

ABSTRACT
Rifampin (RFP), a first-line anti-tuberculosis drug, often induces cholestatic liver injury and hyperbilirubinemia which limits its clinical use. Multidrug resistance-associated protein 2 (MRP2) localizes to the hepatocyte apical membrane and plays a pivotal role in the biliary excretion of bilirubin glucuronides. RFP is discovered to reduce MRP2 expression in liver cells. 4-Phenylbutyrate (4-PBA), a drug used to treat ornithine transcarbamylase deficiency (DILI), is reported to alleviate RFP-induced liver cell injury. However, the underlying mechanism still remains unclear. In the current study, we discovered that RFP induced HepG2 cell viability reduction, apoptosis and MRP2 ubiquitination degradation. Administration of 4-PBA alleviated the effect of RFP on HepG2 cell viability reduction, apoptosis and MRP2 ubiquitination degradation. In mechanism, 4-PBA suppressed RPF-caused intracellular Ca2+ disorder and endoplasmic reticulum (ER) stress, as well as the increases of Clathrin and adapter protein 2 (AP2). ER stress marker protein C/EBP homologous protein took part in the modulation of AP2 and clathrin. Besides, 4-PBA reduced the serum bilirubin level in RFP-induced cholestasis mouse model, along with raised the MRP2 expression in liver tissues. These findings indicated that 4-PBA could alleviate RFP-induced cholestatic liver injury and thereby decreased serum total bilirubin concentration via inhibiting ER stress and ubiquitination degradation of MRP2, which provides new insights into the mechanism of 4-PBA in the treatment of RFP-induced cholestasis and liver damage.
KEYWORDS: Cholestasis, rifampicin, 4-Phenylbutyrate, multidrug resistance-associated protein 2, ubiquitination, endoplasmic reticulum stress
Graphical abstract
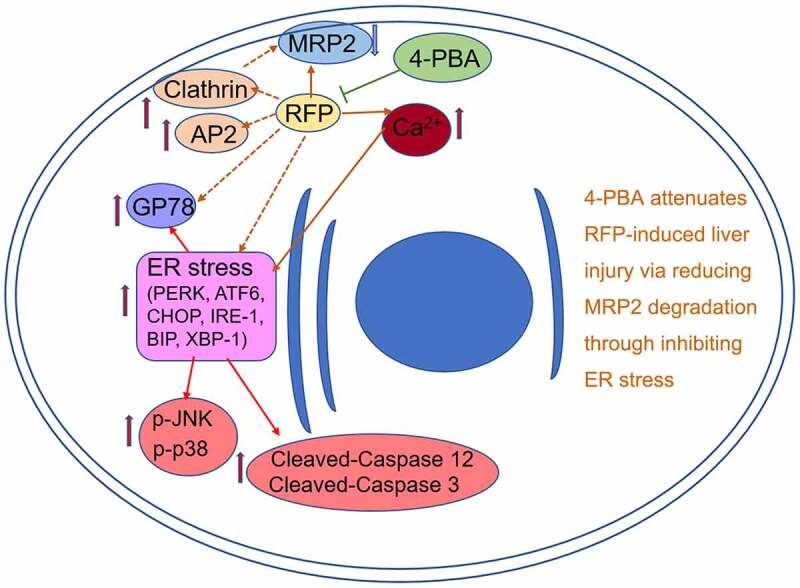
Introduction
The liver, lying between adsorption and systematic circulations, is a very important organ in drug metabolism and elimination, and thus, the liver is also susceptible to drug toxicity. Drug-induced liver injury (DILI), including damage to hepatocytes and other liver cells, is one of the most challenging liver diseases faced by hepatologists [1]. It has become a main reason for acute live failure worldwide [2–4]. Rifampicin (RFP) is a first-line drug widely used for tuberculosis (TB) and has been shown to cause serious liver injury [5], including cholestasis and hyperbilirubinemia [6,7].
Recent study supported that oxidative stress might contribute to RFP-induced hepatotoxicity, which has been a longstanding concern in the treatment of TB [8]. Meanwhile, it has been shown that RFP induced hepatotoxicity is closely related to endoplasmic reticulum (ER) stress and multidrug resistance-associated protein 2 (MRP2) [9,10]. ER stress, caused by the accumulation of unfolded or misfolded proteins in ER, has been implicated in the pathogenesis of DILI and RFP-induced liver injury [10,11]. MRP2, located on the canalicular membrane of hepatocytes, belongs to the ATP-binding cassette transporter family and plays a key role in the biliary excretion of a wide variety of organic anions, such as glutathione, bilirubin, leukotrienes, and sulfated or glucuronidated bile acids [12]. Previous study demonstrated that RFP-induced oxidative stress led to endocytosis and ubiquitination of MRP2 in hepatic epithelial cells, which further induced cholestasis [9].
4-Phenylbutyrate (4-PBA) is an PDA approved drug currently used for the treatment of ornithine transcarbamylase deficiency. Earlier literature reported that 4-PBA could activate MRP2 function by modulating its ubiquitination and reducing total bilirubin concentration in serum [13]. In addition, 4-PBA has been found to contribute to protein folding and thereby alleviate ER stress [14,15]. Recent study discovered that 4-PBA inhibited RFP-activated RNA-dependent protein kinase-like ER kinase (PERK)/activating transcription factor 4 (ATF4)/C/EBP homologous protein (CHOP) signal pathway to alleviate hepatic L02 cell injury [16]. However, the direct connection between 4-PBA with RFP-induced ER stress and ubiquitination of MRP2 in hepatotoxicity is still unclear.
Therefore, we hypothesized that 4-PBA could attenuate RFP-induced liver injury via inhibition of RFP initiated ER stress and ubiquitin-proteasome degradation of MRP2. To verify this hypothesis, human hepatoma HepG2 cells were subjected to RFP and/or 4-PBA treatment, cell viability, apoptosis, MRP2 expression, intracellular calcium disorder and ER stress were tested. Moreover, RFP-induced cholestasis mouse model was established to further confirm the beneficial effects of 4-PBA on RFP-caused liver damage.
