

In this review, Hopkins et al. review the major classes of DNA repair and damage signaling defects in cancer, the genomic instability that they give rise to, and therapeutic strategies to exploit the resulting vulnerabilities. They also discuss the impacts of DNA repair defects on both targeted therapy and immunotherapy, and highlight emerging principles for targeting DNA repair defects in cancer therapy.
Keywords: ATM, ATR, BRCA, cancer, DNA damage, DNA repair, genomic instability, immunotherapy, PARP, targeted therapy
Abstract
DNA repair and DNA damage signaling pathways are critical for the maintenance of genomic stability. Defects of DNA repair and damage signaling contribute to tumorigenesis, but also render cancer cells vulnerable to DNA damage and reliant on remaining repair and signaling activities. Here, we review the major classes of DNA repair and damage signaling defects in cancer, the genomic instability that they give rise to, and therapeutic strategies to exploit the resulting vulnerabilities. Furthermore, we discuss the impacts of DNA repair defects on both targeted therapy and immunotherapy, and highlight emerging principles for targeting DNA repair defects in cancer therapy.
Human cells rely on a complex network of DNA repair and DNA damage signaling pathways, called the DNA damage response (DDR), to remove DNA lesions and aberrant structures from the genome and maintain genomic stability (Ciccia and Elledge 2010). The DDR pathways deal with not only the DNA damage generated by extrinsic insults, but also genomic problems arising from intrinsic cellular processes. In cancer cells, some of the DDR pathways are commonly compromised, allowing genomic instability to accumulate. On the one hand, genomic instability fuels tumorigenesis. On the other hand, the loss of DDR pathways in cancer cells renders them vulnerable to DNA damage and additional defects in the DDR network. Targeting specific DDR proteins in cancer cells harboring DDR defects or high levels of genomic instability can lead to synthetic lethality, providing a promising strategy for cancer therapy (O'Connor 2015). The DDR defects of cancer cells also affect the immune response to tumors, implicating the DDR as a modulator of immunotherapy (Pilger et al. 2021). In this review, we discuss the major classes of DDR defects in cancer, new developments in targeting the DDR pathways, and new strategies to exploit DDR defects in cancer therapy, providing an updated view of the implications of DDR defects in cancer and cancer treatment.
Defects of DDR pathways in cancer
Mutations in a variety of genes encoding DNA repair and damage signaling proteins have been detected in human cancers. While many of these mutations are likely passenger events without a functional impact, some mutations impair the DDR and may contribute to tumorigenesis. The DDR defects resulting from the mutations can be divided into three classes, as discussed below.
Class I: defects in repair of DSBs and replication-associated DNA damage
Mutations in the tumor suppressor genes BRCA1 and BRCA2 are frequently found in several cancer types, including breast, ovarian, prostate, and pancreatic cancers (Venkitaraman 2002; Mersch et al. 2015). Both BRCA1 and BRCA2 genes encode proteins critical for DNA double-stranded break (DSB) repair through homologous recombination (HR) (Chen et al. 2018a). BRCA1 forms a complex with BARD1, which is rapidly recruited to sites of DNA damage after DSB formation (Fig. 1). At DSBs, BRCA1 regulates the formation of single-stranded DNA (ssDNA) and recruits the PALB2–BRCA2 complex. The PALB2–BRCA2 complex displaces the replication protein A (RPA) and assembles RAD51 filaments, a crucial HR intermediate, on ssDNA (Zhao et al. 2015). BRCA1/2-deficient cells cannot repair collapsed DNA replication forks properly. Both BRCA1 and BRCA2 restrict “long-tract” gene conversion, an error-prone repair process (Willis et al. 2014). BRCA1 also suppresses tandem duplications at stalled forks (Willis et al. 2017). Mutations in other HR genes, such as PALB2, BRIP1, RAD51C, and RAD51D, are also found in cancers (Antoniou et al. 2014; Suszynska et al. 2020). These findings suggest that the genomic instability in HR-deficient cancer cells may arise from defective repair of DSBs and collapsed replication forks.
Figure 1.
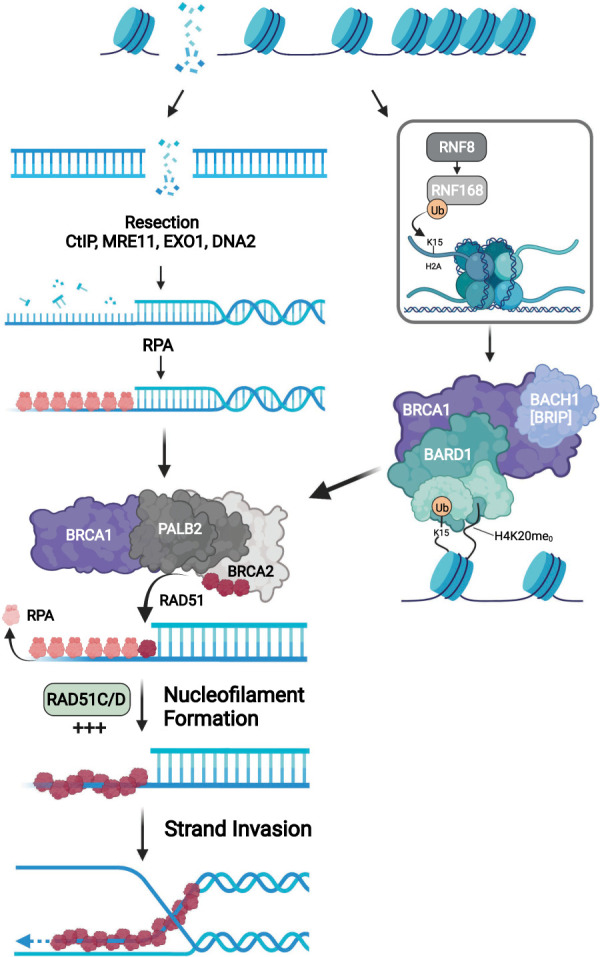
Functions of BRCA1/2 in homologous recombination. The BRCA1–BARD1 complex is recruited to the DSB-flanking chromatin marked by H2A K15ub and H4 K20me0 (Becker et al. 2021; Hu et al. 2021). The BRCA1 at DSBs recruits PALB2–BRCA2 to promote the assembly of RAD51 filaments, which enables D-loop formation following the resection by MRE11-CtIP and EXO1 or MRE11-CtIP and DNA2.
In addition to HR, BRCA1 and BRCA2 are important for protecting stalled replication forks (Fig. 2; Schlacher et al. 2011, 2012). Replication forks undergo a remodeling process upon stress, giving rise to four-way DNA structures called reversed forks (Neelsen and Lopes 2015; Quinet et al. 2017). In cells lacking BRCA1/2, the nascent DNA at reversed forks is increasingly degraded by the MRE11 and EXO1 nucleases (Fig. 2; Lemaçon et al. 2017). The degradation of nascent DNA in BRCA2-deficient cells forces forks to recover through a MUS81- and POLD3-mediated pathway, which is associated with transient DSBs and fork instability (Fig. 2). Several groups reported that BRCA1/2-deficient cells display increased levels of ssDNA gaps in nascent DNA (Fig. 2; Cong et al. 2021; Kang et al. 2021; Paes Dias et al. 2021; Simoneau et al. 2021; Taglialatela et al. 2021). PrimPol, which reprimes for DNA synthesis ahead of stalled DNA polymerases, promotes the formation of ssDNA gaps in BRCA1/2-deficient cells (Quinet et al. 2020; Kang et al. 2021; Taglialatela et al. 2021). Both BRCA1 and BRCA2 are required for the repair of ssDNA gaps after replication (Tirman et al. 2021). The functions of BRCA1/2 at ssDNA gaps may be related to their roles in protecting stalled forks, as BRCA1/2 prevent MRE11-mediated DNA degradation in both contexts. The increase of ssDNA gaps in BRCA1/2-deficient cells elevates replication stress by sequestering RPA and causing fork collapse in a trans cell cycle manner (Cong et al. 2021; Simoneau et al. 2021). Thus, loss of the functions of BRCA1/2 in protecting stalled forks and ssDNA gaps may also contribute to the genomic instability in cancer cells.
Figure 2.

Functions of BRCA1/2 in protecting replication forks. In BRCA1/2-proficient cells, reversed replication forks are protected from nucleolytic degradation by RAD51 in a BRCA1/2-dependent manner. In BRCA1/2-deficient cells, nascent DNA at reversed forks is increasingly degraded. Degradation of nascent DNA in BRCA2-deficient cells leads to MUS81- and POLD3-dependent, error-prone fork restart. Furthermore, ssDNA gaps accumulate during replication in BRCA1/2-deficient cells. BRCA1-deficient cells are defective for protecting ssDNA gaps against the MRE11 nuclease, reducing TS- and TLS-mediated gap repair.
BRCA1 and BRCA2 are also important for suppressing R-loops, a three-stranded polynucleotide structure formed by DNA–RNA hybridization during transcription (Crossley et al. 2019). R-loops are a source of genomic instability because they interfere with DNA replication forks and are cleaved by structure-specific nucleases. BRCA1 suppresses R-loops at transcription termination sites (TTSs) by recruiting the SETX helicase (Hatchi et al. 2015). An increase of R-loops is also detected near transcription start sites (TSSs) in BRCA1 mutation carriers (Chiang et al. 2019). Loss of COBRA1/NELFB, an inhibitor of transcription elongation, reduces R-loops in BRCA1-deficient cells and mitigates mammary tumorigenesis in mice (Zhang et al. 2017). In BRCA2-deficient cells, R-loops are also increased at TSSs due to impaired transcription elongation (Shivji et al. 2018). BRCA2 interacts with the TREX-2 complex, which is important for mRNA processing, to suppress R-loops (Bhatia et al. 2014). The functions of BRCA1/2 in suppressing R-loops may be important in tissues regulated by estrogen because estrogen induces R-loop-associated DNA damage (Stork et al. 2016). BRCA1 is important for maintaining the differentiation state of mammary epithelial cells (Wang et al. 2019c), suggesting a tissue- and cell type-specific function.
