

ABSTRACT
Aging is an important risk factor for osteoarthritis (OA). Butorphanol is a preoperative sedative and analgesic that possesses anti-inflammatory activity. However, the effect of butorphanol on OA has not been reported. Here we aimed to explore the effect of butorphanol tartrate on the cellular senescence of human chondrocyte-articular (HC-A) cells in response to tumor necrosis factor-α (TNF-α) stimulation. Butorphanol tartrate attenuated the TNF-α-caused cellular senescence of HC-A cells, with decreased positive senescence-associated-β-galactosidase (SA-β-gal) staining and elevated telomerase activity. Butorphanol tartrate prevented TNF-α-caused cell cycle arrest in the G0/G1 phase in HC-A cells and decreased p21 expression. The TNF-α-induced production of interleukin (IL)-6 and IL-8 in HC-A cells were mitigated by butorphanol tartrate. In addition, butorphanol tartrate reduced p-NF-κB p65/total p65 and p-STAT3/STAT3 ratios in HC-A cells cultured with TNF-α. Taken together, butorphanol tartrate protected HC-A cells from TNF-α-caused cellular senescence through inactivation of NF-κB and STAT3. These results imply that butorphanol tartrate might be used as a potential agent for the treatment of aging-related OA.
KEYWORDS: Butorphanol tartrate, osteoarthritis (OA), human chondrocyte-articular (HC-A) cells, senescence, inflammation, NF-κB, STAT3
1. Introduction
Osteoarthritis (OA) represents the most ordinary joint disease mainly occurring in aging individuals [1]. OA prevalently affects joints and causes impaired mobility, resulting in heavy socioeconomic costs worldwide [2]. Current treatments and suggestions for OA patients include acupuncture, total joint replacement surgery, low-impact aerobic exercise, and weight loss. These approaches are attempted to reduce stiffness, alleviate pain, as well as maintain the fundamental function capacity and improve life quality [3]. However, there are no effective intervention approaches to prevent the progression of OA. Recently, nonsteroidal anti-inflammatory drugs (NSAIDs) such as celecoxib, etoricoxib, and nimesulide have been widely used as a second-line treatment for OA. However, the side effects of NSAIDs are noticeable. Headaches, heart failure, stroke, and hypertension are common side effects during treatment with NSAIDs, which might be caused by their inhibition of cyclooxygenase-2 (COX-2) [4]. Therefore, drugs with fewer side effects are still necessary for the treatment of OA
Several risk factors including joint malalignment, obesity, genetics, and aging are associated with OA. Amongst these factors, increasing age is considered the primary risk factor of OA. Age-associated mitochondrial dysfunction and oxidative stress might induce cell senescence in both articular cartilage and bone [5]. Chondrocytes are the sole cell type present within articular cartilage and play a central role in senescence. During OA, a rise in the number of senescent chondrocytes and the senescence-associated secretory phenotype (SASP) have been found in joint tissues, which are associated with cartilage degradation [6–8]. Thus, preventing the senescence of chondrocytes may enhance the effectiveness of current therapeutic approaches for patients with OA.
Butorphanol is a preoperative sedative and analgesic which has been used as a supplement to balanced anesthesia and to alleviate post-anesthesia shaking [9]. Currently, butorphanol was found to possess anti-inflammatory and antioxidant functions and exert a potential therapeutic property for inflammation-related diseases such as hypoxia neural injury [10], sepsis-caused brain and myocardial dysfunction, and edema [11]. Interestingly, it has been reported that butorphanol tartrate is effective for the alleviation of pain in microcrystalline sodium urate-induced arthritis in green-cheeked conures [12]. Hardie et al. [13] proved that butorphanol is effective for the treatment of chronic painful OA in cats. However, the effect of butorphanol on cartilage degradation in OA has not been reported. Here, we aimed to explore the effect of butorphanol tartrate on the cellular senescence of chondrocytes in response to inflammatory cytokine TNF-α stimulation.
2. Materials and methods
2.1. Human chondrocytes culture and treatment
Human Chondrocyte-articular (HC-A) cells were commercially purchased from Sciencell, USA. Cells were identified using alcian blue staining. HC-A cells were seeded in DMEM/F-12, supplemented with 10% FCS and 1% penicillin-streptomycin (Sigma-Aldrich, USA) and cultured for 2–3 days after plating. HC-A cells were serum-starved overnight and cultured with different concentrations (2 or 4 μM) of butorphanol tartrate and TNF-α (20 ng/ml) for 14 days.
2.2. MTT assay
Cells were treated with 0.5, 1, 2, 4, 20, 40 μM butorphanol tartrate for 24 hours. Cell viability of HC-A was assessed by incubating with 5 mg/ml MTT solution for 4 hours at 37°C. Then formazan dye was dissolved by incubation with DMSO for 10 minutes with gentle shaking after removal of the medium. The absorbance values at 570 nm were measured using a Microplate Reader (Bio-Rad, USA) [14].