Material and methods
Cell culture
HepG2 was obtained from Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China) and growth in RPMI-1640 medium (Thermo Fisher Scientific, Waltham, MA) containing 10% fetal bovine serum (Thermo Fisher Scientific) and 1% penicillin-streptomycin solution (Thermo Fisher Scientific). Both RFP and 4-PBA were purchased from Sigma-Aldrich (MO, USA).
Cell counting Kit-8 (CCK-8) assay
CCK-8 assay was carried out to test HepG2 cell viability [17]. Briefly, HepG2 cells were seeded 96-well plate with 5 × 103 cells/well for overnight. Then, cells were treated by RFP (50 μM) [9] and/or 4-PBA (0.1 mM, 0.5 mM, 1 mM) for 48 h. 10 μL CCK-8 kit solution (BioVision, Milpitas, CA) was added into the each well of the plate for 1 h. Following shaking, the absorbance of each well was detected at 450 nm.
PI/Annexin V double staining
HepG2 cell apoptosis was measured via PI/Annexin V double staining [18]. Briefly, HepG2 cells were seeded into 24-well plate with 2 × 104 cells/well overnight and treated by RFP (50 μM) and/or 4-PBA (1 mM) for 48 h. Then, cells in each group were collected, washed with phosphate buffer saline (PBS) and stained using Annexin-V-FITC solution and PI solution (Abcam, Cambridge, MA) for 15 min protected from light. Following 400 μL 1 × Binding Buffer adding, HepG2 cell apoptosis was analyzed using flow cytometer (BD, Franklin Lakes, NJ).
qRT-PCR
Total RNAs were extracted from cells or tissues using Trizol reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized using PrimeScriptTM 1st Strand cDNA Synthesis Kit (Takara Biotechnology, Beijing, China). The relative mRNA expressions of MRP2, adapter protein 2 (AP2), cathrin were detected using Takara PCR Amplification Kit (Takara Biotechnology) with GAPDH expression as internal control. The primer sequences were as follows: 5’-CCCTGCTGTTCGATATACCAATC-3’ (MRP2 Forward), 5’-TCGAGAGAATCCAGAATAGGGAC-3’ (MRP2 Reverse), 5’-GGTGAACCCCAACGAAGTCT-3’ (AP2 Forward), 5’-GGGATCGGAATGTTGTCGGT-3’ (AP2 Reverse), 5’-ATTCTGCCAATTCGTTTTCAGGA-3’ (Clathrin Forward), 5’-GCTTTCAGTGCAATTACTTTGCT-3’ (Clathrin Reverse), 5’-GGAGCGAGATCCCTCCAAAAT-3’ (GAPDH Forward) and 5’-GGCTGTTGTCATACTTCTCATGG-3’ (GAPDH Reverse).
Calcium disorder detection
HepG2 cells were cultivated in 24-well plate treated with RFP (50 uM) and/or 4-PBA (1 mM) for 48 h. Then, cells in each group were collected and stained using 5 µmol/L fluo-3/AM (Dojindo Laboratories, Kumamoto, Japan) for 30 min at 37°C in the dark [19]. After rinsed three times with PBS to remove excess fluorescent dye, the results were analyzed using flow cytometer (BD, Franklin Lakes, NJ).
Immunofluorescence assay
1 × 105 HepG2 cells were cultivated on cover glass in 6-well plate overnight and treated by RFP (50 uM) and or 4-PBA (1 mM) for 48 h. Then, cells were washed with PBS, fixed with 4% paraformaldehyde in room temperature, permeated with 0.5% Triton X-100 for 10 min, blocked with bovine serum albumin for 1 h, incubated with anti-MRP2 antibody (#4446, Cell Signaling Technology, MA, USA) overnight at 4°C and incubated with second antibody for 2 h at room temperature protected from light [20]. DAPI was utilized for staining nucleus. Results were observed under fluorescence microscope.
Western blotting
Total proteins in cells and tissues were separated using NP40 lysis buffer (pH = 7.4, 150 mM NaCl, 1 mM EDTA, 20 mM Tris·HCl, 1% NP-40, protease inhibitor and phosphatase inhibitor cocktail). Protein concentrations were measured by Bradford assay. Soluble proteins (30–40 μg) were subjected to SDS-polyacrylamide gel electrophoresis [21]. Then, the protein was transferred onto nitrocellulose membrane. The membrane was blocked with 5% milk for 1 h, incubated with primary antibody overnight at 4°C and incubated with second antibody for 2 h at room temperature. Electrochemiluminescence was carried out to obtain protein band using an imager (Bio-Rad, Philadelphia, PA). GAPDH was used as an internal control. Antibodies against binding immunoglobulin protein (GRP78 BIP), CHOP, p-p38, MRP2 and activating transcription factor 6 (ATF6) were purchased from Cell Signaling Technology (MA, USA). Antibodies against Cleaved-Caspase 12, Cleaved-Caspase 3 and PERK were purchased from Santa Cruz Biotechnology (CA, USA). Antibodies against X-box binding protein 1 (XBP-1), inositol-requiring kinase 1 (IRE-1) and p-JNK were purchased from Millipore (MA, USA).
In vitro ubiquitination assay
In vitro ubiquitination assay was performed to detect the ubiquitination of MRP2 after relevant stimulation [22]. HepG2 cells were seeded into 24-well plate and subjected to relevant treatment. Then, cells were treated by 20 μM MG132 (S1748, Beyotime Biotechnology, Shanghai, China) for 10 h to inhibit degradation of ubiquitinated proteins. Following splitting cells using RIPA Lysis Buffer (Beyotime Biotechnology), the lysate supernatant was harvested for co-immunoprecipitation with antibodies against MRP2 or IgG. After incubated with Protein A/G Agarose, centrifugation and precipitation Protein A/G Agarose, the protein in supernatant was underwent Western blotting.
