

ABSTRACT
The presence of cervical lymph node metastases has been considered as the most important adverse prognostic factor for patients with laryngeal squamous cell carcinoma (LSCC). However, the underlying mechanisms remain to be fully revealed. In this study, we explored the expression profile of Foxhead box D1 (FOXD1), its association with epithelial-to-mesenchymal transition (EMT), and its downstream targets in LSCC. Bioinformatic analysis was performed based on the LSCC subset of The Cancer Genome Atlas-Head and Neck Squamous Cell Carcinoma (TCGA-HSNC) and Chromatin immunoprecipitation (ChIP)-seq data from Cistrome Data Browser. LSCC cell lines AMC-HN-8 and TU212 were used for in vitro studies. Results showed that FOXD1 upregulation was associated with poor prognosis of LSCC. FOXD1 knockdown reduced N-cadherin and Vimentin expression but increased E-cadherin expression in AMC-HN-8 cells. Its overexpression showed opposite effects in TU212 cells. FOXD1 could bind to the promoter of ZNF532 and activate its transcription. ZNF532 overexpression enhanced the invasion of both AMC-HN-8 and TU212 cells. In comparison, its knockdown significantly impaired their invasion. ZNF532 knockdown nearly abrogated the alterations of EMT markers caused by FOXD1 overexpression. Its overexpression largely rescued the phenotypes caused by FOXD1 knockdown. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed that ZNF532 correlated genes are largely enriched in extracellular matrix regulations. LSCC patients with high ZNF532 expression (top 50%) had a significantly worse progression-free survival. In summary, this study confirmed that FOXD1 promotes partial-EMT of LSCC cells via transcriptionally activating the expression of ZNF532.
KEYWORDS: Laryngeal squamous cell carcinoma, FOXD1, ZNF532, epithelial-to-mesenchymal transition
Graphical abstract

Introduction
Laryngeal squamous cell carcinoma (LSCC), which arises from the larynx, is a common subtype of head and neck squamous cell carcinoma (HNSCC) [1]. Currently, the primary management strategies for LSCC are surgery and/or radiotherapy [2]. For the patients with early-stage tumors (T1‐2N0M0), radiotherapy alone might be appropriate [2]. The 5-year relative survival rate for localized LSCC is around 77%. However, the rate significantly drops to 44.7% when patients have lymph node metastasis [3]. Therefore, the presence of cervical lymph node metastases has been considered as the most important adverse prognostic factor [4].
Epithelial-to-mesenchymal transition (EMT) enables neoplastic cells to obtain the plasticity and motility for dissemination [5–7]. It is a plastic and reversible process, with a middle hybrid epithelial/mesenchymal or partial-EMT (p-EMT) status in the transition [8]. Both epithelial and mesenchymal marker proteins are expressed in cells with a p-EMT phenotype [8]. Previous studies confirmed that EMT increases LSCC recurrence risk and is associated with significantly shorter disease-free survival (DFS) [5]. Cadherin switching, which refers to a correlation between decreased E-cadherin and elevated N-cadherin expression, is associated with positive lymph nodes in head and neck cancer [9]. One previous single-cell study confirmed that cells with p-EMT spatially localize to the leading edge of primary HNSCCs [10]. Therefore, it is necessary to explore the triggers of EMT or p-EMT of LSCC tumor cells.
Forkhead box D1 (FOXD1) has been identified as one of the mediators of gene expression program changes during the reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) [11]. One recent study indicated that FOXD1 is generally upregulated at the transcriptional level in HNSCC [12]. HNSCC is a group of heterogeneous squamous cell carcinomas derived from different anatomic sites. The subtypes have different genetic backgrounds and epigenetic alterations [13,14]. Some subtypes have distinct genetic features [15,16]. One recent single-cell study confirmed that malignant proliferating cells of the LSCC tumor edge specifically express TPX2, CKS1B, and MKI67 [17]. Cells from the leading edge of primary tongue SCC tumors show a p-EMT program [10]. Therefore, subgroup analysis might be appropriate to focus on the functions of a specific gene. In oral SCC (OSCC), FOXD1 binds to the promoter of a long non-coding RNA (lncRNA) Cytoskeleton Regulator RNA (CYTOR) and activates its transcription, thereby promoting the EMT of OSCC cells [18]. Besides, it promotes the EMT and cancer stem cell properties of OSCC cells via transcriptional activating SNAI2/Slug [19]. However, its functional regulation in LSCC is not clear.
In this study, we hypothesized that FOXD1 might transcriptionally activate the expression of some EMT-related genes. Therefore, we aimed to explore the expression profile of FOXD1, its association with EMT, and its downstream targets in LSCC.
Materials and methods
Bioinformatic analysis
Gene expression profile and survival data of patients with primary LSCC in TCGA-HNSC were extracted from the TCGA pan-cancer dataset, using the UCSC Xena Browser (https://xenabrowser.net/) [20]. The LSCC subset was identified by using the anatomic information of each patient. Progression-free survival (PFS) and disease-specific survival (DSS) data were extracted for survival analysis. Kaplan–Meier (K-M) survival analysis was performed between LSCC patients with high (top 50%) and low (bottom 50%) FOXD1 expression.
Gene set enrichment analysis (GSEA)
GSEA was performed according to the median expression of FOXD1 in LSCC, using GSEA software (v.4.1.0). The h.all.v7.4.symbols.gmt [Hallmarks] in Molecular Signatures Database (MSigDB) was selected as the reference gene set. Gensets with normalized (NOM) p values <0.05 and FDR q value <0.1 were used as the cutoff criteria [21].
