

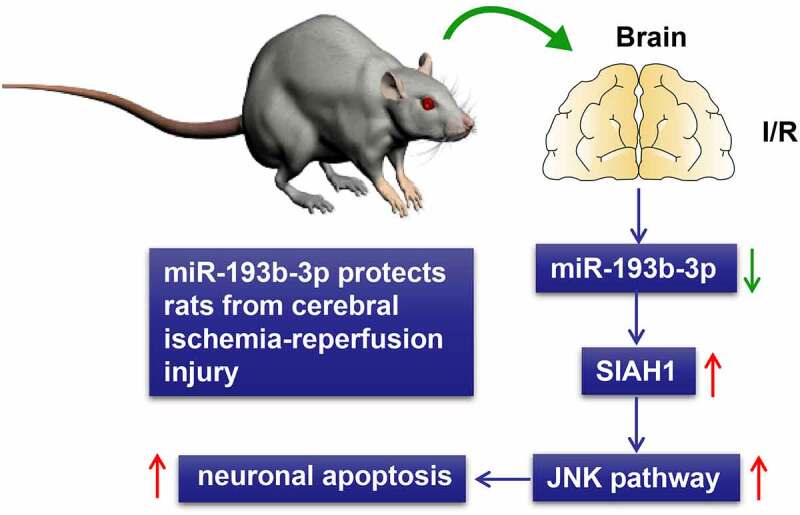
ABSTRACT
Ischemic stroke is one of the major causes of death and disability among adults. This study sought to explore the mechanism of microRNA (miR)-193b-3p in rats with cerebral ischemia-reperfusion (I/R) injury. The cerebral I/R injury models of rats were established using the suture-occluded method. The pathological changes were observed, and oxidative stress (OS) and mitochondrial function indexes in rat brain tissue were examined. The levels of miR-193b-3p and seven in absentia homolog 1 (SIAH1) were detected. miR-193b-3p agomir or antagomir was injected into the lateral ventricle of I/R rats to overexpress or inhibit miR-193b-3p expression. The targeting relationship between miR-193b-3p and SIAH1 was verified. The effect of SIAH1 overexpression on brain injury in I/R rats was investigated by injecting the lentivirus vector into the lateral ventricle. The phosphorylation level of Jun N-terminal kinase (JNK) was identified. miR-193b-3p was lowly expressed in I/R rats. Overexpression of miR-193b-3p alleviated the pathological damage of I/R rats and limited the OS and mitochondrial damage. miR-193b-3p targeted SIAH1. Overexpression of SIAH1 partially reversed the protection of miR-193b-3p overexpression against cerebral I/R injury. p-JNK was up-regulated in I/R rats and overexpression of miR-193b-3p inhibited p-JNK. Overall, overexpression of miR-193b-3p targeted SIAH1 to inhibit the activation of the JNK pathway and protect rats against cerebral I/R injury.
KEYWORDS: Cerebral ischemia-reperfusion injury, miR-193b-3p, SIAH1, oxidative stress, mitochondrial damage, JNK pathway
Graphical Abstract
Introduction
Ischemic stroke is considered as the leading cause of death and disability and is characterized by high rates of morbidity, recurrence, disability, and mortality [1,2]. Ischemic stroke is induced by the reduction of blood flow to the brain due to the blockage of blood vessels in the brain, which leads to oxidative stress (OS) of neurons, energy metabolism failure, mitochondrial apoptosis, and cell apoptosis [3,4]. Many studies have noted the essential role of OS and neuronal mitochondrial dysfunction in the pathogenesis of ischemic stroke [1,5]. Reperfusion therapy is critical to the recovery of ischemic tissue [6]. However, studies have indicated that reperfusion of ischemic areas aggravates brain tissue damage, leading to more serious brain dysfunction, known as cerebral ischemia-reperfusion (I/R) injury [7,8]. Meanwhile, reperfusion exacerbates neuronal damage, including reactive oxygen species (ROS) bursts and mitochondrial dysfunction, which are vital in pathophysiological processes [9]. To date, the mechanism of brain I/R injury has not been thoroughly elucidated [10]. Consequently, it is of paramount significance to identify the regulatory mechanism of OS and mitochondrial damage of neurons during cerebral I/R injury for the pathogenesis and treatment of ischemic stroke.
MicroRNAs (miRNAs) are endogenous single-stranded RNA fragments, usually about 20–22 nucleotides in length, which negatively regulate the protein expression of target genes by binding to complementary sequences of the 3ʹUTR of the target mRNA [11]. Accumulating evidence in previous studies has found the close correlation of numerous miRNAs with cerebral stroke. For example, tretenolic acid B alleviates cerebral I/R injury by regulating miR-10a and the PI3K/Akt/mTOR pathway [12], and inhibition of miR-125b relieves cerebral I/R injury by targeted regulation of CK2α/NADPH oxidase signal transduction [13]. miR-484 attenuates neuronal apoptosis induced by cerebral I/R injury in mice by targeting apoptosis promoter BCL2L13 [7]. Recent studies have uncovered that miR-193b-3p can alleviate the focal cerebral I/R injury in rats by downregulating 5-lipoxygenase [14]. Meanwhile, miR-193b-3p was identified to lower the neuroinflammation of early brain injury in rats after subarachnoid hemorrhage [15]. It is noteworthy that miR-193b-3p may be a potential new target for the treatment of cerebral I/R injury, and it is worth further considering its specific mechanism.