2.3. Senescence associated-β-galactosidase (SA-β-gal) staining
After the indicated treatments, HC-A cells were taken for SA-β-gal staining using the previous method [15]. Briefly, HC-A cells were fixed with 2% formaldehyde and 0.2% glutaraldehyde for 15 minutes and then incubated with SA-β-gal (Sigma, USA) overnight at 37°C. Thereafter, cells were observed under an inverted microscope (×100 magnification) and the number of SA-β-gal-positive cells was calculated.
2.4. Telomerase activity detection
Protein lysates of HC-A cells were prepared from RIPA lysis buffer and quantified using the Bradford method. To detect telomerase activity, equal amounts of cell extracts were used for telomeric repeat amplification protocol (TRAP) reaction with a PCR-based TRAP ELISA kit (Boehringer Mannheim, Mannheim, Germany) following the manufacturer’s instructions.
2.5. Cell cycle analysis
HC-A cells were plated at 1.8 × 104 cells per well in a 24-well plate and fixed in 70% ethanol overnight at 4°C and cell cycle distribution was analyzed using propidium iodide (PI) staining as previously described [16]. Finally, the ratios of the cells in the G0/G1, G2/M, and S phases were analyzed using a FACS Calibur flow cytometer (Becton, USA) and FlowJo software (Treestar, USA).
2.6. Real-time PCR (RT-PCR)
Total RNA was isolated from HC-A cells using Trizol reagent (Invitrogen, USA) and used for the synthesis of first-strand cDNA with a high-capacity cDNA Reverse Transcription Kit (Applied Biosystems, USA). RT-PCR was then performed with an SYBR Green qPCR Master Mix (Applied Biosystems, USA). The expression levels of IL-6, IL-8, and p21 were calculated using the 2−ΔΔCt method [17]. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the reference gene [18].
2.7. Western blot analysis
HC-A cells were lysed in RIPA lysis buffer, followed by the quantification of protein content using the Bradford method. The same amounts of protein were subjected to 10% SDS-PAGE and then transferred to PVDF membranes for further Western blot analysis (Millipore, USA). Briefly, the primary antibodies (1:1000; Abcam, UK) of p21, p65, p-p65, STAT3, and p-STAT3 were incubated with the membranes for 12 hours at 4°C. Then the Goat anti Rabbit IgG (1:3000; Abcam, UK) was added to the membranes for 1 hour incubation at 37°C. Lastly, the membranes were exposed to an ECL chromogenic agent (Abcam, UK). β-actin was used as an internal control.
2.8. ELISA
The contents of IL-6 and IL-8 in the supernatants of HC-A cells were assessed with ELISA kits (R&D Systems, USA) as described in the instructions. Finally, the absorbance at 450 nm was measured.
2.9. Statistical analysis
Results were expressed as mean± standard error of the mean (S.E.M.). The statistical analyses of experimental data were performed using the SPSS software version 13.0 (SPSS, USA) with the analysis of variance (ANOVA), followed by Bonferroni’s post-hoc test. P< 0.05 was used to indicate a statistically significant difference.
3. Results
In this study, we attempted to determine the protective effects of butorphanol tartrate against TNF-α–induced cellular senescence in HC-A cells. Firstly, we tested the cytotoxicity of butorphanol tartrate in HC-A cells to choose the suitable concentrations of butorphanol tartrate for this study. Then we examined the telomerase and SA-β-gal activities to confirm the effect of butorphanol tartrate on cellular senescence. We further detected the effects of butorphanol tartrate on cell cycle arrest in the G0/G1 phase and the expression of p21. To clarify the underlying mechanism, we investigated the effect of butorphanol tartrate on the NF-κB and STAT3 signaling pathways.
3.1. Cytotoxicity of butorphanol tartrate in HC-A cells
The cytotoxicity of butorphanol tartrate (molecular structure in Figure 1a) in HC-A cells was evaluated after exposure to 0, 0.5, 1, 2, 4, 20, 40 μM butorphanol tartrate for 24 hours. MTT assay proved that cell viability of HC-A cells was decreased by 8% and 15% after treatment with 20 and 40 μM butorphanol tartrate, whereas there was no significant change in cell viability after treatment with 0.5, 1, 2, 4 μM butorphanol tartrate (Figure 1b). Thus, the concentrations of 0.5, 1, 2, 4 μM were considered safe, and 2 and 4 μM were applied in the following experiments.
Figure 1.