Cell culture and treatment
LSCC cell lines AMC-HN-8 and TU177 were purchased from YBIO (Shanghai, China), while TU212 and HaCaT (a human epidermal keratinocyte line) were obtained from Mingzhou Biotechnology (Ningbo, China). These cell lines were cultured following the methods introduced previously [22]. Lentiviral FOXD1 and ZNF532 shRNAs were generated based on the pLKO.1-puro backbone, with the following shRNA sequences: shFOXD1#1, 5ʹ-GCCCTTCTCCATCGAGAGCAT-3ʹ; shFOXD1#2, 5ʹ-TTGTTAATAACGCTATGTTAG-3ʹ; shFOXD1#3, 5ʹ-CGTATATCGCGCTCATCACTA-3ʹ; shZNF532#1, 5ʹ-CGGAAGTTGGAAGAACCAGTT-3ʹ; and shZNF532#2, 5ʹ-GCTGCAATTCCACGAACACAT-3ʹ. Lentiviral FOXD1 (NM_004472) expression vector (FOXD1-OE) and ZNF532 (NM_018181) expression vector (ZNF532-OE) were generated based on the pLenti-CMV backbone. Lentiviruses for infection were produced by co-transfecting the expression vector with the packaging plasmids pMDLg/pRRE and pRSV-Rev and the envelope plasmid pMD2.G into 293 T cells using FuGENE 6 transfection reagent, following the methods described previously [23]. Cells were infected with lentivirus in the presence of polybrene (8 µg/ml). 48 h after infection, the infected cells were selected with 0.7 µg/ml of puromycin (Sigma) for another 48 h to eliminate the non-infected cells.
Western blot assay
Total protein from cell samples was extracted using RIPA Lysis Buffer (Beyotime, Wuhan, China). Protein concentrations were determined using a BCA Protein Assay Kit (TaKaRa, Dalian, China). 20 mg proteins were loaded to each lane and separated by SDS-PAGE. After separation, proteins were transferred to the nitrocellulose (NC) membrane. Then, the membranes were blocked and incubated in primary antibody buffer at 4°C overnight. After that, membranes were washed and incubated in secondary antibody buffer containing HRP-conjugated IgG (Proteintech, Wuhan, China). BeyoECL Star (Beyotime, Shanghai, China) substrate was applied to develop the protein band signals. The band signals were quantified using ImageJ software (NIH, USA) [24], via measuring the integrated optical density from three independent experiments.
The following primary antibodies were used, including anti-FOXD1 (1:1000, PA5-27,142; Thermo Fisher Scientific, Waltham, MA, USA); anti-N-cadherin (1:2000, 22,018-1-AP, Proteintech); anti-Vimentin (1:2000, 10,366-1-AP, Proteintech); anti-E-cadherin (1:5000, 20,874-1-AP, Proteintech); and anti-ZNF532 (1:1000, A305-442A, Bethyl Laboratories, Montgomery, TX, USA).
qRT-PCR
Cell samples were homogenized. Total RNA was extracted using RNAiso Plus reagent (TaKaRa, Beijing, China), following the manufacturer’s instruction [25]. Then, cDNA was synthesized using the PrimeScript RT reagent Kit (TaKaRa) and was subjected to qRT-PCR assay using the SYBR Green Real-Time PCR assay kit (Thermo Fisher Scientific). Relative gene expression was analyzed by the 2−ΔΔCt formula based on three technic repeats. GAPDH expression was used for normalization. The following primers were used: FOXD1, F, 5ʹ- GATCTGTGAGTTCATCAGCGGC-3ʹ; R, 5ʹ-TGACGAAGCAGTCGTTGAGCGA-3ʹ; ZNF532, F, 5ʹ- AGGACGAGACATCACTGGCTAC-3ʹ; R, 5ʹ-GTGCTGATGGATTCTCTGGTGTG-3ʹ; GAPDH, F, 5ʹ- GTCTCCTCTGACTTCAACAGCG-3ʹ; R, 5ʹ- ACCACCCTGTTGCTGTAGCCAA-3ʹ;
Transwell assay of invasion
Cell invasion capability was conducted using sterile 6.5 mm Transwell with 8.0 µm pore polycarbonate membrane inserts (#3422, Corning, Cambridge, MA, USA) [26]. In brief, the polycarbonate membrane was coated with reconstituted basement membrane Matrigel (1 mg/mL; BD Biosciences, San Jose, CA, USA). The lower chambers were filled with culture medium containing 10% FBS. 1 × 104 cells with FOXD1 or ZNF532 knockdown or overexpression in 200 μL serum-free medium was inoculated into the upper chamber. After 24 h incubation at 37°C, cells were fixed with paraformaldehyde and stained with 0.1% crystal violet. The numbers of invading cells were counted from five randomly selected visual fields using compound light microscopy (200× magnification).
Immunofluorescence (IF) staining
AMC-HN-8 and TU212 cells were seeded into 24 cells plates with coverslips. At approximately 30% of confluence, cell slides were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked with goat serum, and then incubated with anti-ZNF532 (1:1000, HPA015322, Sigma-Aldrich, Burlington, MA, USA) at 4°C overnight. After washing with PBS, the cells were incubated with Alexa Fluor 488 conjugated secondary antibody (1:1000, srbAF488-1, Proteintech, Wuhan, China). Nuclei were counterstained with 4ʹ6-diamidino-2-phenylindole (DAPI) [27]. IF images were captured using an Olympus IX83 inverted microscope (Olympus, Tokyo, Japan).