Genes in the Fanconi anemia (FA) pathway are also mutated in cancers, particularly in acute myeloid leukemia (AML) and squamous cell carcinoma (Niraj et al. 2019). To date, 22 FA genes have been identified, and some of them are also HR genes. The FA pathway is important for repairing DNA interstrand cross-links (ICLs). Upon collision of replication forks with ICLs, the FA core complex monoubiquitinates the FANCD2–FANCI (ID) complex, enabling it to bind DNA stably (Shakeel et al. 2019; Wang et al. 2021b). The ID complex recruits additional FA proteins to remove ICLs and allow completion of repair through HR. Furthermore, the FA pathway repairs DNA–protein cross-links (DPCs). Formaldehyde, a cellular metabolite generated by histone demethylases, is a source of DPCs. Loss of the aldehyde dehydrogenase ALDH2 and alcohol dehydrogenase ADH5 increases formaldehyde and DNA damage in mice (Dingler et al. 2020). Loss of ALDH2 induces spontaneous DNA damage in FANCD2−/− mice, suggesting that the FA pathway protects cells against DPCs (Langevin et al. 2011). FA proteins are also implicated in HR (Adamson et al. 2012), replication stress response (Gari et al. 2008), R-loop suppression (García-Rubio et al. 2015; Schwab et al. 2015), and maintenance of the epithelial state (Wang et al. 2019c). Collectively, the cancer-associated mutations in HR and FA pathways may compromise the ability of cells to cope with intrinsic DNA damage, affecting their genomic integrity, RNA biogenesis, and differentiation states.
Class II: defects in DNA damage signaling and checkpoints
Mutations in DNA damage checkpoint genes ATM, ATR, CHK1, and CHK2 are also found in cancers. For example, ATM mutations are detected in a wide range of cancers, including colorectal, uterine, prostate, and lung cancers (Jette et al. 2020). ATM and its downstream kinase CHK2 are important for signaling DSBs (Shiloh and Ziv 2013). In response to DSBs, ATM is required for the checkpoint responses in G1, S, and G2/M phases of the cell cycle. NBS1, MRE11, and RAD50, which are involved in ATM activation at DSBs, are also mutated in cancers (McPherson et al. 2020). ATM and CHK2 phosphorylate p53 after DNA damage, and CHK2 is a regulator of apoptosis (Hirao et al. 2002). Upon DSBs, ATM phosphorylates NEMO and promotes activation of the NF-κB pathway (Wu et al. 2006). ATM is also activated by reactive oxygen species (ROS), protecting cells from oxidative DNA damage (Lee and Paull 2021). Thus, loss of ATM in cancer cells may lead to multiple defects in checkpoints and apoptosis, allowing cancer cells to continue proliferation and accumulate chromosomal alterations.
Compared with the ATM–CHK2 pathway, the ATR–CHK1 pathway plays a more important role in the response to DNA replication stress (Marechal and Zou 2013; Saldivar et al. 2017). In S phase, ATR and CHK1 stabilize DNA replication forks and prevent excessive firing of replication origins. ATR also alleviates replication stress by promoting dNTP synthesis (Buisson et al. 2015). Even during the unperturbed cell cycle, ATR ensures proper S–G2 and G2–M transitions and accurate chromosome segregation in mitosis (Kabeche et al. 2018; Lemmens et al. 2018; Saldivar et al. 2018). In response to high replication stress, ATR and CHK1 mediate S-phase and G2/M checkpoint responses. Loss of ATR leads to a range of defects in the cell cycle, including high replication stress, premature entry to mitosis, and mitotic aberrations. Although ATR is a haploid-insufficient tumor suppressor in mice (Fang et al. 2004), ATR mutations are less common than ATM mutations in human cancers because ATR is an essential gene and cancer cells with high levels of genomic instability are especially dependent on ATR signaling for survival.
Class III: elevation of mutation burden
Mutations in mismatch repair (MMR) genes MSH2, MSH6, MLH1, and PMS2 are found in a range of cancers, particularly in colorectal cancer (Lynch and de la Chapelle 2003). While heterozygous mutations in MMR genes cause Lynch syndrome and adult-onset of colorectal cancer, biallelic MMR deficiency (bMMRD) is an autosomal recessive disorder characterized by early onset of cancers. Because MMR plays a critical role in repairing DNA mismatches after DNA replication, loss of MMR results in a drastic increase of mutations in the genome (Jiricny 2013). In addition, MMR prevents DNA replication errors in microsatellite repeats, and loss of MMR leads to microsatellite instability (MSI). Notably, cancer cells with MSI are dependent on the WRN helicase for survival (Chan et al. 2019). WRN suppresses DNA secondary structures at expanded microsatellite repeats, preventing cleavage by the MUS81 nuclease and formation of DSBs (van Wietmarschen et al. 2020). Defects in other DNA repair pathways also contribute to mutations in cancer cells. For example, loss of nucleotide excision repair (NER) compromises repair of UV damage and increases the risk of skin cancer, which is high in UV signature mutations. Loss of MUTHY, a base excision repair (BER) protein, increases single-base substitutions in tumors.
The proofreading activities of DNA polymerases are also critical for suppressing mutations. Mutations in the proofreading domains of DNA polymerases ε and δ (POLE and POLD1) are found in a number of cancers, including colorectal and endometrial cancers (Mur et al. 2020). POLE and POLD1 mutations drastically increase mutation burden in tumors (Campbell et al. 2017). In mouse models, Pole and Pold1 mutants promote tumorigenesis (Albertson et al. 2009; Li et al. 2018). Interestingly, modeling a cancer mutation of POLE in yeast generates a hyperactive polymerase that bypasses DNA mismatches and hairpins, suggesting that POLE/POLD1 mutations may affect replication fidelity in multiple ways (Xing et al. 2019).
It should be noted that DNA repair can be altered in cancer cells by mechanisms other than mutations, such as gene expression, RNA processing, protein modifications, and protein degradation. Furthermore, DNA repair can be affected by other pathways, such as the cell cycle, DNA replication, nucleotide metabolism, chromatin modulation, transcription, and RNA biogenesis. The DDR defects in cancer cells caused by indirect mechanisms can also contribute to genomic instability and affect the therapeutic response.
Targeting the DDR in cancer therapy
Ionizing radiation and genotoxic chemotherapies have been long used to exploit the DNA repair defects in tumors. For example, cisplatin is effective in ovarian cancer probably because HR deficiency is common in this cancer type. Compared with traditional radiotherapy and chemotherapy, targeting the DDR proteins indispensable in cancer cells provides a potentially more selective and less toxic therapeutic approach.
PARP inhibitors
One of the best characterized examples of synthetic lethality in cancer cells is the selective killing of HR-deficient cancer cells by PARP inhibitor (PARPi) (Bryant et al. 2005; Farmer et al. 2005; Fong et al. 2009). To date, several PARPis are approved or under trials for the treatment of breast, ovarian, prostate, and pancreatic cancers (Table 1). Inhibition of PARP1/2 by PARPi impairs BER and increases DNA single-stranded breaks (SSBs) in the genome, which are converted to DSBs during DNA replication. Cells with HR defects are unable to repair replication-associated DSBs and therefore are sensitive to PARPi. The efficacy of PARPi is associated with the trapping of PARP1/2 on chromatin (Murai et al. 2012). Loss of the chromatin remodeler ALC1/CHD1L leads to persistent BER intermediates, increases PARP1 trapping, and renders HR-deficient cancer cells more sensitive to PARPi (Blessing et al. 2020; Juhász et al. 2020; Hewitt et al. 2021; Verma et al. 2021). Loss of RNaseH2, which is required for ribonucleotide excision repair (RER), leads to topoisomerase 1 (TOP1)-mediated cleavage of misincorporated ribonucleotides and trapping of PARP1, rendering cells hypersensitive to PARPi (Zimmermann et al. 2018). Inhibition of DNPH1, which suppresses genomic incorporation of 5-hydroxymethyl deoxyuridine (hmdU), increases BER intermediates and PARP1 trapping and further sensitizes BRCA-deficient cancer cells to PARPi (Fugger et al. 2021).
Table 1.
DDR inhibitors in clinical use or trials

PARP1 is also important at replication forks. PARP1 is required for the proper maturation of Okazaki fragments on the lagging strand (Hanzlikova et al. 2018). PARP1-mediated PARylation inhibits RECQ1, which resolves reversed forks (Berti et al. 2013). Inhibition of PARP1/2 reduces fork reversal and increases repriming by PrimPol, generating ssDNA gaps on the leading strand (Genois et al. 2021). The induction of ssDNA gaps by PARPi may be critical for the killing of BRCA1/2-deficient cells. One study suggests that PARPi generates more ssDNA gaps in BRCA1/2-deficient cells, driving RPA exhaustion and replication catastrophe (Fig. 3A; Cong et al. 2021). In contrast, another study shows that PARPi induces ssDNA gaps similarly in BRCA1-deficient and -proficient cells (Fig. 3B; Simoneau et al. 2021). However, the trapping of PARP1 by PARPi prevents the completion of gap repair and allows the gaps to persist. During the ensuing mitosis, more chromosome aberrations are observed in BRCA1/2-deficient cells (Schoonen et al. 2017). During the subsequent S phase, a new round of DNA replication leads to collisions of replication forks with ssDNA gaps and a surge of DSBs. BRCA1/2-deficient cells fail to activate ATR to suppress replication origin firing and to repair DSBs at collapsed forks, providing a plausible explanation of how PARPis selectively kill BRCA1/2 mutant cells over multiple cell cycles (Simoneau et al. 2021).
Figure 3.

Models for the selective killing of BRCA1/2-deficient cells by PARPi. (A) PARPi induces more ssDNA gaps in BRCA1/2-deficient cells, promoting RPA exhaustion and replication catastrophe. (B) PARPi induces ssDNA gaps during replication and prevents complete gap repair, allowing gaps to persist into the next cell cycle and generate DSBs upon collisions with replication forks. BRCA1/2-deficient cells fail to repair collapsed forks and mount a checkpoint response, leading to progressive accumulation of DSBs over multiple cell cycles and cell death.
ATR, CHK1, and WEE1 inhibitors
Inhibitors of ATR, CHK1, and WEE1 have shown efficacy in cancer cells with specific DDR defects or high genomic instability (Table 1). The ATR–CHK1 pathway is critical for S-phase and G2/M checkpoints. Even in the absence of extrinsic DNA damage, inhibition of ATR or CHK1 in cancer cells elevates CDK1/2 activities, increases firing of replication origins, and reduces replication fork stability (Petermann et al. 2006; Buisson et al. 2015). Similarly, WEE1 inhibitor (WEE1i) also elevates CDK1/2 activities, increases origin firing, and overrides the G2/M checkpoint (Moiseeva et al. 2019). ATR, CHK1, and WEE1 inhibitors exacerbate the replication stress in cancer cells and impair checkpoint responses. In addition, both ATR and CHK1 are important for HR (Sorensen et al. 2005; Buisson et al. 2017b). Thus, ATR and CHK1 inhibitors not only induce replication-associated DSBs, but also prevent DSB repair. ATR inhibitor (ATRi) induces mitotic catastrophe if cells enter mitosis with high levels of DSBs. In cancer cells under high replication stress, ATRi induces excessive ssDNA, leading to RPA exhaustion and replication catastrophe (Toledo et al. 2013). All these effects of ATR, CHK1, and WEE1 inhibitors may contribute to the synthetic lethality in cancer cells.