On a separate note, seven in absentia homolog 1 (SIAH1) protein plays a key role in apoptosis and tumorigenesis [16]. Studies have shown that SIAH1 is involved in the regulatory process of OS and mitochondrial damage [17–19]. Meanwhile, studies have shown that SIAH1 leads to the death of neurons and other types of cells by activating the JNK signaling pathway [20].
So far, there is no research on the regulatory effect of miR-193b-3p on cerebral I/R injury via the SIAH1/JNK axis. Accordingly, we speculated that miR-193b-3p could protect against cerebral I/R injury in rats by targeting SIAH1 to inhibit the activation of the JNK pathway. Therefore, this study aims to explore the protective effect and mechanism of miR-193b-3p on I/R rats, so as to provide a new target for I/R treatment and offer a new theoretical reference for the role of miRNAs in neuronal injury.
Materials and Methods
Ethics statement
All animal experimental procedures were approved by the Animal Ethics Committee, and the surgery was performed under general anesthesia to minimize the pain of included experimental animals.
Animal treatment and grouping
A total of 144 male Sprague Dawley rats (10 weeks old, weighing 250 ± 20 g) provided by Changzhou Cavens Experimental Animal Co., Ltd. (Jiangsu, China) were given free drinking water and standard diet and kept in separate cages at 24 ± 1°C under the conditions of 12 hours light/dark cycles. After one week of adaptive feeding, the cerebral I/R injury model of rats was established using the suture-occluded method according to the literature [12]. The procedure of the surgery was as follows: the rats were anesthetized by an intraperitoneal injection of 3% sodium pentobarbital (0.1 mL/100 g). A small incision was made between the right ear and the right eye of each rat, and a Doppler flowmeter (FLPI, MOOR, UK) probe was fixed at the incision to detect the changes in blood flow. Then, another small incision was made in the carotid artery to separate the carotid artery, and a special nylon suture for the mouse (Cinontech Co., Ltd., Beijing, China) was inserted, leading to middle artery occlusion and cerebral ischemia in the rats. After the sutures were fixed, the wound was sutured. The suture was pulled out 2 hours later to restore blood flow. Then follow-up experiments were conducted 24 hours after reperfusion. The entire surgery was performed under sterile conditions. For the sham group (N = 18), no suture was inserted, and other operations were the same as those of the I/R group.
Twenty-four hours before I/R surgery, 144 rats were evenly assigned as follows: sham group, I/R group, I/R + miR-193b-3p group, I/R + miR-negative control (NC) group, I/R + ant-miR-193b-3p group, I/R + ant-miR-NC group, I/R + miR-193b-3p + overexpression (oe)-SIAH1 group, I/R + miR-193b-3p + oe-NC group. Rats in the sham group and I/R group were injected with sterile enzyme-free double distilled water (ddH2O) (5 μL) through the lateral ventricle; rats in the I/R + miR-193b-3p group and I/R + miR-NC group were injected with miR-193b-3p agomir (5 μL) or its control (5 μL) through the lateral ventricle; rats in the I/R + ant-miR-193b-3p group and I/R + ant-miR-NC group were injected with miR-193b-3 antagomir (5 μL) or its control (5 μL) through the lateral ventricle; rats in the I/R + miR-193b-3p + oe-SIAH1 group and I/R + miR-193b-3p + oe-NC group were injected with miR-193b-3p agomir (5 μL) and overexpressed SIAH1 lentivirus (oe-SIAH1) (5 μL) or empty control of lentivirus (5 μL) through the lateral ventricle. miR-193b-3p agomir and miR-193b-3p antagomir and their corresponding controls were designed and synthesized by Guangzhou RiboBio (Guangzhou, China), and the injection was operated following the provided product instructions. SIAH1 overexpressed lentivirus and its control were designed and packaged by Genechem (Shanghai, China) and operated in strict accordance with provided product instructions.
Sample collection
After cerebral ischemia for 2 hours and reperfusion for 24 hours, the rats were anesthetized and euthanized by inhalation of ether. The rat tissues used to make paraffin sections for hematoxylin and eosin (HE) staining and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling assay (TUNEL) staining were first infused with normal saline until the right atrium outflow became clear. Then, the rats were perfused using 4% paraformaldehyde and the intact brain tissue was collected, fixed with 4% paraformaldehyde at 4°C for 24 hours. The rats used for other tests were perfused with saline until the right atrium outflow became clear, and the brain tissue was removed for the detection of relevant indicators. Six rats in each group were subjected to HE staining and TUNEL staining, six rats were subjected to 2, 3, 5-triphenyltetrazolium chloride (TTC) staining, and the remaining 6 rats in each group were homogenized in liquid nitrogen for reverse transcription quantitative polymerase chain reaction (RT-qPCR), the measurement of ROS, superoxide dismutase (SOD), malondialdehyde (MDA), mitochondrial membrane potential (MMP) and adenosine triphosphate (ATP), and the extraction of total protein for Western blot (WB).
