

In this review Sutherland et al. discuss recent discoveries on the mechanisms of small cell lung cancer (SCLC) plasticity and how these processes may impinge on antitumor immunity.
Keywords: neuroendocrine, plasticity, lung cancer, small cell, heterogeneity
Abstract
Small cell lung cancer (SCLC) is a rapidly growing, highly metastatic, and relatively immune-cold lung cancer subtype. Historically viewed in the laboratory and clinic as a single disease, new discoveries suggest that SCLC comprises multiple molecular subsets. Expression of MYC family members and lineage-related transcription factors ASCL1, NEUROD1, and POU2F3 (and, in some studies, YAP1) define unique molecular states that have been associated with distinct responses to a variety of therapies. However, SCLC tumors exhibit a high degree of intratumoral heterogeneity, with recent studies suggesting the existence of tumor cell plasticity and phenotypic switching between subtype states. While SCLC plasticity is correlated with, and likely drives, therapeutic resistance, the mechanisms underlying this plasticity are still largely unknown. Subtype states are also associated with immune-related gene expression, which likely impacts response to immune checkpoint blockade and may reveal novel targets for alternative immunotherapeutic approaches. In this review, we synthesize recent discoveries on the mechanisms of SCLC plasticity and how these processes may impinge on antitumor immunity.
Small cell lung cancer (SCLC) is known as a neuroendocrine lung cancer that is highly aggressive and metastatic with dismal patient outcomes (Fig. 1; Rudin et al. 2019; Poirier et al. 2020). In recent years, our understanding of SCLC has undergone a series of shapeshifting transformations. For decades, SCLC was treated in the clinic and in the laboratory as a single disease, with a slow-growing appreciation for heterogeneity in tumor cell morphology, gene expression, viral tropism, and other characteristics. More recently, SCLC has been stratified based on expression of MYC family members and lineage-defining transcription factors ASCL1, NEUROD1, and POU2F3 (and initially YAP1, which has recently been called into question) (Rudin et al. 2019; Poirier et al. 2020). These molecular stratifications are important because they correlate with therapeutic responsiveness in preclinical and clinical studies. However, recent findings suggest that SCLC exhibits remarkable subtype plasticity, bringing our notion of SCLC full circle—from one disease, to a disease of multiple subtypes, and (potentially) back to one highly plastic disease. In this review, we (1) provide a brief historical overview of our changing perspective of SCLC, (2) discuss our current understanding of the therapeutic relevance of SCLC molecular subsets, (3) synthesize recent findings on mechanisms and factors regulating SCLC plasticity and how these mechanisms may impact response to immunotherapy, and (4) speculate on future directions in SCLC research.
Figure 1.

Basic clinical background on small cell lung cancer (SCLC) (in the text box; left) with illustrated sites of metastatic spread (right).
While many timely SCLC reviews have been written by our colleagues (Rudin et al. 2019, 2021; Poirier et al. 2020; Ko et al. 2021; Schwendenwein et al. 2021), here we focus specifically on the molecular subtypes of SCLC and the emerging evidence supporting their relationships to drug sensitivities and immune response, and their highly plastic nature.
Early studies documenting phenotypic heterogeneity in SCLC
Following years of optimizing the growth of human SCLC cell lines in culture in the 1970s, Carney et al. (1985) and Gazdar et al. (1985) published back-to-back studies characterizing 50 newly created SCLC cell lines. This detailed characterization found that 70% of cell lines exhibited “classic” morphology with tightly packed, neurosphere-like aggregates and high expression of proteins associated with neuroendocrine fate. The remaining 30% of cell lines were termed “variant,” a fraction of which grew in the classic morphology but harbored reduced neuroendocrine (NE) marker expression, and the remaining of which exhibited a more loosely attached morphology, resembling undifferentiated large cell carcinomas. The latter “variant” lines with reduced NE markers, the investigators presciently observed, tended to harbor MYC amplifications and numerous hallmarks of aggressive behavior: The MYC-high cell lines were more often derived from patients after treatment, grew faster, exhibited radio resistance, and were associated with poor response to therapy and shorter survival times (Radice et al. 1982; Little et al. 1983; Johnson et al. 1987). MYCN and MYCL amplifications and overexpression (and, in the case of MYCL, genomic rearrangements) were also observed in cell lines in a mutually exclusive manner with MYC (Nau et al. 1985, 1986; Wistuba et al. 2001; Kim et al. 2006). These studies represented some of the first examples of SCLC heterogeneity. Through comprehensive gene expression analyses of human SCLC tumors and cell lines, we now know that the non-NE SCLCs are enriched for MYC and markers that signal activation of Notch/Rest, Hippo/Yap1, TGF-β, and epithelial–mesenchymal transition (EMT) pathways (Zhang et al. 2018). The biological and clinical significance of these findings is becoming increasingly appreciated over time.
With the emergence of whole-genome sequencing, gene expression, and methylation profiling, we learned that loss of TP53 and RB1 function are near-universal events in SCLC (Peifer et al. 2012; Rudin et al. 2012; George et al. 2015; Poirier et al. 2015). Notch receptor loss-of-function (LOF) alterations are found in ∼25% of SCLC and are mutually exclusive with alterations in chromatin remodeling genes CREBBP and EP300. In contrast to the personalized treatment approaches achieved with kinase alterations in lung adenocarcinoma, these common genomic alterations in SCLC did not immediately implicate targeted therapy approaches for specific genetic subsets. Instead, SCLC began to be stratified by expression of ASCL1 (a lineage-specific developmental transcription factor driving NE fate) and NEUROD1 (another basic helix–loop–helix transcription factor important for brain development) (Borges et al. 1997; Cau et al. 1997; Ito et al. 2000; Neptune et al. 2008; Osborne et al. 2013; Borromeo et al. 2016). Poirier et al. (2013) discovered that a relatively high NEUROD1/ASCL1 ratio correlated with sensitivity to infection with Seneca Valley virus (SVV), which preferentially inhibited growth of this subset of tumors. The receptor for SVV infection was later determined to be anthrax toxin receptor 1 (ANTXR1) (Miles et al. 2017). The SVV study represents one of the first examples of molecular subtype stratification of SCLC for therapeutic purposes in a preclinical setting.
Heterogeneity in therapeutic responses: ASCL1 vs. MYC
In the last decade, examples abound elucidating SCLC subtype-specific therapeutic responses. Multiple groups found that MYC-high subtypes of SCLC correspond with ASCL1-low samples, and this subset of SCLC responds to Aurora A/B kinase inhibition in cell lines and animal models (Hook et al. 2012; Sos et al. 2012; Helfrich et al. 2016; Cardnell et al. 2017; Mollaoglu et al. 2017; Dammert et al. 2019). More recently, CRISPR-mediated activation of specific Myc family members elegantly demonstrated that Myc, as opposed to Mycn or Mycl, promotes sensitivity to AURKA inhibition (Brägelmann et al. 2017; Dammert et al. 2019). These findings parallel results in clinical trials of chemotherapy-relapsed SCLC patients who received paclitaxel with or without the AURKA inhibitor alisertib, where patients with MYC-high tumors had significantly improved progression-free survival when receiving alisertib (Owonikoko et al. 2020).
In contrast to the MYC-associated vulnerabilities, Delta-like-ligand 3 (DLL3) tends to be expressed most highly in ASCL1-high SCLC, consistent with its identification as an ASCL1 target gene (Saunders et al. 2015; Borromeo et al. 2016; Zhang et al. 2018). DLL3 is a Notch pathway inhibitory ligand, and its high surface expression in the ASCL1 subtype is consistent with the mutual antagonism observed between ASCL1 and the Notch pathway (Chen et al. 1997; Morrison et al. 2000; Ball 2004; Somasundaram et al. 2005; Lim et al. 2017), discussed in more detail later. The DLL3 targeted antibody drug conjugate rovalpituzumab tesirine (Rova-T) has been evaluated in clinical trials and was more effective against DLL3-expressing tumors, albeit limited by toxicities (Saunders et al. 2015; Rudin et al. 2017; Morgensztern et al. 2019). Additional DLL3 targeting approaches are currently under development (Owen et al. 2019).
In addition to the two clinical examples above, preclinical studies have stratified SCLC molecular subsets based on unique cell death phenotypes. BH3 profiling, a functional assay that measures mitochondrial depolarization in response to stimulation with various BH3 peptides, demonstrated that MYC-high SCLC is more apoptotically primed than MYC-low cell lines (Dammert et al. 2019). This observation is consistent with higher basal levels of apoptosis in Myc-high compared with Myc-low tumors in genetically engineered mouse models (GEMMs) (Mollaoglu et al. 2017). BH3 profiling also identified MCL1 as an apoptotic vulnerability in MYC-high cell lines, but this remains to be evaluated in preclinical GEMMs, hampered by the reduced affinity of the MCL1-specific inhibitor S63845 to murine MCL1 (Kotschy et al. 2016; Brennan et al. 2018). In contrast, multiple groups have noted that the prosurvival protein BCL2 is enriched in the classic ASCL1-high (MYC-low) subset of SCLC (Augustyn et al. 2014; Poirier et al. 2015; Borromeo et al. 2016; Cardnell et al. 2017; Lochmann et al. 2018; Dammert et al. 2019). Indeed, BCL2 has been shown to be an ASCL1 target gene (Augustyn et al. 2014; Borromeo et al. 2016) that can be repressed by MYC expression (Dammert et al. 2019), providing rationale to revisit the efficacy of BCL2 inhibitors in the ASCL1-high setting. A more recent study suggests that the machinery required for extrinsic apoptosis and necroptosis is lowly expressed in SCLC, but a subset of SCLC is enriched for ferroptosis pathway genes (Bebber et al. 2021). SCLC cells that are ASCL1-low (often expressing multiple EMT markers) tend to be more sensitive to ferroptosis induction (Bebber et al. 2021), reminiscent of studies suggesting mesenchymal fate is predictive of ferroptosis sensitivity in multiple cancer types (Hangauer et al. 2017; Viswanathan et al. 2017; Jiang et al. 2021). Recent studies also suggest SCLC can undergo pyroptosis (Wu et al. 2021). How SCLC molecular subsets are wired for sensitivity to diverse forms of cell death and how cell death processes impact immune response should be important areas of further study.
In addition to differences in cell death regulation, MYC-high/ASCL1-low SCLC tends to be more proliferative and glycolytic with unique metabolic vulnerabilities (Mollaoglu et al. 2017; Huang et al. 2018a; Cargill et al. 2021). Unbiased metabolite profiling experiments on cell lines and murine tumors have identified key metabolic differences in ASCL1-high versus ASCL1-low SCLC subtypes. For example, Huang et al. (2018a, 2021) found that purine pathway metabolites, particularly guanosine nucleotides, are highly enriched in ASCL1-low cell lines. ASCL1-low cell lines and tumors have higher expression of IMPDH1 and IMPDH2, known MYC target genes that are key for guanine nucleotide biosynthesis (Mannava et al. 2008). Increased IMPDH expression is associated with dependency on these enzymes, conferring sensitivity to mycophenolic acid (MPA) in vitro and mizoribine in vivo, both of which are used in clinical settings as immunosuppressants (Dayton et al. 1992). Metabolite profiling studies were also key to the discovery that Myc-high/Ascl1-low tumors are exquisitely sensitive to depletion of exogenous arginine with pegylated arginine deiminase (ADI-PEG20) in GEMMs and human xenografts (Chalishazar et al. 2019). Taken together, MYC-high/ASCL1-low SCLC appears to be highly proliferative and glycolytic, but primed for cell death and/or ferroptosis specifically, suggesting this aggressive level of tumor growth confers unique therapeutic vulnerabilities.
Inactivating mutations in MAX, the protein product of which is a heterodimer of MYC family members, are found in ∼6% of SCLC (Romero et al. 2014). MAX was found to be a tumor suppressor whose loss leads to derepression of serine and one-carbon pathway genes (Augert et al. 2020). Recent studies suggest that MAX-deficient SCLC is predominantly of the ASCL1 subtype and does not depend on any MYC family member (Llabata et al. 2021). These studies beg the question of whether a non-MYC family member state of SCLC is phenotypically and/or metabolically distinct from other molecular subsets.
Despite the generally higher rate of proliferation and cell cycle vulnerabilities associated with MYC-high/ASCL1-low SCLC, a recent study showed that it is the NE-high ASCL1+ subgroup of SCLC that has high replication stress, which responds preferentially well to ATR inhibition in clinical trials (Thomas et al. 2021). Other preclinical studies have suggested that SCLC is vulnerable to ATR and CHK1 kinase inhibition (Cardnell et al. 2017; Doerr et al. 2017; Sen et al. 2017; Dammert et al. 2019), but some studies suggest that MYC-high SCLC is the most sensitive. Thus, there remain outstanding questions as to how SCLC subsets are metabolically wired to drive proliferation, and how this wiring creates therapeutic vulnerabilities. Given that metabolic and proliferative characteristics of in vivo tumors are not easily mimicked in a dish (Muir et al. 2018), it will be crucial to elucidate these vulnerabilities further in physiologically relevant systems.