Chromatin immunoprecipitation (ChIP)-qPCR assay
ChIP was performed using ChIP Assay Kit (Beyotime, Shanghai, China), following the manufacturer’s instructions. In brief, cell samples were lysed, cross-linked in 1% formaldehyde, and then quenched. Chromatin fragmentation was performed by sonication. Proteins were immunoprecipitated in ChIP dilution buffer, using anti-FOXD1 antibody (PA5-27,142; Thermo Fisher Scientific) or nonspecific rabbit IgG control (Proteintech). Cross-linking was reversed overnight at 65°C. DNA samples were isolated using phenol/chloroform/isoamyl alcohol and then were subjected to qRT-PCR assay [28]. The following primers were used for ChIP-qPCR assay, for amplicon 1: F, 5ʹ-GTGCTGCTTTGCTGTCATTT-3ʹ; R, 5ʹ-AGGTTTCATGGATTTGGGATCT-3ʹ; for amplicon 2: F, 5ʹ-TTGTGTTCCGCAGGCTATG-3ʹ; R, 5ʹ-AGCTAGGTCTTTAAATTCTAGAAGGG-3ʹ.
Dual-luciferase assay
DNA fragments of wild-type (WT) and mutant ZNF532 gene promoters were chemically synthesized and subcloned into the firefly luciferase reporter vector (pGL3-basic), between the KpnI and HindIII sites to construct expression vectors. The recombinant plasmids were transiently transfected into AMC-HN-8 with or without FOXD1 knockdown or into TU212 cells with or without FOXD1 overexpression. 48 h post-transfection, the cells were collected and lysed to measure luciferase activities, using a Promega Glomax 20/20 luminometer. pRL-TK expressing renilla luciferase was utilized as an internal control for transfection efficiency, following the method introduced previously [29]. All the experiments were repeated three times independently, in triplicate.
Functional enrichment analysis of ZNF532 correlated genes in LSCC
Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO, including GO: biological process, GO: cellular component, and GO: molecular function) databases were utilized to explore the functional enrichment analysis of ZNF532 correlated genes (|Spearman’s r|>0.5, p < 0.001) in primary LSCC cases in TCGA-HNSC, using an R package ‘clusterProfiler’ [30]. Only the terms/pathways with a false discovery rate (FDR) <0.05 were considered significantly enriched in this work. GO terms and KEGG pathways with q value (corrected p value) ≤0.05 were considered as significantly enriched.
Statistical analysis
Statistical analysis was performed on GraphPad Prism 8.1. All values are shown as mean ± standard deviation (SD). Welch’s unpaired t-test was used to assess the difference between the two groups. For multiple group comparison, one-way analysis of variance followed by Dunnett’s post hoc test was performed. p value less than 0.05 was considered statistically different.
Results
In the current study, we tried to explore the downstream transcriptional target of FOXD1 in modulating the EMT of LSCC cells. A systematic bioinformatic screening was conducted. Subsequent cellular and molecular studies were then performed to validate the transcriptional regulation of FOXD1.
FOXD1 expression was associated with unfavorable survival of LSCC
To characterize the expression of FOXD1 in LSCC, we extracted RNA-seq data of tumors arising from the larynx and corresponding adjacent (adj.) normal tissues from TCGA-HNSC. Briefly, 116 LSCC and 12 adj. normal tissues were identified (Figure 1a). FOXD1 expression was significantly higher in the tumor group, compared to the normal group (Figure 1a). In combination with survival data (PFS and DSS) extracted from TCGA-HNSC, we observed that LSCC patients with high FOXD1 expression (top 50%) had significantly worse PFS and DSS, compared to the cases with low FOXD1 expression (Figure 1b-c).
Figure 1.
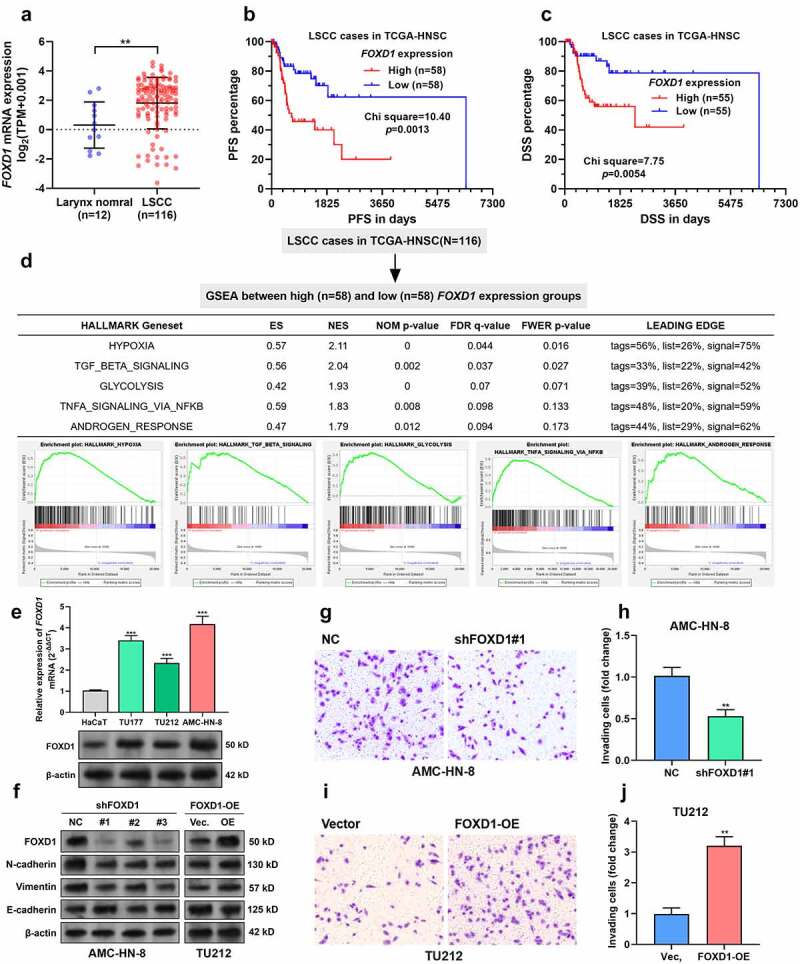
FOXD1 expression was associated with unfavorable survival of LSCC.
