be involved in regulating the downstream functions of PFKFB3. This finding, together with previous data,34,35 demonstrates that key glycolytic enzymes could regulate the functions of immune cells by regulating downstream signaling pathways. Thus, glycolysis metabolism and JAK1 signaling appear to be highly integrated processes, and the PFKFB3-JAK1 axis may provide a mechanistic explanation for the IL-32γ induced PFKFB3 dependent PD-L1 expression. However, further studies are necessary to elucidate the complete mechanisms through which PFKFB3 regulates JAK1 and which specific STAT molecules downstream of JAK1 mediate IL-32γ-induced expression of PD-L1. Unfortunately, as mice do not have the homologous gene of human IL-32, the above experiment cannot be verified in vivo.
In conclusion, our results proved that IL-32γ secreted from MM cells promoted the PD-L1 expression of macrophages by up-regulating PFKFB3 dependent glycolysis, which suppressed the activation and function of CD8 + T cells, thereby promoting MM progression. Moreover, PR3 was essential for the effects stated above induced by IL-32γ. In addition, the PFKFB3-JAK1 axis might mediate the PD-L1 expression in IL-32γ stimulated macrophages. Thus, IL-32 may act as a potential prognostic biomarker for MM patients, and targeting IL-32 signaling might be used as a potential strategy for treating myeloma. (Figure 8(g)).
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China [81800201, 81800202, and 81872322]; National Major Scientific and Technological Special Project for ”Significant New Drug Development” [2018ZX09733-003]; Zhejiang Key Research and Development Project [2020C03014].
Funding Statement
This work was supported by the National Natural Science Foundation of China [81800201, 81800202, and 81872322]; National Major Scientific and Technological Special Project for ”Significant New Drug Development” [2018ZX09733-003]; Zhejiang Key Research and Development Project [2020C03014].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available in Tumor Immune Single-cell Hub (TISCH) at http://tisch.comp-genomics.org/. These data were derived from the following resources available in the public domain: http://tisch.comp-genomics.org/search-gene/?genesearch=CD274. The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials or from the corresponding authors upon reasonable request.
Author contributions
Yang Liu designed the study, analyzed the data, and wrote the manuscript. Haimeng Yan, Huiyao Gu, and Enfan Zhang designed and supervised research. Jingsong He and Donghua He provided patient samples. Wen Cao, Jianwei Qu, Ruyi Xu, Liqin Cao, Jinna Zhang, and Yifan Hou discussed the results and commented on the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Kumar SK, Rajkumar V, Kyle RA, van Duin M, Sonneveld P, Mateos MV, Gay F, Anderson KC.. Multiple myeloma. Nat Rev Dis Primers. 2017;3(1):17046. doi: 10.1038/nrdp.2017.46. [DOI] [PubMed] [Google Scholar]
- 2.Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019;40(4):310–15. doi: 10.1016/j.it.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Hong JT, Son DJ, Lee CK, Yoon DY, Lee DH, Park MH. Interleukin 32, inflammation and cancer. Pharmacol Ther. 2017;174:127–137. doi: 10.1016/j.pharmthera.2017.02.025. [DOI] [PubMed] [Google Scholar]
- 4.Zahoor M, Westhrin M, Aass KR, Moen SH, Misund K, Psonka-Antonczyk KM, Giliberto M, Buene G, Sundan A, Waage A, et al. Hypoxia promotes IL-32 expression in myeloma cells, and high expression is associated with poor survival and bone loss. Blood Adv. 2017;1(27):2656–2666. doi: 10.1182/bloodadvances.2017010801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin X, Yang L, Wang G, Zi F, Yan H, Guo X, Chen J, Chen Q, Huang X, Li Y, et al. Interleukin-32α promotes the proliferation of multiple myeloma cells by inducing production of IL-6 in bone marrow stromal cells. Oncotarget. 2017;8(54):92841–92854. doi: 10.18632/oncotarget.21611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan H, Dong M, Liu X, Shen Q, He D, Huang X, Zhang E, Lin X, Chen Q, Guo X, et al. Multiple myeloma cell-derived IL-32γ increases the immunosuppressive function of macrophages by promoting indoleamine 2,3-dioxygenase (IDO) expression. Cancer Lett. 2019;446:38–48. doi: 10.1016/j.canlet.2019.01.012. [DOI] [PubMed] [Google Scholar]
- 7.Van den Bossche J, O’Neill LA, Menon D. Macrophage Immunometabolism: where Are We (Going)?. Trends Immunol. 2017;38(6):395–406. doi: 10.1016/j.it.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Muri J, Fitzgerald G, Gorski T, Gianni-Barrera R, Masschelein E, D’Hulst G, Gilardoni P, Turiel G, Fan Z, et al. Endothelial lactate controls muscle regeneration from ischemia by inducing M2-like macrophage polarization. Cell Metab. 2020;31(6):1136–1153.e1137. doi: 10.1016/j.cmet.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen DP, Ning WR, Jiang ZZ, Peng ZP, Zhu LY, Zhuang SM, Kuang DM, Zheng L, Wu Y. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3-PD-L1 axis in human hepatocellular carcinoma. J Hepatol. 2019;71(2):333–343. doi: 10.1016/j.jhep.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 10.Lauterbach MA, Hanke JE, Serefidou M, Mangan MSJ, Kolbe CC, Hess T, Rothe M, Kaiser R, Hoss F, Gehlen J, et al. Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via ATP-Citrate Lyase. Immunity. 2019;51(6):997–1011.e1017. doi: 10.1016/j.immuni.2019.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Su P, Wang Q, Bi E, Ma X, Liu L, Yang M, Qian J, Yi Q. