

Abstract
Mutations in the Aminoadipate-Semialdehyde Synthase (AASS) gene encoding α-aminoadipic semialdehyde synthase lead to hyperlysinemia-I, a benign metabolic variant without clinical significance, and hyperlysinemia-II with developmental delay and intellectual disability. Although both forms of hyperlysinemia display biochemical phenotypes of questionable clinical significance, an association between neurologic disorder and a pronounced biochemical abnormality remains a challenging clinical question. Here, we report that Aass mutant male and female mice carrying the R65Q mutation in α-ketoglutarate reductase (LKR) domain have an elevated cerebral lysine level and a normal brain development, whereas the Aass mutant mice carrying the G489E mutation in saccharopine dehydrogenase (SDH) domain exhibit elevations of both cerebral lysine and saccharopine levels and a smaller brain with defective neuronal development. Mechanistically, the accumulated saccharopine, but not lysine, leads to impaired neuronal development by inhibiting the neurotrophic effect of glucose-6-phosphate isomerase (GPI). While extracellular supplementation of GPI restores defective neuronal development caused by G498E mutation in SDH of Aass. Altogether, our findings not only unravel the requirement for saccharopine degradation in neuronal development, but also provide the mechanistic insights for understanding the neurometabolic disorder of hyperlysinemia-II.
SIGNIFICANCE STATEMENT The association between neurologic disorder and a pronounced biochemical abnormality in hyperlysinemia remains a challenging clinical question. Here, we report that mice carrying the R65Q mutation in lysine α-ketoglutarate reductase (LKR) domain of aminoadipate-semialdehyde synthase (AASS) have an elevated cerebral lysine levels and a normal brain development, whereas those carrying the G489E mutation in saccharopine dehydrogenase (SDH) domain of AASS exhibit an elevation of both cerebral lysine and saccharopine and a small brain with defective neuronal development. Furthermore, saccharopine impairs neuronal development by inhibiting the neurotrophic effect of glucose-6-phosphate isomerase (GPI). These findings demonstrate saccharopine degradation is essential for neuronal development.
Keywords: AASS, GPI, hyperlysinemia, neuronal development, saccharopine
Introduction
Lysine is an essential amino for growth, development and reproduction in animals. Lysine is catabolized through two distinct routes, namely saccharopine pathway and pipecolate pathway. The saccharopine pathway is initiated by ε-deamination of lysine, while the pipecolate pathway is conversion of α-amino group of lysine to α-keto acid. Traditionally, the saccharopine pathway (ε-deamination of lysine) is a major degradative pathway for lysine in extracerebral tissue and embryonic brain (Rao et al., 1992; Blemings et al., 1994), whereas the pipecolate pathway (α-deamination of lysine) is increasing during brain development and predominates in the adult brain (Chang, 1976; Rao et al., 1992; Sauer et al., 2011; Posset et al., 2015). However, lysine is able to synthesize glutamate, the most significant excitatory neurotransmitter, by the saccharopine pathway in adult mouse brain (Papes et al., 2001), suggesting the occurrence and importance of the saccharopine pathway in postnatal mammalian brain. In the saccharopine pathway, the first two reactions are catalyzed by enzymatic activities known as lysine α-ketoglutarate reductase (LKR), which condenses lysine and α-ketoglutarate to form saccharopine, and saccharopine dehydrogenase (SDH), by which the saccharopine is subsequently oxidized to generate glutamate and α-aminoadipate semialdehyde (Fig. 1A). LKR and SDH, respectively, locate in the N-terminal and the C-terminal portions of α-aminoadipate semialdehyde synthase (AASS), which is a bifunctional mitochondrial enzyme encoded by Aass gene (Sacksteder et al., 2000). Although the expression and activity of LKR/SDH in the developing and adult brain are still controversial (Rao et al., 1992; Posset et al., 2015; Pena et al., 2017; Leandro and Houten, 2020), mounting evidences suggest that the saccharopine pathway is the predominant degradative pathway for lysine in brain (Pena et al., 2017; Crowther et al., 2019; Leandro and Houten, 2020). Moreover, the lysine degradation pathways display species-specific activities (Grove and Henderson, 1968; Ghadimi et al., 1971; Dancis and Hutzler, 1982; Zaar et al., 1986; Mihalik and Rhead, 1989). So far, it is unclear whether LKR/SDH-initiated saccharopine pathway for lysine degradation plays an important role in brain development.
Figure 1.
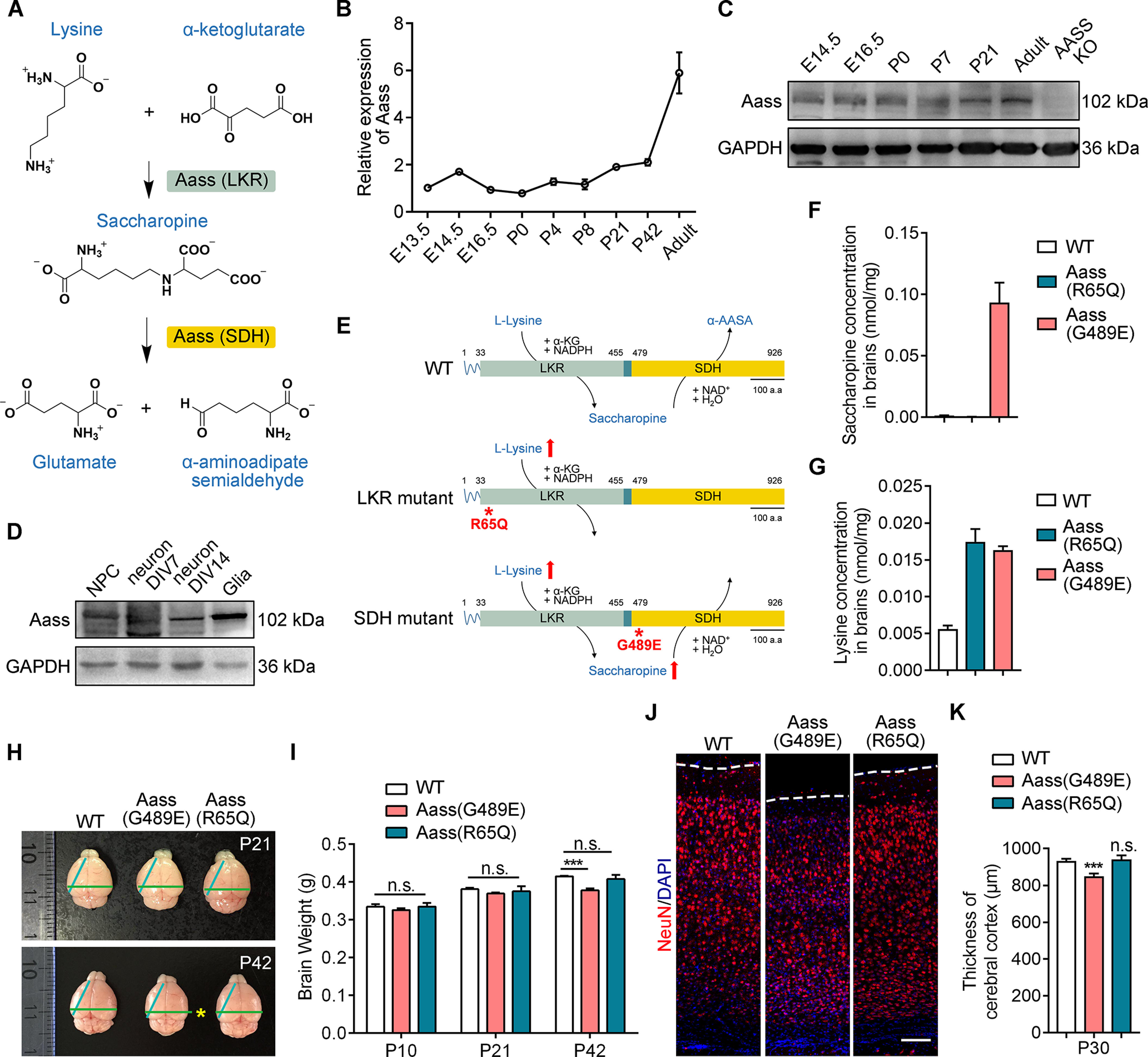
SDH mutation, but not LKR mutation, leads to reduced brain size. A, Graphic description of mitochondrial lysine degradation. B, Real-time PCR analysis showing the relative expression of Aass in cerebral cortex at different developmental stages from E13.5 to adult. Aass mRNA levels were normalized to Gapdh. The values of E13.5 were set to 1. C, Western blotting showing the expression of AASS in cerebral cortex at different developmental stages from E14.5 to adult. D, Western blotting showing the expression of Aass in the cultured NPCs, neurons (DIV7 and DIV14), and glial cells. E, Schematic illustration of full-length Aass, R65Q, and G489E mutations. F, G, Saccharopine (F) and lysine (G) levels in the brains of WT, Aass (R65Q), and Aass (G489E) mice. n = 3 brains. H, Representative images of brains dissected from WT, Aass (G489E), and Aass (R65Q) mice at P21 and P42. I, Quantification of brain weight of WT, Aass (G489E), and Aass (R65Q) mice at P10, P21, and P42. One-way ANOVA, F(2,15) = 2.675, WT versus Aass (G489E), ***p < 0.0001; WT versus Aass (R65Q), p = 0.8061. WT, n = 6 brains; Aass (G489E), n = 7 brains; Aass (R65Q), n = 5 brains. J, Coronal sections of WT, Aass (G489E), and Aass (R65Q) brains stained with Neuronal nuclear antigen (NeuN) (red) at P30. Nuclei were stained with DAPI (blue). Scale bars: 100 μm. K, Quantitative analysis of the thickness of cerebral cortex from WT, Aass (G489E), and Aass (R65Q) brains. One-way ANOVA, WT versus Aass (G489E), F(2,8) = 2.102, ***p = 0.0003; WT versus Aass (R65Q), p = 0.9219. WT, n = 4 brains; Aass (G489E), n = 4 brains; Aass (R65Q), n = 3 brains. Data are presented as mean ± SEM; n.s., no significance, p > 0.05, ***p < 0.001.
Mutations in AASS have been known to result into hyperlysinemia, an autosomal recessive inborn error of lysine degradation, which can be grouped into two subtypes. Hyperlysinemia-I is caused by the mutations in LKR and affected individuals have elevated lysine levels but probably without clinical symptoms (Dancis et al., 1983). Hyperlysinemia-II (also known as saccharopinuria) is caused by mutations that primarily affect SDH activity and patients with hyperlysinemia-II have a marked elevation of both lysine and saccharopine, mostly accompanying by neurologic damage and intellectual disability (Carson et al., 1968; Simell et al., 1972; Cederbaum et al., 1979; Vianey-Liaud et al., 1986; Houten et al., 2013). However, the association between the biochemical and clinical phenotypes in both forms of hyperlysinemia is still questionable. In our previous study, we generated two Aass knock-in mouse models (Zhou et al., 2019). The first model harbors a mutation in the LKR domain, while the second one has a mutation in the SDH domain that leads to impaired SDH activity with preserved LKR function. The LKR mutant mice have no detectable clinical phenotype only with elevation of lysine levels, whereas the SDH mutant mice display mitochondrial damage and functional impairment in the liver, leading to liver hypertrophy, postnatal developmental retardation and death probably because of accumulation of saccharopine (Zhou et al., 2019). Although the link between abnormal saccharopine accumulation and mitochondrial dynamics and function in liver has been established, an association between neurologic disorder and a pronounced biochemical abnormality in hyperlysinemia remains a challenging clinical question.
Materials and Methods
Mice
Mice were housed in the animal facility at Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, on a 12/12 h light/dark reverse cycle with lights on at 8 A.M. and provided with food and water ad libitum. All procedures and husbandry were performed according to protocols approved by the Institutional Animal Care and Use Committee at Institute of Genetics and Developmental Biology, Chinese Academy of Sciences. Aass (R65Q) and Aass (G489E) knock-in mice were generated by Viewsolid Biotech. Mice were bred onto the C57/B6 background for at least six generations. Mice used for experiments in this study were littermates or age matched but not disaggregated by sex.
Antibodies
Primary antibodies used in this study: mouse anti-NeuN (Abcam, ab104224), mouse anti-Map2 (Sigma, M1406), rabbit anti-GPI (ABclonal, A6916), rabbit anti-Aass (Abcam, ab154800), mouse anti-Satb2 (Abcam, ab51502), rat anti-Ctip2 (Abcam, ab18465), rabbit anti-Tbr1 (Abcam, ab31940), rabbit anti-cleaved caspase-3 (CST, 9661), rat anti-bromodeoxyuridine (BrdU; Abcam, ab6326), mouse anti-Pax6 (Invitrogen, 42–6600), rabbit anti-Tbr2 (Abcam, ab15894), rabbit anti-Ki67 (Abcam, ab21700), rabbit anti-CS (ABclonal, A5713), mouse anti-GAPDH (ABclonal, AC036). Secondary antibodies used in this study: donkey anti-mouse 488 (Invitrogen, A21202), donkey anti-mouse 568 (Invitrogen, A10037), donkey anti-rat 488 (Invitrogen, A21208), donkey anti-rat 594 (Invitrogen, A21209), donkey anti-rabbit 488 (Invitrogen, A21206), donkey anti-rabbit 568 (Invitrogen, A10042), donkey anti-rabbit 647 (Invitrogen, A31573), goat anti-rabbit horseradish peroxidas (HRP) (Abclonal, AS014), goat anti-mouse HRP (Abclonal, AS003).