A. FOXD1 expression between primary LSCC (n = 116) and corresponding adj. normal tissues (n = 12). Data were extracted from TCGA-HNSC.B-C. The differences in PFS (B) and DSS (C) between LSCC patients with high (top 50%) and low (bottom 50%) FOXD1 expression were compared. D. GSEA was conducted between LSCC patients with high (top 50%) and low (bottom 50%) FOXD1 expression. The group separation (top) and results (bottom) were provided. E. FOXD1 mRNA (top) and protein (bottom) in an immortalized human epidermal cell line HaCaT and three LSCC cell lines, TU177, TU212 and AMC-HN-8. F. AMC-HN-8 and TU212 were subjected to lentiviral mediated FOXD1 knockdown or overexpression, respectively. 48 h after puromycin selection, cells were subjected to Western blot assays to detect the expression of FOXD1, N-cadherin, Vimentin, and E-cadherin. G-J. Representative image and quantitation (n = 3) of invading AMC-HN-8 cells with or without FOXD1 knockdown (G-H) or TU212 cells with or without FOXD1 overexpression (I-J). **, p < 0.01; ***, p < 0.001.
FOXD1 upregulation was associated with p-EMT of LSCC cells
Then, we performed GSEA to check the difference in gene set enrichment between high and low FOXD1 expression groups by setting NOM p < 0.05 and FDR q < 0.1 as the threshold. Results showed that LSCC with high FOXD1 expression had elevated genes enriched in hypoxia, transforming growth factor (TGF)-β signaling, glycolysis, tumor necrosis factor-α (TNF-α) signaling via NF-κB and androgen response (Figure 1d). Interestingly, hypoxia-induced EMT [31], TGF-β-induced EMT [32], and TNF-α/nuclear factor-kappa B (NF-κB)-mediated EMT [33] have been observed in LSCC. Therefore, we hypothesized that FOXD1 might also act as an important modulator of EMT in LSCC.
Using three LSCC cell lines, TU177, TU212 and AMC-HN-8, and an immortalized human epidermal cell line HaCaT, we confirmed FOXD1 upregulation at both mRNA and protein levels in the LSCC cell lines (Figure 1e). Among the three LSCC cell lines, AMC-HN-8 had the highest, while TU212 had the lowest FOXD1 expression (Figure 1e). Therefore, AMC-HN-8 were subjected to lentiviral-mediated FOXD1 knockdown. Meanwhile, TU212 were infected for FOXD1 overexpression (Figure 1f). Western blot data showed that FOXD1 knockdown reduced N-cadherin and Vimentin expression but increased E-cadherin expression in AMC-HN-8 cells (Figure 1f). In comparison, FOXD1 overexpression enhanced N-cadherin and Vimentin expression but reduced E-cadherin expression in TU212 cells (Figure 1f). These findings suggest that FOXD1 can induce p-EMT in LSCC tumor cells. In addition, transwell assay results indicated that FOXD1 knockdown weakened the invasion capability of AMC-HN-8 cells (Figure 1g-h). In contrast, FOXD1 overexpression stimulated the invasion of TU212 cells (Figure 1i-j).
FOXD1 transcriptionally activates ZNF532 expression in LSCC cells
Since FOXD1 is a transcriptional factor, we further explored its downstream transcriptional targets. FOXD family members shared some common binding motif (such as 5ʹ-GTAAACA-3ʹ). ChIP-seq data (GSE93323) [34] in Cistrome Data Browser were used to explore the potential FOXD binding site in epithelial cells. Fifteen genes with the highest scores of potential promoter-binding sites were identified (Figure 2a). Then, the correlation between the expression of these genes and FOXD1 was assessed in primary LSCC cases (Figure 2b). The following three criteria were applied to identify the high potential target genes: 1. significantly upregulated in LSCC compared to adj. normal tissues; 2. positively correlated with FOXD1 expression (Spearman’s r > 0.2); 3. With high-potential FOXD1 binding sites in the gene promoter regions (threshold score >0.9 in Jaspar database). After the screening, ZNF532 was identified as a high potential target. IF staining confirmed the nucleus accumulation of ZNF532 in AMC-HN-8 and TU212 cells (Figure 2c). QRT-PCR and Western blot assays confirmed that FOXD1 knockdown decreased ZNF532 expression (Figures 2d and f). In comparison, FOXD1 overexpression enhanced ZNF532 expression (Figures 2e-f). In the promoter of ZNF532, there is a 100% match FOXD1 binding site (−1163 to −1154, 5ʹ-GTAAACAA-3ʹ) (Figure 2g). To verify the binding of FOXD1 to the ZNF532 promoter, we design two sets of primers for ChIP-qPCR assay, among which amplicon 1 covers the predicted binding sites while amplicon 2 does not (Figure 2h). ChIP-qPCR results confirmed a significantly higher level of amplicon 1 in the samples immunoprecipitated by anti-FOXD1 (Figure 2i-j). However, this trend was not observed in amplicon 2 (Figure 2i-j). Then, pGL3-basic plasmids carrying integrated wild-type ZNF532 promoter (pGL3-WT) or mutant ZNF532 promoter (−1163 to −1154, 5ʹ-GTCCCCAA-3ʹ) (pGL3-WT) were constructed. Dual-luciferase assay showed that FOXD1 knockdown significantly suppressed the luciferase activity of pGL3-WT, but not pGL3-MT in AMC-HN-8 cells (Figure 2k). In comparison, FOXD1 overexpression significantly increased the luciferase activity of pGL3-WT, but not pGL3-MT in TU212 cells (Figure 2l).