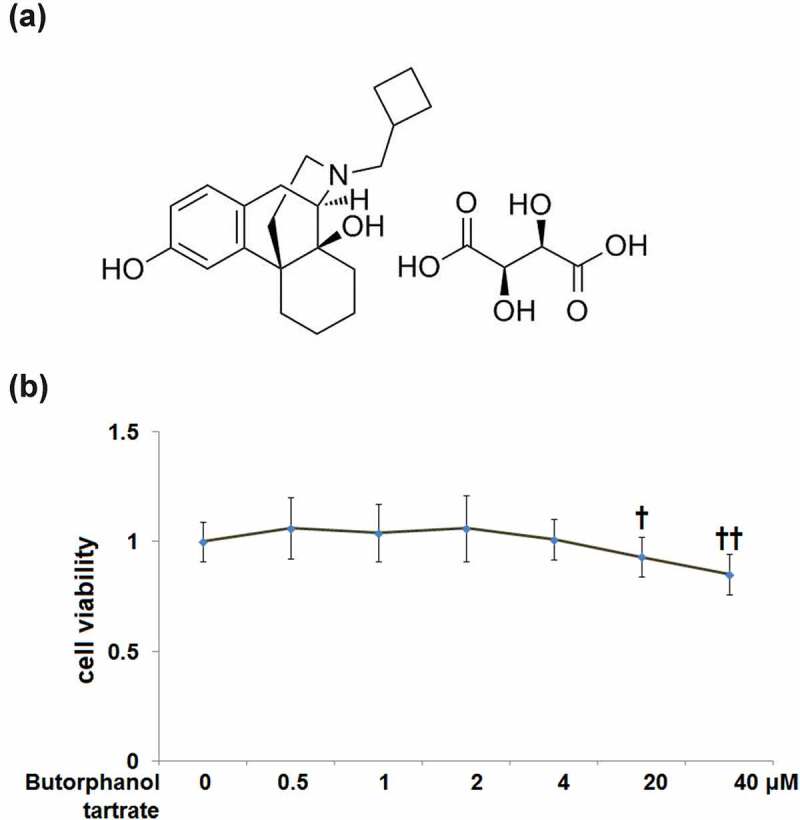
Cytotoxicity of Butorphanol tartrate in human HC-A chondrocytes. (a) Molecular structure of Butorphanol tartrate; (b). Cells were treated with Butorphanol tartrate at varying concentrations (0, 0.5, 1, 2, 4, 20, 40 μM) for 24 hours, the cell viability was determined (†, ††, P < 0.05, 0.01 vs. Control group).
3.2. The effects of butorphanol tartrate on cellular senescence induced by TNF-α in HC-A cells
Cellular senescence evaluated using SA-β-gal staining is shown in Figure 2a. The percentage of SA-β-gal-positive cells in TNF-α-treated HC-A cells was significantly increased to 29.6%±3.32%, as compared to control cells (5.4%±0.5%). Treatment with 2 or 4 μM butorphanol tartrate respectively reduced the percentage of SA-β-gal-positive cells to 18.3 ± 1.95% and 9.5%±1.25% (Figure 2b).
Figure 2.
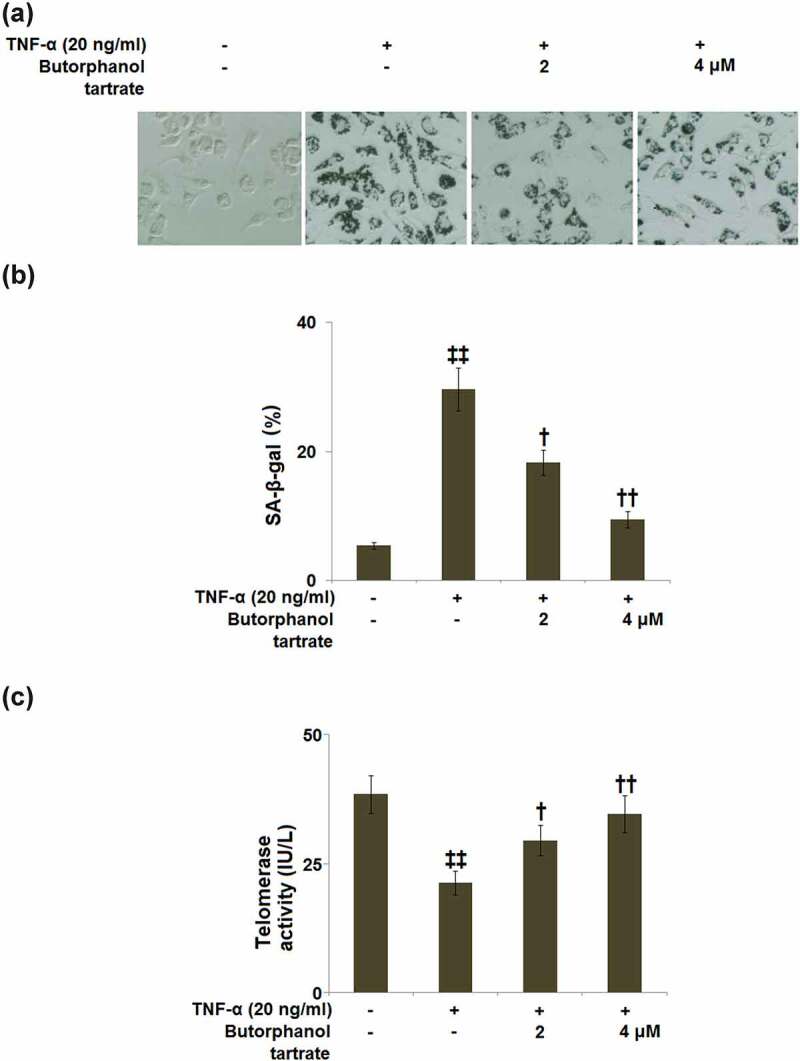
The effects of Butorphanol tartrate on cellular senescence induced by TNF-α in HC-A chondrocytes. The cells were treated with Butorphanol tartrate (2, 4 μM) and TNF-α (20 ng/ml) for 14 days. (a). Representative pictures of the SA-β-gal staining results in the sham group and experimental groups; (b). Quantification of the SA-β-gal staining; (c). Telomerase activity (‡‡, P < 0.01 vs. Control group; †, ††, P < 0.05, 0.01 vs. TNF-α group).