Saccharopine and lysine measurement
Saccharopine and lysine measurement were performed as previously described (Zhou et al., 2019). Briefly, the brain tissue (50 mg) form mouse of either sex was collected, snap-frozen in liquid nitrogen, homogenized, and ultrasonicated in 500 µl ddH2O. The homogenates were then lyophilized and extracted with 1.5 ml of 70% aqueous ethanol (v/v). 3, 4-dihydroxyphenylalanine (DOPA; 2.5 μm) was used as an internal standard. After shaking for 1 min, the extract was centrifuged at 12 000 rpm for 10 min at 4°C. The extract was derivatized with a Waters AccQ-Tag derivation kit for analysis of lysine and saccharopine. The derivatives were analyzed with a UPLC-MS/MS system consisting of an Agilent 1290 Infinity LC pump and a 6495 triple quadruple mass spectrometer (Agilent).
Primary neurons isolation
Primary cortical neurons were isolated from Aass (G489E) or Aass (R65Q) heterozygous mice at embryonic day (E)16.5. Primary hippocampal neurons were isolated from postnatal day (P)0 neonate mice of either sex. The neurons were isolated as described previously (Guo et al., 2018). Briefly, Cerebral cortices or hippocampus were dissected in ice-cold HBSS (Invitrogen, 14175079) and incubated in 0.05% trypsin-EDTA (Invitrogen, 15400-054) for 20 min at 37°C. Dissociated cells were plated onto poly-L-lysine-coated glass coverslips and grown in Neurobasal medium (Invitrogen, 21103049) containing 1% B27 supplement (Invitrogen, 17504044), 2 mm L-glutamine (Invitrogen, 25 030–081), and 1% Antibiotic-Antimycotic (Invitrogen, 15240062) in a 5% CO2 atmosphere at 37°C. Saccharopine, lysine, recombinant GPI (Abcam, ab208311), or 6-PG (Sigma, P7877) was added to the neuronal culture medium at different concentrations. After treatments, the neurons were fixed and examined at day in vitro (DIV)14.
Immunocytochemistry and immunohistochemistry
The primary neurons were fixed in 4% paraformaldehyde (PFA) for 20 min at room temperature, and preblocked with TBS++ (TBS containing 3% bovine serum albumin and 0.3% Triton X-100) for 1 h at room temperature. Subsequently, the neurons were incubated with primary antibodies diluted in TBS++ overnight at 4°C. After being washed five times with TBS, the neurons were incubated with appropriate fluorochrome-conjugated secondary antibodies for 1 h and then washed five times. All neurons were counterstained with 4',6-diamidino-2-phenylindole (DAPI) (Sigma, D9542). After staining, coverslips were mounted onto glass sections and maintained at 4°C in the dark until analysis. Images were captured using confocal microscopy.
For preparing of brain slices, mice of either sex were euthanized by intraperitoneal injection of Avertin and then transcardially perfused with saline followed by 4% PFA. Brains were dissected out, postfixed overnight in 4% PFA, and then equilibrated in 30% sucrose; 20-μm frozen brain sections were generated using a Leica cryostat, and 100- or 40-μm brain sections were generated using a sliding microtone and stored in a −20°C freezer as floating sections in 96-well plates filled with cryoprotectant solution (glycerol, ethylene glycol, and 0.1 m phosphate buffer, pH 7.4, 1:1:2 by volume). The brain sections were washed with TBS, and then antigen retrieval was performed in 0.01 m sodium citrate buffer (pH 6.0) at 98°C for 5 min. Sections were then preblocked with TBS++ for 1 h at room temperature and followed by immunostaining as above. Images were captured using confocal microscopy.
Protein extraction
Whole-cell or brains from mouse of either sex were lysed in ice-cold RIPA buffer (50 mm Tris, 150 mm NaCl, 1 mm EDTA, 1% NP40, 1% sodium deoxycholate and 0.1% SDS) with 1 mm PMSF (Lablead, P0754) and Complete Protease Inhibitor Cocktail (Roche, 11697498001) at a ratio of 100 mg:800 μl. Lysates were centrifuged at 12,000 rpm, 4°C for 10 min to pellet debris. The supernatants were resolved on SDS-PAGE gels, transferred to PVDF membranes and then blotted with the indicated antibodies overnight at 4°C. After being washed 3 times in TBS with 0.1% Tween 20, the membranes were incubated with HRP-conjugated secondary antibody for 1 h at room temperature. The signals were detected using ECL Prime Western Blot Detection reagent (GE Healthcare).
Analysis of the morphology of dendrite
For dendritic analysis on 100-μm-thick floating brain sections, the green fluorescence protein (GFP)-labeled neurons were imaged with a LSM 710 confocal (Zeiss) or a Nikon A1 confocal. Z stacks of the dendrites of neurons were captured at 1-μm intervals, and analyzed by ImageJ software (NIH) using the neurite tracing and Sholl analysis plug-ins, respectively. Data were extracted for Sholl analysis and total dendritic length from each neuron. For in vitro experiments, the MAP2-labeled cultured neurons were imaged with a LSM 710 confocal (Zeiss) or a Nikon A1 confocal, and images were analyzed by ImageJ software as above. The exact value of n was described in the figure legends.
BrdU administration
For analysis of cell proliferation and differentiation in embryonic neurogenesis, Aass (G489E) or Aass (R65Q) pregnant mice were injected with BrdU (200 mg/kg body weight) and analyzed at 2, 24, or 48 h after injection.
Drug affinity-responsive target stability analysis (DARTS)
DARTS analysis was performed as described previously (Lomenick et al., 2009; Chin et al., 2014). Brains from mice of either sex were homogenized and lysed in HEPES buffer (40 mm HEPES, pH 8.0, 120 mm NaCl, 10% glycerol, 0.5% Triton X-100, 10 mm β-glycerophosphate, 50 mm NaF, 0.2 mm Na3VO4, and protease inhibitors) on ice. Lysed brains were centrifuged at 14,000 rpm, 10 min at 4°C to pellet debris, and supernatant was collected for DARTS analysis. The lysates were aliquoted to ensure that the amount of protein in each sample is equal. TNC buffer (50 mm Tris-HCl, pH 8.0, 50 mm NaCl, and 10 mm CaCl2) was added to the brain lysate to a final concentration of 3 μg/μl. The lysates were incubated with vehicle (PBS), saccharopine, or lysine for 1 h on ice and then 50 min at room temperature. Pronase digestions were performed for 30 min at room temperature and stopped by adding SDS loading buffer and heating at 70°C for 10 min. Samples were resolved on SDS–PAGE, and Western blotting was conducted with antibodies against GPI and CS
Enzymatic assay of GPI
The brain tissue (50 mg) form mouse of either sex were homogenized with ice-cold GPI assay buffer (200 µl) from kit. Centrifuge the samples at 13,000 × g for 5 min to remove insoluble material. The enzymatic activity of GPI was determined using a GPI colorimetric assay kit (Sigma, MAK103) according to the manufacturer's instructions.
Glycolysis stress test and mitochondrial stress test
The primary cortical neurons were isolated from wild-type (WT), Aass (G489E), or Aass (R65Q) mice at E16.5 and cultured for 14 d. Oxygen consumption rate (OCR) of neurons was measured in XF media (Seahorse Bioscience, 102353-100) containing 25 mm glucose, 1 mm sodium pyruvate, and 2 mm L-glutamine (pH7.4) using Agilent Seahorse XF Cell Mito Stress Test kit (Seahorse Bioscience, 103015-100) with the XF-96 Extracellular Flux Analyzer (Seahorse Bioscience) following the manufacturer's instructions. Extracellular acidification rate (ECAR) of neurons was measured in XF media containing 2 mm L-glutamine (pH7.4) using XF Glycolysis Stress Test kit (Seahorse Bioscience, 103020-100) with the XF-96 Extracellular Flux Analyzer following the manufacturer' s instructions
RNA isolation and real-time PCR
Total RNA was isolated from mouse neocortex at different stages (E13.5 to adult) using TRIzol (Invitrogen, 15596-018) based on the manufacture protocol. The first-strand cDNA was generated by reverse transcription with oligo (dT) primer or random hexamers (Promega, A5001). To quantify the expression of Aass, G6PDX or PDG, Real-time PCR was performed with gene-specific primers and SYBR Premix EX Taq (CWBIOTECH, CW0682A) using a Bio-Rad Real-Time PCR System (CFX96). Endogenous Gapdh was amplifed as the internal control.
Primers used in this study were as following: Aass forward: CTGGAAGCTGCAGAATGGTT and reverse: CCTTTGGCTTCAATTTCACC; G6pdx forward: AGCCTGGCGTATCTTCACAC and reverse: TGTGAGGGTTCACCCACTTG; Pgd forward: GCTGTCATGGGCCAGAACTT and reverse: TTCACAAGCAGGATGACCCG; Gapdh forward: AATGGGAAGCTTGTCATCAACG and reverse: GAAGACACCAGTAGACTCCACGACATA.
ELISA
The culture medium of primary neurons was collected at DIV14, and centrifuged at 1000 rpm for 5 min. The supernatant was concentrated with Amicon Ultra-15 30K Centrifugal Filter Unit at 4000 × g for 50 min at 4°C. The amount of GPI in culture medium was determined using a GPI ELISA kit (Abcam, ab171575) according to the manufacturer's instructions.
Transmission electron microscopy (TEM) analysis
Mice of either sex were deeply anesthetized and transcardially perfused with 2.5% glutaraldehyde and 1% PFA in 0.1 m phosphate buffer (pH 7.4). The brains were isolated, cut, and fixed overnight at 4°C with 0.1 m phosphate buffer containing 2.5% glutaraldehyde. Fixed samples were rinsed with PBS and further fixed with 1% OsO4 for 2 h at 4°C. The samples were rinsed with distilled water and electron-stained with 2% uranyl acetate, then dehydrated by sequential incubation in an acetone series (30%, 50%, 70%, 80%, 90%, 95%, 100%, and 100%, 10 min each). Samples were infiltrated, embedded, cut, stained, and observed as described previously (Zhou et al., 2019).
Statistical analysis
Statistical analysis was performed using an analysis of variance (ANOVA) and Student's t test, unless specified, with the aid of SPSS version 25 and GraphPad software. A two-tailed, unpaired t test was used to compare two conditions. A one-way ANOVA was used for comparison among multiple experimental conditions. Sholl analysis was conducted using a multivariate ANOVA with SPSS statistical software. The data were presented as the mean ± SEM; n.s. p > 0.05, *p < 0.05, **p < 0.01, and ***p < 0.001. Exact values of n, statistical results, and significance are shown in figure legends.
Results
SDH mutation of Aass leads to accumulated saccharopine and reduced brain size
To confirm the existence of the saccharopine pathway in the mammalian brain (Pena et al., 2017; Crowther et al., 2019; Leandro and Houten, 2020), we firstly investigated the expression pattern of Aass in the mouse brain from the embryonic to the adult stage. The Aass mRNA was constantly expressed during embryonic and early postnatal brain development, but started to increase from P21 to adult stage (Fig. 1B). Moreover, protein levels of Aass were displayed the similar pattern as its mRNA (Fig. 1C). We next examined the Aass protein expression in neural progenitor cells (NPCs), primary neurons and astrocytes. In contrast to the highest expression levels of Aass in the astrocytes, it was relatively low expressed in both NPCs and early cultured primary neurons (DIV7), but significantly increased in DIV14 neurons (Fig. 1D). Thus, these data confirm the previous findings that Aass-involved saccharopine pathway exists in both developing and adult mouse brain (Pena et al., 2017; Crowther et al., 2019; Leandro and Houten, 2020).
To investigate the requirement for Aass in the mouse brain, we analyzed Aass mutant mice carrying the R65Q mutation in LKR or the G489E mutation in SDH (Zhou et al., 2019; Fig. 1E). In comparison to WT mice, Aass (R65Q) homozygous mice had greatly elevated levels of lysine but not saccharopine in the brain at P42; however, Aass (G489E) homozygous mice had significantly elevated levels of both lysine and saccharopine in the brain at P42 (Fig. 1F,G), confirming the occurrence of saccharopine pathway in the mammalian brain at early adulthood.