ATRi has shown efficacy in ATM-deficient tumors (Villaruz et al. 2016; Rafiei et al. 2020). Both ATM and ATR contribute to the S-phase and G2/M checkpoints. Furthermore, ATM and ATR function in concert in DSB signaling (Shiotani and Zou 2009). The loss of ATM in cancer cells may render them increasingly reliant on ATR for damage signaling and checkpoint responses. Notably, ATM-deficient cancer cells are not highly sensitive to PARPi, highlighting the difference between ATRi and PARPi (Rafiei et al. 2020). The functional redundancy between ATM and ATR, rather than an indispensable role of ATM in HR, likely underlies the sensitivity of ATM-deficient cells to ATRi. BRCA1/2-deficient cancer cells are sensitive to ATRi (Kim et al. 2017; Yazinski et al. 2017). In this context, ATRi may suppress residual HR activity, induce replication-associated DSBs, and prevent repair of ssDNA gaps and DSBs, leading to selective killing of cancer cells. In nonsmall cell lung cancer (NSCLC), low expression of ERCC1, which regulates the XPF nuclease in NER and FA pathways, is associated with a better response to cisplatin (Lord et al. 2002). Loss of ERCC1 is synthetically lethal with ATR inhibition (Mohni et al. 2014), suggesting that cancer cells with defects in HR and FA pathways may be generally sensitive to ATRi.
ATRi also selectively kills cancer cells under high replication stress. Cancer cells overexpressing Cyclin E harbor high replication stress and are sensitive to ATRi and CHK1i (Toledo et al. 2011). Loss of ARID1A or SMARCA4/BRG1, two components of SWI/SNF chromatin remodeling complexes, alters chromatin compaction, elevates replication stress, and confers ATRi sensitivity (Williamson et al. 2016; Gupta et al. 2020; Kurashima et al. 2020). Cancer cells reliant on the alternative lengthening of telomeres (ALT) pathway display high replication stress at telomeres and are sensitive to ATRi (Flynn et al. 2015). In cancer cells expressing the APOBEC3A/B cytidine deaminases, APOBEC3A/B generate replication stress by increasing abasic sites, conferring ATRi sensitivity (Green et al. 2017; Buisson et al. 2017a). Hotspot mutations in RNA splicing factors such as SF3B1, U2AF1, and SRSF2 are prevalent in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Some of these splicing factor mutants induce aberrant R-loop accumulation, rendering cells sensitive to ATRi (Nguyen et al. 2018; Chen et al. 2018b; Singh et al. 2020). Cancer cells with reduced RNaseH2 are also sensitive to ATRi, presumably because misincorporated ribonucleotides induce SSBs and interfere with replication (Wang et al. 2019a). A mutation signature in cancer cells was recently linked to low RNaseH2 expression and TOP1-mediated RER (Reijns et al. 2022), providing a potential biomarker for the use of ATRi and PARPi in therapy.
Notably, ATRi and PARPi display a strong synergy in BRCA1/2-deficient cancer cells (Kim et al. 2017, 2020; Yazinski et al. 2017; Shah et al. 2021; Roulston et al. 2022). Because both ATRi and PARPi induce ssDNA and both ATRi and PARPi sensitivities associate with high ssDNA levels (Buisson et al. 2015; Cong et al. 2021), the synergy between ATRi and PARPi may arise from ssDNA accumulation. Alternatively, ATRi may increase replication origin firing and the collision of replication forks with PARPi-induced ssDNA gaps, enhancing the effects of PARPi (Simoneau et al. 2021). ATRi may also reduce the stability of replication forks in response to PARPi and repair of PARPi-induced ssDNA gaps and DSBs.
Other DDR inhibitors and opportunities to exploit genomic instability
ATM inhibitor (ATMi) and DNA-PKcs inhibitor (DNA-PKi) sensitize cancer cells to radiation and chemotherapy (Table 1; Durant et al. 2018; Fok et al. 2019). ATMi and DNA-PKi display a synergy in inhibiting cell proliferation, suggesting that loss of both ATM and DNA-PK is synthetic lethal (Wang et al. 2021a). DNA-PKi prevents the reappearance of CHK1 phosphorylation after ATRi treatment (Buisson et al. 2015), suggesting that DNA-PKi may enhance the effects of ATRi in cancer cells.
POLθ is required for DSB repair through microhomology-mediated end joining (MMEJ), an error-prone repair pathway (Yu and McVey 2010; Mateos-Gomez et al. 2015). HR-deficient cells are hypersensitive to POLθ loss because DSB repair is increasingly dependent on MMEJ in the absence of HR (Ceccaldi et al. 2015). Inhibitors of the ATPase or polymerase activity of POLθ selectively kill BRCA1/2-deficient cancer cells and enhance the effects of PARPi (Table 1; Fig. 4; Zatreanu et al. 2021; Zhou et al. 2021). REV1 forms a complex with the translesion synthesis (TLS) DNA polymerase POLζ and enables its localization to DNA lesions. A REV1 inhibitor (REV1i) that disrupts the interaction of REV1 with POLζ (Chatterjee et al. 2020) enhances the effects of cisplatin in cancer cells (Yoon et al. 2021). REV1i also inhibits the repair of ssDNA gaps in BRCA1/2-deficient cells, leading to cytotoxicity (Fig. 4; Taglialatela et al. 2021).
Figure 4.

Selective killing of BRCA1/2-deficient cells by different DDR targeted drugs. Both PARPi and REV1i render the ssDNA gaps in BRCA1/2-deficient cells persistent, promoting DSBs or replication catastrophe. POLθi blocks MMEJ and prevents alternative repair of DSBs in HR-defective cells, rendering DSBs persistent in BRCA1/2-deficient cells.
Additional inhibitors have been developed to target the HR pathway or BRCA-deficient tumors. A RAD51 inhibitor displays efficacy in chronic lymphoblastic leukemia (CLL) expressing the AID cytidine deaminase (Lamont et al. 2013), presumably because it attenuates the repair of AID-induced DSBs (Table 1). An inhibitor of TRDMT1, an RNA methyltransferase involved in the transcription-coupled HR, sensitizes cancer cells to PARPi and ATRi (Zhu et al. 2021). Furthermore, a RAD52 inhibitor induces synthetic lethality in BRCA2-deficient cells (Yang et al. 2021), which is likely attributed to the indispensable HR function of RAD52 in the absence of BRCA2 (Feng et al. 2011). The deubiquitinase USP1 is required for the deubiquitination of PCNA and FANCD2, which are involved in TLS, template switching (TS), FA, and HR pathways (Huang et al. 2006; Oestergaard et al. 2007). The protection of replication forks in the absence of BRCA1 requires USP1, and a USP1 inhibitor selectively kills BRCA1-deficient cells (Table 1; Lim et al. 2018). In TNBC, an inhibitor of CDK12/13, suppressors of intronic polyadenylation, reduces expression of HR genes and enhances the effects of PARPi and chemotherapy (Quereda et al. 2019). In leukemia cells overexpressing the protein arginine methyltransferase PRMT5, an inhibitor of PRMT5 impairs HR by altering the splicing of repair regulators and synergizes with PARPi to induce cell death (Table 1; Hamard et al. 2018). As discussed above, an inhibitor of DNPH1 synergizes with PARPi in BRCA-deficient cells by increasing BER-generated SSBs and PARP trapping (Fugger et al. 2021). Loss of ALC1 also promotes PARP trapping and enhances the effects of PARPi in BRCA-deficient cells (Blessing et al. 2020; Juhász et al. 2020; Hewitt et al. 2021; Verma et al. 2021), making ALC1 inhibitors potential therapeutics for BRCA-deficient tumors (Abbott et al. 2020).
Several other inhibitors are generated to exploit the replication stress in cancer cells. An RPA inhibitor increases replication stress and synergizes with chemotherapy to suppress tumor growth in mice (Glanzer et al. 2014), possibly by enhancing replication catastrophe. Similar to WEE1, the PKMYT1 kinase restricts CDK1 activity, and a PKMYT1 inhibitor induces synthetic lethality in cancer cells harboring Cyclin E amplification by causing unscheduled CDK1 activation (Table 1; Gallo et al. 2021). Moreover, loss of the WRN helicase increases replication stress at expended microsatellite repeats in MMR-deficient cancer cells, leading to synthetic lethality (van Wietmarschen et al. 2020). Inhibitors of WRN are potentially useful for the treatment of MMR-deficient tumors (Sommers et al. 2019).
A few other potential therapeutic targets have emerged in various contexts of DNA repair defects or genomic instability, although specific inhibitors of these targets are not yet available. APEX2 and CIP2A were identified as synthetic lethal hits in BRCA1/2-deficient cells through CRISPR–Cas9 screens (Mengwasser et al. 2019; Álvarez-Quilón et al. 2020; Adam et al. 2021). Loss of FANCM induces cell death in ALT+ cancer cells (Pan et al. 2019). Loss of HMCES reduces the survival of cancer cells overexpressing the APOBEC3A cytidine deaminase (Mehta et al. 2020; Biayna et al. 2021).
Overcoming resistance to DDR targeted drugs
Although PARPis show efficacy in cancer patients carrying BRCA1/2 mutations, patients inevitably acquire resistance to these drugs over time. A number of PARPi resistance mechanisms have been observed in cell lines, mouse tumor models, and patient samples, including reversion mutations of BRCA1/2 genes (Sakai et al. 2008; Weigelt et al. 2017), up-regulation of drug efflux pumps (Rottenberg et al. 2008), stabilization of truncated BRCA1 protein (Johnson et al. 2013), and loss of PARP1 (Pettitt et al. 2013). Loss of the 53BP1–RIF1–Shieldin pathway increases DNA end resection in BRCA1-deficient cells, conferring resistance to PARPi (Bunting et al. 2010; Jaspers et al. 2013; Xu et al. 2015; Dev et al. 2018; Ghezraoui et al. 2018; Gupta et al. 2018; Mirman et al. 2018; Noordermeer et al. 2018). Loss of the CTC1–STN1–TEN1 (CST) complex, which is recruited by Shieldin, also leads to PARPi resistance (Barazas et al. 2018). Furthermore, loss of other inhibitors of DNA end resection, such as HELB, DYNLL1, and ZPET, also increase PARPi resistance (Tkáč et al. 2016; He et al. 2018; Moquin et al. 2019). When BRCA2-deficient cells regain the ability to protect stalled replication forks, they become resistant to PARPi (Ray Chaudhuri et al. 2016). Finally, loss of SLFN11 confers PARPi resistance (Lok et al. 2017), possibly due to the lack of SLFN11-mediated replication inhibition.