HE staining
As previously described [21], the rat brain tissues were made into paraffin sections about 5 µm thick using a paraffin slicer (XY-973, Xinyu Biotechnology, Shanghai, China). After conventional dewaxing and hydration with xylene and anhydrous ethanol, the sections were stained with HE staining solution (DH0006, Leagene Biotech. Co., Ltd., Beijing, China) and stained with hematoxylin for 5 minutes and with eosin for 2 minutes. After dehydration with anhydrous ethanol and xylene, the sections were sealed with neutral gum. The operation was strictly in accordance with the provided instructions. The sealed sections were observed under the microscope (Olympus, Tokyo, Japan) and photographed.
TUNEL staining
As previously mentioned [22], after conventional dewaxing and hydration, the prepared paraffin sections were stained with TUNEL kits (G001-1, YAJI Biotechnology, Shanghai, China). After fully rinsing with phosphate buffer saline (PBS), the sections were re-stained with 4’,6-diamidino-2-phenylindole dihydrochloride and sealed with neutral gum. The operation was conducted strictly following the provided instructions. The sections were observed under a fluorescence microscope (IX51, Olympus, Tokyo, Japan) and photographed. The excitation wavelength was set to 543 nm and the emission wavelength was set to 571 nm. ImagePro Plus 6.0 (Media Cybernetics, Inc., MD, USA) was used to analyze the percentage of TUNEL-positive cells.
TTC staining
As previously described [22], the intact brain tissue of rats was collected and frozen at −20°C for 15–20 minutes until the brain tissue was slightly hard, and then made into coronal sections at 2 mm thickness. The sections were evenly stained in dark conditions for 20 minutes with 2% TTC staining solution (YT8072, YITA Biotechnology Co., Ltd., Beijing, China) and then fixed with 4% paraformaldehyde for 2 hours. After the sections were photographed, the infarct volume was calculated using ImagePro Plus 6.0. Percentage of infarct volume (%) = cerebral infarct volume/total volume × 100%.
RT-qPCR
The total RNA content was extracted from rat brain tissue homogenate using total RNA extraction kits (ZC-0021A, Zhuocai Biotechnology, Shanghai, China). After the RNA concentration was detected using an ultramicro ultraviolet spectrophotometer (SMA1000, Merinton, Beijing, China), the total RNA was transcribed into cDNA using the fluorescent quantitative RNA reverse transcription kit (AB-4366596, Invitrogen, USA). The SYBR® Premix Ex TaqTM II (RR820A, Takara, Japan) was used for RT-qPCR detection on Mx3000P Real-time PCR System (401,423, Agilent, CA, USA). The reaction conditions were as follows: pre-denaturation at 95°C for 10 minutes, and 40 cycles of denaturation at 95°C for 15 seconds, annealing at 63°C for 20 seconds, and extension at 72°C for 30 seconds. With U6 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as internal references, the data were analyzed based on the 2−ΔΔCt method. The primer sequences of each gene are shown in Table 1.
ROS detection
As previously described [23], the rat brain tissue homogenate was collected and centrifuged at 1200 g for 10 minutes. The supernatant was collected and the protein concentration was detected using the bicinchoninic acid assay (BCA) kit (Beyotime Biotechnology, Shanghai, China), and the ROS concentration was examined using the 2’,7’-dichlorofluorescein diacetate (DCFH-DA) test kit (AAT-15204, Qiyuan Biotechnology, Shanghai, China). DCFH-DA with the final concentration of 10 μmol/L was added to the supernatant and incubated at 37°C in the dark for 1 hour. Fluorescence microscopy was used to observe the fluorescence intensity. The excitation wavelength was 485 nm and the emission wavelength was 525 nm. ImagePro Plus 6.0 software was employed to analyze the average fluorescence intensity, and the results were expressed as fluorescence intensity/mg protein.
Detection of SOD and MDA
As previously described [23], the brain tissue homogenate of rats was collected and centrifuged at 10,000 g for 10 min at 4°C. The supernatant was collected, and the protein concentration was determined. The SOD and MDA detection kits (YITA Biotechnology) were used to measure the levels of SOD and MDA in strict accordance with the instructions of the kits.
MMP detection
As mentioned previously [24], the rat brain tissue homogenate was collected, and the mitochondria were separated and purified using the mitochondrial extraction kit (SM0020, Solarbio, Beijing, China) according to the operation instructions. The MMP was measured by JC-1 staining solution (G009-2, Yaji). The JC-1 staining solution with the final concentration of 10 μg/mL was added to the samples and incubated at 37°C for 30 minutes for fluorescence microscope analysis. The excitation wavelength was 488 nm, and the emission wavelength was 590 nm and 525 nm. The green/red fluorescence ratio was analyzed with the help of ImagePro Plus 6.0 software.