Telomerase activity of HC-A cells was examined using a TRAP reaction. In the TNF-α-induced HC-A cells, the telomerase activity (21.3 ± 2.3 IU/L) was significantly lower than that in control HC-A cells (38.4 ± 3.6 IU/L). In contrast to this, butorphanol tartrate (2, 4 μM) treatment elevated the telomerase activity to 29.5 ± 2.9 and 34.6 ± 3.6 IU/L, respectively (Figure 2c).
3.3. The effects of butorphanol tartrate on cell cycle arrest in the G0/G1 phase in TNF-α-treated HC-A cells
As compared to control cells, TNF-α stimulation was found to induce G0/G1 arrest in HC-A cells, as evidenced by an obviously increased percentage of G0/G1-phase cells (62.5 ± 6.1% vs. 41.5 ± 4.3%; Figure 3). In cells treated with butorphanol tartrate (2, 4 μM), the percentage of those in the G0/G1-phase was decreased to 54.5 ± 5.6% and 45.2 ± 4.6%, respectively, implying that butorphanol tartrate reversed the TNF-α-induced G0/G1arrest in HC-A cells (Figure 3).
Figure 3.
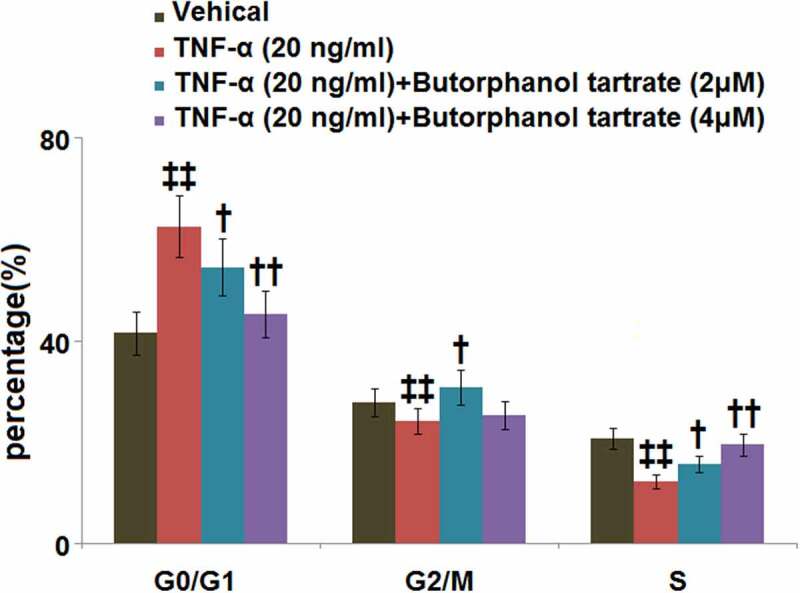
The effects of Butorphanol tartrate on cell cycle arrest in the G0/G1 phase in TNF-α-treated HC-A chondrocytes. The cells were treated with Butorphanol tartrate (2, 4 μM) and TNF-α (20 ng/ml) for 14 days. The percentage of G0/G1-phase was shown in the results (‡‡, P < 0.01 vs. Control group; †, ††, P < 0.05, 0.01 vs. TNF-α group).
3.4. The effect of butorphanol tartrate on the expression of p21 in HC-A cells against TNF-α
The expression of the senescence-associated protein, p21 was subsequently detected. RT-PCR demonstrated that the p21 mRNA level was increased by 2.8-fold in HC-A cells cultured with TNF-α (20 ng/ml), whereas HC-A cells expressed lower mRNA levels of p21 in the presence of 2 or 4 μM butorphanol tartrate (Figure 4a). Western blot analysis showed that the protein level of p21 was significantly higher in HC-A cells cultured with TNF-α (20 ng/ml), which was attenuated by 2 or 4 μM butorphanol tartrate (Figure 4b).