The Aass (R65Q) homozygous mice had a normal brain size and weight compared with WT mice (Fig. 1H,I). Although Aass (G489E) homozygous mice exhibited comparable brain development at P10 and P21, the brain size and weight were significantly decreased at P42 (Fig. 1H,I, one-way ANOVA, WT vs Aass (G489E), ***p < 0.0001; WT vs Aass (R65Q), p = 0.8061). Furthermore, a significant reduction of cerebral cortex thickness was observed in Aass (G489E) mice, but not in Aass (R65Q) mice at P30 (Fig. 1J,K, one-way ANOVA, WT vs Aass (G489E), ***p = 0.0003; WT vs Aass (R65Q), p = 0.9219). However, Aass (R65Q) and Aass (G489E) mice exhibited the normal layer distributions of Satb2+, Ctip2+, and Tbr1+ postmitotic projection neurons in the cerebral cortex at P30 compared with WT mice (Fig. 2A), suggesting the decreased brain size in Aass (G489E) mice was unlikely caused by abnormal neuronal patterning. In addition, there was no elevation of caspase-3 activation and TdT-mediated dUTP nick end labeling (TUNEL) signals in the cerebral cortex of Aass (R65Q) and Aass (G489E) mice (Fig. 2B,C), excluding the involvement of apoptosis in the brain with R65Q mutation in LKR or the G489E mutation in SDH.
Figure 2.
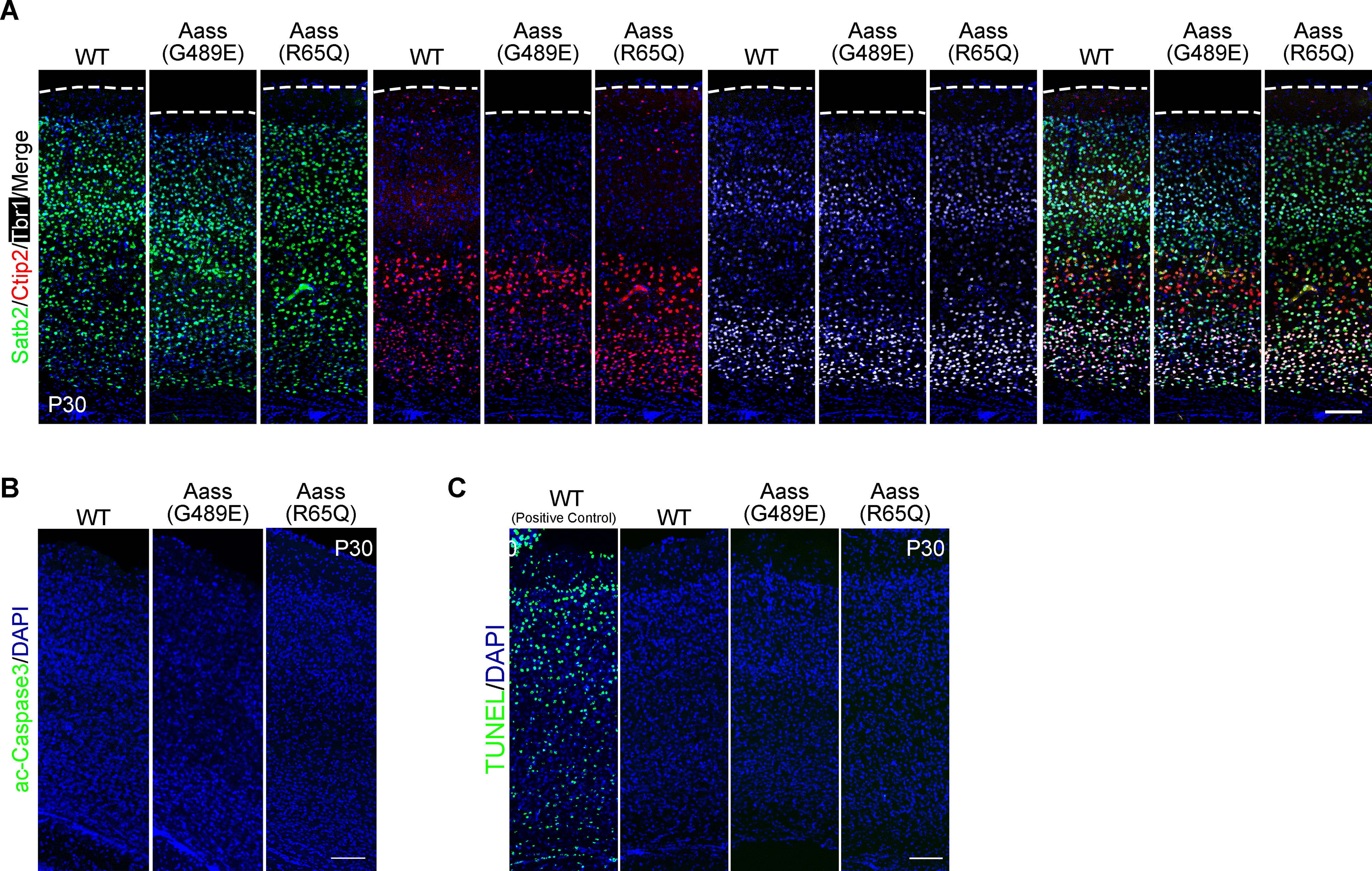
Mutations in LKR or SDH do not affect brain lamination and cell survival. A, Immunostaining images of coronal sections of WT, Aass (G489E), and Aass (R65Q) brains at P30. Different markers were used to label the cortex layers. Satb2, projection neuron marker (green); Ctip2, Layer V (red); Tbr1, Layer VI (white). Nuclei were stained with DAPI (blue). Scale bars: 100 μm. B, Coronal sections of WT, Aass (G489E), and Aass (R65Q) brains stained with active-Caspase 3 (green) at P30. Nuclei were stained with DAPI (blue). Scale bars: 100 μm. C, Cell death detection using TUNEL staining (green) in the coronal sections of WT, Aass (G489E), and Aass (R65Q) brains at P30. Nuclei were stained with DAPI (blue). Scale bars: 100 μm.
Accumulated saccharopine by SDH mutation impairs neuronal development
The decreased brain size could be caused by defects in proliferation and differentiation of NPCs in embryonic brain or by a reduction in dendrites and synaptic connections of neuron in postnatal brain (Kulkarni and Firestein, 2012; Carl, 2016; Guarnieri et al., 2018). We assessed embryonic neurogenesis by BrdU pulse-labeling assay. WT, Aass (R65Q), and Aass (G489E) mice had a similar number of BrdU+Pax6+ radial glial cells and BrdU+Tbr2+ progenitor cells in the ventricular zone/subventricular zone of cerebral cortex 2 h after BrdU injection at E13.5 (Fig. 3A–C, one-way ANVOVA, p > 0.05). There were similar number of Ki67-BrdU+ among BrdU+ in cerebral cortex of Aass (R65Q) and Aass (G489E) mice 24 h after BrdU injection at E13.5 (Fig. 3D,E, one-way ANVOVA, p > 0.05), suggesting that R65Q mutation in LKR or the G489E mutation in SDH did not affect cell cycle exit of NPCs. Moreover, WT, Aass (R65Q), and Aass (G489E) mice had a comparable number of BrdU+Tbr1+ postmitotic neurons in cerebral cortex 48 h after BrdU administration at E13.5 (Fig. 3F,G, one-way ANVOVA, p > 0.05). Therefore, these data suggest that the decreased brain size in Aass (G489E) mice was unlikely caused by defects in proliferation and differentiation of NPCs during embryonic brain development.
Figure 3.
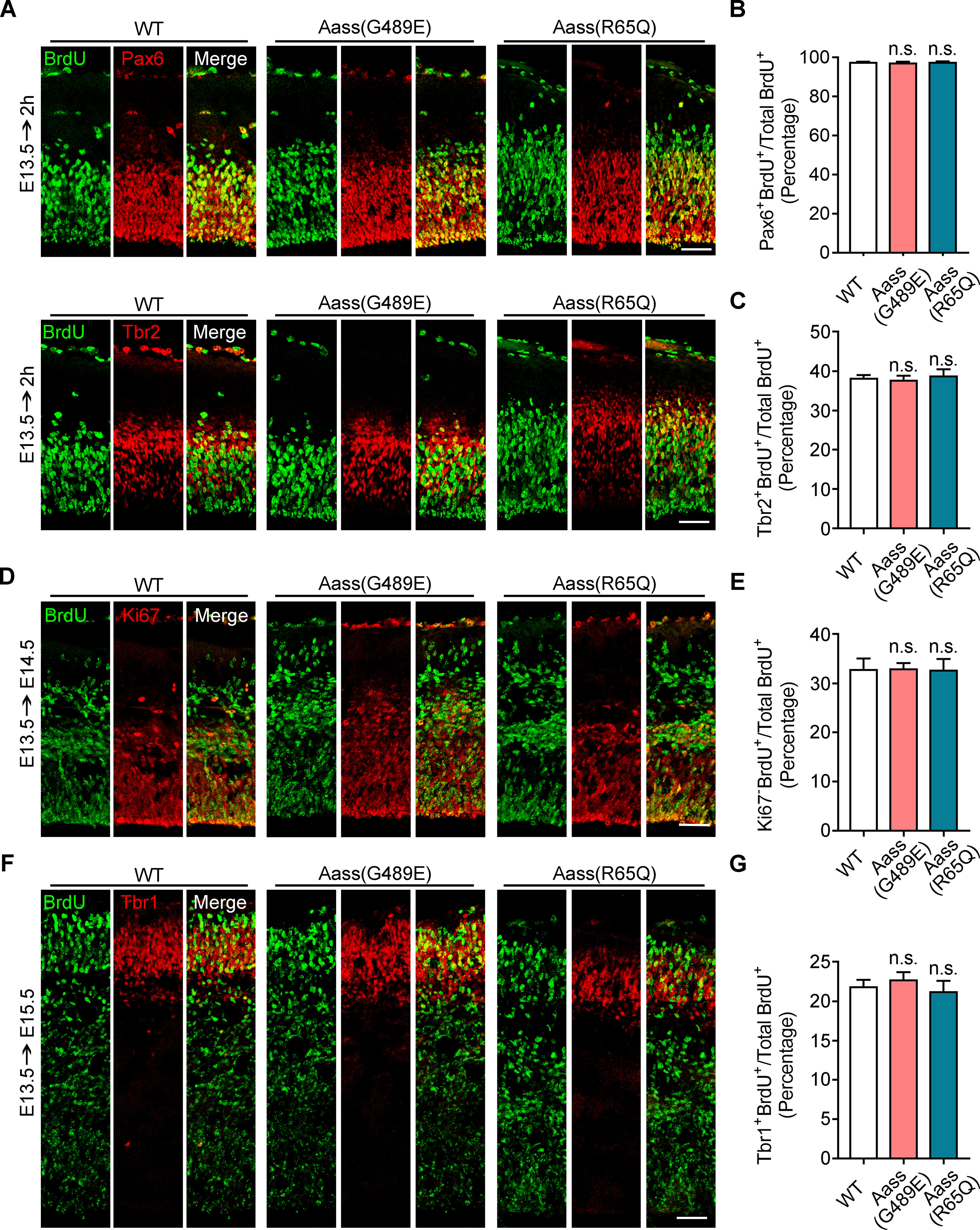
Mutations in LKR or SDH have no significant effect on the proliferation or differentiation of NPCs during embryonic brain development. A, Coronal sections of E13.5 WT, Aass (G489E), and Aass (R65Q) co-stained with anti-BrdU (green) and apical progenitor marker Pax6 (red) or intermediate progenitor marker Tbr2 (red) antibodies. Pregnant mouse were injected with BrdU at E13.5, and the brains of pups were harvested 2 h later. B, C, Quantification analysis of the fractions of Pax6+/BrdU+ cells (B) or Tbr2+/BrdU+ (C) among total BrdU+ cells. B, One-way ANVOVA, F(2,6) = 0.5960, WT versus Aass (G489E), p = 0.9496; WT versus Aass (R65Q), p = 0.9986. C, One-way ANVOVA, F(2,6) = 0.1216, WT versus Aass (G489E), p = 0.7101; WT versus Aass (R65Q), p = 0.5779. n = 3 brains. Scale bars: 100 μm. D, Coronal sections of E14.5 WT, Aass (G489E), and Aass (R65Q) co-stained with anti-BrdU (green) and cell division marker Ki67 (red) antibodies. E, Quantification analysis of the fractions of Ki67-/BrdU+ cells among total BrdU+ cells. One-way ANVOVA, F(2,6) = 0.4138, WT versus Aass (G489E), p = 0.9980; WT versus Aass (R65Q), p = 0.9988. n = 3 brains. Scale bars: 100 μm. F, Coronal sections of E15.5 WT, Aass (G489E), and Aass (R65Q) co-stained with anti-BrdU (green) and newly generated projection neurons marker Tbr1 (red) antibodies. G, Quantification analysis of the fractions of Tbr1+/BrdU+ cells among total BrdU+ cells. One-way ANVOVA, F(2,6) = 0.6478, WT versus Aass (G489E), p = 0.8953; WT versus Aass (R65Q), p = 0.9421. n = 3 brains. Scale bars: 100 μm. Data are presented as mean ± SEM; n.s., no significance, p > 0.05.