ATP detection
According to the previous literature [24], the APT levels in isolated and purified mitochondria were detected with a mitochondrial ATP test kit (KA0806, Abnova Corporation, Taiwan, China) following the operating instructions.
Dual-luciferase report experiment
The targeted binding sites between miR-193b-3p and the 3ʹUTR of SIAH1 were predicted on bioinformatics software TargetScan (www.targetscan.org). As previously mentioned [25], the 3ʹUTR wild type (WT) and mutant (MUT) plasmids of SIAH1 were amplified and cloned into the pmir-Glo vector (HH-LUC-016, HedgehogBio, Shanghai, China). miR-193b-3p mimics and miR-193b-3p NC were co-transfected into HEK-293 T cells (A2001, QuaCell Biotech Co., Ltd., Guangdong, China) with SIAH1-WT and SIAH-1-MUT plasmids, respectively. After culture for 48 hours, the relative activity of dual-luciferase [firefly-luciferase (F-LuC)/Renilla luciferase (R-Luc)] in cells was detected under dark conditions.
WB
As described previously [24], the total protein was extracted from rat brain tissue homogenates using Radio Immunoprecipitation Assay lysate (Beyotime), and the protein concentration was measured using BCA protein detection kits. After the protein was denatured by boiling, the protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride (PVDF) membrane by the wet electrotransfer method. The PVDF membrane was removed, sealed with 5% skim milk and Tris-buffered saline with 0.1% Tween 20 (TBST) and incubated at room temperature for 1 hour. After sealing, the membrane was placed in the incubation box and added with rabbit anti-mouse primary antibodies: anti-SIAH1 (1:1000, ab203198, Abcam), anti-JNK1/2/3 (1:20,000, ab179461, Abcam), anti-p-JNK1/2/3 (1:6000, ab124956, Abcam), ad anti-GAPDH (1:1000, ab9485, Abcam), and incubated overnight at 4°C. The membranes were washed 3 times with TBST, 10 minutes each time. The secondary antibody horseradish peroxidase-labeled goat anti-rabbit IgG-H&L (1:10,000, AB175781, Abcam) was added to the membranes and incubated at room temperature for 1 hour. The membranes were washed 3 times with TBST for 10 minutes each time. After the membranes were developed by enhanced chemiluminescence, the images were obtained. The gray value of the band was analyzed by software ImagePro Plus 6.0, and GAPDH was used as the internal reference.
Statistical analysis
SPSS 22.0 (IBM Corp. Armonk, NY, USA) was used for statistical analyses. Measurement data were expressed as mean ± standard deviation. One-way analysis of variance (ANOVA) was used for data comparison among groups, and Tukey’s multiple test was used for post hoc test. P value was obtained from a bilateral test and a value of P < 0.05 indicated statistical significance.
Results
A study has shown that miR-193b-3p has a protective effect on neuronal injury [14]. Nonetheless, the mechanism of miR-193b-3p has not been fully elucidated. By successfully establishing the rat brain I/R injury model, our study clarified that miR-193b-3p was lowly expressed in the brain tissue of I/R rats. Meanwhile, overexpression of miR-193b-3p can partially alleviate the pathological damage, OS, and mitochondrial damage of brain tissues in I/R rats. Additionally, we found that overexpression of miR-193b-3p can suppress the activation of the JNK pathway to inhibit neuronal injury and protect rats from cerebral I/R injury by targeting SIAH1, which provides new insights into the pathogenesis and therapeutic targets of I/R.
Overexpression of miR-193b-3p alleviated pathological damage of brain tissue in I/R rats
We established the rat brain I/R injury models using the suture-occluded method. Firstly, the pathological changes of brain tissues were assessed by HE, TTC, and TUNEL staining. The results revealed that some neurons in the brain tissues of rats in the I/R group lost their intact cellular structure, and the cells and nuclear cytoplasm were contracted, resulting in obvious infarcted areas in the brain tissues (Figure 1(a)) and increased neuronal apoptosis (Figure 1(b), all P < 0.01). The rat models of brain I/R injury were successfully established. Subsequent RT-qPCR found that miR-193b-3p was lowly expressed in the I/R group (Figure 1(c), p< 0.01).
Figure 1.

Overexpression of miR-193b-3p alleviated pathological damage in I/R rats. The miR-193b-3p agomir or miR-193b-3p antagomir was injected into the lateral ventricle of rats to induce overexpression or downregulation of miR-193b-3p. The rat brain I/R injury model was established using the suture method. After 2 h of ischemia and 24 h of reperfusion, the rat brain tissues were collected for relevant detection. (A) HE staining was used to observe the changes of neuron structure and TTC staining was used to analyze the infarct volume of rat brain; (B) TUNEL staining was used to detect the level of neuronal apoptosis in rats; (C) The level of miR-193b-3p in rat brain tissue was detected by RT-qPCR; Measurement data were expressed as mean ± standard deviation, N = 6, one-way ANOVA was used for data comparison among groups, and Tukey’s multiple test was used for post hoc test. P was obtained from a bilateral test, ** P < 0.01, *** P < 0.001.