Cell transfection
Cell transfection was carried out to alter CHOP expression in HepG2 cells [23]. To silence CHOP expression in HepG2 cells, siRNA specifically targeting CHOP (CHOP-siRNA: 5’- UCAUAUUCUGAAUCUCAUCCU-3’) and control siRNA (CHOP-siNC: 5’- CAGUACUUUUGUGUAGUACAA −3’) were synthesized by Beyotime Biotechnology and transfected into cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to manufacturer’s protocol. To overexpress CHOP in HepG2 cells, full-length sequence of CHOP was inserted into pcDNA3.0 plasmid and named as CHOP-plasmid. Empty pcDNA3.0 plasmid was utilized as negative control and named as Control-plasmid. CHOP-plasmid and Control-plasmid were also transfected into cells using Lipofectamine 2000. Transfection efficiencies were tested via Western blotting.
Animal experiments
RFP-induced cholestasis mouse model was established to further explore the effects of 4-PBA. Briefly, 15 Pathogen-free male ICR mice were purchased from Institute of Laboratory Animal Sciences (Beijing, China). All experimental protocols were approved by the Institutional Committee for Animal Care and Use. After housed in pathogen-free environment, all mice were stochastically divided into Control, RFP and 4-PBA+RFP groups with five mice in each group. 442.5 mg/kg/day RFP [24] and/or 240 mg/kg/day 4-PBA [25] were given for mice via gavage for 2 consecutive weeks.
Total bilirubin assay
After experiments, all mice were anesthetized by isoflurane and sacrificed by cervical dislocation. Blood was collected and centrifuged at 10,000 rpm for 10 min to collect serum. The concentration of total bilirubin in serum was measured using Bilirubin assay kit (Abcam, Cambridge, MA) [26] according to manufacturer’s protocol.
Statistical analysis
All experiments were repeated three times. Graphpad prism 5 software was utilized for statistical analysis. Data were presented as means ± standard deviation (SD). Differences between groups were analyzed using one-way ANOVA. P < 0.05 was considered as statistically significant.
Results
4-PBA reduced RFP-induced toxicity and apoptosis of HepG2 cells
Firstly, the influences of RFP and/or 4-PBA treatment on HepG2 cell viability and apoptosis were tested. The results of CCk8 assay in Figure 1(a) indicated that 50 uM RFP had significant toxicity on HepG2 cells, which notably lowered the viability of HepG2 cells (P < 0.001). 0.1, 0.5 or 1 mM 4-PBA single treatment had no significant effects on HepG2 cell viability, which was able to obviously alleviate RFP-induced decrease of HepG2 cell viability in a dose-dependent manner (P < 0.05 or P < 0.01). 1 mM 4-PBA treatment was chosen for subsequent experiments. Figure 1(b) showed that RFP gradually elevated HepG2 cells apoptosis (P < 0.001), which was reduced by 1 mM 4-PBA treatment (P < 0.001). These outcomes represented that 4-PBA could reduce RFP-induced toxicity and apoptosis of HepG2 cells.
Figure 1.
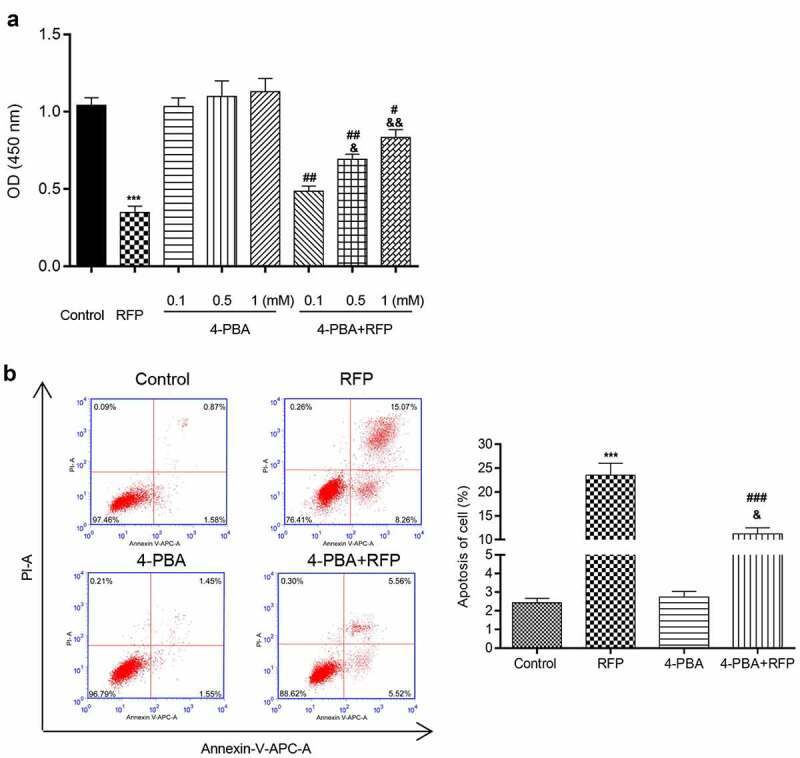
4-PBA reduced RFP-induced toxicity and apoptosis of HepG2 cells.
(a) HepG2 cells were treated by RFP (50 uM) and/or 4-PBA (0.1 mM, 0.5 mM, 1 mM) for 48 h. Cell viability was detected via CCK-8 assay. (b) HepG2 cells were treated by RFP (50 uM) and/or 4-PBA (1 mM) for 48 h. Cell apoptosis was checked by PI/Annexin V double staining. ***P < 0.001 vs. control group; #P< 0.05, ##P< 0.01, ###P< 0.001 vs. RFP group; &P< 0.05, &&P< 0.01 vs. 4-PBA group.
4-PBA attenuated the suppression of MRP2 by RFP
To further investigate whether MRP2 participated in the protective effect of 4-PBA on RFP-resulted HepG2 cell damage, the expression of MRP2 was measured after RFP and/or 4-PBA treatment. qRT-PCR assay showed that RFP and/or 4-PBA treatment did not affect the mRNA level of MRP2 in HepG2 cells (Figure 2(a)). However, the results of Western blotting displayed that RFP treatment decreased the protein level of MRP2 in HepG2 (Figure 2(b), p < 0.001), while 4-PBA partially abolished the effects of RPF on MRP2 protein level (P < 0.05). Immunofluorescent staining showed similar results, which indicated that 4-PBA significantly reversed the RPF-caused decrease of MRP2 expression in HepG2 cells (Figure 2(c), p < 0.001). Moreover, considering that ubiquitination modification is one of the main reasons for intracellular protein degradation, we checked the ubiquitination of MRP2 in HepG2 cells after RFP and/or 4-PBA treatment. The results showed that RFP promoted the ubiquitination of MRP2 in HepG2 cells, while 4-PBA decreased the RFP-resulted ubiquitination of MRP2 (Figure 2(d)). Glycoprotein 78 (GP78) is a key E3 ubiquitin ligase and plays pivotal roles in ER-associated degradation. We discovered that RFP increased the protein expression of GP78 in HepG2 cells (Figure 2(e), p < 0.001). Compared to RFP group, the GP78 expression in 4-PBA+RFP group was decreased (P < 0.001). Collectively, these outcomes suggested that RFP triggered the ubiquitination degradation of MRP2 might be via raising GP78 expression, which was attenuated by administration of 4-PBA.