Next, we investigated the effect of R65Q mutation in LKR or the G489E mutation in SDH on neuronal dendritic development. We then crossed Aass (R65Q) and Aass (G489E) mice with thymocyte antigen 1 (Thy1)-GFP transgenic mice, respectively. Quantification analysis showed that the total length and complexity of basal dendritic arbors in Layer II and Layer V pyramidal neurons of Aass (G489E) mice were significantly reduced at P42 (Fig. 4A–E, the dendritic length of Layer II neurons, Student's t test, **p = 0.0066; Sholl analysis of Layer II neurons, univariate ANOVA, **p = 0.008; the dendritic length of Layer V neurons, Student's t test, **p = 0.0057; Sholl analysis of Layer V neurons, univariate ANOVA, **p = 0.007). However, the dendritic arborization of these pyramidal neurons in Aass (R65Q) mice was indistinguishable from WT mice (Fig. 4F–J, the dendritic length Layer II neurons or Layer V neurons, Student's t test, p > 0.05; Sholl analysis of Layer II neurons or Layer V neurons, univariate ANOVA, p > 0.05), suggesting that mutation in SDH, but not LKR, leads to impaired dendritic development, thereby accounts for a smaller brain.
Figure 4.
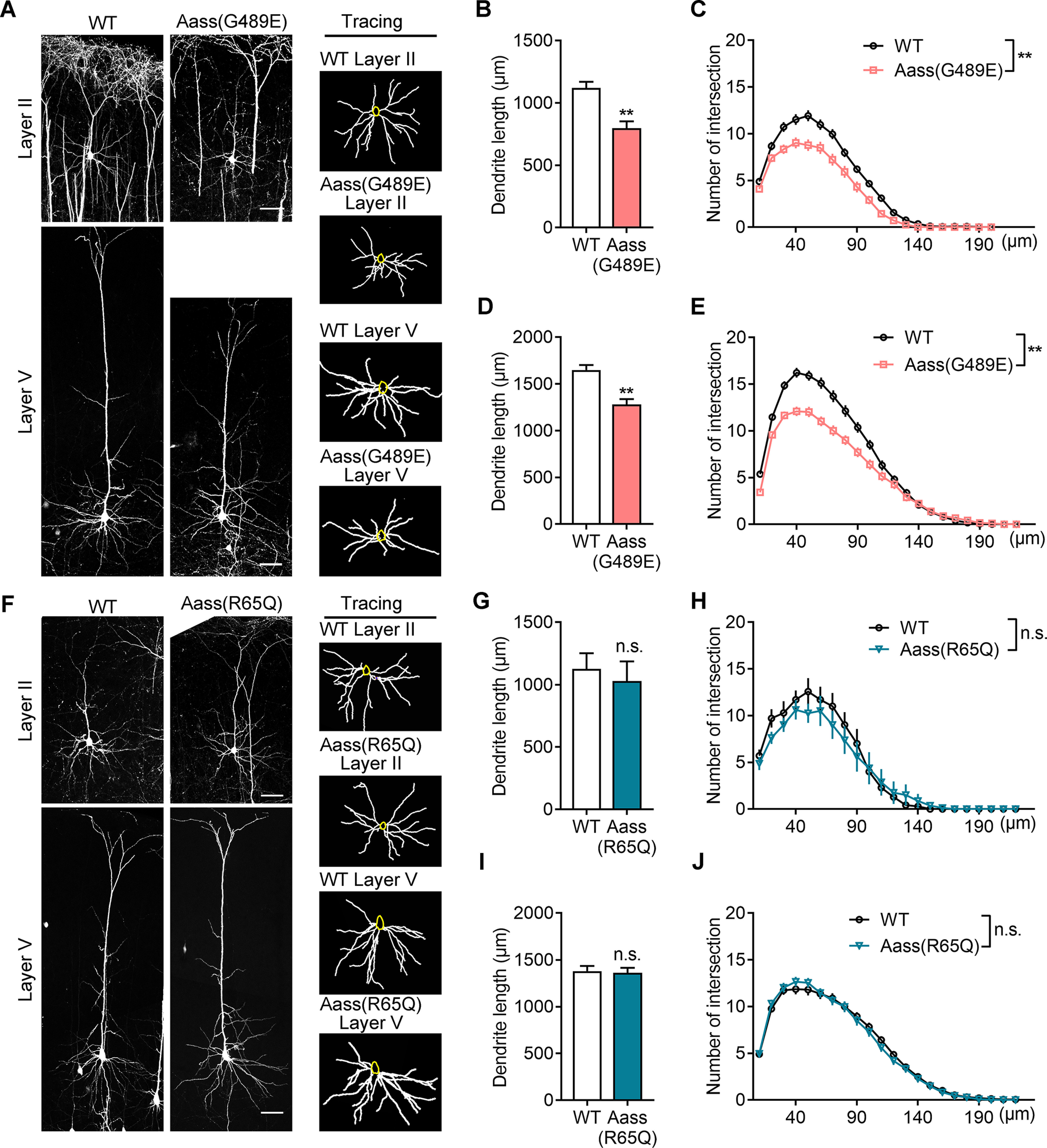
SDH mutation, but not LKR mutation, leads to impaired dendritic development. A, Representative images (left) of Thy1-GFP-labeled pyramidal neurons in Layer II and Layer V of WT, Aass (G489E) somatosensory cortex at P42. The basal dendrites of pyramidal neurons were reconstructed using the ImageJ software (right). Scale bars: 50 μm. B, C, Quantification of the total basal dendritic length (B, Student's t test, **p = 0.0066) and the basal dendritic complexity (C, univariate ANOVA, F(1,5) = 18.442, **p = 0.008) of Layer II pyramidal neurons in A. WT, 51 neurons, n = 4 brains; Aass (G489E), 43 neurons, n = 3 brains. D, E, Quantification of the total basal dendritic length (D, Student's t test, **p = 0.0057) and the basal dendritic complexity (E, univariate ANOVA, F(1,6) = 15.863, **p = 0.007) of Layer V pyramidal neurons in A. WT, 41 neurons, n = 4 brains; Aass (G489E), 44 neurons, n = 4 brains. F, Representative images (left) of Thy1-GFP-labeled pyramidal neurons in Layer II and Layer V of WT, Aass (R65Q) somatosensory cortex at P42. The basal dendrites of pyramidal neurons were reconstructed using the ImageJ software (right). Scale bars: 50 μm. G, H, Quantification of the total basal dendritic length (G, Student's t test, p = 0.1124) and the basal dendritic complexity (H, univariate ANOVA, F(1,5) = 4.139, p = 0.098) of Layer II pyramidal neurons in F. WT, 53 neurons, n = 4 brains; Aass (R65Q), 38 neurons, n = 3 brains. I, J, Quantification of the total basal dendritic length (I, Student's t test, p = 0.7302) and the basal dendritic complexity (J, univariate ANOVA, F(1,5) = 0.125, p = 0.783) of Layer V pyramidal neurons in F. WT, 47 neurons, n = 4 brains; Aass (R65Q), 41 neurons, n = 3 brains. Data are presented as mean ± SEM; n.s., no significance, p > 0.05, **p < 0.01.
To prove the defective neuronal phenotypes further, we performed primary neuronal culture from Aass (R65Q) and Aass (G489E) mice. Quantification analysis revealed that the dendritic length and complexity of DIV14 primary neurons from Aass (G489E) mice, but not Aass (R65Q) mice, were significantly reduced compared with WT control (Fig. 5A–F, WT vs Aass (G489E), the dendritic length, Student's t test, **p = 0.0077, Sholl analysis, univariate ANOVA, **p = 0.004; WT vs Aass (R65Q), the dendritic length, Student's t test, p > 0.05, Sholl analysis, univariate ANOVA, p > 0.05), suggesting a cell-autonomous effect of G489E mutation in SDH on neuronal development likely by accumulation of saccharopine. To consolidate these findings, we found that treatment with 30 μm saccharopine, a concentration closed to the pathologic levels in hyperlysinemia-II patients, significantly reduced the total dendritic length and complexity of primary neurons (Fig. 5G–I, the dendritic length, one-way ANVOVA, **p = 0.0032; Sholl analysis, univariate ANOVA, **p = 0.008). While treatment with 1 mm lysine, a concentration closed to the pathologic levels in hyperlysinemia-I patients, had no obvious effect on the total dendritic length and complexity of primary neurons (Fig. 5J–L, the dendritic length, one-way ANVOVA, p > 0.05; Sholl analysis, univariate ANOVA, p > 0.05).
Figure 5.
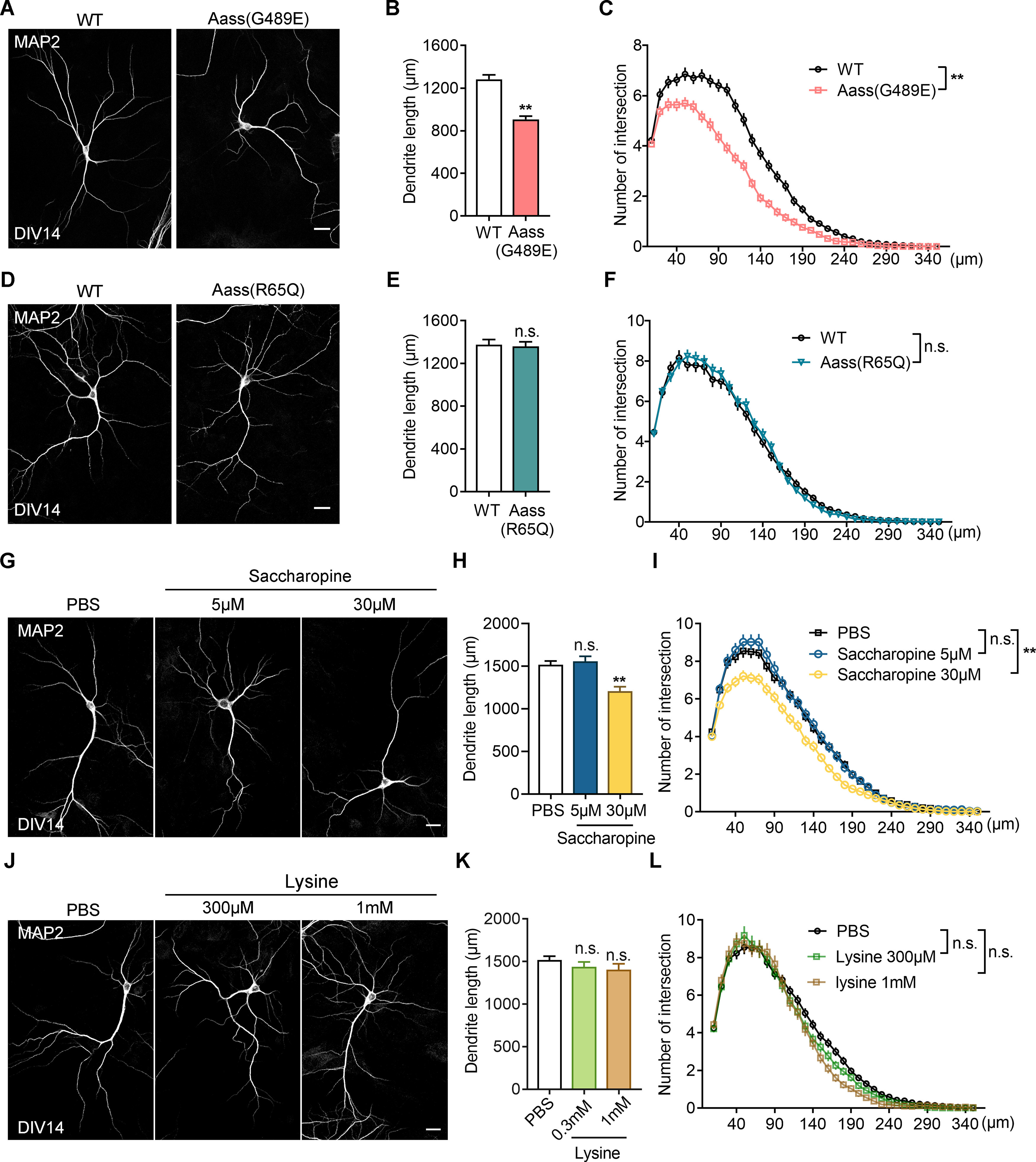
Saccharopine, but not lysine, impairs neuronal dendritic development. A, Representative images of primary neurons dissected from WT or Aass (G489E) brains. The neurons stained with MAP2 antibodies at DIV14. Scale bars: 20 μm. B, C, Quantitative analysis of the total basal dendritic length (B, Student's t test, **p = 0.0077) and the basal dendritic complexity (C, univariate ANOVA, F(1,4) = 34.191, **p = 0.004) of cultured neurons in A. WT, 141 neurons, n = 3 brains; Aass (G489E), 110 neurons, n = 3 brains. D, Representative images of primary neurons dissected from WT or Aass (R65Q) brains. The neurons stained with MAP2 antibodies at DIV14. Scale bars: 20 μm. E, F, Quantitative analysis of the total basal dendritic length (E, Student's t test, p = 0.5507) and the basal dendritic complexity (F, univariate ANOVA, F(1,4) = 0.002, p = 0.996) of cultured neurons in D. WT, 80 neurons, n = 3 brains; Aass (R65Q), 95 neurons, n = 3 brains. G, Representative images of primary neurons dissected from WT brains and treated with saccharopine from DIV4 to DIV14. The neurons were stained with MAP2 antibodies at DIV14. Scale bars: 20 μm. H, Quantitative analysis of the total basal dendritic length of PBS-treated or saccharopine-treated neurons. One-way ANOVA, F(2,6) = 0.1702. PBS versus saccharopine 5 μm, p = 0.8149; PBS versus saccharopine 30 μm, **p = 0.0032. I, Sholl analysis of the dendritic branches of PBS, saccharopine-treated neurons. Univariate ANOVA, PBS versus saccharopine 5 μm, F(1,4) = 0.196, p = 0.681; PBS versus saccharopine 30 μm, F(1,4) = 23.284, **p = 0.008. J, Representative images of primary neurons dissected from WT brains and treated with lysine from DIV4 to DIV14. The neurons were stained with MAP2 antibodies at DIV14. Scale bars: 20 μm. K, Quantitative analysis of the total basal dendritic length of PBS or lysine-treated neurons. One-way ANOVA, F(2,6) = 0.1139. PBS versus lysine, 300 μm, p = 0.7185; lysine, 1 mm, p = 0.7605. L, Sholl analysis of the dendritic branches of PBS-treated or lysine-treated neurons. Univariate ANOVA, PBS versus lysine 300 μm, F(1,4) = 0.931, p = 0.389; PBS versus lysine 1 m, F(1,4) = 0.565, p = 0.494. PBS, 122 neurons; saccharopine 5 μm, 97 neurons; saccharopine 30 μm, 110 neurons; lysine, 300 μm, 88 neurons; lysine, 1 mm, 54 neurons; n = 3 independent experiments. Data are presented as mean ± SEM; n.s., no significance, p > 0.05, **p < 0.01.