A revised and evolving classification scheme based on lineage-related transcription factors
The field has recently converged on finer resolution of SCLC molecular subsets by gene expression analysis, defined by ASCL1 (SCLC-A), NEUROD1 (SCLC-N), POU2F3 (SCLC-P), or (initially) YAP1 (SCLC-Y). The majority of SCLCs express ASCL1, estimated at ∼70% of cases by gene expression (Rudin et al. 2019) and protein analyses (Baine et al. 2020). NEUROD1 is expressed in ∼11% of SCLC cases by gene expression (Rudin et al. 2019) but is detected in ∼45% of cases at the protein level (Baine et al. 2020), likely because bulk gene expression underestimates the frequency of samples with sparsely positive cells. When analyzed at the protein level, the majority of NEUROD1+ tumor samples also have ASCL1+ cells, while ∼17% of SCLC cases are NEUROD1-predominant (Baine et al. 2020). Both SCLC-A and SCLC-N subtypes tend to be NE-high and express MYCL, with SCLC-N having distinct neuronal signatures compared with SCLC-A (Borromeo et al. 2016; Zhang et al. 2018; Chan et al. 2021; Patel et al. 2021). While most studies have not carefully sought to determine whether ASCL1 and NEUROD1 are coexpressed at the protein level in the same tumor cells, single-cell transcriptional approaches have suggested that a minority subset of tumor cells (7%–9%) expresses both markers simultaneously in mouse and patient tumors (Ireland et al. 2020; Chan et al. 2021). The co-occurrence of ASCL1 and NEUROD1 in individual tumor cells could portend subtype plasticity and the presence of transitional states and/or could reflect the inadequacy of individual markers to define a single subtype, discussed in more detail later.
POU2F3 is a master driver of the tuft or brush cell lineage, a rare cell type in the lung and other tissues that exhibits chemosensory functions (Montoro et al. 2018; Plasschaert et al. 2018; Huang et al. 2018b). POU2F3 expression is observed in ∼16% of SCLC cases by gene expression and in ∼7% of cases at the protein level, which tend to be mutually exclusive with ASCL1 and NEUROD1. SCLC-P tumors have a low neuroendocrine gene expression program, consistent with their low expression of ASCL1 and NEUROD1. SCLC-P tumor cell lines depend on POU2F3 for viability, and this tumor subset has been shown to preferentially depend on IGF-1R and PARP signaling (Huang et al. 2018b; Gay et al. 2021) and may be enriched for MCL1 expression (Yasuda et al. 2020), reiterating the notion that distinct SCLC subtypes harbor unique therapeutic vulnerabilities. Further studies will be needed to determine whether POU2F3 co-occurs with other subtype markers in the same tumor cells and whether it can arise during subtype evolution.
Importantly, GEMM tumors that express high levels of ASCL1, such as those from the Rb1fl/fl;p53fl/fl (RP), Rb1fl/fl; p53fl/fl;Ptenfl/fl (RPP), Rb1fl/fl;p53fl/fl;Rbl2fl/fl (RPR2), and Rb1fl/fl;p53fl/fl;Rlf-Mycl fusion mice, are highly neuroendocrine and express or amplify Mycl (Dooley et al. 2011; Huijbers et al. 2014; McFadden et al. 2014; Semenova et al. 2016; Mollaoglu et al. 2017; Ciampricotti et al. 2021), similar to human tumors. In contrast, tumors driven by Myc (Rb1fl/fl;p53fl/fl;LSL-MycT58A/T58A [RPM] mice) often exhibit reduced ASCL1 and Mycl and can express NEUROD1, POU2F3, and/or YAP1 in a context-specific manner (Mollaoglu et al. 2017; Ireland et al. 2020). Thus, these molecular subtype states appear to be conserved between mice and humans, although the mechanisms that lead to these states are not yet fully understood. Interestingly, these studies demonstrate that NE cells have the capacity to give rise to ASCL1+, NEUROD1+, and YAP1+ subtypes, given specific genetic conditions. In contrast, POU2F3+ tumors arose in the presence of stabilized mutant MYC from an unknown cell of origin using CMV-Cre (Ireland et al. 2020), potentially a tuft cell, but this requires further study to definitively conclude. It is important to note that this finding does not exclude the possibility that SCLC-P tumors could arise from a NE cell of origin, given the proper (epi)genetic conditions (Ireland et al. 2020; Olsen et al. 2021; Chen et al. 2022). Although NE cells are recognized as a major cell of origin for SCLC (Park et al. 2011; Sutherland et al. 2011), it has been demonstrated that other lung cells, including club and alveolar type II (AT2) cells, can give rise to NE-high SCLC in the context of high MYC and loss of Rb1 and p53 (Olsen et al. 2021; Chen et al. 2022). These findings emphasize that SCLC may arise from multiple cell lineages in a context-specific manner and that adult differentiated cells can exhibit a high degree of cellular plasticity under potent genetic conditions.
Recent studies have called into question the existence or relevance of YAP1 expression in SCLC (Baine et al. 2020; Gay et al. 2021). YAP1 was originally determined to represent ∼2% of SCLC cases by gene expression analysis (Rudin et al. 2019). However, when SCLC cell lines were taken into consideration, the frequency of YAP1+ SCLC has been estimated to be as high as ∼10% (Ireland et al. 2020), and a recent study of human tissue also reported YAP1 protein in ∼10% of SCLC (Owonikoko et al. 2021). SCLC-Y was noted to be non-NE and express multiple markers associated with EMT, including vimentin (VIM), SNAI2, and CD44 (Zhang et al. 2018; Rudin et al. 2019; Ireland et al. 2020). Importantly, recent analyses suggest that YAP1 mRNA expression fails to distinguish a clear subset of tumors that is distinct from the ASCL1, NEUROD1, and POU2F3 subsets (Gay et al. 2021). Consistent with that notion, protein analyses of 174 tumor samples found low expression of YAP1 that did not appear to be exclusive of other subtypes (Baine et al. 2020). As YAP1 appears to be more commonly expressed in SCLC cell lines than tumors, this calls into question whether microenvironmental or mechano–transduction signals may induce or select for YAP1 (Sun and Irvine 2016). YAP1 can also be present in stromal cells, which makes it difficult to distinguish from tumor cells with bulk analysis approaches. However, YAP1 can be expressed in late stage tumors in multiple mouse models of SCLC (Ireland et al. 2020; Wu et al. 2021) and has been detected in human SCLC circulating tumor cell-derived xenografts (CDX) at variable proportions (Pearsall et al. 2020). These data suggest that YAP1 could be the result of stromal contamination and/or could represent a late stage of SCLC progression. Toward the latter possibility, recent functional studies show that ectopic expression of YAP1 or TAZ (WWTR1) can promote a non-NE fate (Horie et al. 2016; Wu et al. 2021; Jin et al. 2022). Conversely, recent studies suggest that YAP1 may serve as a prognostic biomarker for limited-stage disease and tumors exhibiting a T-cell inflamed phenotype (Tlemsani et al. 2020; Owonikoko et al. 2021). Finally, to further complicate the issue, YAP1 has been associated with RB1 proficiency in cell line studies (McColl et al. 2017; Tlemsani et al. 2020), questioning whether these cells are large cell neuroendocrine, which share morphological similarities with variant SCLC (Sonkin et al. 2019). Hence, the controversy surrounding the clinical significance of YAP1 will be an active area of investigation to resolve these issues and to evaluate its potential implications for immunotherapy response, discussed in more detail below.
In the midst of the development and refinement of the SCLC molecular classification scheme (Rudin et al. 2019), numerous studies began to suggest that tumor heterogeneity can change, particularly during the course of chemotherapy treatment. CDX and patient-derived xenograft (PDX) models provide a unique means to interrogate changes in tumor composition of individual human tumors before, during, and after a given therapy. Specifically, CDX/PDX models have been used to identify molecular and phenotypic changes associated with chemoresistance in SCLC. With these and other models, multiple studies demonstrated that MYC expression and the non-NE SCLC phenotype increase following chemotherapy treatment, while NE identity decreases (Drapkin et al. 2018; Wagner et al. 2018; Chalishazar et al. 2019; Simpson et al. 2020; Stewart et al. 2020; Gay et al. 2021; Huang et al. 2021). For example, in a cohort of 19 SCLC PDX models, a MYC transcriptional signature correlated with resistance to etoposide and platinum (Drapkin et al. 2018). In a matched pair of CDX, the percentage of MYC+ cells increased with tumor progression, concordant with increased expression of Notch signaling and EMT markers (Simpson et al. 2020). These findings are consistent in GEMMs, where high expression of MYC or MYCN and low expression of NE markers correspond with poor response to chemotherapy (Mollaoglu et al. 2017; Grunblatt et al. 2020). In human cell lines, MYC protein levels also increased (while MYCL decreased) following selection for chemoresistance (Wagner et al. 2018; Chalishazar et al. 2019; Huang et al. 2021). ASCL1 was also found to be significantly reduced in chemotherapy-resistant cell lines and in a cohort of human tissue samples following chemotherapy relapse (Wagner et al. 2018). Taken together, these findings suggest that ASCL1+ tumor cells are more vulnerable to chemotherapy and potentially outcompeted by non-NE or MYC-high cells during treatment, and/or that ASCL1+ cells can dedifferentiate to a non-NE phenotype as a mechanism of acquired resistance. Indeed, MYC and ASCL1 have been identified in distinct superenhancers (Christensen et al. 2014; Borromeo et al. 2016), suggesting that epigenetic and transcriptional states may evolve during chemotherapy resistance. Moreover, in a panel of eight CDX/PDX models, intratumoral heterogeneity (as defined by the average distance between the normalized expression profiles of each cell and all other cells in a sample) increased in chemotherapy-relapsed samples (Stewart et al. 2020). This study further supports the provocative notion that pre-existing heterogeneity allows selection to occur during therapy and/or that cells have the capacity for plasticity during resistance (Fig. 2). Importantly, the underlying mechanisms that promote transcriptional heterogeneity and/or plasticity are still relatively undefined and a major focus of current investigations.
Figure 2.

Intratumoral heterogeneity and mechanisms of plasticity in SCLC. (Left) SCLC cells within individual human tumors are classified as NE-high (SCLC-A and SCLC-N subtypes) or non-NE SCLC (SCLC-Y and SCLC-P subtypes). Non-NE tumor cells are immunomodulatory and have an increased response to immune checkpoint blockade (ICB). (Top right) SCLC cells demonstrate subtype plasticity. SCLC-A cells can evolve to SCLC-N and SCLC-Y. It remains unknown whether (1) SCLC-P can evolve to or from the other subtypes or (2) non-NE cells can convert back to a NE-high phenotype. (Middle right) Molecular mechanisms implicated in driving NE to non-NE SCLC cell fate transition.
Mechanisms of SCLC plasticity
Plasticity is a crucial mechanism that endows tumor cells with the capacity to convert to a distinct cell identity in response to processes, including external cues and/or stress, and has been implicated in drug resistance (Boumahdi and de Sauvage 2020). One of the first demonstrations of SCLC plasticity originated from studies of the RP GEMM. Calbo et al. (2011) described the coexistence of NE and non-NE tumor cell populations in murine SCLC. Akin to the human SCLC cell lines generated by Gazdar et al. (1985), NE cells exhibited a nonadherent growth pattern when cultured in vitro, characterized by high expression of Mycl and Ascl1. Conversely, non-NE cells derived from the RP model displayed a propensity for adherent growth and exhibited phospho-ERK signaling and mesenchymal-like characteristics, including expression of CD44 and VIM (Calbo et al. 2011; Kwon et al. 2015; Zhang et al. 2018). Importantly, genomic studies of NE and non-NE clonal cell lines derived from a single tumor shared genomic aberrations, suggesting that SCLC tumor cells have the capacity to undergo phenotypic switching. Interestingly, enforced expression of oncogenic KrasV12 drove the dedifferentiation of NE cells to a more non-NE state (Table 1; Calbo et al. 2011). Ras is a master driver of mitogen-activated protein kinase (MAPK) signaling, and early studies suggested that MAPK activation can promote cell cycle arrest and a reduction in NE markers in a small number of SCLC cell lines (Ravi et al. 1998, 1999). Recent studies have confirmed that hyperactivation of RAS, MEK, or ERK can suppress NE differentiation in SCLC and inhibit the growth of NE SCLC cells (Caeser et al. 2021; Inoue et al. 2021). Ras/Raf/Mek/Erk pathway mutations are conspicuously low in SCLC compared with lung adenocarcinoma, consistent with an incompatibility of Ras activation with neuroendocrine fate. While MAPK activation clearly facilitates lung tumor growth of the adenocarcinoma lineage, NE-high SCLC appears to suppress MAPK signaling through mechanisms that are still not fully understood.
Table 1.