Figure 2.
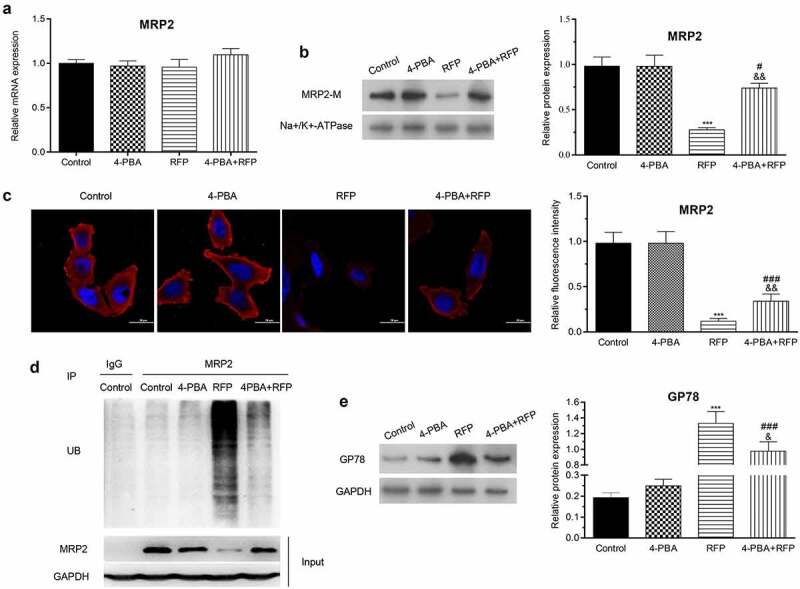
4-PBA attenuated the suppression of MRP2 by RFP.
HepG2 cells were treated by RFP (50 uM) and/or 4-PBA (1 mM) for 48 h. (a and b) mRNA and protein expression of MRP2 were detected through qRT-PCR assay and Western blotting, respectively. (c) Immunofluorescence assay was carried out to further test MRP2 expression. (d) The ubiquitination of MRP2 was checked by in vitro ubiquitination assay. (e) The protein level of GP78 was evaluated via Western blotting. ***P< 0.001 vs. control group; #P< 0.05, ###P< 0.001 vs. RFP group; &P< 0.05, &&P< 0.01 vs. 4-PBA group.
4-PBA improved RFP-induced intracellular calcium disorder
Previous literature reported that intracellular calcium disorder contributed to the decrease of MRP2 in liver cells [9]. Following RFP and/or 4-PBA treatment, the intracellular calcium concentration was monitored by Fluo-3/AM staining. Results in Figure 3(a) showed that RFP significantly increased intracellular calcium concentration (indicated by the increase of fluorescence emission, P < 0.001), while 4-PBA partially abolished the increase of intracellular calcium concentration caused by FRP in HepG2 cells (P < 0.001). These outcomes represented that 4-PBA could reduce RFP-induced intracellular calcium disorder.
Figure 3.
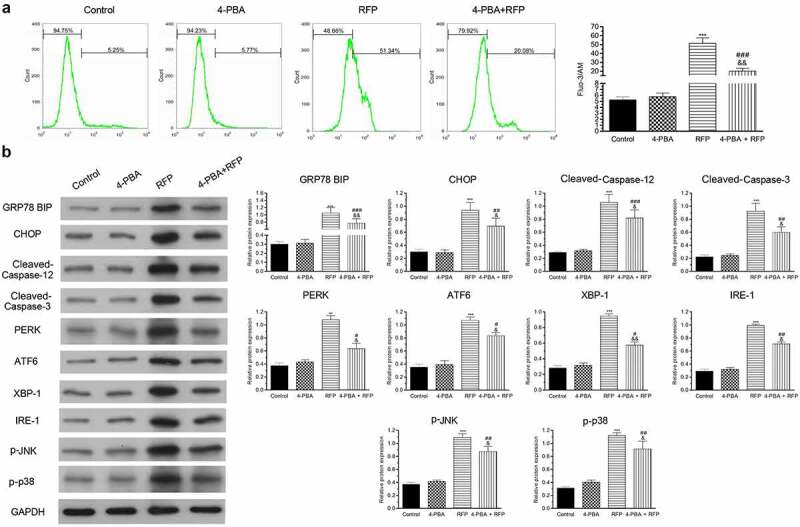
4-PBA improved RFP-induced intracellular calcium disorder and ER stress response.
HepG2 cells were treated by RFP (50 uM) and/or 4-PBA (1 mM) for 48 h. (a) The intracellular calcium concentration was tested via fluo-3/AM staining. (b) The protein levels of GRP78 BIP, CHOP, Cleaved-Caspase-12, Cleaved-Caspase-3, PERK, ATF6, XBP-1, IRE-1, p-JNK and p-p38 in cells were evaluated via Western blotting. **P< 0.01, ***P< 0.001 vs. control group; #P< 0.05, ##P< 0.01, ###P< 0.001 vs. RFP group; &P< 0.05, &&P< 0.01 vs. 4-PBA group.