Using cell lines and patient-derived xenograft models, it was shown that ATRi can effectively overcome the PARPi resistance of BRCA1/2-deficient cancer cells (Kim et al. 2017, 2020; Yazinski et al. 2017). Notably, multiple PARPi resistance mechanisms are observed in single-cell-derived cell lines, suggesting that different mechanisms can operate in the same cells to confer resistance (Yazinski et al. 2017). ATRi inhibits the restored RAD51 loading and fork protection in resistant cells (Yazinski et al. 2017; Parmar et al. 2019). ATRi may also override the regained checkpoint of resistant cells, leading to more collisions between replication forks and ssDNA gaps (Simoneau et al. 2021). Finally, ATRi overcomes the PARPi resistance of SLFN11-deficient cells (Murai et al. 2016). Besides ATRi, POLθi overcomes the PARPi resistance in BRCA1/2-deficient cells by blocking MMEJ (Zatreanu et al. 2021; Zhou et al. 2021). REV1i overcomes the PARPi resistance in BRCA1-deficient cells by inhibiting ssDNA gap repair (Taglialatela et al. 2021). Inhibitors of DNPH1 and USP1 overcome the PARPi resistance in BRCA-deficient cells by increasing SSBs and destabilizing replication forks, respectively (Lim et al. 2018; Fugger et al. 2021). Furthermore, CDK12 inhibition overcomes the PARPi resistance in TNBC by reducing the expression of HR genes (Johnson et al. 2016).
The resistance to ATRi is less understood. Loss of CDC25A, which reduces CDK1/2 activities, confers ATRi resistance (Ruiz et al. 2016). Loss of the mediator protein MED12 renders cells resistant to ATRi by stabilizing replication forks (Schleicher et al. 2020). Finally, loss of Cyclin C and CDK8, two components of the mediator complex, reduces R-loops and leads to ATRi resistance (Lloyd et al. 2021).
Targeting DDR defects with immunotherapy
The production of neoantigens by cancer cells is a key determinant for the antitumor immunity. Cancer cells defective in MMR accumulate high levels of mutations, increasing tumor mutation burden (TMB) (Ballhausen et al. 2020). Tumors with MMR deficiency (MMR-D) or high MSI (MSI-H) are more responsive to immune checkpoint inhibitors (ICIs) (Le et al. 2015). Pembrolizumab, an anti-PD-1 monoclonal antibody, has been approved by the FDA for the treatment of advanced colorectal cancer with MMR-D or MSI-H. Defects in MLH1, which regulates the EXO1 nuclease during MMR, lead to unrestricted EXO1 activity at DSBs and activation of the cGAS–STING pathway (Guan et al. 2021). The cGAS–STING-mediated interferon response enhances the response of MLH1-deficient tumors to immunotherapy independently of changes in neoantigens (Lu et al. 2021). A recent study on MMR-D colorectal tumors revealed multiple spatially organized hubs of interacting malignant and immune cells, providing insights into how MMR-D malignant cells elicit the immune response (Pelka et al. 2021). POLE and POLD1 mutations in tumors are also associated with responses to immunotherapy (Wang et al. 2019b; Li et al. 2020). Thus, a subset of class III repair defects in cancer cells can lead to high TMB and an interferon response, providing an opportunity for immunotherapy.
ICIs have been tested in BRCA1/2-deficient tumors. A recent study suggested that the combination of BRCA2 mutations and TMB is a potential predictor of immunotherapy response (Zhou and Li 2021). Another study showed that BRCA2-deficient tumors are enriched for adaptative and innate immunity (Samstein et al. 2021). In mouse models of BRCA1-deficent ovarian cancer and TNBC, PARPi enhanced the effects of ICIs by eliciting the cGAS–STING pathway (Ding et al. 2018; Pantelidou et al. 2019). PARPi enhances infiltration and activation of CD8+ T cells through the cGAS–STING pathway in tumor cells and paracrine activation of dendritic cells. Combinations of PARPi and ICIs have produced promising results in BRCA1/2-deficient tumors in trials (Vikas et al. 2020). The combinations of PARPi and ICIs are effective in some patients without BRCA1/2 mutations, suggesting that this strategy may exploit the genomic instability from different sources.
In response to DSBs, PD-L1 expression is up-regulated in an ATR-dependent manner (Sato et al. 2017). ATRi reduces the expression of PD-L1 in cancer cells, sensitizing them to T-cell-mediated cell killing (Sun et al. 2018; Vendetti et al. 2018). ATRi also enhances the radiation-induced inflammatory tumor microenvironment (Dillon et al. 2019; Sheng et al. 2020). When treated with ATRi, irradiated cancer cells undergo mitosis in the presence of DSBs, increasing micronuclei and activation of the cGAS–STING pathway (Chen et al. 2020). The RNA-induced and RIG-I-mediated interferon response is also activated in irradiated cells upon ATR inhibition (Chen et al. 2020; Feng et al. 2020). Collapse of replication forks may give rise to AT-rich DNA fragments, which are transcribed into RNA to trigger the RIG-I pathway (Feng et al. 2020). Collectively, ATRi may potentiate the tumor-immune microenvironment in multiple ways, enhancing the efficacy of combined radioimmunotherapy.
Perspectives
Although a number of DNA repair and damage signaling pathways have been extensively studied, we still lack a complete understanding of how these pathways function in various oncogenic contexts. Why are certain DDR pathways lost in specific cancer types? Are the consequences of DDR defects distinct in different tissues and cell types? Our understanding of the interplays between oncogenic stress and the DDR is still limited. Does the oncogenic stress in cancer cells determine which DDR pathways to lose? Does the loss of DDR pathways in cancer cells determine which oncogenic events to acquire? Understanding the different sources of genomic instability in cancer cells and the effects of DDR defects in different oncogenic contexts is critical for developing specific strategies to induce synthetic lethality. It is also important to note that the wiring of DDR pathways can be distinct in various tissues and cell types and under different selective pressures. The loss of specific DDR pathways in cancer cells or the selective pressure during tumor evolution and cancer therapy could reshape the wiring of the DDR network. A better understanding of the rewiring of the DDR network in cancer cells is important for targeting DDR pathways and overcoming the resistance to DDR targeted drugs. Finally, it is important to investigate how the DDR defects in cancer cells affect tumor microenvironments, and how tumor microenvironments impact the DDR in cancer cells. It is conceivable that future studies using patient samples, in vivo models, and single-cell analyses will profoundly improve our understanding of the genomic instability in tumors and the efficacy of DDR targeted therapy.
Acknowledgments
We apologize to those authors whose work could not be cited due to space constrains. L.L. is supported by the National Institutes of Health (NIH; GM118833). L.Z. is the James and Patricia Poitras Endow Chair for Cancer Research and is supported by the NIH (CA263934). Figures were created with BioRender.com.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.349431.122.
Competing interest statement
The authors declare no competing interests.
References
- Abbott JM, Zhou Q, Esquer H, Pike L, Broneske TP, Rinaldetti S, Abraham AD, Ramirez DA, Lunghofer PJ, Pitts TM, et al. 2020. First-in-class inhibitors of oncogenic CHD1L with preclinical activity against colorectal cancer. Mol Cancer Ther 19: 1598–1612. 10.1158/1535-7163.MCT-20-0106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam S, Rossi SE, Moatti N, De Marco Zompit M, Xue Y, Ng TF, Álvarez-Quilón A, Desjardins J, Bhaskaran V, Martino G, et al. 2021. The CIP2A–TOPBP1 axis safeguards chromosome stability and is a synthetic lethal target for BRCA-mutated cancer. Nat Cancer 2: 1357–1371. 10.1038/s43018-021-00266-w [DOI] [PubMed] [Google Scholar]
- Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. 2012. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol 14: 318–328. 10.1038/ncb2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertson TM, Ogawa M, Bugni JM, Hays LE, Chen Y, Wang Y, Treuting PM, Heddle JA, Goldsby RE, Preston BD. 2009. DNA polymerase ε and δ proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci 106: 17101–17104. 10.1073/pnas.0907147106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez-Quilón A, Wojtaszek JL, Mathieu MC, Patel T, Appel CD, Hustedt N, Rossi SE, Wallace BD, Setiaputra D, Adam S, et al. 2020. Endogenous DNA 3′ blocks Are vulnerabilities for BRCA1 and BRCA2 deficiency and are reversed by the APE2 nuclease. Mol Cell 78: 1152–1165.e8. 10.1016/j.molcel.2020.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou AC, Foulkes WD, Tischkowitz M. 2014. Breast-cancer risk in families with mutations in PALB2. N Engl J Med 371: 1651–1652. 10.1056/NEJMoa1400382 [DOI] [PubMed] [Google Scholar]
- Ballhausen A, Przybilla MJ, Jendrusch M, Haupt S, Pfaffendorf E, Seidler F, Witt J, Hernandez Sanchez A, Urban K, Draxlbauer M, et al. 2020. The shared frameshift mutation landscape of microsatellite-unstable cancers suggests immunoediting during tumor evolution. Nat Commun 11: 4740. 10.1038/s41467-020-18514-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barazas M, Annunziato S, Pettitt SJ, de Krijger I, Ghezraoui H, Roobol SJ, Lutz C, Frankum J, Song FF, Brough R, et al. 2018. The CST complex mediates end protection at double-strand breaks and promotes PARP inhibitor sensitivity in BRCA1-deficient cells. Cell Rep 23: 2107–2118. 10.1016/j.celrep.2018.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JR, Clifford G, Bonnet C, Groth A, Wilson MD, Chapman JR. 2021. BARD1 reads H2A lysine 15 ubiquitination to direct homologous recombination. Nature 596: 433–437. 10.1038/s41586-021-03776-w [DOI] [PubMed] [Google Scholar]
- Berger M, Wortmann L, Buchgraber P, Lücking U, Zitzmann-Kolbe S, Wengner AM, Bader B, Bömer U, Briem H, Eis K, et al. 2021. BAY-8400: a novel potent and selective DNA-PK inhibitor which shows synergistic efficacy in combination with targeted α therapies. J Med Chem 64: 12723–12737. 10.1021/acs.jmedchem.1c00762 [DOI] [PubMed] [Google Scholar]
- Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. 2013. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20: 347–354. 10.1038/nsmb.2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia V, Barroso SI, García-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A. 2014. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511: 362–365. 10.1038/nature13374 [DOI] [PubMed] [Google Scholar]
- Biayna J, Garcia-Cao I, Álvarez MM, Salvadores M, Espinosa-Carrasco J, McCullough M, Supek F, Stracker TH. 2021. Loss of the abasic site sensor HMCES is synthetic lethal with the activity of the APOBEC3A cytosine deaminase in cancer cells. PLoS Biol 19: e3001176. 10.1371/journal.pbio.3001176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blessing C, Mandemaker IK, Gonzalez-Leal C, Preisser J, Schomburg A, Ladurner AG. 2020. The oncogenic helicase ALC1 regulates PARP inhibitor potency by trapping PARP2 at DNA breaks. Mol Cell 80: 862–875.e6. 10.1016/j.molcel.2020.10.009 [DOI] [PubMed] [Google Scholar]