Figure 2.
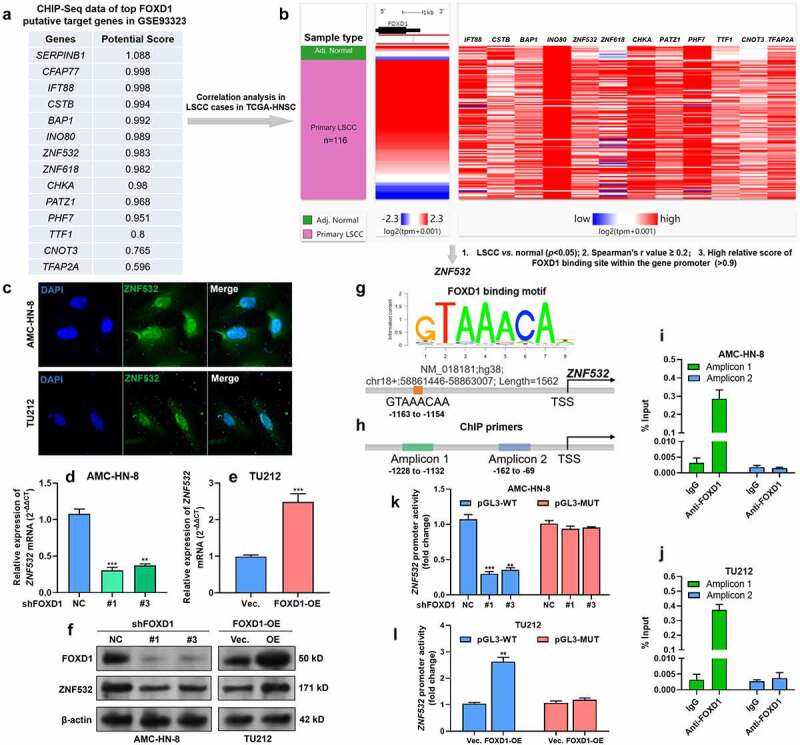
FOXD1 upregulation was associated with EMT of LSCC cells.
A. Top genes with potential FOXD binding sites in epithelial cells (GSE93323) in Cistrome Data Browser. B. A heatmap showing the correlation between the expression of FOXD1 and the predicted FOXD1 promoter binding genes in LSCC cases in TCGA-HNSC.C. IF staining of ZNF532 expression in AMC-HN-8 and TU212 cells. D-F. QRT-PCR (D-E) and Western blot (F) analysis of ZNF532 expression in AMC-HN-8 and TU212 cells with FOXD1 knockdown or overexpression. G. A schematic diagram showing the binding motif of FOXD1 and its potential binding site in the promoter region of ZNF532. H. A schematic diagram showing the position of amplicons designed for ChIP-qPCR analysis. I-J. ChIP-qPCR showing the relative enrichment (% of input) of ZNF532 promoter fragments in the DNA samples immunoprecipitated by anti-FOXD1 in AMC-HN-8 (I) and TU212 (J) cells. K-L. Dual-luciferase assay was performed to analyze the relative luciferase activity of pGL3-WT and pGL3-MT reporter plasmids in AMC-HN-8 with FOXD1 knockdown (K) and TU212 cells with FOXD1 overexpression (L). **, p < 0.01; ***, p < 0.001.
ZNF532 is a downstream effector of FOXD1 in modulating the EMT of LSCC cells
Then, we performed transwell assays to explore the influence of ZNF532 expression on LSCC invasion. ZNF532 overexpression enhanced the invasion of AMC-HN-8 and TU212 cells (Figure 3a-b). In comparison, its knockdown significantly impaired the invasion of these two cell lines (Figure 3a-b). Therefore, we infer that ZNF532 might be an important downstream effector of FOXD1 in promoting p-EMT of LSCC cells. To validate this hypothesis, we conducted Western blot assays to check the alteration of EMT markers. In AMC-HN-8 cells, ZNF532 overexpression did not alter FOXD1 protein expression (Figure 3c) but increased the expression of N-cadherin, Vimentin, and Slug (Figure 3c-f). Although FOXD1 shRNA could reduce the expression of mesenchymal markers (Figure 1f), it could not reverse ZNF532-induced upregulation of the mesenchymal markers (Figure 3c-f). ZNF532 overexpression significantly reduced the expression of N-cadherin (Figure 3c and g). Although FOXD1 shRNA slightly diminished the effect of ZNF532 overexpression, it could not reverse ZNF532-induced downregulation of N-cadherin (Figure 3c and g). In TU212 cells, ZNF532 shRNA reduced the expression of mesenchymal markers and increased the expression of E-cadherin (Figure 3h-l). FOXD1 overexpression could not reverse ZNF532 shRNA-induced downregulation of mesenchymal markers (Figure 3h-k). Although FOXD1 overexpression abrogated ZNF532 shRNA-induced upregulation of E-cadherin, it could not reduce E-cadherin expression compared to the control group (Figures 3h and l).
Figure 3.

ZNF532 is a downstream effector of FOXD1 in modulating the p-EMT of LSCC cells.