4-PBA repressed RFP-mediated ER stress response
ER is the main intracellular Ca2+ reservoir. Intracellular calcium disorder can lead to ER stress. We next checked the ER stress sensors including inositol-requiring enzyme-1α (IRE-1), ATF6) protein kinase RNA-like ER kinase (PERK), ER stress-initiated pro-apoptotic factors (Cleaved-Caspase12, Cleaved-Caspase 3), CHOP, ER stress-responsive marker (GRP78 BIP, XBP-1), p-JNK and p-p38 protein level in HepG2 cells after RFP and 4-PBA treatment. Our results suggested that RFP increased the levels of above indicated proteins (Figure 3(b), p < 0.001), but 4-PBA relieved the influence of RFP (P < 0.05, P < 0.01 or P < 0.001). These outcomes represented that RPF induced ER stress response, which was repressed by 4-PBA administration.
4-PBA alleviated RFP-caused increase of AP2 and clathrin
Clathrin and AP2 complex play important role in renal tubular calcium reabsorption and endosomal-lysosomal degradation pathway [27]. Subsequent experiments were carried out to check whether RFP and/or 4-PBA treatment affect clathrin and AP2 expression in HepG2 cells. qRT-PCR and WB results indicated that RFP enhanced AP2 and clathrin at both mRNA and protein levels (Figure 4(a,b), p < 0.01 or P < 0.001). 4-PBA treatment alleviated the increases of AP2 and clathrin levels caused by RFP (P < 0.05, P < 0.01 or P < 0.001). Besides, to explore whether CHOP took part in the effects of RFP and/or 4-PBA on AP and clathrin in HepG2 cells, CHOP was silenced or overexpressed in hepG2 cells. Results illustrated that CHOP-siRNA transfection notably reduced the CHOP mRNA level in HepG2 (Figure 4(c), p < 0.001), which remarkably suppressed RFP-resulted increases of AP2 and clathrin expressions (P < 0.01), as well as the effect of 4-PBA (P < 0.05 or P < 0.001). CHOP-plasmid transfection had contrary influence (Figure 4(d), p < 0.001), which increased the RFP-induced increases of AP2 and clathrin expressions (P < 0.05). 4-PBA treatment also abolished the effects of RFP on AP2 and clathrin expressions in CHOP-plasmid transfected HepG2 cells (Figure 4(d), p < 0.05 or P < 0.01). These outcomes represented that RFP induced ER stress response via up-regulation of AP2 and clathrin which was attenuated by 4-PBA.
Figure 4.
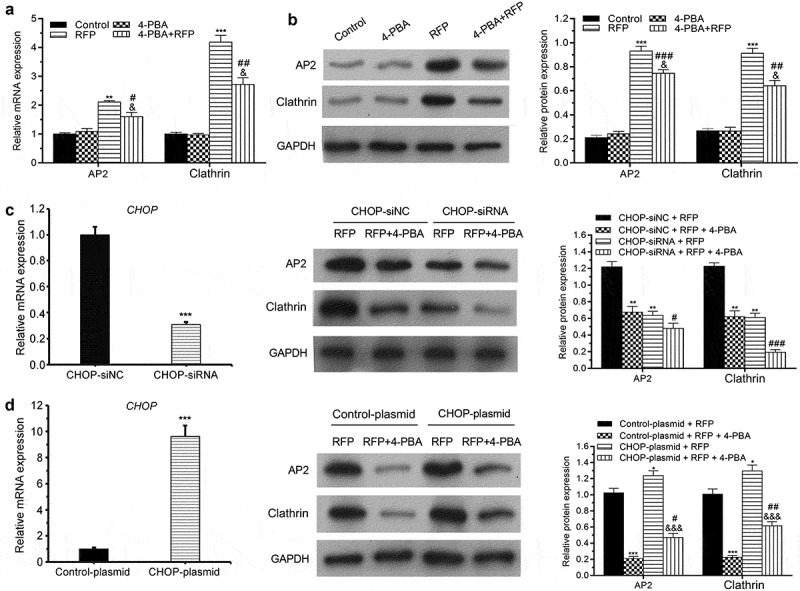
4-PBA alleviated RFP-caused increase of AP2 and clathrin.
(a and b) HepG2 cells were treated by RFP (50 uM) and/or 4-PBA (1 mM) for 48 h. The mRNA and protein expressions of AP2 and clathrin in cells were tested via qRT-PCR and Western blotting. (c and d) Following CHOP-siRNA (or CHOP-plasmid) transfection and/or RFP (50 uM) or 4-PBA (1 mM) treatment for 48 h, the CHOP mRNA expression was tested via qRT-PCR assay and the protein expressions of AP2 and clathrin in HepG2 cells were tested via Western blotting. *P< 0.05, **P< 0.01, ***P< 0.001 vs. control group; #P< 0.05, ##P< 0.01, ###P< 0.001 vs. RFP group; &P< 0.05, &&&P< 0.001 vs. 4-PBA group.
4-PBA protected mice against RFP-induced cholestatic liver injury
Finally, RFP-induced cholestasis mouse model was established to further confirm the beneficial effects of 4-PBA on RFP-caused liver damage. Serum bilirubin level was detected by ELISA. The results showed that RFP elevated bilirubin secretion (Figure 5(a), p < 0.001), while 4-PBA alleviated the increase of bilirubin level in serum (P < 0.05). The protein expression and ubiquitination of MRP2 in the liver tissue were also detected. Results showed that RFP treatment decreased MRP2 protein level and promoted the ubiquitination of MRP2 in liver tissue (Figure 5(b,c), p < 0.001 on MRP2 protein level), while 4-PBA attenuated the effects of RFP on MRP2 (P < 0.01 on MRP2 protein level). Next, the impact of different treatments on the protein level of GRP78 BIP, CHOP, Cleaved-Caspase 12, Cleaved-Caspase 3, PERK, ATF6, XBP-1, IRE-1, p-JNK and p-p38 in liver tissues were checked. Results in Figure 5(d) illustrated that RFP enhanced the levels of abovementioned proteins in liver cells (P < 0.05, P < 0.01 or P < 0.001), which implied that RFP induced ER stress response in liver. However, this effect was attenuated by 4-PBA (P < 0.05). These outcomes represented that 4-PBA could alleviate RFP-induced cholestatic liver injury via regulating bilirubin secretion, MRP expression and ER stress response.
Figure 5.