Mechanisms driving SCLC plasticity and their effect on tumor cell immunogenicity

Notch pathway activity is a recurrent mechanism implicated in lineage switching in SCLC (Lim et al. 2017; Ireland et al. 2020; Patel et al. 2021; Wu et al. 2021). ASCL1 and the Notch pathway have a mutually antagonistic relationship in lung development and cancer. For example, HES1, a highly conserved target of Notch signaling, can act as a transcriptional repressor of ASCL1 expression (Chen et al. 1997). Consistently, during lung development, Hes1 or Notch1/2/3 knockout expands NE progenitor cells (Ito et al. 2000), while Ascl1 loss ablates NE cells (Borges et al. 1997; Kiyokawa and Morimoto 2020). Notch signaling can also promote the proteasomal degradation of ASCL1 in NE cells (Sriuranpong et al. 2002). Together, these studies demonstrate a critical role for ASCL1/Notch mutual antagonism during lung development. ASCL1 transcriptionally induces expression of Notch-activating ligand Dll1; DLL1 then acts on neighboring cells to promote Notch signaling, which represses ASCL1 in those neighboring cells (Nelson et al. 2009; Shimojo et al. 2016; Kiyokawa and Morimoto 2020)—a pattern-forming process termed “lateral inhibition.” Lateral inhibition leads to a “salt and pepper”-like pattern of cell identities, which are observed during lung development (Morimoto et al. 2010) and in tumors from mouse models of SCLC (Lim et al. 2017). In SCLC, Notch directly activates expression of HES1 and the neuronal transcriptional repressor REST. REST silences NE target genes and promotes a non-NE switch in tumor cells (Table 1; Lim et al. 2017). Thus, Notch signaling is highly associated with the non-NE, ASCL1-low SCLC state. Consistently, SCLC tumors with NOTCH LOF tend to express ASCL1 and have a NE-high phenotype (Lim et al. 2017; Ireland et al. 2020). SCLC tumors with active Notch signaling also express EMT markers, like VIM and ZEB1/2, YAP1/TAZ, and MYC—potential drivers of SCLC plasticity, discussed below.
Recent studies demonstrate that MYC is sufficient to promote the conversion of classic SCLC to a variant morphology and to drive SCLC sequentially from an ASCL1+ to a NEUROD1+ to a YAP1+ state from a NE cell of origin (Mollaoglu et al. 2017; Ireland et al. 2020). Single-cell transcriptional profiling of RPM tumors in vitro and in vivo suggested that MYC promotes subtype evolution (Ireland et al. 2020), with similar results in human cell lines (Table 1; Patel et al. 2021). Mechanistically, MYC directly activates pro-Notch factors, induces REST expression, and converts cells from an NE-high to an NE-low state (Ireland et al. 2020). Consistently, human NE-low SCLC is more likely to be NOTCH wild type and express MYC (Ireland et al. 2020). Functional studies suggest that MYC depends on Notch (Ireland et al. 2020) or REST in a Notch-independent manner (Patel et al. 2021) for SCLC subtype plasticity; discrepancies in these two studies may imply that genetic or environmental context may determine how MYC promotes plasticity.
Mechanisms driving SCLC plasticity may co-opt normal processes that occur during lung development and injury repair. In normal lungs, a specific subpopulation of Notch2+ NE cells (termed “NE stem cells”) responds to injury with proliferation, outward migration, and subsequent differentiation into other lung cell fates—a process that is restrained by Rb1 and p53 (Ouadah et al. 2019). Loss of Rb1/p53 can promote self-renewal of Notch2+ NE stem cells, even in the absence of injury. Following injury, Notch signaling is necessary and sufficient to initiate deprogramming, which is critical for differentiation into other cell fates. Constitutive Notch is not sufficient to induce transit amplification or differentiation of deprogrammed cells into alternate lung cell fates, like club or AT2 cells, suggesting a secondary signal is required. Given the ability of MYC to drive cell cycle entry and non-NE fate in SCLC, the secondary signal may impinge on MYC during lung injury response, although this role for MYC remains unexplored.
Notch pathway ligands and receptors are transcriptional targets of the paralogs YAP1 and TAZ (WWTR1) (Totaro et al. 2018), transcriptional coactivators and downstream effectors of the Hippo signaling pathway. YAP1/TAZ are negatively regulated by Hippo kinases and have been implicated in SCLC plasticity and tumor progression (Zhang et al. 2018; Ireland et al. 2020; Wu et al. 2021). YAP1 positively correlates with expression of TAZ, NOTCH1/2/3, and REST in human SCLC cell lines (Tlemsani et al. 2020). In SCLC tumors of RPP mice with constitutively activated forms of YAP1 (i.e., YAP-S127A or YAP-5SA), NE differentiation was lost and non-NE markers, including Notch2, Vim, and Notch targets HES1 and REST, were expressed (Table 1; Wu et al. 2021), suggesting that YAP1/TAZ may act through Notch to promote NE dedifferentiation. Consistently, SCLC tumors of RPP GEMMs with genetic deletion of Yap1 and Taz lacked HES1 expression. YAP1/TAZ may also have a Notch-independent role in driving non-NE fate of SCLC, as YAP1 activation in RPP GEMM tumors prevented NE differentiation, even when Notch activity was blocked via γ-secretase inhibition or Rbpj knockout (Wu et al. 2021). Whether YAP1/TAZ act through MYC or Ras to induce non-NE SCLC fate is yet to be reported.
Increasing evidence supports a key role for epigenetic regulators in SCLC plasticity. Initially identified as a mechanism driving chemoresistance (Gardner et al. 2017), the histone methyltransferase (HMT) EZH2 has been implicated in controlling NE cell fate. EZH2 is the enzymatic subunit of the Polycomb-repressive complex 2 (PRC2), involved in gene repression through catalyzing trimethylation of histone H3K27. Consistent with an emerging role for EZH2 in the maintenance of NE cell fate, pharmacological inhibition of EZH2 in NE-high SCLC cell lines triggered cellular adherence and the transition to a non-NE phenotype (Table 1; Mahadevan et al. 2021). Interestingly, the cell fate change induced by EZH2 was concomitant with derepression of MHC-I expression (Burr et al. 2019; Mahadevan et al. 2021), implicating EZH2 in immune cell evasion, as discussed in greater detail below. Truncating mutations in the H3K4 HMT gene KMT2D (also known as MLL2) have been identified in primary SCLC tumors and cell lines (Augert et al. 2017). Human SCLC cell lines harboring either homozygous or heterozygous mutations in KMT2D displayed a reduction in histone H3K4 monomethylation, a histone mark that has been associated with active enhancers (Heintzman et al. 2007). The functional role of KMT2D in SCLC tumorigenesis and tumor cell plasticity remains unexplored.
The histone acetyltransferases (HATs) CREBBP (also known as CBP) and its paralog, EP300 (also known as p300), are commonly mutated in SCLC (George et al. 2015; Augert et al. 2017). CREBBP/EP300 have been implicated in cell cycle control and transcriptional control of enhancers (Jin et al. 2011; Attar and Kurdistani 2017), but whether they promote NE versus non-NE SCLC fate remains uncertain. In one study, conditional loss of Crebbp/Ep300 in RP GEMMs significantly reduced H3K27Ac levels and increased sensitivity to HDAC inhibition (Jia et al. 2018). In this study, Crebbp/Ep300 loss led to a decrease in epithelial markers such as CDH1 (encoding E-cadherin), associated with NE-high SCLC, and a gain in mesenchymal markers that are associated with non-NE SCLC, including ZEB1 and VIM (Jia et al. 2018). In contrast, another study showed that H3K27Ac levels are reduced in NE-high SCLC and that ERK-mediated activation of CREBBP/EP300 is important for non-NE lineage transformation (Table 1; Inoue et al. 2021). Interestingly, CREBBP/EP300 genomic alterations are mutually exclusive with Notch pathway mutations, such that it is tempting to speculate that Notch could signal through CREBBP/EP300 to promote non-NE fates. Further studies are warranted to reconcile these disparate findings and conclusively delineate the contribution of CREBBP/EP300 to SCLC cell fate.
Notably, multiple epigenetic factors implicated in SCLC cell fate alter Notch activity. Inactivation of the histone demethylases LSD1/KDM1A and KDM5A, both of which demethylate H3K4, induces NE dedifferentiation via down-regulation of ASCL1 (Mohammad et al. 2015; Takagi et al. 2017; Augert et al. 2019; Oser et al. 2019). LSD1/KDM1A and KDM5A have been shown to act synergistically to repress Notch and promote ASCL1 expression (Oser et al. 2019), suggesting independent or parallel mechanisms of action (Table 1). Additionally, histone deacetylase inhibitors like Trichostatin A can induce NE dedifferentiation in SCLC by activating Notch and REST (Augert et al. 2019; Wu et al. 2021). Finally, while genes encoding chromatin remodelers CHD7 and PBRM1 (whose protein product is part of the SWI/SNF remodeling complex) are also found altered in SCLC (George et al. 2015; Augert et al. 2017), their impact on SCLC cell fate and plasticity is relatively unexplored. Together, these studies emphasize that changes in chromatin structure are undoubtedly critical for SCLC plasticity. However, it is still unclear how the epigenetic regulators discussed above impact specific SCLC-A, SCLC-N, SCLC-P, and SCLC-Y states. Whether and how plasticity drivers, including Ras, Notch, Myc, and Yap1/Taz, impinge on these epigenetic regulators in SCLC are also largely unknown.
It is likely that additional molecular mechanisms underlie lineage plasticity. NFIB, a transcription factor with an oncogenic role in SCLC (Semenova et al. 2016; Wu et al. 2016), drives a global increase in chromatin accessibility that augments the metastatic capacity of SCLC tumor cells (Denny et al. 2016). Also, in the developing lung, INSM1 is a key regulator of pulmonary NE cell differentiation (Jia et al. 2015), while in SCLC cell lines, a reciprocal relationship between INSM1 and YAP1 has been demonstrated (McColl et al. 2017; Tlemsani et al. 2020). INSM1 can inhibit Notch activity by repressing Hes1 (Jia et al. 2015), and INSM1 may also be inhibited by Notch in a mutually inhibitory relationship (Fujino et al. 2015). While ASCL1 knockout prevents tumorigenesis in classic SCLC models (Borromeo et al. 2016), ASCL1 knockout in the context of MYC-driven tumors leads to a neural crest and/or mesenchymal stem-like state with activation of many of the pathways associated with non-NE tumors, including Notch/Rest, TGF-β, and Hippo/Yap1 (Olsen et al. 2021). In addition, ASCL1 loss leads to induction of SOX9 (Olsen et al. 2021), which is associated with non-NE SCLC fate and can act downstream from MAPK signaling (Inoue et al. 2021). SOX9 mediates lineage plasticity in other tissues (Christin et al. 2020; Nouri et al. 2020) but remains relatively unexplored in SCLC. Computational methods have been used to predict candidate regulators of lineage plasticity and cell fate changes in SCLC (Wooten et al. 2019; Chan et al. 2021; Chauhan et al. 2021). These studies support a role for Ras/Mapk, Notch, Myc, Yap1/Taz, and epigenetic factors as determinants of SCLC plasticity, and more factors remain to be explored. Moving forward, unbiased -omic methodologies may represent powerful approaches to identify key drivers and mechanisms of subtype switching.
Although multiple mechanisms that drive NE dedifferentiation of SCLC toward a non-NE phenotype have been identified, the full extent of SCLC plasticity is still not well understood. For example, while Notch activation has been shown to promote an NE-to-non-NE transition in SCLC, studies suggest that Notch blockade cannot reverse cells back to an NE-high state (Lim et al. 2017; Ireland et al. 2020; Wu et al. 2021). In fact, little to no evidence exists to date to support the occurrence of a non-NE-to-NE transition in SCLC. However, other non-NE, epithelial cancers can undergo NE transformation in the context of resistance to targeted therapies. Specifically, EGFR-driven lung adenocarcinoma is known to convert to SCLC during resistance to tyrosine kinase inhibition (Niederst et al. 2015; Oser et al. 2015; Quintanal-Villalonga et al. 2021). Analysis of these samples suggest that adeno-to-SCLC conversion has the capacity to give rise to all four subtypes, albeit YAP1 expression was higher in the lung adenocarcinoma component (Quintanal-Villalonga et al. 2021). Prostate adenocarcinoma treated with androgen deprivation therapy can also convert to neuroendocrine prostate carcinoma (NEPC) that shares many similarities to SCLC, including expression of ASCL1 and NEUROD1 (Cejas et al. 2021; Kaarijärvi et al. 2021). Whether non-NE subtypes of SCLC can use similar mechanisms of plasticity to transition to an NE-high state remains an open question. Moreover, whether the SCLC cell of origin dictates or constrains the directionality of SCLC plasticity is yet to be investigated. Advanced lineage tracing approaches could be used to gain a deeper understanding of tumor evolution during disease progression and in response to chemotherapy. Specifically, CRISPR- or barcode-based lineage tracing paired with single-cell technologies, such as those recently used in the context of hematopoietic malignancies (Fennell et al. 2022), could be used to identify nongenetic mechanisms that govern SCLC plasticity and drug resistance. Combining epigenetic drugs with additional therapies may hold promise for treating SCLC and will undoubtedly be an area of increased focus in the coming years.