A-B. Representative image (A) and quantitation (B) (n = 3) of invading AMC-HN-8 and TU212 cells with FOXD1 overexpression or knockdown. C-L. Representative image (C and H) and quantitation (D-G and H-L) of the expression of mesenchymal markers (N-cadherin, Vimentin, and Slug) and epithelial marker (E-cadherin) in AMC-HN-8 cells with ZNF532 overexpression alone or in combination with FOXD1 knockdown (C-G) and TU212 cells with ZNF532 knockdown alone or in combination with FOXD1 overexpression (H-L). The protein levels between the first column and other experimental columns were compared by Welch’s unpaired t-test. Quantitation was performed based on the results of three biological repeats. *, p < 0.05; **, p < 0.01; ***, p < 0.001. FC: fold change.
ZNF532 expression was correlated with poor survival of LSCC
Only limited studies explored the role of ZNF532 in carcinogenesis [35–37]. It is generally considered as a potential transcriptional regulator [36]. Therefore, its regulatory effects might be largely determined by its downstream effectors. By performing correlation analysis, we identified the top genes (n = 156, Supplementary Table 1) highly co-expressed (|Spearman’s r|>0.5, p < 0.001) with ZNF532 (Figure 4a). Then, these genes were subjected to GO and KEGG analysis. Results showed that these genes are largely enriched in extracellular matrix regulations, such as collagen-containing extracellular matrix, extracellular structure organization, and extracellular matrix organization (Figure 4b, Table 1). These findings imply that ZNF532 might have an i+mportant role in regulating extracellular matrix, which is closely associated with tumor cell invasion. Survival analysis showed that LSCC patients with high ZNF532 expression (top 50%) also had significantly worse PFS (p = 0.035) (Figure 4c). However, no significant difference (p = 0.34) was observed in DSS (Figure 4d).
Figure 4.

ZNF532 expression was correlated with poor survival of LSCC.
A. A heatmap showing the genes highly correlated with ZNF532 expression (|Spearman’s r|≥0.5, p < 0.001) in primary LSCC cases (n = 116) in TCGA-HNSC.B. The bubble map showing the enrichment of ZNF532 correlated genes in GO and KEGG terms. Y-axis represents pathways, and X-axis represents gene ratio (gene ratio = amount of ZNF532 correlated genes enriched in the pathway/amount of all genes in background gene set). The color depends on the p-value, and the size of bubbles represents the number of enriched genes. C-D. The differences in PFS (C) and DSS (D) between LSCC patients with high (top 50%) and low (bottom 50%) ZNF532 expression were compared.
Table 1.
The enrichment of GO/KEGG terms of ZNF532 correlated genes in 116 primary LSCC tumors
| Ontology | Pathway ID | Description | Gene Ratio | Bg Ratio | p value | p. adjust | Q value |
|---|---|---|---|---|---|---|---|
| BP | GO:0030198 | extracellular matrix organization | 29/147 | 368/18,670 | 5.93e-21 | 1.46e-17 | 1.23e-17 |
| BP | GO:0043062 | extracellular structure organization | 29/147 | 422/18,670 | 2.61e-19 | 3.20e-16 | 2.69e-16 |
| BP | GO:0030199 | collagen fibril organization | 11/147 | 54/18,670 | 3.54e-13 | 2.90e-10 | 2.43e-10 |
| CC | GO:0062023 | collagen-containing extracellular matrix | 30/153 | 406/19,717 | 5.48e-21 | 1.39e-18 | 1.26e-18 |
| CC | GO:0005581 | collagen trimer | 10/153 | 87/19,717 | 1.41e-09 | 1.79e-07 | 1.62e-07 |
| CC | GO:0098644 | complex of collagen trimers | 6/153 | 19/19,717 | 4.94e-09 | 3.32e-07 | 3.01e-07 |
| MF | GO:0005201 | extracellular matrix structural constituent | 23/142 | 163/17,697 | 2.26e-22 | 7.49e-20 | 6.69e-20 |
| MF | GO:0030020 | extracellular matrix structural constituent conferring tensile strength | 9/142 | 41/17,697 | 3.01e-11 | 4.98e-09 | 4.45e-09 |
| MF | GO:0005518 | collagen binding | 8/142 | 67/17,697 | 6.17e-08 | 6.80e-06 | 6.08e-06 |
| KEGG | hsa04512 | ECM-receptor interaction | 9/74 | 88/8076 | 8.86e-08 | 1.42e-05 | 1.31e-05 |
| KEGG | hsa04974 | Protein digestion and absorption | 9/74 | 103/8076 | 3.49e-07 | 2.79e-05 | 2.57e-05 |
| KEGG | hsa04510 | Focal adhesion | 11/74 | 201/8076 | 1.83e-06 | 9.77e-05 | 9.00e-05 |
Discussion
Emerging studies showed that FOXD1 upregulation has critical roles in promoting cancer progression via enhancing EMT, including HNSCC [18,19,38]. Bioinformatic analysis in the current study showed that FOXD1 upregulation was associated with poor prognosis of LSCC. GSEA data imply that high FOXD1 expression might be positively correlated with EMT activity of LSCC cells. Our subsequent molecular studies confirmed that FOXD1 promotes p-EMT and invasion of LSCC cells. These findings imply that FOXD1 might exert similar regulations in HNSCC cells.