Therefore, these results suggest that SDH mutation of Aass blocks saccharopine degradation, thereby resulting in accumulated saccharopine and impaired neuronal development in the brain, which may contribute to the neurologic disorders seen in hyperlysinemia-II patients.
Glucose-6-phosphate isomerase (GPI) is a molecular target of saccharopine
Our previous study showed that the abnormal accumulation of saccharopine results in defective mitochondrial dynamics and function in the hepatocyte of Aass (G489E) mice (Zhou et al., 2019). Proper mitochondrial dynamics is crucial for neurodevelopment (Flippo and Strack, 2017a, b; Khacho and Slack, 2018). Surprisingly, unlike the greatly enlarged and damaged mitochondrial in the liver (Zhou et al., 2019), the mitochondria in the brain of Aass (G489E) mice were indistinguishable from those in WT and Aass (R65Q) mice by TEM analysis (Fig. 6A–C, Student's t test, p > 0.05). Mitochondrial functions delineate the bioenergetic cellular state (Barbato et al., 2020). We then assessed mitochondrial oxygen consumption rate (OCR) in mouse primary neurons using the Seahorse XF96 extracellular flux analyzer. There were no significant alterations in OCR by analyzing the bioenergetic parameters, such as basal respiration, proton leak, Adenosine triphosphate (ATP) production, nonmitochondrial oxygen, max respiration and spare respiration, in the primary neurons isolated from WT, Aass (G489E), and Aass (R65Q) mice (Fig. 6D–G). Therefore, accumulation of saccharopine by SDH mutation of Aass has no significant effects on mitochondrial dynamics and function in neurons.
Figure 6.
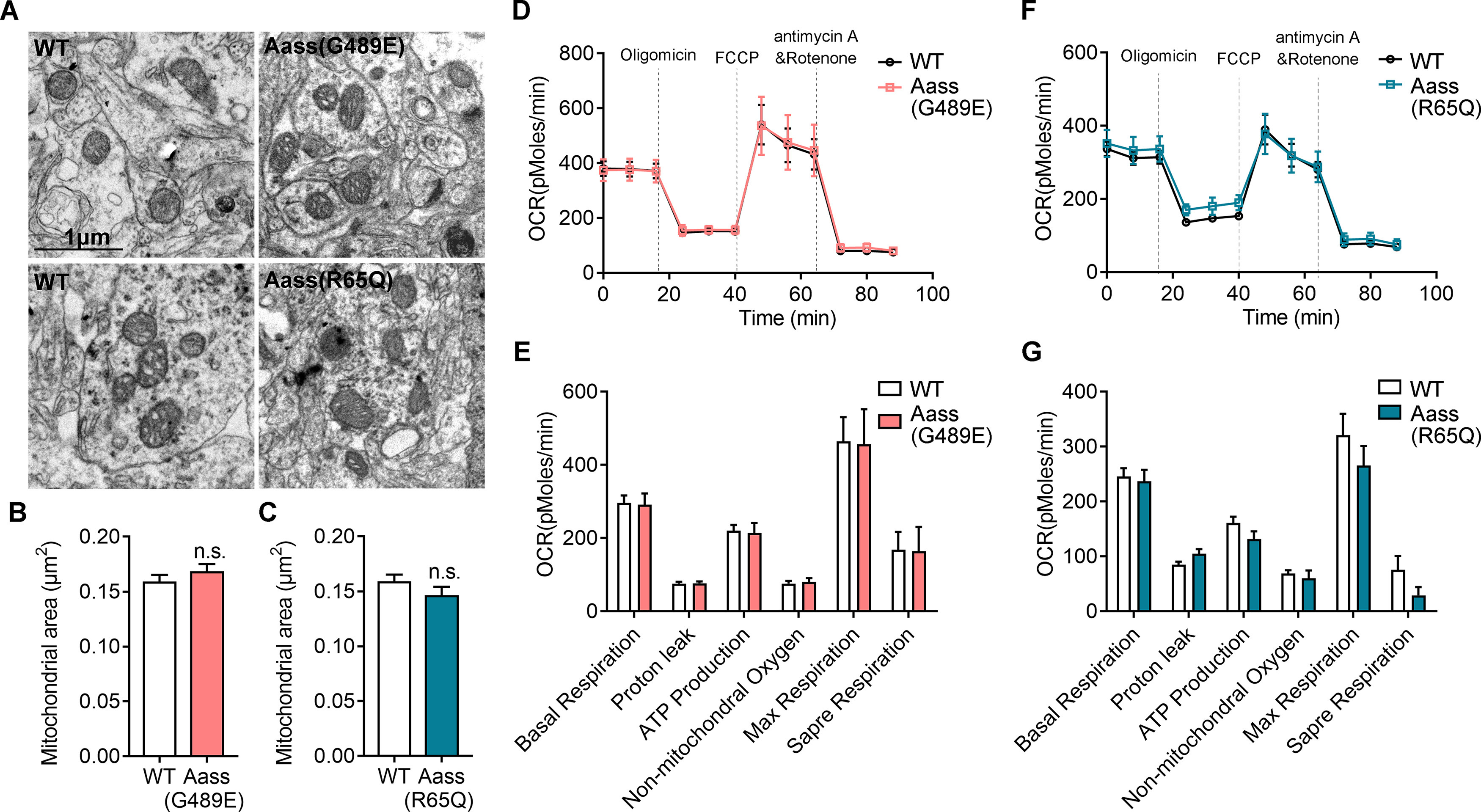
Accumulation of saccharopine in Aass (G489E) mice do not affect mitochondrial function. A, TEM images of mitochondria in the brains of WT, Aass (G489E), and Aass (R65Q) mice. Scale bars: 1 μm. B, C, The average sizes of mitochondria in brains of WT, Aass (G489E) (B, Student's t test, p = 0.5065) and Aass (R65Q) mice (C, Student's t test, p = 0.2993). WT, 517 mitochondria, n = 4 brains; Aass (G489E), 404 mitochondria, n = 3 brains; Aass (R65Q), 181 mitochondria, n = 3 brains. D–G, Representative profile of mitochondria stress test showing the OCR of the cultured neurons isolated from WT, Aass (G489E) (D), or Aass (R65Q) (F) cortex. Graphs showing basal respiration, protein leak, ATP production, nonmitochondrial oxygen, max respiration and spare respiration of cortex neurons of Aass (G489E) (E) or Aass (R65Q) (G) mice compared with cortex neurons of WT littermates. WT, n = 3 brains; Aass (G489E), n = 3 brains; Aass (R65Q), n = 3 brains. Data are presented as mean ± SEM, n.s., no significance, p > 0.05.
Next, we took advantage of an unbiased biochemical screening approach, DARTS (Lomenick et al., 2009; Fig. 7A) to investigate the mechanism underlying accumulated saccharopine in neuronal development. Mass spectrometry identified GPI among the most abundant and enriched proteins present in the saccharopine-treated sample (Fig. 7B,C). Furthermore, proteolysis analysis verified that saccharopine displayed a concentration-dependent proteolytic protection of GPI (Fig. 7D,E), but not citrate synthase (CS; Fig. 7F,G), which was also presented in the initial DARTS screening, suggesting that GPI is potential molecular target of saccharopine. While lysine treatment did not display a concentration-dependent proteolytic protection of GPI and CS (Fig. 7D–G). Furthermore, the total protein levels of GPI (Fig. 7H,I) and CS (Fig. 7J,K) were not significantly affected in the brain of Aass (R65Q) and Aass (G489E) mice, suggesting that saccharopine did not impair the stability of its molecular targets.
Figure 7.
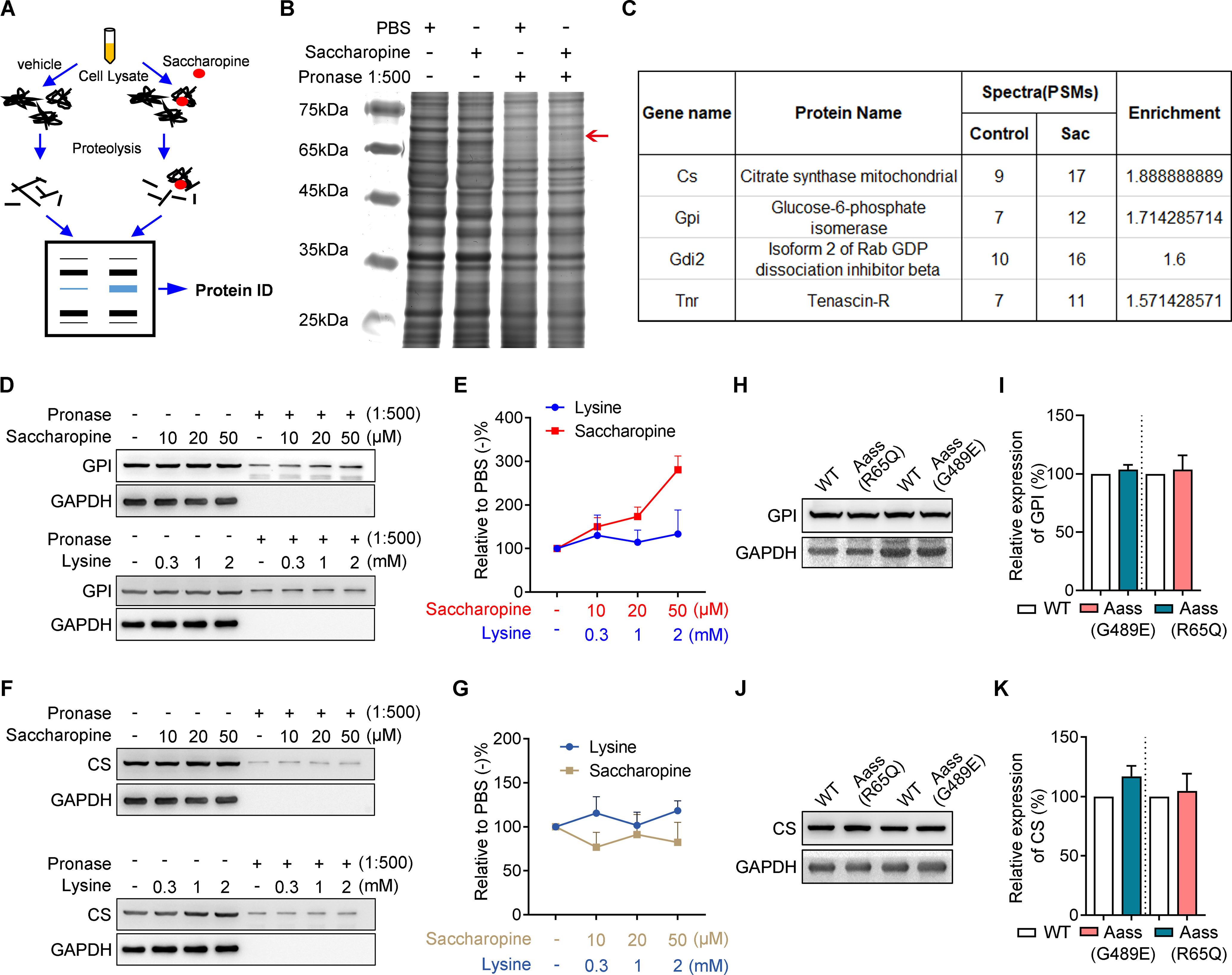
Identification of putative saccharopine molecular target by DARTS assay. A, Scheme of DARTS. B, Sliver staining of SDS-PAGE gel in DARTS assay. The cerebral cortex lysates were treated with saccharopine, followed by Pronase digestion and silver staining. Red arrow, protected bands. C, Enrichment proteins in the protected band from B were revealed by mass spectrometry analysis. D, Western blotting analyzes the enrichment of GPI in DARTS assay. The brain lysates treated with saccharopine or lysine, digested with Pronase and then analyzed by Western blotting. E, Normalized grayscale value of GPI in D. The protein levels of GPI were normalized to PBS-treated groups. Data from three independent experiments. F, Western blotting analyzes the enrichment of CS in DARTS assay. The brain lysates treated with saccharopine or lysine, digested with Pronase and then analyzed by Western blotting. G, Normalized grayscale value of CS in F. The protein levels of CS were normalized to PBS-treated groups. Data from 3 independent experiments. H, Western blotting showing the expression of GPI in brain lysates. I, Quantitative analysis of the greyscale of GPI in H. Protein levels were normalized to GAPDH. The ratio in WT was set to 100%. WT, n = 4 brains; Aass (G489E), n = 4 brains; Aass (R65Q), n = 4 brains. J, Western blotting showing the expression of CS in brain lysates. K, Quantitative analysis of the greyscale of CS in J. Protein levels were normalized to GAPDH. The ratio in WT was set to 100%. WT, n = 4 brains; Aass (G489E), n = 4 brains; Aass (R65Q), n = 4 brains. Data are presented as mean ± SEM.