- Brehmer D, Beke L, Wu T, Millar HJ, Moy C, Sun W, Mannens G, Pande V, Boeckx A, van Heerde E, et al. 2021. Discovery and pharmacological characterization of JNJ-64619178, a novel small-molecule inhibitor of PRMT5 with potent antitumor activity. Mol Cancer Ther 20: 2317–2328. 10.1158/1535-7163.MCT-21-0367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917. 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- Buisson R, Boisvert JL, Benes CH, Zou L. 2015. Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S phase. Mol Cell 59: 1011–1024. 10.1016/j.molcel.2015.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Lawrence MS, Benes CH, Zou L. 2017a. APOBEC3A and APOBEC3B activities render cancer cells susceptible to ATR inhibition. Cancer Res 77: 4567–4578. 10.1158/0008-5472.CAN-16-3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, Hardy EJ, Dellaire G, Haas W, Xia B, et al. 2017b. Coupling of homologous recombination and the checkpoint by ATR. Mol Cell 65: 336–346. 10.1016/j.molcel.2016.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141: 243–254. 10.1016/j.cell.2010.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, Davidson S, Edwards M, Elvin JA, Hodel KP, et al. 2017. Comprehensive analysis of hypermutation in human cancer. Cell 171: 1042–1056.e10. 10.1016/j.cell.2017.09.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O'Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, et al. 2015. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 518: 258–262. 10.1038/nature14184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EM, Shibue T, McFarland JM, Gaeta B, Ghandi M, Dumont N, Gonzalez A, McPartlan JS, Li T, Zhang Y, et al. 2019. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568: 551–556. 10.1038/s41586-019-1102-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee N, Whitman MA, Harris CA, Min SM, Jonas O, Lien EC, Luengo A, Vander Heiden MG, Hong J, Zhou P, et al. 2020. REV1 inhibitor JH-RE-06 enhances tumor cell response to chemotherapy by triggering senescence hallmarks. Proc Natl Acad Sci 117: 28918–28921. 10.1073/pnas.2016064117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Feng W, Lim PX, Kass EM, Jasin M. 2018a. Homology-directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Annu Rev Cancer Biol 2: 313–336. 10.1146/annurev-cancerbio-030617-050502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen JY, Huang YJ, Gu Y, Qiu J, Qian H, Shao C, Zhang X, Hu J, Li H, et al. 2018b. The augmented R-loop is a unifying mechanism for myelodysplastic syndromes induced by high-risk splicing factor mutations. Mol Cell 69: 412–425.e6. 10.1016/j.molcel.2017.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Harding SM, Natesan R, Tian L, Benci JL, Li W, Minn AJ, Asangani IA, Greenberg RA. 2020. Cell cycle checkpoints cooperate to suppress DNA- and RNA-associated molecular pattern recognition and anti-tumor immune responses. Cell Rep 32: 108080. 10.1016/j.celrep.2020.108080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HC, Zhang X, Li J, Zhao X, Chen J, Wang HT, Jatoi I, Brenner A, Hu Y, Li R. 2019. BRCA1-associated R-loop affects transcription and differentiation in breast luminal epithelial cells. Nucleic Acids Res 47: 5086–5099. 10.1093/nar/gkz262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, Colombo N, Weberpals JI, Clamp A, Scambia G, et al. 2017. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390: 1949–1961. 10.1016/S0140-6736(17)32440-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, Okamoto A, Moore KN, Efrat Ben-Baruch N, Werner TL, et al. 2019. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med 381: 2403–2415. 10.1056/NEJMoa1909707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong K, Peng M, Kousholt AN, Lee WTC, Lee S, Nayak S, Krais J, VanderVere-Carozza PS, Pawelczak KS, Calvo J, et al. 2021. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol Cell 81: 3227. 10.1016/j.molcel.2021.07.015 [DOI] [PubMed] [Google Scholar]
- Crossley MP, Bocek M, Cimprich KA. 2019. R-Loops as cellular regulators and genomic threats. Mol Cell 73: 398–411. 10.1016/j.molcel.2019.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei W, Ostermaier M, et al. 2018. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol 20: 954–965. 10.1038/s41556-018-0140-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon MT, Bergerhoff KF, Pedersen M, Whittock H, Crespo-Rodriguez E, Patin EC, Pearson A, Smith HG, Paget JTE, Patel RR, et al. 2019. ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin Cancer Res 25: 3392–3403. 10.1158/1078-0432.CCR-18-1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH, et al. 2018. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep 25: 2972–2980.e5. 10.1016/j.celrep.2018.11.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingler FA, Wang M, Mu A, Millington CL, Oberbeck N, Watcham S, Pontel LB, Kamimae-Lanning AN, Langevin F, Nadler C, et al. 2020. Two aldehyde clearance systems Are essential to prevent lethal formaldehyde accumulation in mice and humans. Mol Cell 80: 996–1012.e9. 10.1016/j.molcel.2020.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant ST, Zheng L, Wang Y, Chen K, Zhang L, Zhang T, Yang Z, Riches L, Trinidad AG, Fok JHL, et al. 2018. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci Adv 4: eaat1719. 10.1126/sciadv.aat1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Tsao CC, Goodman BK, Furumai R, Tirado CA, Abraham RT, Wang XF. 2004. ATR functions as a gene dosage-dependent tumor suppressor on a mismatch repair-deficient background. EMBO J 23: 3164–3174. 10.1038/sj.emboj.7600315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, Powell SN. 2011. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci 108: 686–691. 10.1073/pnas.1010959107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Tubbs A, Zhang C, Tang M, Sridharan S, Wang C, Jiang D, Su D, Zhang H, Chen Z, et al. 2020. ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J 39: e104036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, Bersani F, Pineda JR, Suva ML, Benes CH, et al. 2015. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347: 273–277. 10.1126/science.1257216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fok JHL, Ramos-Montoya A, Vazquez-Chantada M, Wijnhoven PWG, Follia V, James N, Farrington PM, Karmokar A, Willis SE, Cairns J, et al. 2019. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat Commun 10: 5065. 10.1038/s41467-019-12836-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al. 2009. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361: 123–134. 10.1056/NEJMoa0900212 [DOI] [PubMed] [Google Scholar]
- Fugger K, Bajrami I, Silva Dos Santos M, Young SJ, Kunzelmann S, Kelly G, Hewitt G, Patel H, Goldstone R, Carell T, et al. 2021. Targeting the nucleotide salvage factor DNPH1 sensitizes BRCA-deficient cells to PARP inhibitors. Science 372: 156–165. 10.1126/science.abb4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo D, Young JTF, Fourtounis J, Martino G, Álvarez-Quilón A, Bernier C, Duffy NM, Papp R, Roulston A, Stocco R, et al. 2021. CCNE1 amplification is synthetic-lethal with PKMYT1 kinase inhibition. bioRxiv 10.1101/2021.04.08.438361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Rubio ML, Pérez-Calero C, Barroso SI, Tumini E, Herrera-Moyano E, Rosado IV, Aguilera A. 2015. The Fanconi anemia pathway protects genome integrity from R-loops. PLoS Genet 11: e1005674. 10.1371/journal.pgen.1005674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Décaillet C, Delannoy M, Wu L, Constantinou A. 2008. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci 105: 16107–16112. 10.1073/pnas.0804777105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genois MM, Gagné JP, Yasuhara T, Jackson J, Saxena S, Langelier MF, Ahel I, Bedford MT, Pascal JM, Vindigni A, et al. 2021. CARM1 regulates replication fork speed and stress response by stimulating PARP1. Mol Cell 81: 784–800.e8. 10.1016/j.molcel.2020.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart SV, Kellner WA, Thompson C, Pappalardi MB, Zhang XP, Montes de Oca R, Penebre E, Duncan K, Boriack-Sjodin A, Le B, et al. 2018. Activation of the p53–MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci Rep 8: 9711. 10.1038/s41598-018-28002-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar-Lele M, Sanchiz-Calvo M, Fueyo-Marcos E, et al. 2018. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560: 122–127. 10.1038/s41586-018-0362-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanzer JG, Liu S, Wang L, Mosel A, Peng A, Oakley GG. 2014. RPA inhibition increases replication stress and suppresses tumor growth. Cancer Res 74: 5165–5172. 10.1158/0008-5472.CAN-14-0306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, McCormick C, Lorusso D, Hoskins P, Freyer G, et al. 2019. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 381: 2391–2402. 10.1056/NEJMoa1910962 [DOI] [PubMed] [Google Scholar]
- Green AM, Budagyan K, Hayer KE, Reed MA, Savani MR, Wertheim GB, Weitzman MD. 2017. Cytosine deaminase APOBEC3A sensitizes leukemia cells to inhibition of the DNA replication checkpoint. Cancer Res 77: 4579–4588. 10.1158/0008-5472.CAN-16-3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Lu C, Jin Q, Lu H, Chen X, Tian L, Zhang Y, Ortega J, Zhang J, Siteni S, et al. 2021. MLH1 deficiency-triggered DNA hyperexcision by exonuclease 1 activates the cGAS–STING pathway. Cancer Cell 39: 109–121.e5. 10.1016/j.ccell.2020.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A, et al. 2018. DNA repair network analysis reveals Shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 173: 972–988.e23. 10.1016/j.cell.2018.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Concepcion CP, Fahey CG, Keshishian H, Bhutkar A, Brainson CF, Sanchez-Rivera FJ, Pessina P, Kim JY, Simoneau A, et al. 2020. BRG1 loss predisposes lung cancers to replicative stress and ATR dependency. Cancer Res 80: 3841–3854. 10.1158/0008-5472.CAN-20-1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamard PJ, Santiago GE, Liu F, Karl DL, Martinez C, Man N, Mookhtiar AK, Duffort S, Greenblatt S, Verdun RE, et al. 2018. PRMT5 regulates DNA repair by controlling the alternative splicing of histone-modifying enzymes. Cell Rep 24: 2643–2657. 10.1016/j.celrep.2018.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, Caldecott KW. 2018. The importance of poly(ADP-ribose) Polymerase as a sensor of unligated Okazaki fragments during DNA replication. Mol Cell 71: 319–331.e3. 10.1016/j.molcel.2018.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML, et al. 2015. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell 57: 636–647. 10.1016/j.molcel.2015.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YJ, Meghani K, Caron MC, Yang C, Ronato DA, Bian J, Sharma A, Moore J, Niraj J, Detappe A, et al. 2018. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 563: 522–526. 10.1038/s41586-018-0670-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt G, Borel V, Segura-Bayona S, Takaki T, Ruis P, Bellelli R, Lehmann LC, Sommerova L, Vancevska A, Tomas-Loba A, et al. 2021. Defective ALC1 nucleosome remodeling confers PARPi sensitization and synthetic lethality with HRD. Mol Cell 81: 767–783.e11. 10.1016/j.molcel.2020.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao A, Cheung A, Duncan G, Girard PM, Elia AJ, Wakeham A, Okada H, Sarkissian T, Wong JA, Sakai T, et al. 2002. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol Cell Biol 22: 6521–6532. 10.1128/MCB.22.18.6521-6532.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Botuyan MV, Zhao D, Cui G, Mer E, Mer G. 2021. Mechanisms of BRCA1–BARD1 nucleosome recognition and ubiquitylation. Nature 596: 438–443. 10.1038/s41586-021-03716-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D'Andrea AD. 2006. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol 8: 339–347. [DOI] [PubMed] [Google Scholar]
- Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, et al. 2013. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov 3: 68–81. 10.1158/2159-8290.CD-12-0049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jette NR, Kumar M, Radhamani S, Arthur G, Goutam S, Yip S, Kolinsky M, Williams GJ, Bose P, Lees-Miller SP. 2020. ATM-deficient cancers provide new opportunities for precision oncology. Cancers (Basel) 12: 687. 10.3390/cancers12030687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin MH, Oh DY. 2019. ATM in DNA repair in cancer. Pharmacol Ther 203: 107391. 10.1016/j.pharmthera.2019.07.002 [DOI] [PubMed] [Google Scholar]
- Jiricny J. 2013. Postreplicative mismatch repair. Cold Spring Harb Perspect Biol 5: a012633. 10.1101/cshperspect.a012633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes JW, Balazs A, Barratt D, Bista M, Chuba MD, Cosulich S, Critchlow SE, Degorce SL, Di Fruscia P, Edmondson SD, et al. 2021. Discovery of 5-{4-[(7-ethyl-6-oxo-5,6-dihydro-1,5-naphthyridin-3-yl)methyl]piperazin-1-yl}-N-methylpyridine-2-carboxamide (AZD5305): a PARP1–DNA trapper with high selectivity for PARP1 over PARP2 and other PARPs. J Med Chem 64: 14498–14512. 10.1021/acs.jmedchem.1c01012 [DOI] [PubMed] [Google Scholar]
- Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, Wang Y, Capelletti M, Sarosiek KA, Moreau LA, et al. 2013. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci 110: 17041–17046. 10.1073/pnas.1305170110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, Primack B, Cao S, Bernhardy AJ, Coulson R, et al. 2016. CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep 17: 2367–2381. 10.1016/j.celrep.2016.10.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhász S, Smith R, Schauer T, Spekhardt D, Mamar H, Zentout S, Chapuis C, Huet S, Timinszky G. 2020. The chromatin remodeler ALC1 underlies resistance to PARP inhibitor treatment. Sci Adv 6: eabb8626. 10.1126/sciadv.abb8626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeche L, Nguyen HD, Buisson R, Zou L. 2018. A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science 359: 108–114. 10.1126/science.aan6490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Z, Fu P, Alcivar AL, Fu H, Redon C, Foo TK, Zuo Y, Ye C, Baxley R, Madireddy A, et al. 2021. BRCA2 associates with MCM10 to suppress PRIMPOL-mediated repriming and single-stranded gap formation after DNA damage. Nat Commun 12: 5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, George E, Ragland R, Rafail S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown E, Simpkins F. 2017. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin Cancer Res 23: 3097–3108. 10.1158/1078-0432.CCR-16-2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, Medvedev S, Kinose Y, Devins K, Verma P, et al. 2020. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun 11: 3726. 10.1038/s41467-020-17127-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurashima K, Kashiwagi H, Shimomura I, Suzuki A, Takeshita F, Mazevet M, Harata M, Yamashita T, Yamamoto Y, Kohno T, et al. 2020. SMARCA4 deficiency-associated heterochromatin induces intrinsic DNA replication stress and susceptibility to ATR inhibition in lung adenocarcinoma. NAR Cancer 2: zcaa005. 10.1093/narcan/zcaa005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont KR, Hasham MG, Donghia NM, Branca J, Chavaree M, Chase B, Breggia A, Hedlund J, Emery I, Cavallo F, et al. 2013. Attenuating homologous recombination stimulates an AID-induced antileukemic effect. J Exp Med 210: 1021–1033. 10.1084/jem.20121258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. 2011. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475: 53–58. 10.1038/nature10192 [DOI] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. 2015. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372: 2509–2520. 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Paull TT. 2021. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat Rev Mol Cell Biol 22: 796–814. 10.1038/s41580-021-00394-2 [DOI] [PubMed] [Google Scholar]
- Lee JM, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, Swisher EM, Harrell MI, Trepel JB, Lee MJ, et al. 2018. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol 19: 207–215. 10.1016/S1470-2045(18)30009-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leijen S, van Geel RM, Sonke GS, de Jong D, Rosenberg EH, Marchetti S, Pluim D, van Werkhoven E, Rose S, Lee MA, et al. 2016. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol 34: 4354–4361. 10.1200/JCO.2016.67.5942 [DOI] [PubMed] [Google Scholar]
- Lemaçon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, et al. 2017. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun 8: 860. 10.1038/s41467-017-01180-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens B, Hegarat N, Akopyan K, Sala-Gaston J, Bartek J, Hochegger H, Lindqvist A. 2018. DNA replication determines timing of mitosis by restricting CDK1 and PLK1 activation. Mol Cell 71: 117–128.e3. 10.1016/j.molcel.2018.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HD, Cuevas I, Zhang M, Lu C, Alam MM, Fu YX, You MJ, Akbay EA, Zhang H, Castrillon DH. 2018. Polymerase-mediated ultramutagenesis in mice produces diverse cancers with high mutational load. J Clin Invest 128: 4179–4191. 10.1172/JCI122095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HD, Lu C, Zhang H, Hu Q, Zhang J, Cuevas IC, Sahoo SS, Aguilar M, Maurais EG, Zhang S, et al. 2020. A PoleP286R mouse model of endometrial cancer recapitulates high mutational burden and immunotherapy response. JCI Insight 5: e138829. 10.1172/jci.insight.138829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KS, Li H, Roberts EA, Gaudiano EF, Clairmont C, Sambel LA, Ponnienselvan K, Liu JC, Yang C, Kozono D, et al. 2018. USP1 is required for replication fork protection in BRCA1-deficient tumors. Mol Cell 72: 925–941.e4. 10.1016/j.molcel.2018.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee KH, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, et al. 2018. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med 379: 753–763. 10.1056/NEJMoa1802905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RL, Urban V, Muñoz-Martínez F, Ayestaran I, Thomas JC, de Renty C, O'Connor MJ, Forment JV, Galanty Y, Jackson SP. 2021. Loss of cyclin C or CDK8 provides ATR inhibitor resistance by suppressing transcription-associated replication stress. Nucleic Acids Res 49: 8665–8683. 10.1093/nar/gkab628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, de Stanchina E, Teicher BA, Riaz N, Powell SN, et al. 2017. PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin Cancer Res 23: 523–535. 10.1158/1078-0432.CCR-16-1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord RV, Brabender J, Gandara D, Alberola V, Camps C, Domine M, Cardenal F, Sanchez JM, Gumerlock PH, Taron M, et al. 2002. Low ERCC1 expression correlates with prolonged survival after cisplatin plus gemcitabine chemotherapy in non-small cell lung cancer. Clin Cancer Res 8: 2286–2291. [PubMed] [Google Scholar]
- Lu C, Guan J, Lu S, Jin Q, Rousseau B, Lu T, Stephens D, Zhang H, Zhu J, Yang M, et al. 2021. DNA sensing in mismatch repair-deficient tumor cells is essential for anti-tumor immunity. Cancer Cell 39: 96–108.e6. 10.1016/j.ccell.2020.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HT, de la Chapelle A. 2003. Hereditary colorectal cancer. N Engl J Med 348: 919–932. 10.1056/NEJMra012242 [DOI] [PubMed] [Google Scholar]
- Marechal A, Zou L. 2013. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5: a012716. 10.1101/cshperspect.a012716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A. 2015. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 518: 254–257. 10.1038/nature14157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson MT, Holub AS, Husbands AY, Petreaca RC. 2020. Mutation spectra of the MRN (MRE11, RAD50, NBS1/NBN) break sensor in cancer cells. Cancers 12: 3794. 10.3390/cancers12123794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta KPM, Lovejoy CA, Zhao R, Heintzman DR, Cortez D. 2020. HMCES maintains replication fork progression and prevents double-strand breaks in response to APOBEC deamination and abasic site formation. Cell Rep 31: 107705. 10.1016/j.celrep.2020.107705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengwasser KE, Adeyemi RO, Leng Y, Choi MY, Clairmont C, D'Andrea AD, Elledge SJ. 2019. Genetic screens reveal FEN1 and APEX2 as BRCA2 synthetic lethal targets. Mol Cell 73: 885–899.e6. 10.1016/j.molcel.2018.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK, Litton JK. 2015. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 121: 269–275. 10.1002/cncr.29041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, de Lange T. 2018. 53BP1–RIF1–shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 560: 112–116. 10.1038/s41586-018-0324-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohni KN, Kavanaugh GM, Cortez D. 2014. ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res 74: 2835–2845. 10.1158/0008-5472.CAN-13-3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva TN, Qian C, Sugitani N, Osmanbeyoglu HU, Bakkenist CJ. 2019. WEE1 kinase inhibitor AZD1775 induces CDK1 kinase-dependent origin firing in unperturbed G1- and S-phase cells. Proc Natl Acad Sci 116: 23891–23893. 10.1073/pnas.1915108116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moquin DM, Genois MM, Zhang JM, Ouyang J, Yadav T, Buisson R, Yazinski SA, Tan J, Boukhali M, Gagne JP, et al. 2019. Localized protein biotinylation at DNA damage sites identifies ZPET, a repressor of homologous recombination. Genes Dev 33: 75–89. 10.1101/gad.315978.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munster P, Mita M, Mahipal A, Nemunaitis J, Massard C, Mikkelsen T, Cruz C, Paz-Ares L, Hidalgo M, Rathkopf D, et al. 2019. First-in-human phase I study of a dual mTOR kinase and DNA-PK inhibitor (CC-115) in advanced malignancy. Cancer Manag Res 11: 10463–10476. 10.2147/CMAR.S208720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mur P, García-Mulero S, Del Valle J, Magraner-Pardo L, Vidal A, Pineda M, Cinnirella G, Martín-Ramos E, Pons T, López-Doriga A, et al. 2020. Role of POLE and POLD1 in familial cancer. Genet Med 22: 2089–2100. 10.1038/s41436-020-0922-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. 2012. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 72: 5588–5599. 10.1158/0008-5472.CAN-12-2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, Pommier Y. 2016. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 7: 76534–76550. 10.18632/oncotarget.12266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Lopes M. 2015. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16: 207–220. 10.1038/nrm3935 [DOI] [PubMed] [Google Scholar]