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res. 2020;80(7):1438–1450. doi: 10.1158/0008-5472.Can-19-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferreira C, Mesquita I, Barbosa AM, Osório NS, Torrado E, Beauparlant C-J, Droit A, Cunha C, Carvalho A, Saha B, et al. Glutamine supplementation improves the efficacy of miltefosine treatment for visceral leishmaniasis. PLoS Negl Trop Dis. 2020;14(3):e0008125. doi: 10.1371/journal.pntd.0008125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms Controlling PD-L1 expression in cancer. Mol Cell. 2019;76(3):359–370. doi: 10.1016/j.molcel.2019.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesokhin AM, Ansell SM, Armand P, C EC, Halwani A, Gutierrez M, M MM, Cohen AD, Schuster SJ, Lebovic D, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase ib study. J Clin Oncol: Off J Am Soci Clin Oncol. 2016;34(23):2698–2704. doi: 10.1200/jco.2015.65.9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribrag V, Avigan DE, Green DJ, Wise-Draper T, Posada JG, Vij R, Zhu Y, Farooqui MZH, Marinello P, Siegel DS. Phase 1b trial of pembrolizumab monotherapy for relapsed/refractory multiple myeloma: KEYNOTE-013. Br J Haematol. 2019;186(3):e41–e44. doi: 10.1111/bjh.15888. [DOI] [PubMed] [Google Scholar]
- 16.Costello C. The future of checkpoint inhibition in multiple myeloma? Lancet Haematol. 2019;6(9):e439–e440. doi: 10.1016/s2352-3026(19)30149-8. [DOI] [PubMed] [Google Scholar]
- 17.Rosenblatt J, Avigan D. Targeting the PD-1/PD-L1 axis in multiple myeloma: a dream or a reality? Blood. 2017;129(3):275–279. doi: 10.1182/blood-2016-08-731885. [DOI] [PubMed] [Google Scholar]
- 18.Lin C, He H, Liu H, Li R, Chen Y, Qi Y, Jiang Q, Chen L, Zhang P, Zhang H, et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut. 2019;68(10):1764–1773. doi: 10.1136/gutjnl-2018-316324. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, Wang F, Li X, Liu Q, Li Y, et al. Endoplasmic Reticulum Stress Causes Liver Cancer Cells to Release Exosomal miR-23a-3p and Up-regulate Programmed Death Ligand 1 Expression in Macrophages. Hepatol (Baltimore, Md). 2019;70(1):241–258. doi: 10.1111/bjh.15888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed cell death ligand 1 (PD-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol Res. 2018;6(10):1260–1273. doi: 10.1158/2326-6066.Cir-17-0537. [DOI] [PubMed] [Google Scholar]
- 21.Zheng Y, Cai Z, Wang S, Zhang X, Qian J, Hong S, Li H, Wang M, Yang J, Yi Q. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood. 2009;114(17):3625–3628. doi: 10.1182/blood-2009-05-220285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novick D, Rubinstein M, Azam T, Rabinkov A, Dinarello CA, Kim SH. Proteinase 3 is an IL-32 binding protein. Proc Natl Acad Sci U S A. 2006;103(9):3316–3321. doi: 10.1073/pnas.0511206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakayama M, Niki Y, Kawasaki T, Takeda Y, Ikegami H, Toyama Y, Miyamoto T. IL-32-PAR2 axis is an innate immunity sensor providing alternative signaling for LPS-TRIF axis. Sci Rep. 2013;3(1):2960. doi: 10.1038/srep02960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–565. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduction Targeted Ther. 2021;6(1):127. doi: 10.1038/s41392-021-00506-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19(6):369–382. doi: 10.1038/s41577-019-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe R, Shirai T, Namkoong H, Zhang H, Berry GJ, Wallis BB, Schaefgen B, Harrison DG, Tremmel JA, Giacomini JC, et al. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J Clin Invest. 2017;127(7):2725–2738. doi: 10.1172/jci92167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206(6):1327–1337. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30(1):36–50. doi: 10.1016/j.cmet.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 30.Yi M, Ban Y, Tan Y, Xiong W, Li G, Xiang B. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 and 4: a pair of valves for fine-tuning of glucose metabolism in human cancer. Mol. Metab. 2019;20:1–13. doi: 10.1016/j.molmet.2018.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol. 2009;86(3):174–179. doi: 10.1016/j.yexmp.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Meldolesi J. Exosomes and ectosomes in intercellular communication. Curr Biol: CB. 2018;28(8):R435–r444. doi: 10.1016/j.cub.2018.01.059. [DOI] [PubMed] [Google Scholar]
- 33.Luo N, Formisano L, Gonzalez-Ericsson PI, Sanchez V, Dean PT, Opalenik SR, Sanders ME, Cook RS, Arteaga CL, Johnson DB, et al. Melanoma response to anti-PD-L1 immunotherapy requires JAK1 signaling, but not JAK2. Oncoimmunology. 2018;7(6):e1438106. doi: 10.1080/2162402x.2018.1438106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu K, Yin N, Peng M, G SE, Shyu A, Li P, Zhang X, H DM, Wang Z, J CK, et al. Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Sci (New York, NY). 2021;371(6527):405–410. doi: 10.1126/science.abb2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, Panteleyev AA, Okkenhaug K, Cantrell DA. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209(13):2441–2453. doi: 10.1084/jem.20112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in Tumor Immune Single-cell Hub (TISCH) at http://tisch.comp-genomics.org/. These data were derived from the following resources available in the public domain: http://tisch.comp-genomics.org/search-gene/?genesearch=CD274. The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials or from the corresponding authors upon reasonable request.