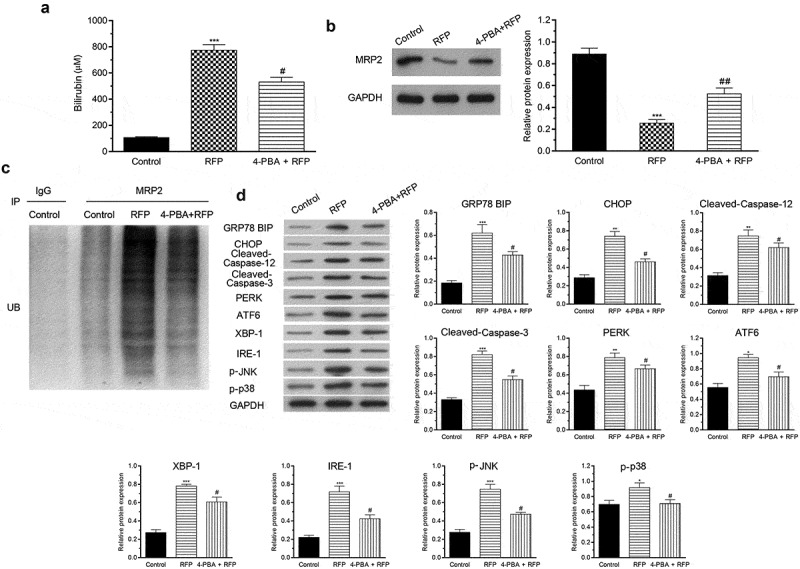
4-PBA protected mice against RFP-induced cholestatic liver injury.
RFP-induced cholestasis mouse model was established and treated by 4-PBA. (a) Serum bilirubin concentration was tested vi ELISA. (b) The protein expression of MRP2 in liver tissues was measured via Western blotting. (c) The ubiquitination of MRP2 in liver tissues was tested via ubiquitination assay. (d) The protein levels of GRP78 BIP, CHOP, Cleaved-Caspase-12, Cleaved-Caspase-3, PERK, ATF6, XBP-1, IRE-1, p-JNK and p-p38 in liver tissues were evaluated via Western blotting. *P< 0.05, **P< 0.01, ***P< 0.001 vs. control group; #P< 0.05, ##P< 0.01 vs. RFP group.
Discussion
Anti-TB drugs are the main cause of DILI, accounting for 58% of all cases of DILI and 5–22% of drug-induced acute liver failure cases [28]. RFP is the most widely used anti-TB drug recommended by the World Health Organization, but it has well-known hepatotoxicity that remains to be an important clinical problem [29]. Previous study discovered that the cholestasis caused by RFP was associated with ER stress and ubiquitination of MRP2 [30]. As a low molecular weight fatty acid, 4-PBA was found to stabilize protein structure and inhibited ER stress through reducing the accumulation of misfolded proteins in the ER [31,32]. Earlier literature has demonstrated that 4-PBA treatment could attenuate RFP-caused liver cell injury via PERK/ATF4/CHOP signal pathway [16]. However, the internal regulatory mechanism is still unknown.
In this study, we observed that 4-PBA ameliorated RFP-induced HepG2 cell viability reduction and apoptosis. Mutation of MRP2 has been demonstrated to cause Dubin-Johnson syndrome, which is characterized by hyperbilirubinemia and elevated bile acid level [33]. Previous study reported that RFP decreased the membrane localization of MRP2 via clathrin-dependent endocytosis and ubiquitin–proteasome degradation induced by oxidative stress in HepG2 cells [9]. In consistent with previous research, herein, we further revealed that RPF lowered MRP2 protein levels but had no significant effect on MRP2 mRNA expression in HepG2 cells. Moreover, we discovered that 4-PBA partially restored the protein expression and membrane localization of MRP2 but without significant change of the MRP2 mRNA level in RFP-stimulated HepG2 cells. Besides, pervious study on cholestatic patients and rat models of cholestasis has shown that endocytic retrieval of MRP2 and decreased MRP2 protein expression without any significant changes in MRP2 mRNA levels [34]. These results suggested that 4-PBA improved RFP-resulted liver cell injury at least in part through reducing endocytic retrieval of MRP2 and raising MRP2 protein level.
Ubiquitination functions as an internalization signal to conjugate targeted protein with ubiquitin ligases for identification and degradation by a proteasome. Among the ubiquitin ligases, GP78, as a E3 ligase, plays pivotal roles in ER-associated ubiquitination degradation [35]. Considering that ER stress also participated in the RFP-induced hepatotoxicity [10], in this study, we detected the GP78 protein level after RFP and 4-PBA treatment. Result discovered that RFP notably upregulated the GP78 protein level in HepG2 cells, while 4-PBA partially reversed the increase of GP78 protein level caused by RFP, which implied that 4-PBA increased MRP2 protein level in HepG2 cells might be by inhibiting GP78-associated ubiquitination degradation.
The acute oxidative stress has been found to elevate Ca2+ and promote endocytic internalization of MRP2, ultimately results in cholestasis [36,37]. Moreover, Ca2+ disorder also can cause ER stress [38]. In the current research, we discovered that RFP increased Ca2+ levels in HepG2 cells, while 4-PBA clearly attenuated the increase of Ca2+ levels. ER stress is the result of the accumulation of unfolded or misfolded proteins in ER [39]. The accumulation of unfolded proteins in ER may directly induce the unfolded protein response through activating three stress sensor proteins, IRE-1, PERK and ATF6 [40,41]. They can combine with CHOP, BIP and XBP-1 to modulate numerous signal pathway, such as MAPK pathway, to regulate redox metabolism, amino acid homeostasis and apoptosis [40]. As mentioned above, 4-PBA can suppress ER stress through helping protein folding and trafficking in the ER [15]. Therefore, we investigated the effects of 4-PBA on the expression of ER stress markers in HepG2 cells after RFP pretreatment. Results indicated that RFP upregulated BIP, XBP-1, CHOP, IRE-1, PERK and ATF6 protein levels, which was accompanied with the increases of p-JNK, p-p38, Cleaved-Caspase 12 and Cleaved-Caspase 3 expressions, suggesting that ER-stress was activated by RFP. 4-PBA reduced the expression levels of these ER stress markers, as well as the p-JNK, p-p38, Cleaved-Caspase 12 and Cleaved-Caspase 3 expressions, indicating 4-PBA could inhibit ER stress. In summary, these results suggested that the reduction of ER stress resulted by 4-PBA contributed to the protective effect of 4-PBA on RFP-induced MRP2 ubiquitination degradation.