- Nguyen HD, Leong WY, Li W, Reddy PNG, Sullivan JD, Walter MJ, Zou L, Graubert TA. 2018. Spliceosome mutations induce R loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Res 78: 5363–5374. 10.1158/0008-5472.CAN-17-3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niraj J, Färkkilä A, D'Andrea AD. 2019. The fanconi anemia pathway in cancer. Annu Rev Cancer Biol 3: 457–478. 10.1146/annurev-cancerbio-030617-050422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Álvarez-Quilón A, Moatti N, Zimmermann M, et al. 2018. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560: 117–121. 10.1038/s41586-018-0340-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor MJ. 2015. Targeting the DNA damage response in cancer. Mol Cell 60: 547–560. 10.1016/j.molcel.2015.10.040 [DOI] [PubMed] [Google Scholar]
- Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, Ohzeki M, Takata M, Sale JE, Patel KJ. 2007. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell 28: 798–809. 10.1016/j.molcel.2007.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paes Dias M, Tripathi V, van der Heijden I, Cong K, Manolika EM, Bhin J, Gogola E, Galanos P, Annunziato S, Lieftink C, et al. 2021. Loss of nuclear DNA ligase III reverts PARP inhibitor resistance in BRCA1/53BP1 double-deficient cells by exposing ssDNA gaps. Mol Cell 81: 4692–4708.e9. 10.1016/j.molcel.2021.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Chen Y, Biju B, Ahmed N, Kong J, Goldenberg M, Huang J, Mohan N, Klosek S, Parsa K, et al. 2019. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci Rep 9: 19110. 10.1038/s41598-019-55537-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. 2019. PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov 9: 722–737. 10.1158/2159-8290.CD-18-1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar K, Kochupurakkal BS, Lazaro JB, Wang ZC, Palakurthi S, Kirschmeier PT, Yang C, Sambel LA, Färkkilä A, Reznichenko E, et al. 2019. The CHK1 inhibitor prexasertib exhibits monotherapy activity in high-grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin Cancer Res 25: 6127–6140. 10.1158/1078-0432.CCR-19-0448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, Bejnood A, Dionne D, Ge WH, Xu KH, et al. 2021. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184: 4734–4752.e20. 10.1016/j.cell.2021.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Maya-Mendoza A, Zachos G, Gillespie DA, Jackson DA, Caldecott KW. 2006. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol 26: 3319–3326. 10.1128/MCB.26.8.3319-3326.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt SJ, Rehman FL, Bajrami I, Brough R, Wallberg F, Kozarewa I, Fenwick K, Assiotis I, Chen L, Campbell J, et al. 2013. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS One 8: e61520. 10.1371/journal.pone.0061520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilger D, Seymour LW, Jackson SP. 2021. Interfaces between cellular responses to DNA damage and cancer immunotherapy. Genes Dev 35: 602–618. 10.1101/gad.348314.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quereda V, Bayle S, Vena F, Frydman SM, Monastyrskyi A, Roush WR, Duckett DR. 2019. Therapeutic targeting of CDK12/CDK13 in triple-negative breast cancer. Cancer Cell 36: 545–558.e7. 10.1016/j.ccell.2019.09.004 [DOI] [PubMed] [Google Scholar]
- Quinet A, Lemaçon D, Vindigni A. 2017. Replication fork reversal: players and guardians. Mol Cell 68: 830–833. 10.1016/j.molcel.2017.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Tirman S, Jackson J, Šviković S, Lemaçon D, Carvajal-Maldonado D, González-Acosta D, Vessoni AT, Cybulla E, Wood M, et al. 2020. PRIMPOL-mediated adaptive response suppresses replication fork reversal in BRCA-deficient cells. Mol Cell 77: 461–474.e9. 10.1016/j.molcel.2019.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafiei S, Fitzpatrick K, Liu D, Cai MY, Elmarakeby HA, Park J, Ricker C, Kochupurakkal BS, Choudhury AD, Hahn WC, et al. 2020. ATM loss confers greater sensitivity to ATR inhibition than PARP inhibition in prostate cancer. Cancer Res 80: 2094–2100. 10.1158/0008-5472.CAN-19-3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratiu JJ, Racine JJ, Hasham MG, Wang Q, Branca JA, Chapman HD, Zhu J, Donghia N, Philip V, Schott WH, et al. 2017. Genetic and small molecule disruption of the AID/RAD51 axis similarly protects nonobese diabetic mice from type 1 diabetes through expansion of regulatory B lymphocytes. J Immunol 198: 4255–4267. 10.4049/jimmunol.1700024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. 2016. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535: 382–387. 10.1038/nature18325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MAM, Parry DA, Williams TC, Nadeu F, Hindshaw RL, Rios Szwed DO, Nicholson MD, Carroll P, Boyle S, Royo R, et al. 2022. Signatures of TOP1 transcription-associated mutagenesis in cancer and germline. Nature 602: 623–631. 10.1038/s41586-022-04403-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RF, Walton MI, Cherry DL, Collins I, Clarke PA, Garrett MD, Workman P. 2020. CHK1 inhibition is synthetically lethal with loss of B-family DNA polymerase function in human lung and colorectal cancer cells. Cancer Res 80: 1735–1747. 10.1158/0008-5472.CAN-19-1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, et al. 2008. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci 105: 17079–17084. 10.1073/pnas.0806092105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulston A, Zimmermann M, Papp R, Skeldon A, Pellerin C, Dumas-Bérube E, Dumais V, Dorich S, Fader LD, Fournier S, et al. 2022. RP-3500: a novel, potent, and selective ATR inhibitor that is effective in preclinical models as a monotherapy and in combination with PARP inhibitors. Mol Cancer Ther 21: 245–256. 10.1158/1535-7163.MCT-21-0615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz S, Mayor-Ruiz C, Lafarga V, Murga M, Vega-Sendino M, Ortega S, Fernandez-Capetillo O. 2016. A genome-wide CRISPR screen identifies CDC25A as a determinant of sensitivity to ATR inhibitors. Mol Cell 62: 307–313. 10.1016/j.molcel.2016.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. 2008. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 451: 1116–1120. 10.1038/nature06633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, Cimprich KA. 2017. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18: 622–636. 10.1038/nrm.2017.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Hamperl S, Bocek MJ, Chung M, Bass TE, Cisneros-Soberanis F, Samejima K, Xie L, Paulson JR, Earnshaw WC, et al. 2018. An intrinsic S/G2 checkpoint enforced by ATR. Science 361: 806–810. 10.1126/science.aap9346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Krishna C, Ma X, Pei X, Lee KW, Makarov V, Kuo F, Chung J, Srivastava RM, Purohit TA, et al. 2021. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat Cancer 1: 1188–1203. 10.1038/s43018-020-00139-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, Nuryadi E, Sekine R, Oike T, Kakoti S, et al. 2017. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 8: 1751. 10.1038/s41467-017-01883-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542. 10.1016/j.cell.2011.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51–BRCA1/2. Cancer Cell 22: 106–116. 10.1016/j.ccr.2012.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleicher EM, Dhoonmoon A, Jackson LM, Clements KE, Stump CL, Nicolae CM, Moldovan GL. 2020. Dual genome-wide CRISPR knockout and CRISPR activation screens identify mechanisms that regulate the resistance to multiple ATR inhibitors. PLoS Genet 16: e1009176. 10.1371/journal.pgen.1009176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonen PM, Talens F, Stok C, Gogola E, Heijink AM, Bouwman P, Foijer F, Tarsounas M, Blatter S, Jonkers J, et al. 2017. Progression through mitosis promotes PARP inhibitor-induced cytotoxicity in homologous recombination-deficient cancer cells. Nat Commun 8: 15981. 10.1038/ncomms15981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, Niedzwiedz W. 2015. The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol Cell 60: 351–361. 10.1016/j.molcel.2015.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PD, Wethington SL, Pagan C, Latif N, Tanyi J, Martin LP, Morgan M, Burger RA, Haggerty A, Zarrin H, et al. 2021. Combination ATR and PARP inhibitor (CAPRI): a phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol Oncol 163: 246–253. 10.1016/j.ygyno.2021.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakeel S, Rajendra E, Alcón P, O'Reilly F, Chorev DS, Maslen S, Degliesposti G, Russo CJ, He S, Hill CH, et al. 2019. Structure of the Fanconi anaemia monoubiquitin ligase complex. Nature 575: 234–237. 10.1038/s41586-019-1703-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng H, Huang Y, Xiao Y, Zhu Z, Shen M, Zhou P, Guo Z, Wang J, Wang H, Dai W, et al. 2020. ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J Immunother Cancer 8: e000340. 10.1136/jitc-2019-000340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y. 2013. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14: 197–210. 10.1038/nrm3546 [DOI] [PubMed] [Google Scholar]