Figure 4.
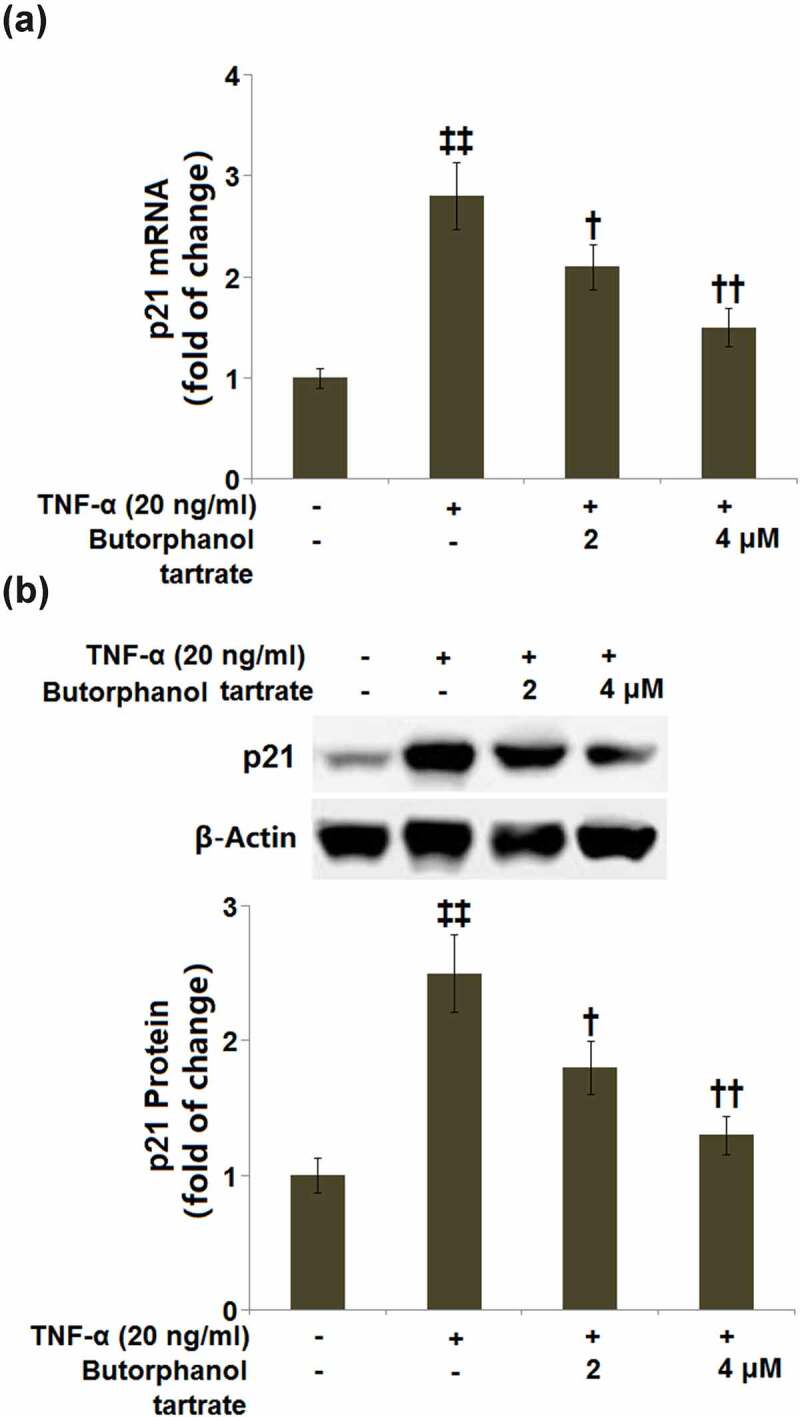
The effects of Butorphanol tartrate on the expression of p21 in HC-A chondrocytes against TNF-α. The cells were treated with Butorphanol tartrate (0, 2, 4 μM) and TNF-α (20 ng/ml) for 24 hours. (a). mRNA of p21; (b). Protein of p21 (‡‡, P < 0.01 vs. Control group; †, ††, P < 0.05, 0.01 vs. TNF-α group).
3.5. The effect of butorphanol tartrate on TNF-α-induced inflammation in HC-A cells
As shown in Figure 5a, the mRNA levels of IL-6 and IL-8 in HC-A cells were significantly induced by 3.6- and 4.1-fold, respectively, after stimulation with TNF-α. The upregulated mRNA levels of IL-6 and IL-8 were neutralized by pretreatment with butorphanol tartrate (2, 4 μM). In a parallel experiment, we also used ELISA to determine the secretion levels of IL-6 and IL-8. In control cells, the secretion levels of IL-6 and IL-8 were 155.2 ± 5.7 pg/ml and 107.6 ± 15.3 pg/ml, while after TNF-α stimulation, the secretion levels of IL-6 and IL-8 were markedly increased to 651.7 ± 77.8 pg/ml and 410.9 ± 54.6 pg/ml, respectively, both of which were neutralized by 2 and 4 μM butorphanol tartrate (Figure 5b).