AP2 and clathrin are the main constituents of endocytic clathrin-coated vesicles, which responsible for the endocytosis of MRP2 in the cholestatic rat liver [42,43]. In this research, RPF enhanced the mRNA and protein levels of AP2 and clathrin in HepG2 cells, while 4-PBA attenuated the enhancements of mRNA and protein levels of AP2 and clathrin. By alter CHOP expression via cell transfection, we discovered that silence of CHOP reduced RFP-induced increases of AP2 and clathrin expressions, as well as the effect of 4-PBA. Overexpression of CHOP had opposite influence. These outcomes suggested that CHOP took part in the modulation of AP2 and clathrin expression in HepG2 cells and indicated that 4-PBA reduced the endocytosis of MRP2 in HepG2 cells might be via suppressing CHOP-mediated AP2 and clathrin expressions.
Finally, RFP-induced cholestasis mouse model was established. We found that 4-PBA reduced the serum bilirubin level in mouse, as well as raised the protein levels of MRP2, ER stress markers, p-JNK, p-p38, Cleaved-Caspase 12 and Cleaved-Caspase 3. These outcomes further verified the beneficial effect of 4-PBA on RFP-caused cholestatic liver injury.
Conclusion
In summary, our findings revealed the protective effect of 4-PBA on RFP-caused cholestatic liver injury. 4-PBA attenuated RFP-induced hepatotoxicity via reducing MRP2 ubiquitination degradation through inhibiting ER stress and clathrin expression. These results suggested that 4-PBA might be a pharmacological therapeutic drug for RFP-induced intrahepatic cholestasis.
Funding Statement
This work was supported by grants from the National Nature Science Foundation of China [No.81470850 and No. 30900678] and the Key Project of Science and Technology of Chongqing [CSTC, 2009BB5159].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Danan G, Teschke R.. RUCAM in drug and herb induced liver injury: the update. International Journal of Molecular Sciences. 2015;17:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Iruzubieta P, Arias-Loste MT, Barbier-Torres L, et al. The need for biomarkers in diagnosis and prognosis of drug-induced liver disease: does metabolomics have any role? Biomed Res Int. 2015;2015. DOI: 10.1155/2015/386186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].David S, Hamilton JP. Drug-induced liver injury. US Gastroenterol Hepatol Rev. 2010;6:73–80. [PMC free article] [PubMed] [Google Scholar]
- [4].Andrade RJ, Chalasani N, Björnsson ES, et al. Drug-induced liver injury. Nat Rev Dis Primers. 2019;5:58. [DOI] [PubMed] [Google Scholar]
- [5].Abera W, Cheneke W, Abebe G. Incidence of antituberculosis-drug-induced hepatotoxicity and associated risk factors among tuberculosis patients in Dawro Zone, South Ethiopia: a cohort study. Int J Mycobacteriol. 2016;5:14–20. [DOI] [PubMed] [Google Scholar]
- [6].Capelle P, Dhumeaux D, Mora M, et al. Effect of rifampicin on liver function in man. Gut. 1972;13:366–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Su Q, Liu Q, Liu J, et al. Study on the associations between liver damage and antituberculosis drug rifampicin and relative metabolic enzyme gene polymorphisms. Bioengineered. 2021;12:11700–11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yew WW, Chang KC, Chan DP. Oxidative stress and first-line antituberculosis drug-induced hepatotoxicity. Antimicrob Agents Chemother. 2018;62:e02637–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Xu B-Y, Tang X-D, Chen J, et al. Rifampicin induces clathrin-dependent endocytosis and ubiquitin–proteasome degradation of MRP2 via oxidative stress-activated PKC-ERK/JNK/p38 and PI3K signaling pathways in HepG2 cells. Acta Pharmacol Sin. 2020;41:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bozaykut P, Sahin A, Karademir B, et al. Endoplasmic reticulum stress related molecular mechanisms in nonalcoholic steatohepatitis. Mech Ageing Dev. 2016;157:17–29. [DOI] [PubMed] [Google Scholar]
- [11].Chen X, Zhang C, Wang H, et al. Altered integrity and decreased expression of hepatocyte tight junctions in rifampicin-induced cholestasis in mice. Toxicol Appl Pharmacol. 2009;240:26–36. [DOI] [PubMed] [Google Scholar]
- [12].Borst P, Zelcer N, van de Wetering K. MRP2 and 3 in health and disease. Cancer Lett. 2006;234:51–61. [DOI] [PubMed] [Google Scholar]
- [13].Hayashi H, Mizuno T, Horikawa R, et al. 4-Phenylbutyrate modulates ubiquitination of hepatocanalicular MRP2 and reduces serum total bilirubin concentration. J Hepatol. 2012;56:1136–1144. [DOI] [PubMed] [Google Scholar]
- [14].Carlisle RE, Brimble E, Werner KE, et al. 4-Phenylbutyrate inhibits tunicamycin-induced acute kidney injury via CHOP/GADD153 repression. PloS One. 2014;9:e84663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kolb P, Ayaub E, Zhou W, et al. The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int J Biochem Cell Biol. 2015;61:45–52. [DOI] [PubMed] [Google Scholar]
- [16].Zhang W, Chen L, Shen Y, et al. Rifampicin-induced injury in L02 cells is alleviated by 4-PBA via inhibition of the PERK-ATF4-CHOP pathway. Toxicol in Vitro. 2016;36:186–196. [DOI] [PubMed] [Google Scholar]
- [17].Ma ZJ, Lu L, Yang JJ, et al. Lariciresinol induces apoptosis in HepG2 cells via mitochondrial-mediated apoptosis pathway. Eur J Pharmacol. 2018;821:1–10. [DOI] [PubMed] [Google Scholar]
- [18].Najafi A, Behnam B, Anani H, et al. Aminoguanidine induced apoptosis in human hepatocarcinoma HepG2 cells. Gene Rep. 2021;25:101329. [Google Scholar]