Tumor heterogeneity and the immune microenvironment
SCLC has a high tumor mutational burden (TMB) (Alexandrov et al. 2013) and harbors a prevalence of C > A transversions—a result of DNA adducts forming between tobacco carcinogens and guanine nucleotides (Pleasance et al. 2010; Alexandrov et al. 2013, 2016; George et al. 2015). While high TMB may correlate with increased neoantigen production (Schumacher and Schreiber 2015) and favorable response to immune checkpoint inhibitors anti-PD-L1/PD1 in NSCLC (Ready et al. 2019), this does not appear to be the case for SCLC (Hellmann et al. 2018; Horn et al. 2018; Paz-Ares et al. 2019). Indeed, the recent approval of immunotherapy (anti-PD-L1 and atezolizumab [Atezo]) in combination with chemotherapy for first-line treatment of SCLC provides only marginal benefit for ∼10%–20% of patients (Horn et al. 2018). Multiple studies have therefore sought to uncover the mechanisms restricting immune cell infiltration in SCLC and whether it can be attributed to an absence of tumor antigens or defects in antigen presentation machinery. Indeed, it was noted in the 1980s that compared with other tumor types, SCLC exhibits remarkably low expression of class 1 MHC antigens, including human leukocyte antigens (HLAs) and β-2-microglobulin (B2M) (Doyle et al. 1985). The identification of additional mechanisms that underpin SCLC's lack of immunogenicity is an area of active investigation. Additionally, the search for therapeutic approaches to warm up the immune landscape of SCLC is a high priority in the field.
The classification of SCLC as an “immune-cold” disease lacking infiltration of cytotoxic immune cells (Busch et al. 2016) has recently been revisited, sparked by the stratification of SCLC into four transcriptional subtypes (Rudin et al. 2019). Using bulk transcriptional profiles from SCLC patient samples, several groups have recently discovered that the immune landscape differs among SCLC subtypes (Best et al. 2020; Dora et al. 2020; Cai et al. 2021; Gay et al. 2021; Roper et al. 2021). Expression of immune-associated genes such as PD-L1 and MHC-I were repressed in SCLC-A tumors and in NE tumors of other tissue origins, consistent with the expression profile of pulmonary NE cells (Sutherland et al. 2011; Cai et al. 2021). However, a subset of ASCL1+ tumors exhibited high HLA expression and high T-cell and NK cell scores (transcriptional signatures used to predict T-cell and NK cell infiltrate in solid tumors), suggesting that additional biomarkers may be required to stratify response to immune checkpoint blockade (ICB) (Best et al. 2020). SCLC-N tumors appear to be the most immune-cold subset of SCLC, lacking expression of HLA- and antigen-presenting genes with the lowest T-cell and NK cell scores of the four subtypes (Best et al. 2020; Chan et al. 2021; Gay et al. 2021). Consistent with NEUROD1's association with low immune infiltration, permissivity for SVV infection (which is enriched in NEUROD1+ cells) was shown to be associated with lower type I interferon (IFN) innate immune gene expression (Fig. 3; Miles et al. 2017).
Figure 3.
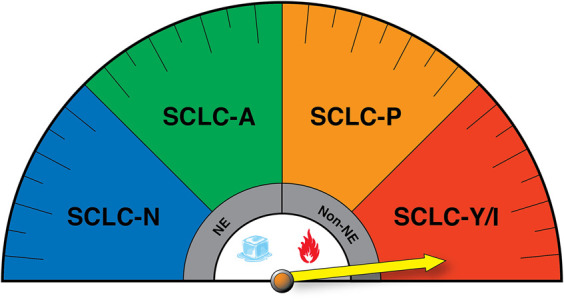
SCLC transcriptional subtypes display distinct immunogenic profiles that may impact response to immune checkpoint blockade (ICB). (NE) Neuroendocrine, (non-NE) nonneuroendocrine. The yellow arrow reflects potential enhanced responsiveness of the SCLC-Y/I subtypes to ICB, while SCLC-N has been suggested to be the most immune-cold.
Interestingly, Gay et al. (2021) recently identified a novel inflamed (I) subtype, denoted SCLC-I, identified based on its low expression of ASCL1, NEUROD1, and POU2F3 and their associated transcriptional signatures. Critically, it was coined “inflamed” due to elevated expression of immune checkpoint molecules and HLAs, as well as high immune cell infiltration (Gay et al. 2021). Up-regulation of IFN-γ response genes and high expression of HLA were similarly reported in an independent SCLC-Y patient cohort (Owonikoko et al. 2021) and in SCLC-Y cell line samples (Tlemsani et al. 2020), adding fuel to the debate over the existence of SCLC-Y and its relationship to the newly annotated SCLC-I subtype. This highlights the need to re-evaluate the nomenclature used to classify SCLC subtypes. For example, while YAP1 mRNA expression alone might be insufficient to define a distinct subtype, use of well-validated transcriptional signatures of the HIPPO pathway may serve as more powerful classification tools and help address discrepancies in the field (McColl et al. 2017; Wang et al. 2018; Pearsall et al. 2020; Zhang et al. 2020b). Nevertheless, translating such a classification scheme to a clinical diagnostic tool remains a significant challenge for the field. Likely, it will be important to use a panel of markers, including NE markers and histopathology, to better define SCLC-A, SCLC-N, and SCLC-P states, rather than the use of a single transcriptional marker (Rekhtman 2022). This may be particularly important for identifying non-NE subtypes; for example, POU2F3 immunostaining in combination with morphological phenotyping may serve as the most robust diagnostic approach for SCLC-P tumors.
The emergence of distinct immune cell profiles per SCLC subtype has raised new questions related to the efficacy of ICB in SCLC patients. Indeed, it is tantalizing to hypothesize that patients harboring SCLC-P and/or SCLC-Y/I subtypes may exhibit more durable responses to ICB due to relatively higher expression of antigen presentation machinery (APM) genes and their low-NE phenotype (Fig. 3). Recent studies by Gay et al. (2021) were the first to directly address this hypothesis in a retrospective analysis of SCLC patients enrolled in IMpower133, the first randomized trial to demonstrate improvements in progression-free and overall survival (OS) when Atezo was combined with platinum chemotherapy in treatment-naive SCLC patients (Horn et al. 2018; Gay et al. 2021). While this study was not powered for subtype stratification, a significant median OS benefit was observed for SCLC-I in the combination treatment arm (EP + Atezo 18.2 mo vs. EP + placebo 10.4 mo). Moreover, SCLC-P tumors showed a trend for OS benefit in the combination ICB arm (EP + Atezo 9.6 mo vs. EP + placebo 6.0 mo), consistent with high expression of HLA and APM genes in this subset. It should be noted, however, that SCLC-P tumors exhibited the poorest outcomes overall regardless of the treatment arm (Gay et al. 2021). This finding urges analysis on a larger number of samples to determine whether SCLC-P truly exhibits increased resistance to therapy compared with other non-NE SCLC tumors. Using gene expression from tumors in an independent cohort of 20 SCLC patients, a direct correlation between Notch pathway activation and improved response to ICB was found (Roper et al. 2021). By some metrics, MYC expression also correlated with clinical benefit to ICB therapy, while MYCL and MYCN displayed no enrichment (Roper et al. 2021). Together, these studies reveal an association between a low-NE differentiation state and enhanced antitumor immunity. To date, however, no clinical trials have stratified patients based on SCLC subtype or degree of NE differentiation. Indeed, stratification could be hampered by the high level of intratumoral heterogeneity observed in patient samples (Baine et al. 2020; Ireland et al. 2020; Stewart et al. 2020), the source of material analyzed, and/or the highly plastic nature of SCLC tumor cells (Ireland et al. 2020; Patel et al. 2021). Given that Notch receptor genes and many epigenetic regulators are genomically altered in SCLC, it would be valuable to determine how these genomic alterations impact plasticity and whether they are predictive of therapeutic responses. Moreover, recent studies that have reconstructed the phylogenetics of SCLC revealed that the majority of somatic alterations (∼80%) represent “trunk” mutations (Chen et al. 2021b; Zhou et al. 2021). It remains unknown whether these alterations could serve as useful neoantigens for immunotherapeutic purposes (Levine et al. 2019).
Given the lack of durable response to ICB in the majority of SCLC patients, several studies have sought to evaluate combination therapies to enhance the efficacy of ICB. Targeting DNA damage response (DDR) pathways with either PARP or CHK1 inhibition led to increased PD-L1 expression on the surface of SCLC tumor cells (Sen et al. 2019). Interestingly, CHK1 inhibition alone was sufficient to induce elevated T-cell infiltration in vivo; however, tumor eradication was only observed when CHK1 inhibition was combined with anti-PD-L1 treatment. CD8+ T-cell recruitment to the tumor microenvironment following DDR inhibition was mediated by activation of a STING–TBK1–IRF3 pathway (Sen et al. 2019). Selective inhibition of CDK7 by YKL-5-124 also caused replication stress and displayed synergy when combined with chemotherapy and ICB (Zhang et al. 2020a). However, in contrast to targeting the DDR, CDK7 inhibition appears to elicit its effects in a STING-independent manner (Zhang et al. 2020a).
The innate immune system has also been implicated in the control of SCLC tumorigenesis. Using a syngeneic RP cell line model, Best et al. (2020) implicated NK cells, but not CD8+ T cells, as key regulators of SCLC metastasis. Indeed, mice genetically engineered to lack NK cells were unable to control the dissemination of RP cells following intravenous injection. Critically, and in line with the emergence of NK cell immunotherapeutic approaches (Huntington et al. 2020), chemical or genetic activation of NK cells ameliorated SCLC metastasis, an effect that synergized with T-cell activation. NK cell-mediated killing, however, does not appear to correlate with low MHC-I expression (Stam et al. 1989), and instead was recently shown to be reliant on expression of the NK cell-activating receptor NKG2D/MICA (Zhu et al. 2021). Interestingly, HDAC inhibition enhances NK cell-mediated killing through the depression of NKG2D expression, highlighting an important role of epigenetic regulation in antitumor immunity (Liu et al. 2018; Zhu et al. 2021). SCLC-P and the inflamed SCLC-I subtype exhibit the highest expression of MICA and harbor a high NK cell score (Cursons et al. 2019; Best et al. 2020; Gay et al. 2021), suggesting that NK cell immunotherapies may be most effective in NE-low SCLC subtypes—an observation that requires further validation in the preclinical and clinical settings.
Recent single-cell transcriptomic analysis has provided unprecedented insights into the phenotype of infiltrating immune cells in 21 SCLC patient samples (Chan et al. 2021). A profibrotic monocytic/macrophage population was shown to be uniquely abundant in SCLC compared with levels seen in lung adenocarcinoma and normal lung tissue. While the functional significance of this macrophage population remains unknown, an association was observed with a rare, prometastatic PLCG2+ tumor cell population, suggesting a potential protumorigenic role of the macrophages. Other studies have explored therapies that harness the anticancer activity of myeloid cells. Blocking antibodies against CD47 display efficacy in preclinical models by inducing macrophage phagocytosis of SCLC tumor cells (Weiskopf et al. 2016). Interestingly, lurbinectedin, recently provisionally approved for the treatment of metastatic platinum-resistant SCLC (Trigo et al. 2020; Singh et al. 2021), demonstrates efficacy in reducing tumor-associated macrophages in preclinical studies (Belgiovine et al. 2017). It is therefore of interest to explore whether the antitumor effects of lurbinectedin are due to effects on the tumor or macrophage compartments or both.
Increasing SCLC immunogenicity through the induction of phenotype switching
The long-standing observation that SCLC exhibits low MHC-I expression (Doyle et al. 1985), despite the lack of genetic alterations in APM components, implies epigenetic and transcriptional mechanisms of APM control. Indeed, recent studies have uncovered a crucial role for the PRC2 complex, comprising core subunits EZH2, EED, and SUZ12, in the transcriptional repression of MHC-I in SCLC (Burr et al. 2019; Mahadevan et al. 2021). In a genome-wide CRISPR–Cas9 screen, Burr et al. (2019) identified sgRNAs targeting EED and SUZ12 as key regulators of MHC-I expression. This appears to be a conserved mechanism in neuroendocrine tumors, as genetic or pharmacological inhibition of EZH2 restored MHC-I cell surface expression in neuroblastoma, Merkel cell carcinoma, and SCLC cell lines (Burr et al. 2019), a finding that was independently validated by Mahadevan et al. (2021). Similar to the SCLC-I subtype, high MHC-I cell surface expression was associated with a low-NE cell phenotype and derepression of the cGAS–STING pathway (Mahadevan et al. 2021). Critically, EZH2 inhibition resulted in increased T-cell-mediated tumor cell killing (Burr et al. 2019) and was shown to synergize with STING agonists to enhance tumor cell clearance in vivo (Mahadevan et al. 2021). EZH2 inhibitors are currently being evaluated in patients with recurrent SCLC (NCT038979798), based on earlier studies implicating EZH2 in chemoresistance (Sato et al. 2013; Gardner et al. 2017).