SDH mutation of Aass leads to reduced extracellular level of GPI without affecting cellular glycolytic function
GPI has been identified as a moonlighting protein based on its ability to perform mechanistically distinct functions between cytoplasm and extracellular space (Fig. 8A). In the cytoplasm, the gene product functions as a glycolytic enzyme that interconverts glucose-6-phosphate (G6P) and fructose-6-phosphate (Harrison, 1974). Extracellularly, GPI (also referred to as neuroleukin) functions as an autosecreted neurotrophic factor that promotes survival of skeletal motor neurons and sensory neurons (Gurney et al., 1986a), and as a lymphokine that induces immunoglobulin secretion (Gurney et al., 1986b). By quantifying the GPI activity, we found that the brain lysis from Aass (G489E) mice exhibited a slightly lower GPI catalytic activity than the WT controls (Fig. 8B, Student's t test, **p = 0.001), while lysates from Aass (R65Q) had similar GPI catalytic activity (Fig. 8C, Student's t test, p = 0.3217) compared with WT mice. Furthermore, we found that saccharopine, but not lysine, dose dependently inhibited GPI activity (Fig. 8D,E, one-way ANOVA; saccharopine, *p < 0.05, **p = 0.0046; lysine, p > 0.05).
Figure 8.
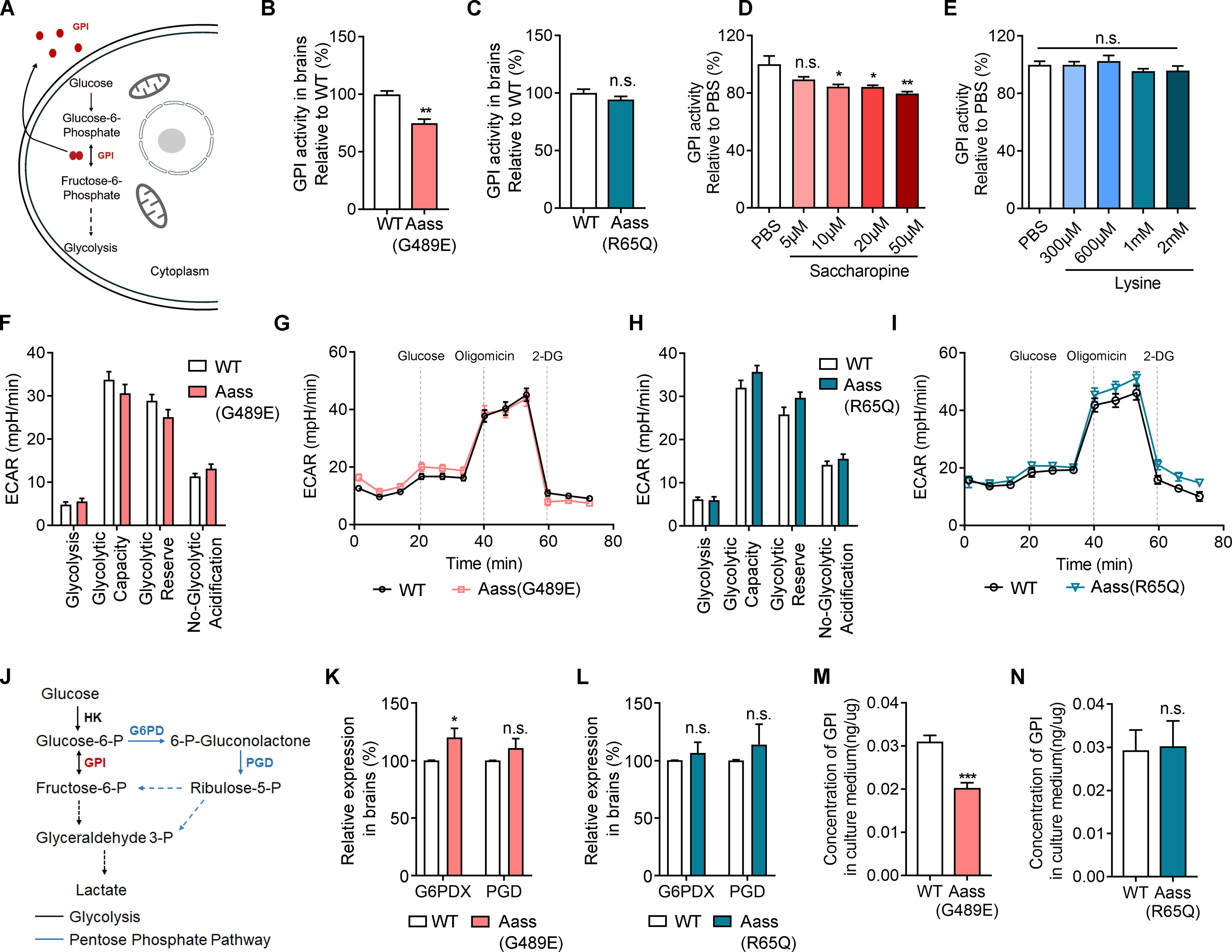
SDH mutation results in reduced extracellular levels of GPI. A, Schematic illustration of GPI involved in glycolysis intracellularly or secreted into extracellular domain in an autocrine manner. B, C, The enzyme catalytic activity of GPI in cerebral cortex of WT, Aass (G489E) (B, Student's t test, **p = 0.001) or Aass (R65Q) mice (C, Student's t test, p = 0.3217). The ratio in WT mice was set to 100%. WT, n = 4 brains; Aass (R65Q), n = 4 brains; Aass (G489E), n = 4 brains. D, E, The enzyme catalytic activity of GPI in WT cortex lysates. The cortex lysates were treated with saccharopine (D) or lysine (E) in different concentrations as indicated. D, One-way ANOVA, PBS versus saccharopine 5 μm, p = 0.1596; PBS versus saccharopine 10 μm, *p = 0.0256; PBS versus saccharopine 20 μm, *p = 0.0242; PBS versus saccharopine 50 μm, **p = 0.0046. E, One-way ANOVA, PBS versus lysine 300 μm, p = 0.9999; PBS versus lysine 600 μm, p = 0.9741; PBS versus lysine 1 mm, p = 0.7953; PBS versus lysine 2 mm, p = 0.8453. F, Graph showing glycolysis, glycolytic capacity, glycolytic reserve, and nonglycolytic acidification of cortex neurons of Aass (G489E) mice compared with cortex neurons of WT littermates. G, Representative profile of glycolysis stress assay showing the ECAR of the cultured neurons isolated from WT and Aass (G489E) cortex. WT, n = 3 brains; Aass (G489E), n = 3 brains. H, Graph showing glycolysis, glycolytic capacity, glycolytic reserve and nonglycolytic acidification of cortex neurons of Aass (R65Q) mice compared with cortex neurons of WT littermates. I, Representative profile of glycolysis stress assay showing the ECAR of the cultured neurons isolated from WT and Aass (R65Q) cortex. WT, n = 3 brains; Aass (R65Q), n = 3 brains. J, Simplified schematic of glycolysis and the PPP. HK, hexokinase; GPI, Glucose-6-phosphate isomerase; G6PD, glucose-6-phosphate dehydrogenase; PGD, 6-phosphogluconate dehydrogenase. K, L, Relative expression profiles of G6PD and PGD in Aass (G489E) (K, Student's t test, G6PDX, *p = 0.0417; PGD, p = 0.2394) or Aass (R65Q) (L, Student's t test, G6PDX, p = 0.2473; PGD, p = 0.1943) cortices compared with their WT littermate cortices at P42. WT, n = 4 brains; Aass (G489E), n = 4 brains; Aass (R65Q), n = 4 brains. M, N, The concentration of secreted GPI in the culture medium of primary neurons dissected from Aass (G489E) (M, Student's t test, ***p = 0.0005) or Aass (R65Q) (N, Student's t test, p = 0.9048) brains compared with their WT littermates by ELISA. WT, n = 4 brains; Aass (G489E), n = 4 brains, Aass (R65Q), n = 4 brains. Data are presented as mean ± SEM, n.s., no significance, p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001.
To assess whether R65Q mutation in LKR or the G489E mutation in SDH affect the glycolytic function, we then directly measured the extracellular acidification rate (ECAR) of mouse primary neurons by using the Seahorse XF96 extracellular flux analyzer. However, there were no significant changes in ECAR by analyzing the key parameters, such as glycolytic flux, including glycolysis, glycolytic capacity, glycolytic reserve and nonglycolytic acidification, in the primary neurons isolated from WT, Aass (G489E), and Aass (R65Q) mice (Fig. 8F–I). The pentose phosphate pathway (PPP), a multienzyme pathway, branches from glycolysis at the first step and begins with a key intermediate molecule of glycolysis, G6P (Fig. 8J). We found that the expression of G6P dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (PGD), two rate-limiting enzymes in PPP, were slightly increased in the brain of Aass (G489E) mice, but no in Aass (R65Q), mice, compared with WT mice (Fig. 8K,L, Student's t test; WT vs Aass (G489E), G6PDX, *p = 0.0417; PGD, p = 0.2394; WT vs Aass (R65Q), p > 0.05), suggesting the glycolytic function might be compensated by PPP pathway in Aass (G489E) mutant with a slightly defective activity of GPI.
GPI crystal structure analyses revealed that the regions important for the enzymatic function correspond to those required for its cytokine function (Sun et al., 1999; Chou et al., 2000). We therefore hypothesized that saccharopine might inhibit the cytokine function of GPI. By measuring the GPI content in the cultured medium of primary neurons, we found that the amount of GPI was comparable in the cultured medium of primary neurons from Aass (R65Q) mice, but significantly decreased in those from Aass (G489E) mice in comparison to WT mice (Fig. 8M,N, Student's t test; WT vs Aass (G489E), ***p = 0.0005; WT vs Aass (R65Q), p > 0.05). Thus, these data not only demonstrate that GPI is released into the extracellular space by cortical neurons, but also suggest that accumulated saccharopine by Aass (G489E) mutation binds to GPI and reduces its extracellular levels.
Supplementation of extracellular GPI rescues the defective neuronal development caused by SDH mutation
Given inhibition of the enzymatic activity of GPI also prohibit its ligand-stimulation effects, we firstly assessed whether inhibition of extracellular GPI function affect neuronal development by treating with 6-phosphogluconate (6PG), a GPI inhibitor. Treatment of 6PG led to significant decreased total dendritic length and complexity of primary neurons (Fig. 9A–C, the dendritic length, one-way ANOVA, PBS vs 6PG 50 μm, **p = 0.0037; Sholl analysis, univariate ANOVA, PBS vs 6PG 50 μm, **p = 0.006). However, by using ECAR to assess the glycolytic function, we found that extracellular addition of 6PG did not change the glycolytic function of primary neurons (Fig. 9D,E). On the other hand, treatment with the recombinant GPI protein was sufficient to promote the total dendritic length and complexity of primary neurons (Fig. 10A–C, the dendritic length, one-way ANOVA, PBS vs GPI 100 ng, **p = 0.01; PBS vs GPI 300 ng, **p = 0.0047; Sholl analysis, univariate ANOVA, PBS vs GPI 100 ng, *p = 0.028; PBS vs GPI 300 ng, **p = 0.01), but had minimal effect on the glycolytic function in neurons (Fig. 10D,E). Thus, these data suggest that manipulation of extracellular function of GPI influences neuronal development without affecting cellular glycolytic function.
Figure 9.