To explore the role of miR-193b-3p in cerebral I/R injury in rats, we used miR-193b-3p agomir and miR-193b-3p antagomir to promote or inhibit miR-193b-3p expression in the brain tissue, respectively. RT-qPCR showed that miR-193b-3p was overexpressed or inhibited in I/R rats (Figure 1(c), all P < 0.01). Furthermore, we evaluated the effect of miR-193b-3b overexpression or downregulation on the pathological injury of brain tissue after cerebral I/R injury in rats by HE, TTC, and TUNEL staining. The results showed that after overexpression of miR-193b-3p, the morphologic changes of neurons in brain tissues of I/R rats were few, the cerebral infarction volume was reduced, and neuronal apoptosis was decreased; inhibition of miR-193b-3b aggravated the morphological damage of neurons in rat brain tissue, increased the cerebral infarction volume, and promoted neuronal apoptosis (Figure 1(a/b), all P < 0.01). These results indicate that the overexpression of miR-193b-3p can partially alleviate the pathological damage of cerebral tissue in rats with cerebral I/R injury, while the results of the downregulation of miR-193b-3p are contrary.
Overexpression of miR-193b-3p inhibited OS and mitochondrial damage of neurons in brain tissues of rats with cerebral I/R damage
Previous studies have shown that cerebral I/R injury is closely related to OS and mitochondrial dysfunction [26,27]. To explore whether the alleviation of miR-193b-3p overexpression on the pathological damage of brain tissue in rats with I/R was related to OS and mitochondrial dysfunction, we measured the levels of ROS, SOD, and MDA, which are related to OS, in the brain neurons of rats in different groups. Compared with the sham group, ROS level in the brain tissue of rats in the I/R group was enhanced (Figure 2(a)), SOD level was decreased (Figure 2(b)), and MDA level was increased (Figure 2(c)), while the overexpression of miR-193b-3p partially reversed the OS of neurons in the I/R group (all P < 0.01). Furthermore, we examined the levels of MMP and ATP in neuron mitochondria in different groups. Compared with the sham group, the MMP and ATP levels of neurons in the I/R group were decreased (Figures 2(d) and (e)), while the overexpression of miR-193b-3p partially reversed the mitochondrial damage of neurons (all P < 0.01). Altogether, overexpression of miR-193b-3p can inhibit OS of neurons and mitochondrial damage in brain tissues of rats with brain I/R injury.
Figure 2.
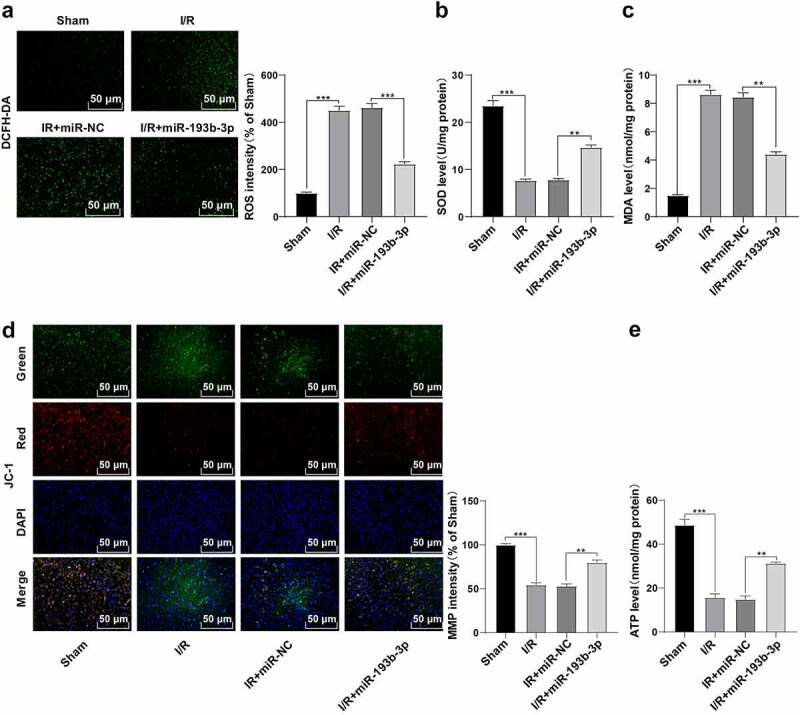
Overexpression of miR-193b-3p inhibits oxidative stress and mitochondrial apoptosis of neurons in the cerebral cortex of rats. The miR-193b-3p agomir was injected into the lateral ventricle of rats to induce the overexpression of miR-193b-3p, and the rat brain I/R injury model was established by suture method. After 2 h of ischemia and 24 h of reperfusion, the rat brain tissues were collected for relevant detection. (A) DCFH-DA fluorescent probe was used to detect ROS levels in neurons of rat brain tissue; (B) SOD level of neurons in rat brain tissue was detected by the kit; (C) MDA level of neurons in rat brain tissue was detected by the kit; (D) JC-1 fluorescence probe was used to detect MMP of neuron mitochondria in rat brain; (E) The level of mitochondrial ATP in rat brain tissue was detected by kits. Measurement data were expressed as mean ± standard deviation, N = 6, one-way ANOVA was used for data comparison among groups, and Tukey’s multiple test was used for the post hoc test. P was obtained from a bilateral test, ** P < 0.01, *** P < 0.001.