Alternative mechanisms of MHC-I regulation associated with subtype switching in SCLC are now emerging. Recently, the RNA-binding protein ZFP36L1 was identified in a CRISPR/Cas9 genomic screen designed to identify the molecular mechanisms underlying LSD1 inhibitor (ORY-1001) sensitivity in SCLC tumor cells (Chen et al. 2021a). Interestingly, high expression of HLA-B and HLA-C is observed in ZFP36L1-activated SCLC tumor cells. Moreover, the phenotype of ZFP36L1-activated cells resembled that of the SCLC-I subtype (Gay et al. 2021), characterized by enrichment of mesenchymal and IFN-γ gene signatures and a loss of NE markers and ASCL1 target genes. Together, these emerging studies offer great promise for the use of therapeutic agents to prime SCLC tumors for immune rejection through inducing NE cell dedifferentiation. It is notable, however, that multiple factors associated with non-NE fate (i.e., Myc, Notch, Yap1, and Ezh2) have been associated with chemotherapy resistance and/or tumor aggressiveness (Gardner et al. 2017; Lim et al. 2017; Mollaoglu et al. 2017; Wagner et al. 2018; Ireland et al. 2020; Wu et al. 2021), warranting caution for implementing therapeutic approaches that promote non-NE fates.
Future directions
Recent advances have transformed our view of SCLC and shed light on approaches that could improve outcomes for patients with specific molecular subsets of SCLC. However, the remarkable transcriptional and epigenetic plasticity in SCLC suggests that defining molecular states is a challenging notion, especially for clinical purposes. Moreover, the capacity for plasticity implies that drug resistance will be a continual obstacle to successful treatment. It is critically important that we improve our understanding of the mechanisms of SCLC plasticity and determine how to harness this knowledge to improve the efficacy of combination therapies and immunotherapy. In addition to the gaps in knowledge discussed previously in this review, we propose additional key areas of study:
An outstanding question is whether early studies identifying MYC-high therapeutic vulnerabilities pertain to all ASCL1-negative subgroups or specific ones; i.e., which therapeutic sensitivities are NEUROD1-specific versus POU2F3-specific or triple-negative ASCL1/NEUROD1/POU2F3-specific? How does C-MYC function differently from other MYC family members like MYCL? Is dosage important? How do MYC family members differ in function? Mechanistic studies in cell lines and animal models will more definitively address these questions.
While modulation of key factors such as Ras, Notch/Rest, Myc, Yap1/Taz, and epigenetic regulators has been shown to impact NE and non-NE fates, how these signals are coordinated and interrelated is not well understood. Deciphering the basic mechanisms of how extracellular signals impinge on these factors and epigenetic regulators such as the PRC2 complex, Crebbp/p300, Lsd1, Kdm5a, Zfp36l1, Ets family members, and others will be important for manipulating cell fate. How these factors control each other from a network perspective and whether they function in the same or parallel and distinct pathways remain unknown.
Since the refinement of the four SCLC molecular subtypes by Rudin et al. (2019), a novel transcriptional subtype based on the expression of ATOH1 has emerged from PDX studies (Westerman et al. 2007; Simpson et al. 2020). ATOH1 is another bHLH proneural transcription factor that is important for inner ear and cerebellar development. ATOH1+ SCLC CDX models appear to exhibit a distinct transcriptional signature when compared with SCLC-A, SCLC-N, and SCLC-P subtypes and correlate with expression of the mesenchymal marker VIM. Whether ATOH1 is expressed in primary human SCLC tumors has been questioned, highlighting the need to better understand this observation.
Vascular endothelial (VE)-cadherin-positive SCLC tumor cells have been frequently observed in SCLC CDX models (Williamson et al. 2016), and the signals and mechanisms that promote the process of vascular mimicry (VM) and how they relate to SCLC molecular subtypes are unknown. VM may be an important process for therapeutic intervention given that VM correlates with poor outcomes and chemotherapy resistance in CDX studies (Williamson et al. 2016).
Given that tumors often harbor cells in multiple subtype states (Baine et al. 2020; Ireland et al. 2020), there is likely extensive cross-talk among populations of cells (Fig. 2) that provide survival advantages in the face of environmental stresses. Cell–cell interactions have been implicated in metastases, but how subtype states interact with each other during hypoxia, nutrient deprivation, and chemotherapy-induced stresses are largely unknown.
Exciting new studies suggest that cell fate or lineage can impact expression of antigen presentation and immune response machinery, providing renewed hope that these mechanisms can be exploited to improve immunotherapy responses. A key area of future investigation concerns how to increase immune cell recognition and response to ICB.
SCLC is thought of as a relatively immune-cold tumor, but single-cell approaches have been crucial in illuminating novel immune populations like the profibrotic monocytic/macrophage population associated with the prometastatic PLCG2+ population (Chan et al. 2021). Immune populations such as these may be important in the pathogenesis of SCLC and/or may be targeted to enhance therapeutic responses.
Acknowledgments
Figures were created with Biorender.com and with the assistance of Peter Maltezos.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.349359.122.
Freely available online through the Genes & Development Open Access option.
Competing interest statement
T.G.O. is named on a patent related to SCLC subtype biomarkers (US11124841B2). K.D.S. and A.S.I. declare no competing interests.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Ju YS, Haase K, Loo PV, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, et al. 2016. Mutational signatures associated with tobacco smoking in human cancer. Science 354: 618–622. 10.1126/science.aag0299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar N, Kurdistani SK. 2017. Exploitation of EP300 and CREBBP lysine acetyltransferases by cancer. Cold Spring Harb Perspect Med 7: a026534. 10.1101/cshperspect.a026534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augert A, Zhang Q, Bates B, Cui M, Wang X, Wildey G, Dowlati A, MacPherson D. 2017. Small cell lung cancer exhibits frequent inactivating mutations in the histone methyltransferase KMT2D/MLL2: CALGB 151111 (alliance). J Thorac Oncol 12: 704–713. 10.1016/j.jtho.2016.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augert A, Eastwood E, Ibrahim AH, Wu N, Grunblatt E, Basom R, Liggitt D, Eaton KD, Martins R, Poirier JT, et al. 2019. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci Signal 12: eaau2922. 10.1126/scisignal.aau2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augert A, Mathsyaraja H, Ibrahim AH, Freie B, Geuenich MJ, Cheng PF, Alibeckoff SP, Wu N, Hiatt JB, Basom R, et al. 2020. MAX functions as a tumor suppressor and rewires metabolism in small cell lung cancer. Cancer Cell 38: 97–114.e7. 10.1016/j.ccell.2020.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustyn A, Borromeo M, Wang T, Fujimoto J, Shao C, Dospoy PD, Lee V, Tan C, Sullivan JP, Larsen JE, et al. 2014. ASCL1 is a lineage oncogene providing therapeutic targets for high-grade neuroendocrine lung cancers. Proc Natl Acad Sci 111: 14788–14793. 10.1073/pnas.1410419111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baine MK, Hsieh MS, Lai WV, Egger JV, Jungbluth AA, Daneshbod Y, Beras A, Spencer R, Lopardo J, Bodd F, et al. 2020. SCLC subtypes defined by ASCL1, NEUROD1, POU2F3, and YAP1: a comprehensive immunohistochemical and histopathologic characterization. J Thorac Oncol 15: 1823–1835. 10.1016/j.jtho.2020.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball DW. 2004. Achaete-scute homolog-1 and notch in lung neuroendocrine development and cancer. Cancer Lett 204: 159–169. 10.1016/S0304-3835(03)00452-X [DOI] [PubMed] [Google Scholar]
- Bebber CM, Thomas ES, Stroh J, Chen Z, Androulidaki A, Schmitt A, Höhne MN, Stüker L, de Pádua Alves C, Khonsari A, et al. 2021. Ferroptosis response segregates small cell lung cancer (SCLC) neuroendocrine subtypes. Nat Commun 12: 2048. 10.1038/s41467-021-22336-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgiovine C, Bello E, Liguori M, Craparotta I, Mannarino L, Paracchini L, Beltrame L, Marchini S, Galmarini CM, Mantovani A, et al. 2017. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br J Cancer 117: 628–638. 10.1038/bjc.2017.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best SA, Hess JB, Souza-Fonseca-Guimaraes F, Cursons J, Kersbergen A, Dong X, Rautela J, Hyslop SR, Ritchie ME, Davis MJ, et al. 2020. Harnessing natural killer immunity in metastatic SCLC. J Thorac Oncol 15: 1507–1521. 10.1016/j.jtho.2020.05.008 [DOI] [PubMed] [Google Scholar]
- Borges M, Linnoila RI, van de Velde HJ, Chen H, Nelkin BD, Mabry M, Baylin SB, Ball DW. 1997. An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature 386: 852–855. 10.1038/386852a0 [DOI] [PubMed] [Google Scholar]
- Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, Girard L, Minna JD, Gazdar AF, Cobb MH, et al. 2016. ASCL1 and NEUROD1 reveal heterogeneity in pulmonary neuroendocrine tumors and regulate distinct genetic programs. Cell Rep 16: 1259–1272. 10.1016/j.celrep.2016.06.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumahdi S, de Sauvage FJ. 2020. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 19: 39–56. 10.1038/s41573-019-0044-1 [DOI] [PubMed] [Google Scholar]
- Brägelmann J, Böhm S, Guthrie MR, Mollaoglu G, Oliver TG, Sos ML. 2017. Family matters: how MYC family oncogenes impact small cell lung cancer. Cell Cycle 16: 1489–1498. 10.1080/15384101.2017.1339849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan MS, Chang C, Tai L, Lessene G, Strasser A, Dewson G, Kelly GL, Herold MJ. 2018. Humanized Mcl-1 mice enable accurate preclinical evaluation of MCL-1 inhibitors destined for clinical use. Blood 132: 1573–1583. 10.1182/blood-2018-06-859405 [DOI] [PubMed] [Google Scholar]
- Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, Azidis-Yates E, Vassiliadis D, Bell CC, Gilan O, et al. 2019. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 36: 385–401.e8. 10.1016/j.ccell.2019.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch SE, Hanke ML, Kargl J, Metz HE, MacPherson D, Houghton AM. 2016. Lung cancer subtypes generate unique immune responses. J Immunol 197: 4493–4503. 10.4049/jimmunol.1600576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caeser R, Hulton C, Costa E, Durani V, Little M, Chen X, Tischfield SE, Asher M, Kombak FE, Chavan SS, et al. 2021. MAPK pathway activation selectively inhibits ASCL1-driven small cell lung cancer. iScience 24: 103224. 10.1016/j.isci.2021.103224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Liu H, Huang F, Fujimoto J, Girard L, Chen J, Li Y, Zhang Y-A, Deb D, Stastny V, et al. 2021. Cell-autonomous immune gene expression is repressed in pulmonary neuroendocrine cells and small cell lung cancer. Commun Biol 4: 314. 10.1038/s42003-021-01842-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calbo J, van Montfort E, Proost N, van Drunen E, Beverloo HB, Meuwissen R, Berns A. 2011. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 19: 244–256. 10.1016/j.ccr.2010.12.021 [DOI] [PubMed] [Google Scholar]
- Cardnell RJ, Li L, Sen T, Bara R, Tong P, Fujimoto J, Ireland AS, Guthrie MR, Bheddah S, Banerjee U, et al. 2017. Protein expression of TTF1 and cMYC define distinct molecular subgroups of small cell lung cancer with unique vulnerabilities to aurora kinase inhibition, DLL3 targeting, and other targeted therapies. Oncotarget 8: 73419–73432. 10.18632/oncotarget.20621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargill KR, Stewart CA, Park EM, Ramkumar K, Gay CM, Cardnell RJ, Wang Q, Diao L, Shen L, Fan YH, et al. 2021. Targeting MYC-enhanced glycolysis for the treatment of small cell lung cancer. Cancer Metab 9: 33. 10.1186/s40170-021-00270-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney DN, Gazdar AF, Bepler G, Guccion JG, Marangos PJ, Moody TW, Zweig MH, Minna JD. 1985. Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res 45: 2913–2923. [PubMed] [Google Scholar]
- Cau E, Gradwohl G, Fode C, Guillemot F. 1997. Mash1 activates a cascade of bHLH regulators in olfactory neuron progenitors. Development 124: 1611–1621. 10.1242/dev.124.8.1611 [DOI] [PubMed] [Google Scholar]