Inhibition of extracellular function of GPI impairs neuronal development without affecting glycolysis. A, Representative images of primary neurons dissected from WT brains. The neurons treated with or without 6PG in different concentrations as indicated from DIV4 to DIV14 and stained with MAP2 antibodies at DIV14. Scale bars: 20 μm. B, C, Quantitative analysis of the total basal dendritic length (B, one-way ANOVA, F(2,6) = 0.2276; PBS vs 6PG 10 μm, p = 0.0999; PBS vs 6PG 50 μm, **p = 0.0037) and the basal dendritic complexity (C, univariate ANOVA, PBS vs 6PG 10 μm, F(1,4) = 3.946, p = 0.118; PBS vs 6PG 50 μm, F(1,4) = 29.193, **p = 0.006) of cultured neurons in (A). PBS, 96 neurons; 6PG 10 μm, 63 neurons; 6PG 50 μm, neurons; n = 3 independent experiments. D, E, Representative profile of glycolysis stress assay (D) showing the ECAR of the cultured neurons treated with or without 6PG (50 μm). Graph in E showing glycolysis, glycolytic capacity, glycolytic reserve and nonglycolytic acidification of neurons in D. Data are presented as mean ± SEM; n.s., no significance, p > 0.05, **p < 0.01.
Figure 10.

Extracellular GPI promotes neuronal dendritic development without affecting glycolysis. A, Representative images of primary neurons dissected from WT brains. The neurons treated with or without GPI in different concentrations as indicated from DIV4 to DIV14, and stained with MAP2 antibodies at DIV14. Scale bars: 20 μm. B, C, Quantitative analysis of the total basal dendritic length (B, one-way ANOVA, F(2,6) = 0.9092; PBS vs GPI 100 ng, *p = 0.01; PBS vs GPI 300 ng, **p = 0.0047) and the basal dendritic complexity (C, univariate ANOVA, PBS vs GPI 100 ng, F(1,4) = 11.258, *p = 0.028; PBS vs GPI 300 ng, F(1,4) = 21.435, **p = 0.01) of cultured neurons in A. PBS, 100 neurons; GPI 100 ng, 85 neurons; GPI 300 ng, 91 neurons; n = 3 independent experiments. D, E, Representative profile of glycolysis stress assay (D) showing the ECAR of the cultured neurons treated with or without GPI (100 ng). Graph in E showing glycolysis, glycolytic capacity, glycolytic reserve, and nonglycolytic acidification of neurons in D. Data are presented as mean ± SEM; n.s., no significance, p > 0.05, *p < 0.05, **p < 0.01.
Next, we assessed whether GPI act as a downstream effector of SDH mutation on neuronal development. By treating primary neurons from Aass (G489E) mice with 6PG, we found that there was no additive effect on the reduction of dendritic length and complexity (Fig. 11A–C, the dendritic length, one-way ANVOVA, WT + PBS vs WT + 6PG, **p = 0.01; WT + PBS vs Aass (G489E) + PBS, *p = 0.0355; WT + PBS versus Aass (G489E) + 6 PG, *p = 0.0115; Aass (G489E) + PBS vs Aass (G489E) + 6 PG, p = 0.9041; Sholl analysis, univariate ANOVA; WT + PBS vs WT + 6PG, *p = 0.012; WT + PBS vs Aass (G489E) + PBS, **p = 0.006; WT + PBS vs Aass (G489E) + 6 PG, **p = 0.01; Aass (G489E) + PBS vs Aass (G489E) + 6 PG, p = 0.055), suggesting SDH mutation and GPI inhibition function in the same regulatory pathway to inhibit neuronal development.
Figure 11.

Supplementation of GPI rescues defective dendritic development caused by SDH mutation. A, Representative images of primary neurons dissected from WT or Aass (G489E) brains. The neurons treated with or without 6PG (50 μm) from DIV7 to DIV14, and stained with MAP2 antibodies at DIV14. Scale bars: 20 μm. B, C, Quantitative analysis of the total basal dendritic length (B) and the basal dendritic complexity (C) of cultured neurons in A. B, One-way ANVOVA, F(3,11) = 0.9085. WT + PBS versus WT + 6PG, **p = 0.01; WT + PBS versus Aass (G489E) + PBS, *p = 0.0355; WT + PBS versus Aass (G489E) + 6 PG, *p = 0.0115; Aass (G489E) + PBS versus Aass (G489E) + 6 PG, p = 0.9041. C, Univariate ANOVA, WT + PBS versus WT + 6PG, F(1,4) = 19.287, *p = 0.012; WT + PBS versus Aass (G489E) + PBS, F(1,5) = 21.306,**p = 0.006; WT + PBS versus Aass (G489E) + 6 PG, F(1,5) = 16.737, **p = 0.01; Aass (G489E) + PBS versus Aass (G489E) + 6 PG, F(1,6) = 5.647, p = 0.055. WT + PBS, 104 neurons, n = 3 brains; WT + 6PG, 58 neurons, n = 3 brains; Aass (G489E) + PBS, 94 neurons n = 4 brains; Aass (G489E) + 6PG, 40 neurons, n = 4 brains. D, Representative images of primary neurons dissected from WT or Aass (G489E) brains. The neurons treated with or without GPI (100 ng) from DIV7 to DIV14, and stained with MAP2 antibodies at DIV14. Scale bars: 20 μm. E, F, Quantitative analysis of the total basal dendritic length (E) and the basal dendritic complexity (F) of cultured neurons in D. E, One-way ANVOVA, F(3,12) = 1.189. WT + PBS versus WT + GPI, *p = 0.038; WT + PBS versus Aass (G489E) + PBS, *p = 0.0224; WT + PBS versus Aass (G489E) + GPI, p = 0.9360; Aass (G489E) + PBS versus Aass (G489E) + GPI, **p = 0.0081. F, Univariate ANOVA, WT + PBS versus WT + GPI, F(1,6) = 17.738, **p = 0.006; WT + PBS versus Aass (G489E) + PBS, F(1,7) = 19.996,**p = 0.003; WT + PBS versus Aass (G489E) + GPI, F(1,7) = 0.740, p = 0.418; Aass (G489E) + PBS versus Aass (G489E) + GPI, F(1,8) = 11.224, **p = 0.01. WT + PBS, 168 neurons, n = 4 brains; WT + GPI, 232 neurons, n = 4 brains; Aass (G489E) + PBS, 207 neurons, n = 5 brains; Aass (G489E) + GPI, 177 neurons, n = 5 brains. Data are presented as mean ± SEM; n.s., no significance, p > 0.05, *p < 0.05, **p < 0.01.
Finally, we hypothesized that supplementation of GPI could rescue the defective dendritic development caused by G498E mutation in SDH. As expected, extracellular addition of GPI was able to restore the reduced dendritic length and complexity caused by G498E mutation in SDH (Fig. 11D–F, the dendritic length, one-way ANVOVA, WT + PBS vs WT + GPI, *p = 0.038; WT + PBS vs Aass (G489E) + PBS, *p = 0.0224; WT + PBS vs Aass (G489E) + GPI, p = 0.9360; Aass (G489E) + PBS vs Aass (G489E) + GPI, **p = 0.0081; Sholl analysis, univariate ANOVA; WT + PBS vs WT + GPI, **p = 0.006; WT + PBS vs Aass (G489E) + PBS, **p = 0.003; WT + PBS vs Aass (G489E) + GPI, p = 0.418; Aass (G489E) + PBS vs Aass (G489E) + GPI, **p = 0.01).
Taken together, our data demonstrate that accumulation of saccharopine, the lysine catabolism intermediate, negatively regulates the brain development in mice carrying the G489E mutation in SDH. Mechanistically, GPI is identified as a molecular target of saccharopine. Through inhibiting the neurotropic function of GPI, accumulated saccharopine impairs neuronal dendritic development in mice carrying the G489E mutation in SDH (Fig. 12). Hence, our study unravels the requirement of saccharopine degradation for neuronal development and elucidates mechanistic insights for understanding the neurologic disorder seen in hyperlysinemia-II (saccharopinuria).
Figure 12.

Working model illustrating that SDH mutation leads to accumulated saccharopine, thereby inhibits neurotrophic function of GPI on neuronal development.
Discussion
It has been long believed that the enzyme activity of LKR/SDH is highly dependent on the stage of brain development, with high levels of activity in embryonic brain and low activity in postnatal and adult brain (Rao et al., 1992). In supporting with this notion, no detectable LKR/SDH activity and low expression of Aass mRNA had been found in adult mouse brain (Rao et al., 1992; Sauer et al., 2011; Posset et al., 2015). However, it is worth noting that several reports offer arguments in favor of the existence of an active Aass-mediated saccharopine pathway in brain. The mRNA and protein of Aass are highly expressed in adult brain (Papes et al., 2001; Pena et al., 2017). The saccharopine pathway has been found to be highly activated in adult brain (Papes et al., 2001; Posset et al., 2015; Pena et al., 2016, 2017; Crowther et al., 2019; Leandro and Houten, 2020). The saccharopine (ε-deamination of lysine) and pipecolate (α-deamination of lysine) pathways lead to the formation of aminoadipic semialdehyde (AASA) that is then oxidized to aminoadipate (AAA) by antiquitin. Isotope tracking analysis reveals that 15N-AAA was detected in the brain of mice injected with α-15N lysine (Pena et al., 2017). Moreover, saccharopine has been found to be elevated in the brain of antiquitin knock-out (KO) mice (∼20 nmol/g; Al-Shekaili et al., 2020), suggesting that the saccharopine pathway is an active route of lysine degradation in the brain. In this study, we found that both protein and mRNA levels of Aass were constantly expressed during embryonic and early postnatal brain development, and conspicuously increased in adult brain. Furthermore, we took advantage of two Aass knock-in mouse models, in which one harbors a mutation (R65Q) in the LKR domain and the other has a mutation (G489E) in the SDH domain. By measuring the amount of lysine and saccharopine in adult brain, we found that Aass (R65Q) mice had greatly elevated levels of lysine but not saccharopine; however, Aass (G489E) mice had greatly elevated levels of both lysine and saccharopine (∼80 nmol/g). Therefore, our study provides direct genetic evidence for supporting the existence and significant activation of saccharopine pathway in postnatal mammalian brain.
Saccharopine is a transient intermediate of lysine metabolism, and is undetectable in plasma and urine of normal individuals. Whereas patients with hyperlysinemia-II abnormally accumulate saccharopine in plasma and urine, accompanying with developmental delay and intellectual disability. Our previous study has shown that the accumulation of saccharopine resulting from SDH mutation causes lethal mitochondrial damage in the liver, leading to postnatal developmental retardation and death (Zhou et al., 2019). However, little is known about the association between abnormal accumulated saccharopine and neurologic symptoms seen in patients with hyperlysinemia-II. In this study, we found that Aass (G489E) mice displayed a smaller brain with defective dendritic arborization of pyramidal neurons in cerebral cortex. Furthermore, treatment with pathologic concentration of saccharopine resulted into impaired dendritic arborization of cultured primary neurons. These data suggested that negative impact of abnormal accumulation of saccharopine on neuronal development directly contributes to the neuropathy in hyperlysinemia-II. However, Aass (R65Q) mice displayed a normal brain with proper neuronal development. In addition, neurons seem to tolerate to the pathologic concentration of lysine seen in hyperlysinemia-I. Thus, these data also support the notion that pharmacological inhibition of LKR may represent an attractive strategy for treatment of various inherited disorders of lysine metabolism, including hyperlysinemia-II, pyridoxine-dependent epilepsy because of antiquitin deficiency and glutaric aciduria type I.
GPI (also known as autocrine motility factor, maturation factor and neuroleukin) is a multifunctional protein involved in glucose metabolism, neuronal survival, axon growth, cell motility and differentiation (Fairbank et al., 2009). Originally GPI acts as a glycolytic enzyme to catalyze the reversible isomerization between G6P and fructose-6-phosphate in the cytoplasm (Harrison, 1974). Later, GPI has been found to be secreted into extracellular space of cancer cells and stimulates oncogenesis and tumor progression (Watanabe et al., 1991, 1996; Funasaka et al., 2007; Araki et al., 2009; Kho et al., 2013, 2014). In physiological condition, extracellular GPI mediates the differentiation of human myeloid cells to terminal monocytic cells (Xu et al., 1996) and promotes the survival and development of neurons (Gurney et al., 1986a; Deng et al., 2014; Tanie et al., 2018). GPI crystal structure analyses revealed that the regions important for the enzymatic function of GPI correspond to those required for its cytokine function (Sun et al., 1999; Chou et al., 2000). In this study, we found that saccharopine bound to and inhibited the activity of GPI. Moreover, we found that saccharopine impaired neuronal dendritogenesis by inhibiting the neurotrophic effect, not the glycolytic function, of GPI. However, supplementation of GPI extracellularly was able to rescue dendritic development of neurons from Aass (G489E) mice. Therefore, our findings not only demonstrate the essential role for saccharopine degradation in neuronal development but also offer the mechanistic insights for understanding the neurometabolic disorder seen in hyperlysinemia.
Footnotes
This work was supported by National Key Research and Development Program of China Grants 2019YFA0802100 and 2021ZD0202302 (to W.G.), National Science Foundation of China Grants 31921002 (to W.G.) and 31800859 (to Y.G.), and the China Postdoctoral Science Foundation Grant 2018M640193 (to Y.G.).
The authors declare no competing financial interests.