- [19].Chen W, Deng Y, Zhang J, et al. Uniaxial repetitive mechanical overloading induces influx of extracellular calcium and cytoskeleton disruption in human tenocytes. Cell Tissue Res. 2015;359:577–587. [DOI] [PubMed] [Google Scholar]
- [20].Hao J, Madigan MC, Khatri A, et al. In vitro and in vivo prostate cancer metastasis and chemoresistance can be modulated by expression of either CD44 or CD147. PLoS One. 2012;7:e40716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li R, Yin F, Guo YY, et al. Knockdown of ANRIL aggravates H2O2-induced injury in PC-12 cells by targeting microRNA-125a. Biomed Pharmacothe. 2017;92:952–961. [DOI] [PubMed] [Google Scholar]
- [22].Yu Y, Chen Y, Zheng YJ, et al. LncRNA TUG1 promoted osteogenic differentiation through promoting bFGF ubiquitination. In Vitro Cell Dev Biol Anim. 2020;56:42–48. [DOI] [PubMed] [Google Scholar]
- [23].Zhao Y, Tian B, Yong W, et al. Kaempferol sensitizes human ovarian cancer cells-OVCAR-3 and SKOV-3 to Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL)-induced apoptosis via JNK/ERK-CHOP pathway and up-regulation of death receptors 4 and 5. Medical Science Monitor International Medical Journal of Experimental & Clinical Research. 2017;23:5096–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kim JH, Nam WS, Kim SJ, et al. Mechanism investigation of rifampicin-induced liver injury using comparative toxicoproteomics in mice. Int J Mol Sci. 2017;18:1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Park HJ, Son HJ, Sul OJ, et al. 4-Phenylbutyric acid protects against lipopolysaccharide-induced bone loss by modulating autophagy in osteoclasts. Biochem Pharmacol. 2018;151:9–17. [DOI] [PubMed] [Google Scholar]
- [26].Shams S, Imran N, Afridi S, et al. In vitro differentiation effect of CCL4-induced liver injured mice serum on bone marrow-derived mesenchymal stem cells toward hepatocytes like cells. Cell Tissue Bank. 2021;22:1–7. [DOI] [PubMed] [Google Scholar]
- [27].Nakatsu F, Ohno H. Adaptor protein complexes as the key regulators of protein sorting in the post-Golgi network. Cell Struct Funct. 2003;28:419–429. [DOI] [PubMed] [Google Scholar]
- [28].Devarbhavi H, Singh R, Patil M, et al. Outcome and determinants of mortality in 269 patients with combination anti-tuberculosis drug-induced liver injury. J Gastroenterol Hepatol. 2013;28:161–167. [DOI] [PubMed] [Google Scholar]
- [29].Huang JH, Zhang C, Zhang DG, et al. Rifampicin-induced hepatic lipid accumulation: association with up-regulation of peroxisome proliferator-activated receptor γ in mouse liver. PLoS One. 2016;11:e0165787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Guo YX, Xu XF, Zhang QZ, et al. The inhibition of hepatic bile acids transporters Ntcp and Bsep is involved in the pathogenesis of isoniazid/rifampicin-induced hepatotoxicity. Toxicol Mech Methods. 2015;25:382–387. [DOI] [PubMed] [Google Scholar]
- [31].Humeres C, Montenegro J, Varela M, et al. 4-Phenylbutyric acid prevent cytotoxicity induced by thapsigargin in rat cardiac fibroblast. Toxicol in Vitro. 2014;28:1443–1448. [DOI] [PubMed] [Google Scholar]
- [32].Ayala P, Montenegro J, Vivar R, et al. Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp Mol Pathol. 2012;92:97–104. [DOI] [PubMed] [Google Scholar]
- [33].Keitel V, Vogt C, Häussinger D, et al. Combined mutations of canalicular transporter proteins cause severe intrahepatic cholestasis of pregnancy. Gastroenterology. 2006;131:624–629. [DOI] [PubMed] [Google Scholar]
- [34].Paulusma CC, Kothe MJ, Bakker CT, et al. Zonal down-regulation and redistribution of the multidrug resistance protein 2 during bile duct ligation in rat liver. Hepatology. 2000;31:684–693. [DOI] [PubMed] [Google Scholar]
- [35].Ying Z, Wang H, Fan H, et al. Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Hum Mol Genet. 2009;18:4268–4281. [DOI] [PubMed] [Google Scholar]
- [36].Basiglio CL, Toledo FD, Boaglio AC, et al. Physiological concentrations of unconjugated bilirubin prevent oxidative stress-induced hepatocanalicular dysfunction and cholestasis. Arch Toxicol. 2014;88:501–514. [DOI] [PubMed] [Google Scholar]
- [37].Sekine S, Ito K, Horie T. Oxidative stress and Mrp2 internalization. Free Radic Biol Med. 2006;40:2166–2174. [DOI] [PubMed] [Google Scholar]
- [38].Krebs J, Agellon LB, Michalak M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: an integrated view of calcium signaling. Biochem Biophys Res Commun. 2015;460:114–121. [DOI] [PubMed] [Google Scholar]
- [39].Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lindholm D, Korhonen L, Eriksson O, et al. Recent insights into the role of unfolded protein response in ER stress in health and disease. Front Cell Dev Biol. 2017;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dai L, Jie S, Bi S, et al. Angiopoietin-2 silence alleviates lipopolysaccharide-induced inflammation, barrier dysfunction and endoplasmic reticulum stress of intestinal epithelial cells by blocking Notch signaling pathway. Bioengineered. 2021;12:8116–8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Miszczuk GS, Barosso IR, Larocca MC, et al. Mechanisms of canalicular transporter endocytosis in the cholestatic rat liver. Biochimica et Biophysica Acta Mol Basis Dis. 2018;1864:1072–1085. [DOI] [PubMed] [Google Scholar]
- [43].Shin J, Nile A, Oh JW. Role of adaptin protein complexes in intracellular trafficking and their impact on diseases. Bioengineered. 2021;12:8259–8278. [DOI] [PMC free article] [PubMed] [Google Scholar]