miR-193b-3p targeted SIAH1
Through much-valuable literature review, we learned that SIAH1 is associated with OS and mitochondrial damage [28,29]. We predicted the binding sites between miR-193b-3p and the 3ʹUTR of SIAH1 using bioinformatics online software TargetScan (www.targetscan.org), and the binding sequence was GGCCAGUG (Figure 3(a)). To explore whether overexpression of miR-193b-3p onOS and the inhibition of mitochondrial damage was related to SIAH1, we further verified the targeting relationship of miR-193b-3p through dual-luciferase assay. Compared with the SIAH1-WT + NC group, the relative ratio of F-luc/R-luc in the SIAH1-WT + mimic group was reduced (Figure 3(b), p < 0.01). Compared with the SIAH1-MUT + NC group, there was no apparent difference in the fluorescence expression of F-Luc/R-Luc in the SIAH1-MUT + mimic group. The results noted a targeting relationship between miR-193b-3p and SIAH1. We further detected the levels of SIAH1 in brain tissues of rats by RT-qPCR and WB. Compared with the sham group, the SIAH1 levels in brain neurons of rats in the I/R group were up-regulated, and after treatment with miR-193b-3p agomir, SIAH1 levels were down-regulated (Figure 3(c), all P < 0.01). Briefly, miR-193b-3p targeted SIAH1.
Figure 3.

miR-193b-3p targeted SIAH1. (A) The targeted binding sites of miR-193b-3p and SIAH1 were predicted by TargetScan; (B) The targeting relationship between mir193B-3p and SIAH1 was verified by dual-luciferase reporter assay; (C) The mRNA and protein levels of SIAH1 in brain tissue of rats in different groups were detected by RT-qPCR and WB; Measurement data were expressed as mean ± standard deviation, N = 6, one-way ANOVA was used for data comparison among groups, and Tukey’s multiple test was used for the post hoc test. P was obtained from a bilateral test, ** P < 0.01, *** P< 0.001.
Overexpression of SIAH1 antagonized the protective effect of overexpression of miR-193b-3p on cerebral I/R injury
To further explore the role of SIAH1 in cerebral I/R injury in rats, we injected both SIAH1-overexpressed lentivirus or empty lentivirus into rats with brain I/R injury that had been treated with miR-193b-3p agomir. The levels of SIAH1 were detected by RT-qPCR and WB, which revealed that SIAH1 was up-regulated in the I/R + miR-193b-3p + oe-SIAH1 group (Figure 4(a), p < 0.001), indicating that the rats were successfully transfected with the overexpressed SIAH1 lentivirus. We then detected OS indicators such as ROS, SOD, and MDA, and mitochondrial function indicators such as MMP and ATP in neurons of rat brain tissue, and found that overexpression of SIAH1 partially annulled the overexpression of miR-193b-3p-inhibited OS and mitochondrial damage of neurons in the brain tissue of I/R rats (Figure 4(b)-F, all P < 0.001). We further detected the pathological changes of rat brain tissue by HE, TTC, and TUNEL staining (Figure 4(g/h)), and found that the overexpression of SIAH1 partially reversed the effect of overexpression of miR-193b-3p on alleviating brain tissue damage in I/R rats (all P < 0.01). Overall, overexpression of SIAH1 antagonizes the protective effect of miR-193b-3p overexpression on cerebral I/R injury.
Figure 4.

Overexpression of SIAH1 antagonizes the protective effect of overexpression of miR-193b-3p on cerebral I/R injury in rats. The SIAH1 was overexpressed in rats by injecting the SIAH1 lentivirus. (A) The level of SIAH1 was detected by RT-qPCR and WB. (B) DCFH-DA fluorescent probe was used to detect ROS levels in rat brain tissue; (C) SOD level of neurons in rat brain tissue was detected by the kit; (D) MDA level of neurons in rat brain tissue was detected by the kit; (E) JC-1 fluorescence probe was used to detect MMP of neuron mitochondria in rat brain tissue. (F) The kit was used to detect the level of mitochondrial ATP of neurons in rat brain tissue; (G) HE staining and TTC staining were used to detect the neuronal damage and cerebral infarct volume of rat brain tissue; (H) TUNEL staining was used to detect neuronal apoptosis in rats. Measurement data were expressed as mean ± standard deviation, N = 6, one-way ANOVA was used for data comparison among groups, and Tukey’s multiple test was used for the post hoc test. P was obtained from a bilateral test, ** P < 0.01, *** P < 0.001.