References
- Al-Shekaili, Petkau TL, Pena I, Lengyell TC, Verhoeven-Duif NM, Ciapaite J, Bosma M, Faassen M, Kema IP, Horvath G, Ross C, Simpson EM, Friedman JM, Karnebeek C, Leavitt BR (2020) A novel mouse model for pyridoxine-dependent epilepsy due to antiquitin deficiency. Hum Mol Genet 29:3266–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Shimura T, Yajima T, Tsutsumi S, Suzuki H, Okada K, Kobayashi T, Raz A, Kuwano H (2009) Phosphoglucose isomerase/autocrine motility factor promotes melanoma cell migration through ERK activation dependent on autocrine production of interleukin-8. J Biol Chem 284:32305–32311. 10.1074/jbc.M109.008250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbato A, Scandura G, Puglisi F, Cambria D, Spina EL, Palumbo GA, Lazzarino G, Tibullo D, Raimondo FD, Giallongo C, Romano A (2020) Mitochondrial bioenergetics at the onset of drug resistance in hematological malignancies: an overview. Front Oncol 10:604143. 10.3389/fonc.2020.604143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blemings KP, Crenshaw TD, Swick RW, Benevenga NJ (1994) Lysine-alpha-ketoglutarate reductase and saccharopine dehydrogenase are located only in the mitochondrial matrix in rat liver. J Nutr 124:1215–1221. 10.1093/jn/124.8.1215 [DOI] [PubMed] [Google Scholar]
- Carl E (2016) Proliferation and differentiation deficits are a major convergence point for neurodevelopmental disorders. Trends Neurosci 39:290–299. [DOI] [PubMed] [Google Scholar]
- Carson NAJ, Scally BG, Neill DW, Carré LJ (1968) Saccharopinuria: a new inborn error of lysine metabolism. Nature 218:679. 10.1038/218679a0 [DOI] [PubMed] [Google Scholar]
- Cederbaum SD, Shaw KN, Dancis J, Hutzler J, Blaskovics JC (1979) Hyperlysinemia with saccharopinuria due to combined lysine-ketoglutarate reductase and saccharopine dehydrogenase deficiencies presenting as cystinuria. J Pediatr 95:220–227. 10.1016/S0022-3476(79)80657-5 [DOI] [PubMed] [Google Scholar]
- Chang YF (1976) Pipecolic acid pathway: the major lysine metabolic route in the rat brain. Biochem Biophys Res Commun 69:174–180. 10.1016/s0006-291x(76)80288-4 [DOI] [PubMed] [Google Scholar]
- Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, Hu E, Whelan SA, Wang JX, Jung G, Solis GM, Fazlollahi F, Kaweeteerawat C, Quach A, Nili M, Krall AS, et al. (2014) The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 510:397–401. 10.1038/nature13264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CC, Sun YJ, Meng M, Hsiao CD (2000) The crystal structure of phosphoglucose isomerase/autocrine motility factor/neuroleukin complexed with its carbohydrate phosphate inhibitors suggests its substrate/receptor recognition. J Biol Chem 275:23154–23160. 10.1074/jbc.M002017200 [DOI] [PubMed] [Google Scholar]
- Crowther LM, Mathis D, Poms M, Plecko B (2019) New insights into human lysine degradation pathways with relevance to pyridoxine-dependent epilepsy due to antiquitin deficiency. J Inherit Metab Dis 42:620–628. 10.1002/jimd.12076 [DOI] [PubMed] [Google Scholar]
- Dancis J, Hutzler J (1982) Comparative rates of metabolism of pipecolic acid in several animal species. Comp Biochem Physiol B 73:1011–1012. 10.1016/0305-0491(82)90351-0 [DOI] [PubMed] [Google Scholar]
- Dancis J, Hutzler J, Ampola MG, Shih VE, van Gelderen HH, Kirby LT, Woody NC (1983) The prognosis of hyperlysinemia: an interim report. Am J Hum Genet 35:438–442. [PMC free article] [PubMed] [Google Scholar]
- Deng L, Shi B, Zhuang Y, Chu J, Shi X, Zhang S, Guo M (2014) Performance and mechanism of neuroleukin in the growth and survival of Sertoli cell-induced neurons in a coculture system. Cell Transplant 23:381–394. 10.3727/096368913X663578 [DOI] [PubMed] [Google Scholar]
- Fairbank M, St-Pierre P, Nabi IR (2009) The complex biology of autocrine motility factor/phosphoglucose isomerase (AMF/PGI) and its receptor, the gp78/AMFR E3 ubiquitin ligase. Mol Biosyst 5:793–801. [DOI] [PubMed] [Google Scholar]
- Flippo KH, Strack S (2017a) An emerging role for mitochondrial dynamics in schizophrenia. Schizophr Res 187:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flippo KH, Strack S (2017b) Mitochondrial dynamics in neuronal injury, development and plasticity. J Cell Sci 130:671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funasaka T, Hu H, Yanagawa T, Hogan V, Raz A (2007) Down-regulation of phosphoglucose isomerase/autocrine motility factor results in mesenchymal-to-epithelial transition of human lung fibrosarcoma cells. Cancer Res 67:4236–4243. [DOI] [PubMed] [Google Scholar]
- Ghadimi H, Chou WS, Kesner L (1971) Biosynthesis of saccharopine and pipecolic acid from L- and DL- 14 C-lysine by human and dog liver in vitro. Biochem Med 5:56–66. 10.1016/0006-2944(71)90075-5 [DOI] [PubMed] [Google Scholar]
- Grove J, Henderson LM (1968) The metabolism of D- and L-lysine in the intact rat, perfused liver and liver mitochondria. Biochim Biophys Acta 165:113–120. 10.1016/0304-4165(68)90195-5 [DOI] [PubMed] [Google Scholar]
- Guarnieri FC, de Chevigny A, Falace A, Cardoso C (2018) Disorders of neurogenesis and cortical development. Dialogues Clin Neurosci 20:255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Chen X, Xing R, Wang M, Zhu X, Guo W (2018) Interplay between FMRP and lncRNA TUG1 regulates axonal development through mediating SnoN-Ccd1 pathway. Hum Mol Genet 27:475–485. [DOI] [PubMed] [Google Scholar]
- Gurney M, Heinrich S, Lee M, Yin H (1986a) Molecular cloning and expression of neuroleukin, a neurotrophic factor for spinal and sensory neurons. Science 234:566–574. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Apatoff BR, Spear GT, Baumel MJ, Antel JP, Bania MB, Reder AT (1986b) Neuroleukin: a lymphokine product of lectin-stimulated T cells. Science 234:574–581. 10.1126/science.3020690 [DOI] [PubMed] [Google Scholar]
- Harrison RAP (1974) The detection of hexokinase, glucosephosphate isomerase and phosphoglucomutase activities in polyacrylamide gels after electrophoresis: a novel method using immobilized glucose 6-phosphate dehydrogenase. Anal Biochem 61:500–507. [DOI] [PubMed] [Google Scholar]
- Houten SM, Brinke Ht, Denis S, Ruiter JP, Knegt AC, Klerk JBD, Augoustides-Savvopoulou P, Häberle J, Baumgartner MR, Coşkun T, Zschocke J, Sass JO, Poll-The BT, Wanders RJ, Duran M (2013) Genetic basis of hyperlysinemia. Orphanet J Rare Dis 8:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khacho M, Slack RS (2018) Mitochondrial dynamics in the regulation of neurogenesis: from development to the adult brain. Dev Dyn 247:47–53. 10.1002/dvdy.24538 [DOI] [PubMed] [Google Scholar]
- Kho DH, Nangia-Makker P, Balan V, Hogan V, Tait L, Wang Y, Raz A (2013) Autocrine motility factor promotes HER2 cleavage and signaling in breast cancer cells. Cancer Res 73:1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kho DH, Zhang T, Balan V, Wang Y, Ha SW, Xie Y, Raz A (2014) Autocrine motility factor modulates EGF-mediated invasion signaling. Cancer Res 74:2229–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni VA, Firestein BL (2012) The dendritic tree and brain disorders. Mol Cell Neurosci 50:10–20. [DOI] [PubMed] [Google Scholar]
- Leandro J, Houten SM (2020) The lysine degradation pathway: subcellular compartmentalization and enzyme deficiencies. Mol Genet Metab 131:14–22. [DOI] [PubMed] [Google Scholar]
- Lomenick B, Hao R, Jonai N, Chin R, Aghajan M, Warburton S, Wang JN, Wu R, Gomez F, Loo JA, Wohlschlegel JA, Vondriska TM, Pelletier J, Herschman HR, Clardy J, Clarke CF, Huang J (2009) Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci USA 106:21984–21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalik SJ, Rhead WJ (1989) L-pipecolic acid oxidation in the rabbit and cynomolgus monkey. Evidence for differing organellar locations and cofactor requirements in each species. J Biol Chem 264:2509–2517. [PubMed] [Google Scholar]
- Papes F, Surpili MJ, Langone F, Trigo JR, Arruda P (2001) The essential amino acid lysine acts as precursor of glutamate in the mammalian central nervous system. FEBS Lett 488:34–38. [DOI] [PubMed] [Google Scholar]
- Pena IA, Marques LA, Laranjeira ABA, Yunes JA, Eberlin MN, Arruda P (2016) Simultaneous detection of lysine metabolites by a single LC–MS/MS method: monitoring lysine degradation in mouse plasma. SpringerPlus 5:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena IA, Marques LA, Laranjeira AB, Yunes JA, Eberlin MN, MacKenzie A, Arruda P (2017) Mouse lysine catabolism to aminoadipate occurs primarily through the saccharopine pathway; implications for pyridoxine dependent epilepsy (PDE). Biochim Biophys Acta Mol Basis Dis 1863:121–128. [DOI] [PubMed] [Google Scholar]
- Posset R, Opp S, Struys EA, Volkl A, Mohr H, Hoffmann GF, Kolker S, Sauer SW, Okun JG (2015) Understanding cerebral L-lysine metabolism: the role of L-pipecolate metabolism in Gcdh-deficient mice as a model for glutaric aciduria type I. J Inherit Metab Dis 38:265–272. [DOI] [PubMed] [Google Scholar]
- Rao VV, Pan X, Chang YF (1992) Developmental changes of L-lysine-ketoglutarate reductase in rat brain and liver. Comp Biochem Physiol B 103:221–224. [DOI] [PubMed] [Google Scholar]
- Sacksteder KA, Biery BJ, Morrell JC, Goodman BK, Geisbrecht BV, Cox RP, Gould SJ, Geraghty MT (2000) Identification of the alpha-aminoadipic semialdehyde synthase gene, which is defective in familial hyperlysinemia. Am J Hum Genet 66:1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer SW, Opp S, Hoffmann GF, Koeller DM, Okun JG, Kolker S (2011) Therapeutic modulation of cerebral L-lysine metabolism in a mouse model for glutaric aciduria type I. Brain 134:157–170. 10.1093/brain/awq269 [DOI] [PubMed] [Google Scholar]
- Simell O, Visakorpi JK, Donner M (1972) Saccharopinuria. Arch Dis Child 47:52–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YJ, Chou CC, Chen WS, Wu RT, Meng M, Hsiao CD (1999) The crystal structure of a multifunctional protein: phosphoglucose isomerase/autocrine motility factor/neuroleukin. Proc Natl Acad Sci USA 96:5412–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanie Y, Tanabe N, Kuboyama T, Tohda C (2018) Extracellular neuroleukin enhances neuroleukin secretion from astrocytes and promotes axonal growth in vitro and in vivo. Front Pharmacol 9:1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vianey-Liaud C, Rolland MO, Divry P, Puthet G, Zabot MT, Cotte J (1986) A new case of hyperlysinaemia with saccharopinuria. J Inherit Metab Dis 9:265–267. 10.1007/BF01799724 [DOI] [Google Scholar]
- Watanabe H, Carmi P, Hogan V, Raz T, Silletti S, Nabi IR, Raz A (1991) Purification of human tumor cell autocrine motility factor and molecular cloning of its receptor. J Biol Chem 266:13442–13448. [PubMed] [Google Scholar]
- Watanabe H, Takehana K, Date M, Shinozaki T, Raz A (1996) Tumor cell autocrine motility factor is the neuroleukin/phosphohexose isomerase polypeptide. Cancer Res 56:2960–2963. [PubMed] [Google Scholar]
- Xu W, Seiter K, Feldman E, Ahmed T, Chiao JW (1996) The differentiation and maturation mediator for human myeloid leukemia cells shares homology with neuroleukin or phosphoglucose isomerase. Blood 87:4502–4506. [PubMed] [Google Scholar]
- Zaar K, Angermuller S, Volkl A, Fahimi HD (1986) Pipecolic acid is oxidized by renal and hepatic peroxisomes. Implications for Zellweger's cerebro-hepato-renal syndrome (CHRS). Exp Cell Res 164:267–271. [DOI] [PubMed] [Google Scholar]
- Zhou J, Wang X, Wang M, Chang Y, Zhang F, Ban Z, Tang R, Gan Q, Wu S, Guo Y, Zhang Q, Wang F, Zhao L, Jing Y, Qian W, Wang G, Guo W, Yang C (2019) The lysine catabolite saccharopine impairs development by disrupting mitochondrial homeostasis. J Cell Biol 218:580–597. [DOI] [PMC free article] [PubMed] [Google Scholar]