One recent study found that the lncRNA FOXD1-AS1 can increase FOXD1 via sponging miR-369-3p and adenosine deaminase RNA specific (ADAR) and reducing their negative influence on FOXD1 mRNA stability in OSCC cells [39]. However, the downstream regulation in LSCC remains poorly understood. Therefore, we explored its downstream targets in LSCC. Via combined use of previous ChIP-seq data (GSE93323), RNA-seq data from TCGA-HNSC, and binding motif prediction data from Jaspar, we identified ZNF532 as a high potential target of FOXD1. Following experimental data confirmed a FOXD1 binding site in the ZNF532 promoter. Via binding to this site, FOXD1 can elevate the transcription of ZNF532.
Very limited studies explored the functional role of ZNF532 in cancer. Previous studies reported that ZNF532 participates in the development of nuclear protein in testis (NUT) squamous carcinoma, and epithelioid and rhabdoid tumor of bone by forming fusion protein with NUT [35,37]. Besides, it is a component of a transcriptional coregulator complex, together with other three zinc finger proteins-ZNF592, ZNF687, and ZMYND8 [35]. This complex is involved in feed-forward regulatory loops that drive tumor cell proliferation [35]. Two recent bioinformatic analyses identified that ZNF532 upregulation was also associated with poor prognosis of hepatocellular carcinoma [40] and rectal adenocarcinoma [41]. In this study, we further explored the functional role of ZNF532 in LSCC cells. Transwell assay confirmed that its expression was positively correlated with invasion capability and the expression of mesenchymal markers. ZNF532 knockdown nearly abrogated the EMT marker alteration caused by FOXD1 overexpression, while its overexpression largely rescued the phenotypes caused by FOXD1 knockdown. These findings imply that it acts as a downstream effector of FOXD1 in modulating the p-EMT of LSCC cells.
GO annotation indicated that ZNF532 has a nucleic acid binding capability. Therefore, it may be involved in transcriptional regulation. However, the detailed binding site and transcriptional regulations remain mysterious. To support future exploration of the potential transcriptional regulation of ZNF532 in LSCC, we identified genes correlated with its expression. GO and KEGG analysis indicated that these co-expressed genes were largely enriched in extracellular matrix regulations, which is closely associated with tumor cell metastasis. Proteases pave the way for invading tumor cells via degrading extracellular matrix, which maintains epithelial cell–cell contact [42]. Then, the degradation of adherens junctions is further enhanced by the transcriptional repression of epithelial-specific genes (such as E-cadherin, Occludin, and Desmoplakin) [42]. These alterations are partially induced by the upregulation of pro-EMT transcription factors such as Snail1, Snail2/Slug and Twist1 [42]. During EMT, some components of epithelial adherens junctions are replaced by mesenchymal proteins such as N-Cadherin, which generates an intracellular environment supporting cell detachment and moving [42]. Therefore, ZNF532 might promote p-EMT via regulating the extracellular matrix. However, future molecular studies are required to validate this hypothesis.
Conclusion
In summary, this study confirmed that FOXD1 promotes p-EMT of LSCC cells partially via transcriptionally activating the expression of ZNF532. Elevated FOXD1 and ZNF532 expression might predict unfavorable survival of LSCC.
Supplementary Material
Funding Statement
The author(s) reported that there is no funding associated with the work featured in this article.
Data availability
The data and material used to support the findings of this study are available from the corresponding author upon request.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Highlights
FOXD1 expression was associated with unfavorable survival of LSCC.
FOXD1 upregulation was associated with EMT of LSCC cells
ZNF532 is a downstream effector of FOXD1 in modulating the EMT of LSCC cells.
ZNF532 expression was correlated with poor survival of LSCC.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- [2].Du Y, Shao S, Lv M, et al. Radiotherapy versus surgery-which is better for patients with T1-2N0M0 glottic laryngeal squamous cell carcinoma? Individualized survival prediction based on web-based nomograms. Front Oncol. 2020;10:1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bradford CR, Ferlito A, Devaney KO, et al. Prognostic factors in laryngeal squamous cell carcinoma. Laryngoscope Investig Otolaryngol. 2020;5(1):74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Adam MA, Pura J, Goffredo P, et al. Presence and number of lymph node metastases are associated with compromised survival for patients younger than age 45 years with papillary thyroid cancer. J Clin Oncol. 2015;33(21):2370–2375. [DOI] [PubMed] [Google Scholar]
- [5].Cappellesso R, Marioni G, Crescenzi M, et al. The prognostic role of the epithelial-mesenchymal transition markers E-cadherin and Slug in laryngeal squamous cell carcinoma. Histopathology. 2015;67(4):491–500. [DOI] [PubMed] [Google Scholar]
- [6].Luo H, Xia X, Kim GD, et al. Characterizing dedifferentiation of thyroid cancer by integrated analysis. Sci Adv. 2021;7(31). DOI: 10.1126/sciadv.abf3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zheng S, Wu R, Deng Y, et al. Dihydroartemisinin represses oral squamous cell carcinoma progression through down-regulating mitochondrial calcium uniporter. Bioengineered. 2022;13(1):227–241 . [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [8].Nieto MA, Huang RY, Jackson RA, et al. Emt: 2016. Cell. 2016;166(1):21–45. [DOI] [PubMed] [Google Scholar]
- [9].Smith A, Teknos TN, Pan Q. Epithelial to mesenchymal transition in head and neck squamous cell carcinoma. Oral Oncol. 2013;49(4):287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Puram SV, Tirosh I, Parikh AS, et al. Single-Cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171(7):1611–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Koga M, Matsuda M, Kawamura T, et al. Foxd1 is a mediator and indicator of the cell reprogramming process. Nat Commun. 2014;5(1):3197. [DOI] [PubMed] [Google Scholar]