miR-193b-3p inhibited the activation of the JNK pathway by targeting SIAH1
A study has shown that SIAH1 mediated neuronal apoptosis by regulating the JNK pathway [20]. We speculated that SIAH1 may regulate the JNK pathway to reduce neuronal apoptosis in rats with cerebral I/R injury. Herein, we detected the levels of JNK and p-JNK in rat brain tissue by WB. The results discovered that the p-JNK/JNK levels in the I/R group were up-regulated compared with that in the sham group, while the overexpression of miR-193b-3p decreased the p-JNK/JNK levels in the IR group. In addition, after the overexpression of miR-193b-3p and the overexpression of SIAH1, the level of p-JNK/JNK was increased again (Figure 5(a/b), all P < 0.05). Conjointly, miR-193b-3p may inhibit the activation of the JNK pathway by targeting SIAH1, thus alleviating OS, mitochondrial damage, and neuronal apoptosis in brain tissues of rats with brain I/R injury.
Figure 5.
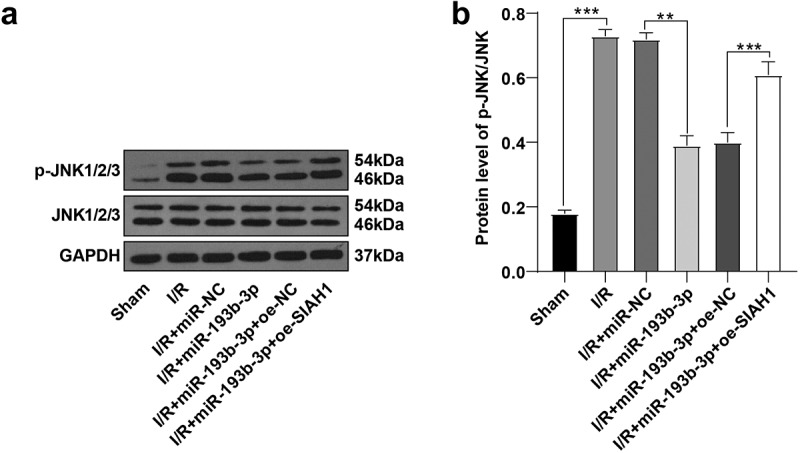
miR-193b-3p inhibits the activation of the JNK pathway by targeting SIAH1. (A-B): The phosphorylation level of JNK1/2/3 in the brain tissues of rats in different groups was detected by WB. Measurement data were expressed as mean /2/3 in the brain tissue. N = 6, one-way ANOVA was used for data comparison among groups, and Tukey’s multiple test was used for the post hoc test. P was obtained from a bilateral test, ** P < 0.01, *** P < 0.001.
Discussion
Ischemic stroke remains one of the leading causes of death and disability worldwide [3]. In recent years, thrombolytic therapy and mechanical thrombectomy have been regarded the main methods for the clinical treatment of ischemic stroke, aiming to open blood vessels and achieve cerebral I/R [8]. However, after blood flow from cerebrovascular to the ischemic area was restored, brain tissue damage was aggravated, and even irreversible damage appeared [30]. miR-193b-3p has a potential neuroprotective effect on focal brain injury induced by I/R [14]. In this study, we elucidated the protection of miR-193b-3p against cerebral I/R through the SIAH1/JNK axis.
I/R causes a series of physiological and pathological changes, including inflammation, OS, cerebral infarct, and neuronal apoptosis [31,32]. We established the rat cerebral I/R injury models using the suture-occluded method. Subsequently, we found that miR-193b-3p was poorly expressed in I/R rats. Likewise, the expression of miR-193b-3p in brain tissues of mice with subarachnoid hemorrhage was lower than that of healthy mice [15]. Afterward, we explored the specific role of miR-193b-3p in cerebral I/R injury by injecting miR-193b-3p agomir or miR-193b-3p antagomir into rats. The histological stainings observed that some neurons in the brain tissue of I/R rats lost the complete cell structure, with contracted nuclear, obvious infarction areas, and increased neuronal apoptosis. After overexpression of miR-193b-3p, the morphological changes of neurons were improved, cerebral infarction volume was reduced, and neuronal apoptosis was decreased, while the pathological results obtained by inhibiting miR-193b-3b using the miR-193b-3p antagomir were contrary. Much in accordance with our discoveries, prior studies have shown that miR-193b-3p can reduce neurobehavioral injury, brain edema, and neurodegeneration induced by subarachnoid hemorrhage, and reduce inflammatory cytokines in the rat brain [15]. Besides, down-regulating the expression of miR-449c-5p can upgrade neurological deficits, reduce brain water content, blood–brain barrier permeability, brain tissue damage, and inflammation in ischemic stroke rats [33]. These findings made it plausible to suggest that overexpression of miR-193b-3p can partially reduce the pathological damage in rats with cerebral I/R injury, while downregulation of miR-193b-3p has the opposite effect.