Figure 5.
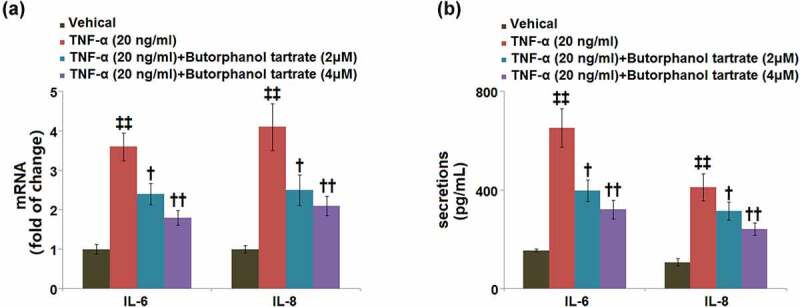
The effects of Butorphanol tartrate on TNF-α-induced inflammation in HC-A chondrocytes. The cells were treated with Butorphanol tartrate (0, 2, 4 μM) and TNF-α (20 ng/ml) for 24 hours. (a) The mRNA level of IL-6 and IL-8; (b) secretions of IL-6 and IL-8 (‡‡, P < 0.01 vs. Control group; †, ††, P < 0.05, 0.01 vs. TNF-α group).
3.6. The effect of butorphanol tartrate on NF-κB and STAT3 in HC-A cells against TNF-α
Western blot analysis showed that TNF-α stimulation upregulated the p-NF-κB p65/total p65 in HC-A cells with a 3.6-fold change. Treatment with butorphanol tartrate (2, 4 μM) markedly reduced the p-NF-κB p65/total p65 ratio (Figure 6). In addition, the TNF-α-induced increase in p-STAT3/STAT3 ratio (2.6-fold change) was mitigated by butorphanol tartrate (2, 4 μM).
Figure 6.
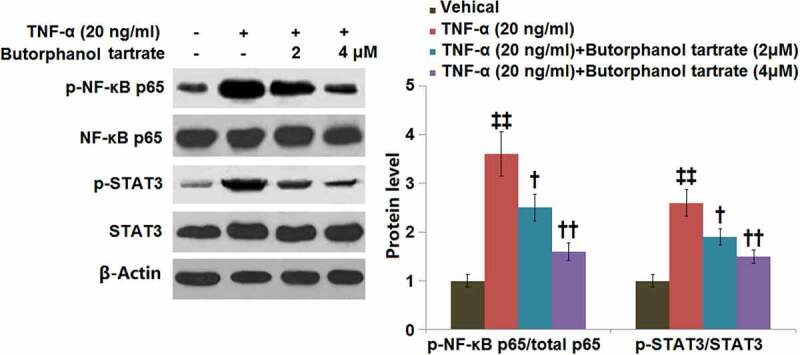
The effects of Butorphanol tartrate on the activation of NF-κB and STAT3 in HC-A chondrocytes against TNF-α. The cells were treated with Butorphanol tartrate (2, 4 μM) and TNF-α (20 ng/ml) for 24 hours. Representative Western blot results of p-NF-κB p65/total p65 and p-STAT3/STAT3 (‡‡, P < 0.01 vs. Control group; †, ††, P < 0.05, 0.01 vs. TNF-α group).
4. Discussion
Articular cartilage is a flexible connective tissue located on the surfaces of synovial joints, mainly composed of chondrocytes, proteoglycans, and collagen. Amongst these components, chondrocytes are the primary cellular components responsible for the production of extracellular matrix, thereby maintaining cartilage structure and function. Currently increasing evidence has shown that aging is the most important risk factor for OA. Aging-associated changes in chondrocytes such as cellular senescence, epigenetic alterations, genomic instability, mitochondrial dysfunction, oxidative stress, protein imbalance, inflammation, altered intercellular and intracellular signaling, and telomere attrition, promote the development of OA. In this study, we focused on the contributions of butorphanol tartrate on the following factors: cellular senescence, telomere attrition, cell cycle arrest, inflammation, and alternations in cell signalings.