- Cejas P, Xie Y, Font-Tello A, Lim K, Syamala S, Qiu X, Tewari AK, Shah N, Nguyen HM, Patel RA, et al. 2021. Subtype heterogeneity and epigenetic convergence in neuroendocrine prostate cancer. Nat Commun 12: 5775. 10.1038/s41467-021-26042-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalishazar MD, Wait SJ, Huang F, Ireland AS, Mukhopadhyay A, Lee Y, Schuman SS, Guthrie MR, Berrett KC, Vahrenkamp JM, et al. 2019. MYC-driven small-cell lung cancer is metabolically distinct and vulnerable to arginine depletion. Clin Cancer Res 25: 5107–5121. 10.1158/1078-0432.CCR-18-4140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JM, Quintanal-Villalonga Á, Gao VR, Xie Y, Allaj V, Chaudhary O, Masilionis I, Egger J, Chow A, Walle T, et al. 2021. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell 39: 1479–1496.e18. 10.1016/j.ccell.2021.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan L, Ram U, Hari K, Jolly MK. 2021. Topological signatures in regulatory network enable phenotypic heterogeneity in small cell lung cancer. Elife 10: e64522. 10.7554/eLife.64522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Thiagalingam A, Chopra H, Borges MW, Feder JN, Nelkin BD, Baylin SB, Ball DW. 1997. Conservation of the Drosophila lateral inhibition pathway in human lung cancer: a hairy-related protein (HES-1) directly represses achaete-scute homolog-1 expression. Proc Natl Acad Sci 94: 5355–5360. 10.1073/pnas.94.10.5355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-Y, Durmaz YT, Li Y, Sabet AH, Vajdi A, Denize T, Walton E, Doench JG, Mahadevan NR, Losman J-A, et al. 2021a. Regulation of neuroendocrine plasticity by the RNA-binding protein ZFP36L1. bioRxiv 10.1101/2021.10.22.465501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Chen R, Jin Y, Li J, Hu X, Zhang J, Fujimoto J, Hubert SM, Gay CM, Zhu B, et al. 2021b. Cold and heterogeneous T cell repertoire is associated with copy number aberrations and loss of immune genes in small-cell lung cancer. Nat Commun 12: 6655. 10.1038/s41467-021-26821-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Guanizo A, Luong Q, Jayasekara WSN, Jayasinghe D, Inampudi C, Szczepny A, Garama DJ, Russell PA, Ganju V, et al. 2022. Lineage-restricted neoplasia driven by Myc defaults to small cell lung cancer when combined with loss of p53 and Rb in the airway epithelium. Oncogene 41: 138–145. 10.1038/s41388-021-02070-3 [DOI] [PubMed] [Google Scholar]
- Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, et al. 2014. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 26: 909–922. 10.1016/j.ccell.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christin JR, Wang C, Chung CY, Liu Y, Dravis C, Tang W, Oktay MH, Wahl GM, Guo W. 2020. Stem cell determinant SOX9 promotes lineage plasticity and progression in basal-like breast cancer. Cell Rep 31: 107742. 10.1016/j.celrep.2020.107742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampricotti M, Karakousi T, Richards AL, Quintanal-Villalonga À, Karatza A, Caeser R, Costa EA, Allaj V, Manoj P, Spainhower KB, et al. 2021. Rlf–Mycl gene fusion drives tumorigenesis and metastasis in a mouse model of small cell lung cancer. Cancer Discov 11: 3214–3229. 10.1158/2159-8290.CD-21-0441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cursons J, Souza-Fonseca-Guimaraes F, Foroutan M, Anderson A, Hollande F, Hediyeh-Zadeh S, Behren A, Huntington ND, Davis MJ. 2019. A gene signature predicting natural killer cell infiltration and improved survival in melanoma patients. Cancer Immunol Res 7: 1162–1174. 10.1158/2326-6066.CIR-18-0500 [DOI] [PubMed] [Google Scholar]
- Dammert MA, Brägelmann J, Olsen RR, Böhm S, Monhasery N, Whitney CP, Chalishazar MD, Tumbrink HL, Guthrie MR, Klein S, et al. 2019. MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat Commun 10: 3485. 10.1038/s41467-019-11371-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayton JS, Turka LA, Thompson CB, Mitchell BS. 1992. Comparison of the effects of mizoribine with those of azathioprine, 6-mercaptopurine, and mycophenolic acid on T lymphocyte proliferation and purine ribonucleotide metabolism. Mol Pharmacol 41: 671–676. [PubMed] [Google Scholar]
- Denny SK, Yang D, Chuang CH, Brady JJ, Lim JS, Grüner BM, Chiou SH, Schep AN, Baral J, Hamard C, et al. 2016. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell 166: 328–342. 10.1016/j.cell.2016.05.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr F, George J, Schmitt A, Beleggia F, Rehkämper T, Hermann S, Walter V, Weber J-P, Thomas RK, Wittersheim M, et al. 2017. Targeting a non-oncogene addiction to the ATR/CHK1 axis for the treatment of small cell lung cancer. Sci Rep 7: 15511. 10.1038/s41598-017-15840-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley AL, Winslow MM, Chiang DY, Banerji S, Stransky N, Dayton TL, Snyder EL, Senna S, Whittaker CA, Bronson RT, et al. 2011. Nuclear factor I/B is an oncogene in small cell lung cancer. Genes Dev 25: 1470–1475. 10.1101/gad.2046711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora D, Rivard C, Yu H, Bunn P, Suda K, Ren S, Lueke Pickard S, Laszlo V, Harko T, Megyesfalvi Z, et al. 2020. Neuroendocrine subtypes of small cell lung cancer differ in terms of immune microenvironment and checkpoint molecule distribution. Mol Oncol 14: 1947–1965. 10.1002/1878-0261.12741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle A, Martin WJ, Funa K, Gazdar A, Carney D, Martin SE, Linnoila I, Cuttitta F, Mulshine J, Bunn P, et al. 1985. Markedly decreased expression of class I histocompatibility antigens, protein, and mRNA in human small-cell lung cancer. J Exp Med 161: 1135–1151. 10.1084/jem.161.5.1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapkin BJ, George J, Christensen CL, Mino-Kenudson M, Dries R, Sundaresan T, Phat S, Myers DT, Zhong J, Igo P, et al. 2018. Genomic and functional fidelity of small cell lung cancer patient-derived xenografts. Cancer Discov 8: 600–615. 10.1158/2159-8290.CD-17-0935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennell KA, Vassiliadis D, Lam EYN, Martelotto LG, Balic JJ, Hollizeck S, Weber TS, Semple T, Wang Q, Miles DC, et al. 2022. Non-genetic determinants of malignant clonal fitness at single-cell resolution. Nature 601: 125–131. 10.1038/s41586-021-04206-7 [DOI] [PubMed] [Google Scholar]
- Fujino K, Motooka Y, Hassan WA, Ali Abdalla MO, Sato Y, Kudoh S, Hasegawa K, Niimori-Kita K, Kobayashi H, Kubota I, et al. 2015. Insulinoma-associated protein 1 is a crucial regulator of neuroendocrine differentiation in lung cancer. Am J Pathol 185: 3164–3177. 10.1016/j.ajpath.2015.08.018 [DOI] [PubMed] [Google Scholar]
- Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, Ni A, Khodos I, de Stanchina E, Nguyen T, et al. 2017. Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell 31: 286–299. 10.1016/j.ccell.2017.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay CM, Stewart CA, Park EM, Diao L, Groves SM, Heeke S, Nabet BY, Fujimoto J, Solis LM, Lu W, et al. 2021. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 39: 346–360.e7. 10.1016/j.ccell.2020.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar AF, Carney DN, Nau MM, Minna JD. 1985. Characterization of variant subclasses of cell lines derived from small cell lung cancer having distinctive biochemical, morphological, and growth properties. Cancer Res 45: 2924–2930. [PubMed] [Google Scholar]
- George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, et al. 2015. Comprehensive genomic profiles of small cell lung cancer. Nature 524: 47–53. 10.1038/nature14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunblatt E, Wu N, Zhang H, Liu X, Norton JP, Ohol Y, Leger P, Hiatt JB, Eastwood EC, Thomas R, et al. 2020. MYCN drives chemoresistance in small cell lung cancer while USP7 inhibition can restore chemosensitivity. Genes Dev 34: 1210–1226. 10.1101/gad.340133.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. 2017. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551: 247–250. 10.1038/nature24297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318. 10.1038/ng1966 [DOI] [PubMed] [Google Scholar]
- Helfrich BA, Kim J, Gao D, Chan DC, Zhang Z, Tan AC, Bunn PA Jr. 2016. Barasertib (AZD1152), a small molecule aurora B inhibitor, inhibits the growth of SCLC cell lines in vitro and in vivo. Mol Cancer Ther 15: 2314–2322. 10.1158/1535-7163.MCT-16-0298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, Rizvi NA, Hirsch FR, Selvaggi G, Szustakowski JD, et al. 2018. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell 33: 853–861.e4. 10.1016/j.ccell.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook KE, Garza SJ, Lira ME, Ching KA, Lee NV, Cao J, Yuan J, Ye J, Ozeck M, Shi ST, et al. 2012. An integrated genomic approach to identify predictive biomarkers of response to the aurora kinase inhibitor PF-03814735. Mol Cancer Ther 11: 710–719. 10.1158/1535-7163.MCT-11-0184 [DOI] [PubMed] [Google Scholar]
- Horie M, Saito A, Ohshima M, Suzuki HI, Nagase T. 2016. YAP and TAZ modulate cell phenotype in a subset of small cell lung cancer. Cancer Sci 107: 1755–1766. 10.1111/cas.13078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, Huemer F, Losonczy G, Johnson ML, Nishio M, et al. 2018. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med 379: 2220–2229. 10.1056/NEJMoa1809064 [DOI] [PubMed] [Google Scholar]
- Huang F, Ni M, Chalishazar MD, Huffman KE, Kim J, Cai L, Shi X, Cai F, Zacharias LG, Ireland AS, et al. 2018a. Inosine monophosphate dehydrogenase dependence in a subset of small cell lung cancers. Cell Metab 28: 369–382.e5. 10.1016/j.cmet.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Klingbeil O, He XY, Wu XS, Arun G, Lu B, Somerville TDD, Milazzo JP, Wilkinson JE, Demerdash OE, et al. 2018b. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev 32: 915–928. 10.1101/gad.314815.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Huffman KE, Wang Z, Wang X, Li K, Cai F, Yang C, Cai L, Shih TS, Zacharias LG, et al. 2021. Guanosine triphosphate links MYC-dependent metabolic and ribosome programs in small-cell lung cancer. J Clin Invest 131: e139929. 10.1172/JCI139929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers IJ, Bin Ali R, Pritchard C, Cozijnsen M, Kwon MC, Proost N, Song JY, de Vries H, Badhai J, Sutherland K, et al. 2014. Rapid target gene validation in complex cancer mouse models using re-derived embryonic stem cells. EMBO Mol Med 6: 212–225. 10.1002/emmm.201303297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington ND, Cursons J, Rautela J. 2020. The cancer–natural killer cell immunity cycle. Nat Rev Cancer 20: 437–454. 10.1038/s41568-020-0272-z [DOI] [PubMed] [Google Scholar]
- Inoue Y, Nikolic A, Farnsworth D, Shi R, Johnson FD, Liu A, Ladanyi M, Somwar R, Gallo M, Lockwood WW. 2021. Extracellular signal-regulated kinase mediates chromatin rewiring and lineage transformation in lung cancer. Elife 10: e66524. 10.7554/eLife.66524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, Conley CC, Chen OS, Guthrie MR, Soltero D, et al. 2020. MYC drives temporal evolution of small cell lung cancer subtypes by reprogramming neuroendocrine fate. Cancer Cell 38: 60–78.e12. 10.1016/j.ccell.2020.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Udaka N, Yazawa T, Okudela K, Hayashi H, Sudo T, Guillemot F, Kageyama R, Kitamura H. 2000. Basic helix–loop–helix transcription factors regulate the neuroendocrine differentiation of fetal mouse pulmonary epithelium. Development 127: 3913–3921. 10.1242/dev.127.18.3913 [DOI] [PubMed] [Google Scholar]
- Jia S, Wildner H, Birchmeier C. 2015. Insm1 controls the differentiation of pulmonary neuroendocrine cells by repressing Hes1. Dev Biol 408: 90–98. 10.1016/j.ydbio.2015.10.009 [DOI] [PubMed] [Google Scholar]
- Jia D, Augert A, Kim DW, Eastwood E, Wu N, Ibrahim AH, Kim KB, Dunn CT, Pillai SPS, Gazdar AF, et al. 2018. Crebbp loss drives small cell lung cancer and increases sensitivity to HDAC inhibition. Cancer Discov 8: 1422–1437. 10.1158/2159-8290.CD-18-0385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Stockwell BR, Conrad M. 2021. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22: 266–282. 10.1038/s41580-020-00324-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY, Ge K. 2011. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J 30: 249–262. 10.1038/emboj.2010.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Zhao Q, Zhu W, Feng Y, Xiao T, Zhang P, Jiang L, Hou Y, Guo C, Huang H, et al. 2022. Identification of TAZ as the essential molecular switch in orchestrating SCLC phenotypic transition and metastasis. Nat Sci Rev 10.1093/nsr/nwab232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BE, Ihde DC, Makuch RW, Gazdar AF, Carney DN, Oie H, Russell E, Nau MM, Minna JD. 1987. myc family oncogene amplification in tumor cell lines established from small cell lung cancer patients and its relationship to clinical status and course. J Clin Invest 79: 1629–1634. 10.1172/JCI112999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaarijärvi R, Kaljunen H, Ketola K. 2021. Molecular and functional links between neurodevelopmental processes and treatment-induced neuroendocrine plasticity in prostate cancer progression. Cancers 13: 692. 10.3390/cancers13040692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Girard L, Giacomini CP, Wang P, Hernandez-Boussard T, Tibshirani R, Minna JD, Pollack JR. 2006. Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene 25: 130–138. 10.1038/sj.onc.1208997 [DOI] [PubMed] [Google Scholar]