- Shiotani B, Zou L. 2009. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell 33: 547–558. 10.1016/j.molcel.2009.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji MKK, Renaudin X, Williams CH, Venkitaraman AR. 2018. BRCA2 regulates transcription elongation by RNA polymerase II to prevent R-loop accumulation. Cell Rep 22: 1031–1039. 10.1016/j.celrep.2017.12.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoneau A, Xiong R, Zou L. 2021. The trans cell cycle effects of PARP inhibitors underlie their selectivity toward BRCA1/2-deficient cells. Genes Dev 35: 1271–1289. 10.1101/gad.348479.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Ahmed D, Dolatshad H, Tatwavedi D, Schulze U, Sanchi A, Ryley S, Dhir A, Carpenter L, Watt SM, et al. 2020. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 34: 2525–2530. 10.1038/s41375-020-0753-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommers JA, Kulikowicz T, Croteau DL, Dexheimer T, Dorjsuren D, Jadhav A, Maloney DJ, Simeonov A, Bohr VA, Brosh RM Jr. 2019. A high-throughput screen to identify novel small molecule inhibitors of the werner syndrome helicase-nuclease (WRN). PLoS One 14: e0210525. 10.1371/journal.pone.0210525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. 2005. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 7: 195–201. 10.1038/ncb1212 [DOI] [PubMed] [Google Scholar]
- Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chédin F, Swigut T, Cimprich KA. 2016. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife 5: e17548. 10.7554/eLife.17548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LL, Yang RY, Li CW, Chen MK, Shao B, Hsu JM, Chan LC, Yang Y, Hsu JL, Lai YJ, et al. 2018. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am J Cancer Res 8: 1307–1316. [PMC free article] [PubMed] [Google Scholar]
- Suszynska M, Ratajska M, Kozlowski P. 2020. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: mutation prevalence and precise risk estimates based on a pooled analysis of ∼30,000 cases. J Ovarian Res 13: 50. 10.1186/s13048-020-00654-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Leuzzi G, Sannino V, Cuella-Martin R, Huang JW, Wu-Baer F, Baer R, Costanzo V, Ciccia A. 2021. REV1-Polζ maintains the viability of homologous recombination-deficient cancer cells through mutagenic repair of PRIMPOL-dependent ssDNA gaps. Mol Cell 81: 4008–4025.e7. 10.1016/j.molcel.2021.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirman S, Quinet A, Wood M, Meroni A, Cybulla E, Jackson J, Pegoraro S, Simoneau A, Zou L, Vindigni A. 2021. Temporally distinct post-replicative repair mechanisms fill PRIMPOL-dependent ssDNA gaps in human cells. Mol Cell 81: 4026–4040.e8. 10.1016/j.molcel.2021.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkáč J, Xu G, Adhikary H, Young JTF, Gallo D, Escribano-Díaz C, Krietsch J, Orthwein A, Munro M, Sol W, et al. 2016. HELB is a feedback inhibitor of DNA end resection. Mol Cell 61: 405–418. 10.1016/j.molcel.2015.12.013 [DOI] [PubMed] [Google Scholar]
- Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR, Fernandez-Capetillo O. 2011. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol 18: 721–727. 10.1038/nsmb.2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. 2013. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103. 10.1016/j.cell.2013.10.043 [DOI] [PubMed] [Google Scholar]
- van Bussel MTJ, Awada A, de Jonge MJA, Mau-Sørensen M, Nielsen D, Schöffski P, Verheul HMW, Sarholz B, Berghoff K, El Bawab S, et al. 2021. A first-in-man phase 1 study of the DNA-dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br J Cancer 124: 728–735. 10.1038/s41416-020-01151-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wietmarschen N, Sridharan S, Nathan WJ, Tubbs A, Chan EM, Callen E, Wu W, Belinky F, Tripathi V, Wong N, et al. 2020. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 586: 292–298. 10.1038/s41586-020-2769-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, Nugent K, Ballew M, Kiesel BF, Beumer JH, Sarkar SN, et al. 2018. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J Clin Invest 128: 3926–3940. 10.1172/JCI96519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaraman AR. 2002. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 108: 171–182. 10.1016/S0092-8674(02)00615-3 [DOI] [PubMed] [Google Scholar]
- Verma P, Zhou Y, Cao Z, Deraska PV, Deb M, Arai E, Li W, Shao Y, Puentes L, Li Y, et al. 2021. ALC1 links chromatin accessibility to PARP inhibitor response in homologous recombination-deficient cells. Nat Cell Biol 23: 160–171. 10.1038/s41556-020-00624-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikas P, Borcherding N, Chennamadhavuni A, Garje R. 2020. Therapeutic potential of combining PARP inhibitor and immunotherapy in solid tumors. Front Oncol 10: 570. 10.3389/fonc.2020.00570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villaruz LC, Jones H, Dacic S, Abberbock S, Kurland BF, Stabile LP, Siegfried JM, Conrads TP, Smith NR, O'Connor MJ, et al. 2016. ATM protein is deficient in over 40% of lung adenocarcinomas. Oncotarget 7: 57714–57725. 10.18632/oncotarget.9757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wang G, Feng X, Shepherd P, Zhang J, Tang M, Chen Z, Srivastava M, McLaughlin ME, Navone NM, et al. 2019a. Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene 38: 2451–2463. 10.1038/s41388-018-0606-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zhao Q, Wang YN, Jin Y, He MM, Liu ZX, Xu RH. 2019b. Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol 5: 1504–1506. 10.1001/jamaoncol.2019.2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Xiang D, Liu B, He A, Randle HJ, Zhang KX, Dongre A, Sachs N, Clark AP, Tao L, et al. 2019c. Inadequate DNA damage repair promotes mammary transdifferentiation, leading to BRCA1 breast cancer. Cell 178: 135–151.e19. 10.1016/j.cell.2019.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Tang M, Chen Z, Nie L, Li S, Xiong Y, Szymonowicz KA, Park JM, Zhang H, Feng X, et al. 2021a. Genetic vulnerabilities upon inhibition of DNA damage response. Nucleic Acids Res 49: 8214–8231. 10.1093/nar/gkab643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang R, Peralta C, Yaseen A, Pavletich NP. 2021b. Structure of the FA core ubiquitin ligase closing the ID clamp on DNA. Nat Struct Mol Biol 28: 300–309. 10.1038/s41594-021-00568-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B, Comino-Méndez I, de Bruijn I, Tian L, Meisel JL, García-Murillas I, Fribbens C, Cutts R, Martelotto LG, Ng CKY, et al. 2017. Diverse BRCA1 and BRCA2 reversion mutations in circulating cell-free DNA of therapy-resistant breast or ovarian cancer. Clin Cancer Res 23: 6708–6720. 10.1158/1078-0432.CCR-17-0544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A, Badham N, Rafiq R, Brough R, Gulati A, et al. 2016. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun 7: 13837. 10.1038/ncomms13837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, Scully R. 2014. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510: 556–559. 10.1038/nature13295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Frock RL, Menghi F, Duffey EE, Panday A, Camacho V, Hasty EP, Liu ET, Alt FW, Scully R. 2017. Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature 551: 590–595. 10.1038/nature24477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. 2006. Molecular linkage between the kinase ATM and NF-κB signaling in response to genotoxic stimuli. Science 311: 1141–1146. 10.1126/science.1121513 [DOI] [PubMed] [Google Scholar]
- Xing X, Kane DP, Bulock CR, Moore EA, Sharma S, Chabes A, Shcherbakova PV. 2019. A recurrent cancer-associated substitution in DNA polymerase ε produces a hyperactive enzyme. Nat Commun 10: 374. 10.1038/s41467-018-08145-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, et al. 2015. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521: 541–544. 10.1038/nature14328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Li Y, Sun R, Li J. 2021. Identification of a RAD52 inhibitor inducing synthetic lethality in BRCA2-deficient cancer cells. Front Pharmacol 12: 637825. 10.3389/fphar.2021.637825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap TA, O'Carrigan B, Penney MS, Lim JS, Brown JS, de Miguel Luken MJ, Tunariu N, Perez-Lopez R, Rodrigues DN, Riisnaes R, et al. 2020. Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol 38: 3195–3204. 10.1200/JCO.19.02404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap TA, Krebs MG, Postel-Vinay S, El-Khouiery A, Soria JC, Lopez J, Berges A, Cheung SA, Irurzun-Arana I, Goldwin A, et al. 2021a. Ceralasertib (AZD6738), an oral ATR kinase inhibitor, in combination with carboplatin in patients with advanced solid tumors: a phase I study. Clin Cancer Res 27: 5213–5224. 10.1158/1078-0432.CCR-21-1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap TA, Tan DSP, Terbuch A, Caldwell R, Guo C, Goh BC, Heong V, Haris NRM, Bashir S, Drew Y, et al. 2021b. First-in-human trial of the oral ataxia telangiectasia and RAD3-related (ATR) inhibitor BAY 1895344 in patients with advanced solid tumors. Cancer Discov 11: 80–91. 10.1158/2159-8290.CD-20-0868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A, et al. 2017. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 31: 318–332. 10.1101/gad.290957.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Johnson RE, Prakash L, Prakash S. 2021. Implications of inhibition of Rev1 interaction with Y family DNA polymerases for cisplatin chemotherapy. Genes Dev 35: 1256–1270. 10.1101/gad.348662.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AM, McVey M. 2010. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res 38: 5706–5717. 10.1093/nar/gkq379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatreanu D, Robinson HMR, Alkhatib O, Boursier M, Finch H, Geo L, Grande D, Grinkevich V, Heald RA, Langdon S, et al. 2021. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat Commun 12: 3636. 10.1038/s41467-021-23463-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chiang HC, Wang Y, Zhang C, Smith S, Zhao X, Nair SJ, Michalek J, Jatoi I, Lautner M, et al. 2017. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat Commun 8: 15908. 10.1038/ncomms15908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Vaithiyalingam S, San Filippo J, Maranon DG, Jimenez-Sainz J, Fontenay GV, Kwon Y, Leung SG, Lu L, Jensen RB, et al. 2015. Promotion of BRCA2-dependent homologous recombination by DSS1 via RPA targeting and DNA mimicry. Mol Cell 59: 176–187. 10.1016/j.molcel.2015.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Li M. 2021. Evaluation of BRCA1 and BRCA2 as indicators of response to immune checkpoint inhibitors. JAMA Netw Open 4: e217728. 10.1001/jamanetworkopen.2021.7728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, Färkkilä A, Kochupurakkal B, Syed A, Shapiro GI, et al. 2021. A first-in-class polymerase θ inhibitor selectively targets homologous-recombination-deficient tumors. Nat Cancer 2: 598–610. 10.1038/s43018-021-00203-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Wang X, Yan W, Yang H, Xiang Y, Lv F, Shi Y, Li HY, Lan L. 2021. Ubiquitination-mediated degradation of TRDMT1 regulates homologous recombination and therapeutic response. NAR Cancer 3: zcab010. 10.1093/narcan/zcab010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Z, Muir M, Fluteau A, Aregger M, McEwan A, et al. 2018. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559: 285–289. 10.1038/s41586-018-0291-z [DOI] [PMC free article] [PubMed] [Google Scholar]