Several phenotypic transformations occur in chondrocytes during cellular senescence. Various senescence markers including SA-β-Gal, telomerase, as well as p53, p21, and p16 proteins are found to be dysregulated in senescent chondrocytes [6]. The cytochemical staining of SA-β-Gal activity is a frequently used technique to detect senescent cells [19]. Here, the SA-β-gal staining showed that treatment with butorphanol tartrate reduced the percentage of SA-β-gal-positive cells in HC-A cells cultured with TNF-α. We also found that butorphanol tartrate elevated the telomerase activity in TNF-α-induced HC-A cells, which might contribute to alleviating the telomere dysfunction. The aging-associated DNA damage accumulation and detrimental oxidative stress in the OA tissue microenvironment alter gene profiles in chondrocytes. One common consequence is increased expression levels of various cell cycle inhibitors, for instance, p21 and p16. These proteins are found to mediate the senescence-related cell cycle arrest [20,21]. We demonstrated that butorphanol tartrate prevented cell cycle arrest in the G0/G1 phase and decreased p21 expression in TNF-α-induced HC-A cells.
In addition to growth arrest, senescent chondrocytes also show features of SASP, which produces increased levels of matrix-degrading enzymes and plenty of pro-inflammatory cytokines, thereby contributing to joint tissue destruction [22,23]. Mounting evidence has suggested that the development of OA is related to low-grade systemic and local inflammation. Aging has sometimes been referred to as ‘inflammaging’ since it is likewise associated with chronic low-grade inflammation [24]. Thus, an age-related increase in the production of pro-inflammatory mediators in joint tissues plays causative roles in disrupting cartilage homeostasis during OA progression [25]. In this study, we found that TNF-α-induced upregulation of IL-6 and IL-8 were attenuated by butorphanol tartrate in HC-A cells.
Increasing evidence has revealed that transcription factors such as NF-κB, RELA, and STAT3 are key mediators in OA progression [26]. Their interactions can regulate the cellular metabolism and inflammation, and drive the secretion of inflammatory cytokines, as well as immune responses in the OA microenvironment [26]. NF-κB is a collection of inducible transcription factors that play critical roles in immune and inflammatory responses [27]. Dysregulation of NF-κB activation has been frequently observed in many inflammatory diseases, including OA [28]. STAT3 is a DNA-binding molecule that has key roles in controlling inflammation and immunity [29]. Tight regulation of STAT3 function is central to health, while inactivation or hyperactivation of STAT3 usually results in human diseases. It has been documented that STAT3 is a core transcription factor that speeds up the progression of OA through the NF-κB signaling pathway [26]. Our results show that butorphanol tartrate caused the inactivation of both NF-κB and STAT3 in TNF-α-induced HC-A cells. The main limitation of the current study is that we only examined the protective effects of butorphanol tartrate against TNF-α- induced cellular senescence in an in vitro chondrocytes model. Animal experiments are one of the important ways to explore treatment of OA. A future study with ideal OA animal models should provide a more complete picture of the underlying mechanisms.
Conclusion
In this study, our results reveal the protective effects of butorphanol tartrate on the cellular senescence of HC-A cells in response to TNF-α stimulation. In view of the signaling mechanism, we found that butorphanol tartrate caused the inactivation of both NF-κB and STAT3 in TNF-α-induced HC-A cells. Collectively, butorphanol tartrate might be used as a potential agent for the treatment of aging-related OA.
Author contribution
Chengyuan Zhang, Shilin Jiang, Feng Yuan, and Ye Lu contributed to the study design. Shilin Jiang and Ye Lu collected and performed the experiments. Chengyuan Zhang and Shilin Jiang performed the statistical analysis. Chengyuan Zhang and Feng Yuan drafted the manuscript and supervised the entire study. All authors critically reviewed the manuscript and approved the final draft.
Consent to publication
All the authors agreed to publish this article.
Ethical statements
The protocol of this study was approved by the ethical committee of Shanghai Municipal Health Commission.
Funding Statement
This study was supported by the “Project of Shanghai Shen Kang Hospital Development Center(SHDC12020118), Project of Science and Technology Commission of Shanghai Municipality (20Y11913300), Shanghai Municipal Health Commission Special Funding for Clinical Research in the Health Industry General Project (201940427), Shanghai Municipal Health Commission Special Funding for Clinical Research in the Health Industry Youth Project (20194Y0279) and Scientific Research Project of Shanghai Sixth People’s Hospital Medical Group.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement /availability of data materials
Data of this study are available upon reasonable request to the corresponding authors.