- [12].Huang J, Liang B, Wang T. FOXD1 expression in head and neck squamous carcinoma: a study based on TCGA, GEO and meta-analysis. Biosci Rep. 2021;41(7). DOI: 10.1042/BSR20210158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Solomon B, Young RJ, Rischin D. Head and neck squamous cell carcinoma: genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin Cancer Biol. 2018;52(Pt 2):228–240. [DOI] [PubMed] [Google Scholar]
- [14].Ledgerwood LG, Kumar D, Eterovic AK, et al. The degree of intratumor mutational heterogeneity varies by primary tumor sub-site. Oncotarget. 2016;7(19):27185–27198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Johnson DE, Burtness B, Leemans CR, et al. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2020;6(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lleras RA, Smith RV, Adrien LR, et al. Unique DNA methylation loci distinguish anatomic site and HPV status in head and neck squamous cell carcinoma. Clin Cancer Res. 2013;19(19):5444–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Song L, Zhang S, Yu S, et al. Cellular heterogeneity landscape in laryngeal squamous cell carcinoma. Int J Cancer. 2020;147(10):2879–2890. [DOI] [PubMed] [Google Scholar]
- [18].Chen S, Yang M, Wang C, et al. Forkhead box D1 promotes EMT and chemoresistance by upregulating lncRNA CYTOR in oral squamous cell carcinoma. Cancer Lett. 2021;503:43–53. [DOI] [PubMed] [Google Scholar]
- [19].Chen Y, Liang W, Liu K, et al. FOXD1 promotes EMT and cell stemness of oral squamous cell carcinoma by transcriptional activation of SNAI2. Cell Biosci. 2021;11(1):154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Goldman MJ, Craft B, Hastie M, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38(6):675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yuan L, Tian X, Zhang Y, et al. LINC00319 promotes cancer stem cell-like properties in laryngeal squamous cell carcinoma via E2F1-mediated upregulation of HMGB3. Exp Mol Med. 2021;53(8):1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Leong WZ, Tan SH, Ngoc PCT, et al. ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Genes Dev. 2017;31(23–24):2343–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Steinau M, Rajeevan MS, Unger ER. DNA and RNA references for qRT-PCR assays in exfoliated cervical cells. J Mol Diagn. 2006;8(1):113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pijuan J, Barcelo C, Moreno DF, et al. In vitro cell migration, invasion, and adhesion assays: from cell imaging to data analysis. Front Cell Dev Biol. 2019;7:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Donaldson JG. Immunofluorescence staining. Curr Protoc Cell Biol. 2001;Chapter 4:Chapter 4:Unit 4 3 10.1002/0471143030.cb0403s00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schoppee Bortz PD, Wamhoff BR, Dalal Y. Chromatin immunoprecipitation (ChIP): revisiting the efficacy of sample preparation, sonication, quantification of sheared DNA, and analysis via PCR. PLoS One. 2011;6(10):e26015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mei X, Din H, Zhao J, et al. Transcription factor Kruppel-like factor 5-regulated N-myc downstream-regulated gene 2 reduces IL-1beta-induced chondrocyte inflammatory injury and extracellular matrix degradation. Bioengineered. 2021;12(1):7020–7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zuo J, Wen J, Lei M, et al. Hypoxia promotes the invasion and metastasis of laryngeal cancer cells via EMT. Med Oncol. 2016;33(2):15. [DOI] [PubMed] [Google Scholar]
- [32].Song D, Wang L, Su K, et al. WISP1 aggravates cell metastatic potential by abrogating TGF-beta-Smad2/3-dependent epithelial-to-mesenchymal transition in laryngeal squamous cell carcinoma. Exp Biol Med (Maywood). 2021;246(11):1244–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhu Y, Yan L, Zhu W, et al. MMP2/3 promote the growth and migration of laryngeal squamous cell carcinoma via PI3K/Akt-NF-kappaB-mediated epithelial-mesenchymal transformation. J Cell Physiol. 2019. [DOI] [PubMed]
- [34].Zheng R, Wan C, Mei S, et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019;47(D1):D729–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shiota H, Elya JE, Alekseyenko AA, et al. “Z4” complex member fusions in NUT carcinoma: implications for a novel oncogenic mechanism. Mol Cancer Res. 2018;16(12):1826–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Alekseyenko AA, Walsh EM, Zee BM, et al. Ectopic protein interactions within BRD4-chromatin complexes drive oncogenic megadomain formation in NUT midline carcinoma. Proc Natl Acad Sci U S A. 2017;114(21):E4184–E4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chien YW, Hsieh TH, Chu PY, et al. Primary malignant epithelioid and rhabdoid tumor of bone harboring ZNF532-NUTM1 fusion: the expanding NUT cancer family. Genes Chromosomes Cancer. 2019;58(11):809–814. [DOI] [PubMed] [Google Scholar]
- [38].Ma Q, Yang T. E2F transcription factor 1/small nucleolar RNA host gene 18/microRNA-338-5p/forkhead box D1: an important regulatory axis in glioma progression. Bioengineered. 2022;13(1):418–430 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ma Y, Han J, Luo X. FOXD1-AS1 upregulates FOXD1 to promote oral squamous cell carcinoma progression. Oral Dis. 2021. DOI: 10.1111/odi.14002 [DOI] [PubMed] [Google Scholar]
- [40].Zhang Q, Wang J, Liu M, et al. Weighted correlation gene network analysis reveals a new stemness index-related survival model for prognostic prediction in hepatocellular carcinoma. Aging (Albany NY). 2020;12(13):13502–13517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yi X, Zhou Y, Zheng H, et al. Prognostic targets recognition of rectal adenocarcinoma based on transcriptomics. Medicine (Baltimore). 2021;100(32):e25909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and material used to support the findings of this study are available from the corresponding author upon request.