- Kiyokawa H, Morimoto M. 2020. Notch signaling in the mammalian respiratory system, specifically the trachea and lungs, in development, homeostasis, regeneration, and disease. Dev Growth Differ 62: 67–79. 10.1111/dgd.12628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Winslow MM, Sage J. 2021. Mechanisms of small cell lung cancer metastasis. EMBO Mol Med 13: e13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong J-N, Moujalled DM, et al. 2016. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538: 477–482. 10.1038/nature19830 [DOI] [PubMed] [Google Scholar]
- Kwon MC, Proost N, Song JY, Sutherland KD, Zevenhoven J, Berns A. 2015. Paracrine signaling between tumor subclones of mouse SCLC: a critical role of ETS transcription factor Pea3 in facilitating metastasis. Genes Dev 29: 1587–1592. 10.1101/gad.262998.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Jenkins NA, Copeland NG. 2019. The roles of initiating truncal mutations in human cancers: the order of mutations and tumor cell type matters. Cancer Cell 35: 10–15. 10.1016/j.ccell.2018.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JS, Ibaseta A, Fischer MM, Cancilla B, O'Young G, Cristea S, Luca VC, Yang D, Jahchan NS, Hamard C, et al. 2017. Intratumoural heterogeneity generated by notch signalling promotes small-cell lung cancer. Nature 545: 360–364. 10.1038/nature22323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little CD, Nau MM, Carney DN, Gazdar AF, Minna JD. 1983. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 306: 194–196. 10.1038/306194a0 [DOI] [PubMed] [Google Scholar]
- Liu Y, Li Y, Liu S, Adeegbe DO, Christensen CL, Quinn MM, Dries R, Han S, Buczkowski K, Wang X, et al. 2018. NK cells mediate synergistic antitumor effects of combined inhibition of HDAC6 and BET in a SCLC preclinical model. Cancer Res 78: 3709–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llabata P, Torres-Diz M, Gomez A, Tomas-Daza L, Romero OA, Grego-Bessa J, Llinas-Arias P, Valencia A, Esteller M, Javierre BM, et al. 2021. MAX mutant small-cell lung cancers exhibit impaired activities of MGA-dependent noncanonical polycomb repressive complex. Proc Natl Acad Sci 118: e2024824118. 10.1073/pnas.2024824118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochmann TL, Floros KV, Naseri M, Powell KM, Cook W, March RJ, Stein GT, Greninger P, Maves YK, Saunders LR, et al. 2018. Venetoclax is effective in small-cell lung cancers with high BCL-2 expression. Clin Cancer Res 24: 360–369. 10.1158/1078-0432.CCR-17-1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan NR, Knelson EH, Wolff JO, Vajdi A, Saigí M, Campisi M, Hong D, Thai TC, Piel B, Han S, et al. 2021. Intrinsic immunogenicity of small cell lung carcinoma revealed by its cellular plasticity. Cancer Discov 11: 1952–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannava S, Grachtchouk V, Wheeler LJ, Im M, Zhuang D, Slavina EG, Mathews CK, Shewach DS, Nikiforov MA. 2008. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 7: 2392–2400. 10.4161/cc.6390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl K, Wildey G, Sakre N, Lipka MB, Behtaj M, Kresak A, Chen Y, Yang M, Velcheti V, Fu P, et al. 2017. Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget 8: 73745–73756. 10.18632/oncotarget.20572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, Bhutkar A, McKenna A, Dooley A, Vernon A, et al. 2014. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 156: 1298–1311. 10.1016/j.cell.2014.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles LA, Burga LN, Gardner EE, Bostina M, Poirier JT, Rudin CM. 2017. Anthrax toxin receptor 1 is the cellular receptor for Seneca valley virus. J Clin Invest 127: 2957–2967. 10.1172/JCI93472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, et al. 2015. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 28: 57–69. 10.1016/j.ccell.2015.06.002 [DOI] [PubMed] [Google Scholar]
- Mollaoglu G, Guthrie MR, Böhm S, Brägelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C, Chalishazar MD, et al. 2017. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to Aurora kinase inhibition. Cancer Cell 31: 270–285. 10.1016/j.ccell.2016.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, et al. 2018. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560: 319–324. 10.1038/s41586-018-0393-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgensztern D, Besse B, Greillier L, Santana-Davila R, Ready N, Hann CL, Glisson BS, Farago AF, Dowlati A, Rudin CM, et al. 2019. Efficacy and safety of rovalpituzumab tesirine in third-line and beyond patients with DLL3-expressing, relapsed/refractory small-cell lung cancer: results from the phase II TRINITY study. Clin Cancer Res 25: 6958–6966. 10.1158/1078-0432.CCR-19-1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto M, Liu Z, Cheng HT, Winters N, Bader D, Kopan R. 2010. Canonical notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. J Cell Sci 123: 213–224. 10.1242/jcs.058669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, Perez SE, Qiao Z, Verdi JM, Hicks C, Weinmaster G, Anderson DJ. 2000. Transient notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell 101: 499–510. 10.1016/S0092-8674(00)80860-0 [DOI] [PubMed] [Google Scholar]
- Muir A, Danai LV, Vander Heiden MG. 2018. Microenvironmental regulation of cancer cell metabolism: implications for experimental design and translational studies. Dis Model Mech 11: dmm035758. 10.1242/dmm.035758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau MM, Brooks BJ, Battey J, Sausville E, Gazdar AF, Kirsch IR, McBride OW, Bertness V, Hollis GF, Minna JD. 1985. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature 318: 69–73. 10.1038/318069a0 [DOI] [PubMed] [Google Scholar]
- Nau MM, Brooks BJ, Carney DN, Gazdar AF, Battey JF, Sausville EA, Minna JD. 1986. Human small-cell lung cancers show amplification and expression of the N-myc gene. Proc Natl Acad Sci 83: 1092–1096. 10.1073/pnas.83.4.1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BR, Hartman BH, Ray CA, Hayashi T, Bermingham-McDonogh O, Reh TA. 2009. Acheate-scute like 1 (Ascl1) is required for normal Delta-like (Dll) gene expression and notch signaling during retinal development. Dev Dyn 238: 2163–2178. 10.1002/dvdy.21848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neptune ER, Podowski M, Calvi C, Cho JH, Garcia JG, Tuder R, Linnoila RI, Tsai MJ, Dietz HC. 2008. Targeted disruption of NeuroD, a proneural basic helix-loop-helix factor, impairs distal lung formation and neuroendocrine morphology in the neonatal lung. J Biol Chem 283: 21160–21169. 10.1074/jbc.M708692200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, et al. 2015. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 6: 6377. 10.1038/ncomms7377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouri M, Massah S, Caradec J, Lubik AA, Li N, Truong S, Lee AR, Fazli L, Ramnarine VR, Lovnicki JM, et al. 2020. Transient Sox9 expression facilitates resistance to androgen-targeted therapy in prostate cancer. Clin Cancer Res 26: 1678–1689. 10.1158/1078-0432.CCR-19-0098 [DOI] [PubMed] [Google Scholar]
- Olsen RR, Ireland AS, Kastner DW, Groves SM, Spainhower KB, Pozo K, Kelenis DP, Whitney CP, Guthrie MR, Wait SJ, et al. 2021. ASCL1 represses a SOX9+ neural crest stem-like state in small cell lung cancer. Genes Dev 35: 847–869. 10.1101/gad.348295.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne JK, Larsen JE, Shields MD, Gonzales JX, Shames DS, Sato M, Kulkarni A, Wistuba II, Girard L, Minna JD, et al. 2013. NeuroD1 regulates survival and migration of neuroendocrine lung carcinomas via signaling molecules TrkB and NCAM. Proc Natl Acad Sci 110: 6524–6529. 10.1073/pnas.1303932110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oser MG, Niederst MJ, Sequist LV, Engelman JA. 2015. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol 16: e165–e172. 10.1016/S1470-2045(14)71180-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oser MG, Sabet AH, Gao W, Chakraborty AA, Schinzel AC, Jennings RB, Fonseca R, Bonal DM, Booker MA, Flaifel A, et al. 2019. The KDM5A/RBP2 histone demethylase represses NOTCH signaling to sustain neuroendocrine differentiation and promote small cell lung cancer tumorigenesis. Genes Dev 33: 1718–1738. 10.1101/gad.328336.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouadah Y, Rojas ER, Riordan DP, Capostagno S, Kuo CS, Krasnow MA. 2019. Rare pulmonary neuroendocrine cells are stem cells regulated by Rb, p53, and Notch. Cell 179: 403–416.e23. 10.1016/j.cell.2019.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DH, Giffin MJ, Bailis JM, Smit M-AD, Carbone DP, He K. 2019. DLL3: an emerging target in small cell lung cancer. J Hematol Oncol 12: 61. 10.1186/s13045-019-0745-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owonikoko TK, Niu H, Nackaerts K, Csoszi T, Ostoros G, Mark Z, Baik C, Joy AA, Chouaid C, Jaime JC, et al. 2020. Randomized phase II study of paclitaxel plus alisertib versus paclitaxel plus placebo as second-line therapy for SCLC: primary and correlative biomarker analyses. J Thorac Oncol 15: 274–287. 10.1016/j.jtho.2019.10.013 [DOI] [PubMed] [Google Scholar]
- Owonikoko TK, Dwivedi B, Chen Z, Zhang C, Barwick B, Ernani V, Zhang G, Gilbert-Ross M, Carlisle J, Khuri FR, et al. 2021. YAP1 expression in SCLC defines a distinct subtype with T-cell-inflamed phenotype. J Thorac Oncol 16: 464–476. 10.1016/j.jtho.2020.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Liang MC, Raiser DM, Zamponi R, Roach RR, Curtis SJ, Walton Z, Schaffer BE, Roake CM, Zmoos AF, et al. 2011. Characterization of the cell of origin for small cell lung cancer. Cell Cycle 10: 2806–2815. 10.4161/cc.10.16.17012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AS, Yoo S, Kong R, Sato T, Sinha A, Karam S, Bao L, Fridrikh M, Emoto K, Nudelman G, et al. 2021. Prototypical oncogene family Myc defines unappreciated distinct lineage states of small cell lung cancer. Sci Adv 7: eabc2578. 10.1126/sciadv.abc2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, Statsenko G, Hochmair MJ, Özgüroğlu M, Ji JH, et al. 2019. Durvalumab plus platinum–etoposide versus platinum–etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet 394: 1929–1939. 10.1016/S0140-6736(19)32222-6 [DOI] [PubMed] [Google Scholar]
- Pearsall SM, Humphrey S, Revill M, Morgan D, Frese KK, Galvin M, Kerr A, Carter M, Priest L, Blackhall F, et al. 2020. The rare YAP1 subtype of SCLC revisited in a biobank of 39 circulating tumor cell patient derived explant models: a brief report. J Thorac Oncol 15: 1836–1843. 10.1016/j.jtho.2020.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer M, Fernández-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, et al. 2012. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 44: 1104–1110. 10.1038/ng.2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, Klein AM, Jaffe AB. 2018. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560: 377–381. 10.1038/s41586-018-0394-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Stephens PJ, O'Meara S, McBride DJ, Meynert A, Jones D, Lin M-L, Beare D, Lau KW, Greenman C, et al. 2010. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 463: 184–190. 10.1038/nature08629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier JT, Dobromilskaya I, Moriarty WF, Peacock CD, Hann CL, Rudin CM. 2013. Selective tropism of seneca valley virus for variant subtype small cell lung cancer. J Natl Cancer Inst 105: 1059–1065. 10.1093/jnci/djt130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier JT, Gardner EE, Connis N, Moreira AL, de Stanchina E, Hann CL, Rudin CM. 2015. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 34: 5869–5878. 10.1038/onc.2015.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier JT, George J, Owonikoko TK, Berns A, Brambilla E, Byers LA, Carbone D, Chen HJ, Christensen CL, Dive C, et al. 2020. New approaches to SCLC therapy: from the laboratory to the clinic. J Thorac Oncol 15: 520–540. 10.1016/j.jtho.2020.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanal-Villalonga A, Taniguchi H, Zhan YA, Hasan MM, Chavan SS, Meng F, Uddin F, Manoj P, Donoghue MTA, Won HH, et al. 2021. Multi-omic analysis of lung tumors defines pathways activated in neuroendocrine transformation. Cancer Discov 11: 3028–3047. 10.1158/2159-8290.CD-20-1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radice PA, Matthews MJ, Ihde DC, Gazdar AF, Carney DN, Bunn PA, Cohen MH, Fossieck BE, Makuch RW, Minna JD. 1982. The clinical behavior of ‘mixed’ small cell/large cell bronchogenic carcinoma compared to ‘pure’ small cell subtypes. Cancer 50: 2894–2902. [DOI] [PubMed] [Google Scholar]