References
- [1].Vina ER, Kwoh CK.. Epidemiology of osteoarthritis: literature update. Curr Opin Rheumatol. 2018;30:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Xia B, Di C, Zhang J, et al. Osteoarthritis pathogenesis: a review of molecular mechanisms. Calcif Tissue Int. 2014;95:495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Felson DT. Clinical practice. Osteoarthritis of the knee. N Engl J Med. 2006;354:841–848. [DOI] [PubMed] [Google Scholar]
- [4].Cadet C, Maheu E; The French AGRHUM Group (Association Geriatric and RHeUMatology) . Non-steroidal anti-inflammatory drugs in the pharmacological management of osteoarthritis in the very old: prescribe or proscribe? Ther Adv Musculoskelet Dis. 2021;13:1759720X211022149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Coryell PR, Diekman BO, Loeser RF. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat Rev Rheumatol. 2021;17:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].McCulloch K, Litherland GJ, Rai TS. Cellular senescence in osteoarthritis pathology. Aging Cell. 2017;16:210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wan M, Gray-Gaillard EF, Elisseeff JH. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res. 2021. 41;9. DOI: 10.1038/s41413-021-00164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Boulestreau J, Maumus M, Jorgensen C, et al. Extracellular vesicles from mesenchymal stromal cells: therapeutic perspectives for targeting senescence in osteoarthritis. Adv Drug Deliv Rev. 2021;175:113836. [DOI] [PubMed] [Google Scholar]
- [9].Vogelsang J, Hayes SR. Butorphanol tartrate (stadol): a review. J Post Anesth Nurs. 1991;6:129–135. [PubMed] [Google Scholar]
- [10].Yang Z, Wang L, Hu Y, et al. Butorphanol protects PC12 cells against OGD/R-induced inflammation and apoptosis. Mol Med Rep. 2020;22:1969–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vachon P, Moreau JP. Butorphanol decreases edema following carrageenan-induced paw inflammation in rats. Contemp Top Lab Anim Sci. 2002;41:15–17. [PubMed] [Google Scholar]
- [12].Paul-Murphy JR, Krugner-Higby LA, Tourdot RL, et al. Evaluation of liposome-encapsulated butorphanol tartrate for alleviation of experimentally induced arthritic pain in green-cheeked conures (Pyrrhura molinae). Am J Vet Res. 2009;70:1211–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hardie EM. Management of osteoarthritis in cats. Vet Clin North Am Small Anim Pract. 1997;27:945–953. [DOI] [PubMed] [Google Scholar]
- [14].Liu YN, Zhang SH, Xue ZW, et al. Bone mesenchymal stem cells-derived miR-223-3p-containing exosomes ameliorate lipopolysaccharide-induced acute uterine injury via interacting with endothelial progenitor cells. Bioengineered. 2021;12(2):10654–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kim KW, Chung HN, Ha KY, et al. Senescence mechanisms of nucleus pulposus chondrocytes in human intervertebral discs. Spine J. 2009;9:658–666. [DOI] [PubMed] [Google Scholar]
- [16].Jayasooriya RG, Kang SH, Kang CH, et al. Apigenin decreases cell viability and telomerase activity in human leukemia cell lines. Food Chem Toxicol. 2012;50:2605–2611. [DOI] [PubMed] [Google Scholar]
- [17].Ren XY, Li A, Ying E, et al. Upregulation of ubiquitin-conjugating enzyme E2T (UBE2T) predicts poor prognosis and promotes hepatocellular carcinoma progression. Bioengineered. 2021;12(1):1530–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang ML, Guo SQ, Zhang Y, et al. Remifentanil attenuates sepsis-induced intestinal injury by inducing autophagy. Bioengineered. 2021;12(2):9575–9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Noren Hooten N, Evans MK. Techniques to induce and quantify cellular senescence. J Vis Exp. 2017. DOI: 10.3791/55533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lotz M, Loeser RF. Effects of aging on articular cartilage homeostasis. Bone. 2012;51:241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rose J, Soder S, Skhirtladze C, et al. DNA damage, discoordinated gene expression and cellular senescence in osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2012;20:1020–1028. [DOI] [PubMed] [Google Scholar]
- [22].Charlier E, Relic B, Deroyer C, et al. Insights on molecular mechanisms of chondrocytes death in osteoarthritis. Int J Mol Sci. 2016;17. DOI: 10.3390/ijms17122146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Aigner T, Soder S, Gebhard PM, et al. Mechanisms of disease: role of chondrocytes in the pathogenesis of osteoarthritis–structure, chaos and senescence. Nat Clin Pract Rheumatol. 2007;3:391–399. [DOI] [PubMed] [Google Scholar]
- [24].Greene MA, Loeser RF. Aging-related inflammation in osteoarthritis. Osteoarthritis Cartilage. 2015;23:1966–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hashimoto M, Nakasa T, Hikata T, et al. Molecular network of cartilage homeostasis and osteoarthritis. Med Res Rev. 2008;28:464–481. [DOI] [PubMed] [Google Scholar]
- [26].Wang F, Guo Z, Yuan Y. STAT3 speeds up progression of osteoarthritis through NF-kappaB signaling pathway. Exp Ther Med. 2020;19:722–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kumar A, Takada Y, Boriek AM, et al. Nuclear factor-kappaB: its role in health and disease. J Mol Med (Berl). 2004;82:434–448. [DOI] [PubMed] [Google Scholar]
- [28].Choi MC, Jo J, Park J, et al. NF-kappaB signaling pathways in osteoarthritic cartilage destruction. Cells. 2019;8. DOI: 10.3390/cells8070734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hillmer EJ, Zhang H, Li HS, et al. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016;31:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data of this study are available upon reasonable request to the corresponding authors.