- Ravi RK, Weber E, McMahon M, Williams JR, Baylin S, Mal A, Harter ML, Dillehay LE, Claudio PP, Giordano A, et al. 1998. Activated Raf-1 causes growth arrest in human small cell lung cancer cells. J Clin Invest 101: 153–159. 10.1172/JCI831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi RK, Thiagalingam A, Weber E, McMahon M, Nelkin BD, Mabry M. 1999. Raf-1 causes growth suppression and alteration of neuroendocrine markers in DMS53 human small-cell lung cancer cells. Am J Respir Cell Mol Biol 20: 543–549. 10.1165/ajrcmb.20.4.3406 [DOI] [PubMed] [Google Scholar]
- Ready N, Hellmann MD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, Borghaei H, Jolivet J, Horn L, Mates M, et al. 2019. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J Clin Oncol 37: 992–1000. 10.1200/JCO.18.01042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekhtman N. 2022. Lung neuroendocrine neoplasms: recent progress and persistent challenges. Mod Pathol 35: 36–50. 10.1038/s41379-021-00943-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero OA, Torres-Diz M, Pros E, Savola S, Gomez A, Moran S, Saez C, Iwakawa R, Villanueva A, Montuenga LM, et al. 2014. MAX inactivation in small cell lung cancer disrupts MYC–SWI/SNF programs and is synthetic lethal with BRG1. Cancer Discov 4: 292–303. 10.1158/2159-8290.CD-13-0799 [DOI] [PubMed] [Google Scholar]
- Roper N, Velez MJ, Chiappori A, Kim YS, Wei JS, Sindiri S, Takahashi N, Mulford D, Kumar S, Ylaya K, et al. 2021. Notch signaling and efficacy of PD-1/PD-L1 blockade in relapsed small cell lung cancer. Nat Commun 12: 3880. 10.1038/s41467-021-24164-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, et al. 2012. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 44: 1111–1116. 10.1038/ng.2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Pietanza MC, Bauer TM, Ready N, Morgensztern D, Glisson BS, Byers LA, Johnson ML, Burris HA III, Robert F, et al. 2017. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol 18: 42–51. 10.1016/S1470-2045(16)30565-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, Heymach JV, Johnson JE, Lehman JM, MacPherson D, et al. 2019. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 19: 289–297. 10.1038/s41568-019-0133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Brambilla E, Faivre-Finn C, Sage J. 2021. Small-cell lung cancer. Nat Rev Dis Primers 7: 3. 10.1038/s41572-020-00235-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Kaneda A, Tsuji S, Isagawa T, Yamamoto S, Fujita T, Yamanaka R, Tanaka Y, Nukiwa T, Marquez VE, et al. 2013. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci Rep 3: 1911. 10.1038/srep01911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, Desai R, Escarpe PA, Hampl J, Laysang A, et al. 2015. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med 7: a136. 10.1126/scitranslmed.aac9459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TN, Schreiber RD. 2015. Neoantigens in cancer immunotherapy. Science 348: 69–74. 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- Schwendenwein A, Megyesfalvi Z, Barany N, Valko Z, Bugyik E, Lang C, Ferencz B, Paku S, Lantos A, Fillinger J, et al. 2021. Molecular profiles of small cell lung cancer subtypes: therapeutic implications. Mol Ther Oncolytics 20: 470–483. 10.1016/j.omto.2021.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova EA, Kwon M-C, Monkhorst K, Song J-Y, Bhaskaran R, Krijgsman O, Kuilman T, Peters D, Buikhuisen Wieneke A, Smit Egbert F, et al. 2016. Transcription factor NFIB is a driver of small cell lung cancer progression in mice and marks metastatic disease in patients. Cell Rep 16: 631–643. 10.1016/j.celrep.2016.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, Redwood AB, Fan YH, Li L, Glisson BS, et al. 2017. CHK1 inhibition in small-cell lung cancer produces single-agent activity in biomarker-defined disease subsets and combination activity with cisplatin or olaparib. Cancer Res 77: 3870–3884. 10.1158/0008-5472.CAN-16-3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L, et al. 2019. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov 9: 646–661. 10.1158/2159-8290.CD-18-1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo H, Isomura A, Ohtsuka T, Kori H, Miyachi H, Kageyama R. 2016. Oscillatory control of Delta-like1 in cell interactions regulates dynamic gene expression and tissue morphogenesis. Genes Dev 30: 102–116. 10.1101/gad.270785.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson KL, Stoney R, Frese KK, Simms N, Rowe W, Pearce SP, Humphrey S, Booth L, Morgan D, Dynowski M, et al. 2020. A biobank of small cell lung cancer CDX models elucidates inter- and intratumoral phenotypic heterogeneity. Nat Cancer 1: 437–451. 10.1038/s43018-020-0046-2 [DOI] [PubMed] [Google Scholar]
- Singh S, Jaigirdar AA, Mulkey F, Cheng J, Hamed SS, Li Y, Liu J, Zhao H, Goheer A, Helms WS, et al. 2021. FDA Approval summary: lurbinectedin for the treatment of metastatic small cell lung cancer. Clin Cancer Res 27: 2378–2382. 10.1158/1078-0432.CCR-20-3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram K, Reddy SP, Vinnakota K, Britto R, Subbarayan M, Nambiar S, Hebbar A, Samuel C, Shetty M, Sreepathi HK, et al. 2005. Upregulation of ASCL1 and inhibition of notch signaling pathway characterize progressive astrocytoma. Oncogene 24: 7073–7083. 10.1038/sj.onc.1208865 [DOI] [PubMed] [Google Scholar]
- Sonkin D, Thomas A, Teicher BA. 2019. Are neuroendocrine negative small cell lung cancer and large cell neuroendocrine carcinoma with WT RB1 two faces of the same entity? Lung Cancer Manag 8: Lmt13. 10.2217/lmt-2019-0005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sos ML, Dietlein F, Peifer M, Schöttle J, Balke-Want H, Müller C, Koker M, Richters A, Heynck S, Malchers F, et al. 2012. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci 109: 17034–17039. 10.1073/pnas.1207310109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriuranpong V, Borges MW, Strock CL, Nakakura EK, Watkins DN, Blaumueller CM, Nelkin BD, Ball DW. 2002. Notch signaling induces rapid degradation of achaete-scute homolog 1. Mol Cell Biol 22: 3129–3139. 10.1128/MCB.22.9.3129-3139.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam NJ, Kast WM, Voordouw AC, Pastoors LB, van der Hoeven FA, Melief CJ, Ploegh HL. 1989. Lack of correlation between levels of MHC class I antigen and susceptibility to lysis of small cellular lung carcinoma (SCLC) by natural killer cells. J Immunol 142: 4113–4117. [PubMed] [Google Scholar]
- Stewart CA, Gay CM, Xi Y, Sivajothi S, Sivakamasundari V, Fujimoto J, Bolisetty M, Hartsfield PM, Balasubramaniyan V, Chalishazar MD, et al. 2020. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat Cancer 1: 423–436. 10.1038/s43018-019-0020-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Irvine KD. 2016. Cellular organization and cytoskeletal regulation of the hippo signaling network. Trends Cell Biol 26: 694–704. 10.1016/j.tcb.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. 2011. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 19: 754–764. 10.1016/j.ccr.2011.04.019 [DOI] [PubMed] [Google Scholar]
- Takagi S, Ishikawa Y, Mizutani A, Iwasaki S, Matsumoto S, Kamada Y, Nomura T, Nakamura K. 2017. LSD1 Inhibitor T-3775440 inhibits SCLC cell proliferation by disrupting LSD1 interactions with SNAG domain proteins INSM1 and GFI1B. Cancer Res 77: 4652–4662. 10.1158/0008-5472.CAN-16-3502 [DOI] [PubMed] [Google Scholar]
- Thomas A, Takahashi N, Rajapakse VN, Zhang X, Sun Y, Ceribelli M, Wilson KM, Zhang Y, Beck E, Sciuto L, et al. 2021. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell 39: 566–579.e7. 10.1016/j.ccell.2021.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlemsani C, Pongor L, Elloumi F, Girard L, Huffman KE, Roper N, Varma S, Luna A, Rajapakse VN, Sebastian R, et al. 2020. SCLC-CellMiner: a resource for small cell lung cancer cell line genomics and pharmacology based on genomic signatures. Cell Rep 33: 108296. 10.1016/j.celrep.2020.108296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totaro A, Castellan M, Di Biagio D, Piccolo S. 2018. Crosstalk between YAP/TAZ and notch signaling. Trends Cell Biol 28: 560–573. 10.1016/j.tcb.2018.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigo J, Subbiah V, Besse B, Moreno V, López R, Sala MA, Peters S, Ponce S, Fernández C, Alfaro V, et al. 2020. Lurbinectedin as second-line treatment for patients with small-cell lung cancer: a single-arm, open-label, phase 2 basket trial. Lancet Oncol 21: 645–654. 10.1016/S1470-2045(20)30068-1 [DOI] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. 2017. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547: 453–457. 10.1038/nature23007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AH, Devarakonda S, Skidmore ZL, Krysiak K, Ramu A, Trani L, Kunisaki J, Masood A, Waqar SN, Spies NC, et al. 2018. Recurrent WNT pathway alterations are frequent in relapsed small cell lung cancer. Nat Commun 9: 3787. 10.1038/s41467-018-06162-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xu X, Maglic D, Dill MT, Mojumdar K, Ng PK-S, Jeong KJ, Tsang YH, Moreno D, Bhavana VH, et al. 2018. Comprehensive molecular characterization of the Hippo signaling pathway in cancer. Cell Rep 25: 1304–1317.e5. 10.1016/j.celrep.2018.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskopf K, Jahchan NS, Schnorr PJ, Cristea S, Ring AM, Maute RL, Volkmer AK, Volkmer J-P, Liu J, Lim JS, et al. 2016. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J Clin Invest 126: 2610–2620. 10.1172/JCI81603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerman BA, Breuer RH, Poutsma A, Chhatta A, Noorduyn LA, Koolen MG, Postmus PE, Blankenstein MA, Oudejans CB. 2007. Basic helix-loop-helix transcription factor profiling of lung tumors shows aberrant expression of the proneural gene atonal homolog 1 (ATOH1, HATH1, MATH1) in neuroendocrine tumors. Int J Biol Markers 22: 114–123. 10.1177/172460080702200205 [DOI] [PubMed] [Google Scholar]
- Williamson SC, Metcalf RL, Trapani F, Mohan S, Antonello J, Abbott B, Leong HS, Chester CPE, Simms N, Polanski R, et al. 2016. Vasculogenic mimicry in small cell lung cancer. Nat Commun 7: 13322. 10.1038/ncomms13322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wistuba II, Gazdar AF, Minna JD. 2001. Molecular genetics of small cell lung carcinoma. Semin Oncol 28: 3–13. 10.1016/S0093-7754(01)90072-7 [DOI] [PubMed] [Google Scholar]
- Wooten DJ, Groves SM, Tyson DR, Liu Q, Lim JS, Albert R, Lopez CF, Sage J, Quaranta V. 2019. Systems-level network modeling of small cell lung cancer subtypes identifies master regulators and destabilizers. PLoS Comput Biol 15: e1007343. 10.1371/journal.pcbi.1007343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Jia D, Ibrahim AH, Bachurski CJ, Gronostajski RM, MacPherson D. 2016. NFIB overexpression cooperates with Rb/p53 deletion to promote small cell lung cancer. Oncotarget 7: 57514–57524. 10.18632/oncotarget.11583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Guo J, Liu Y, Zheng Q, Li X, Wu C, Fang D, Chen X, Ma L, Xu P, et al. 2021. YAP drives fate conversion and chemoresistance of small cell lung cancer. Sci Adv 7: eabg1850. 10.1126/sciadv.abg1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y, Ozasa H, Kim YH, Yamazoe M, Ajimizu H, Yamamoto Funazo T, Nomizo T, Tsuji T, Yoshida H, Sakamori Y, et al. 2020. MCL1 inhibition is effective against a subset of small-cell lung cancer with high MCL1 and low BCL-XL expression. Cell Death Dis 11: 177. 10.1038/s41419-020-2379-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Girard L, Zhang YA, Haruki T, Papari-Zareei M, Stastny V, Ghayee HK, Pacak K, Oliver TG, Minna JD, et al. 2018. Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl Lung Cancer Res 7: 32–49. 10.21037/tlcr.2018.02.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Christensen CL, Dries R, Oser MG, Deng J, Diskin B, Li F, Pan Y, Zhang X, Yin Y, et al. 2020a. CDK7 Inhibition potentiates genome instability triggering anti-tumor immunity in small cell lung cancer. Cancer Cell 37: 37–54.e9. 10.1016/j.ccell.2019.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yang L, Szeto P, Abali GK, Zhang Y, Kulkarni A, Amarasinghe K, Li J, Vergara IA, Molania R, et al. 2020b. The Hippo pathway oncoprotein YAP promotes melanoma cell invasion and spontaneous metastasis. Oncogene 39: 5267–5281. 10.1038/s41388-020-1362-9 [DOI] [PubMed] [Google Scholar]
- Zhou H, Hu Y, Luo R, Zhao Y, Pan H, Ji L, Zhou T, Zhang L, Long H, Fu J, et al. 2021. Multi-region exome sequencing reveals the intratumoral heterogeneity of surgically resected small cell lung cancer. Nat Commun 12: 5431. 10.1038/s41467-021-25787-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Huang Y, Bender ME, Girard L, Kollipara R, Eglenen-Polat B, Naito Y, Savage TK, Huffman KE, Koyama S, et al. 2021. Evasion of innate immunity contributes to small cell lung cancer progression and metastasis. Cancer Res 81: 1813–1826. 10.1158/0008-5472.CAN-20-2808 [DOI] [PMC free article] [PubMed] [Google Scholar]