Compelling evidence exists to support the connection between cerebral I/R injury and OS and mitochondrial dysfunction [5,27]. In our present study, ROS levels were increased, SOD activity was decreased, MDA activity was increased, and mitochondrial MMP and ATP levels were reduced in the brain tissues of I/R rats. Overexpression of miR-193b-3p partially reversed the OS and mitochondrial damage of neurons in I/R rats. miR-193b-3p treatment can reduce neurobehavioral damage, neurodegenerative changes caused by OS and mitochondrial dysfunction in the brain of mice [15,34]. Overexpression of miR-7a-5p reduced mitochondrial fragmentation, OS, and autophagy proteins that promote neuronal death [35]. Altogether, overexpression of miR-193b-3p can inhibit OS and mitochondrial damage of neurons in rats with cerebral I/R injury.
Previous studies have shown the correlation of SIAH1 with OS and mitochondrial damage [17,28]. We predicted the binding site between miR-193b-3p and SIAH1 and verified the targeting relationship between miR-193b-3p and SIAH1. Meanwhile, we observed that SIAH1 was elevated in the neurons of I/R rats and down-regulated after overexpression of miR-193b-3p. Similarly, up-regulation of miR-15b-5p alleviates neurodegeneration caused by neuronal apoptosis by targeting SIAH1 [36]. Hence, miR-193b-3p targeted SIAH1. To further explore the role of SIAH1 in cerebral I/R injury, we overexpressed miR-193b-3p, followed by the overexpression of SIAH1, and found that overexpression of SIAH1 promoted the OS and mitochondrial damage of neurons and aggravated brain tissue injury in I/R rats. Consistently, down-regulation of SIAH1 inhibited apoptosis and decreased cerebral infarct size and hippocampal neuronal apoptosis [37]. Overall, these results strongly support that overexpression of SIAH1 antagonizes the protective effect of miR-193b-3p overexpression on cerebral I/R injury in rats.
There is evidence to suggest that SIAH1 can mediate neuronal apoptosis through the JNK pathway [20]. We discovered that p-JNK/JNK level was up-regulated in I/R rats, declined after overexpression of miR-193b-3p, and increased again after overexpression of miR-193b-3p and SIAH1. These results suggest that miR-193b-3p may inhibit the activation of the JNK pathway by inhibiting SIAH1 levels. Inhibition of the JNK pathway has been reported to block the activation of Caspase-3, thereby reducing cerebral I/R injury [38]. We will perform a functional rescue experiment to intervene in the JNK pathway to further confirm its involvement in cerebral I/R injury.
Conclusion
To conclude, this study mainly found that the overexpression of miR-193b-3p targeted SIAH1 to inhibit the activation of the JNK pathway and protected against the cerebral I/R injury in rats. However, the regulation mechanism of miR-193b-3p in cerebral I/R injury was only studied in animals in this study, and the mechanism of miR-193b-3p was not further verified at the cellular level due to the limited research conditions and time. It is warranted to further study the regulatory mechanism of miR-193b-3p from the epigenetic perspective in our future studies and verify its effect in the in vitro experiments and the clinic setting with the participation of other pathways.
Supplementary Material
Funding Statement
This study was supported by grant from SJTU-STAR Interdisciplinary Research Grant on Medicine and Engineering of Shanghai JiaoTong University (Grant No. GY2020YG31).
Availability of Data and Materials
All the data generated or analyzed during this study are included in this published article.
Authors’ contributions
TY, JW contributed to the study concepts, study design, and definition of intellectual content; KG, FW contributed to the literature research; TY contributed to the manuscript preparation; KG contributed to the manuscript editing and review; FW, JF contributed to the experimental studies and data acquisition; TY, JW contributed to the data analysis and statistical analysis. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
Ethics approval
All animal experimental procedures were approved by the Animal Ethics Committee, and the surgery was performed under general anesthesia to minimize the pain of included experimental animals.
References
- [1].Song G, Zhao M, Chen H, et al. The Role of Nanomaterials in Stroke Treatment: targeting Oxidative Stress. Oxid Med Cell Longev 2021. 8857486 10.1155/2021/8857486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yang B, Li Y, Ma Y, et al. Selenium attenuates ischemia/reperfusion injury-induced damage to the blood-brain barrier in hyperglycemia through PI3K/AKT/mTOR pathway-mediated autophagy inhibition. Int J Mol Med. 2021;47(4):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pacifici F, Rovella V, Pastore D, et al. Polyphenols and Ischemic Stroke: insight into One of the Best Strategies for Prevention and Treatment. Nutrients. 2021;14(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Xu H, Wang E, Chen F, et al. Neuroprotective Phytochemicals in Experimental Ischemic Stroke: mechanisms and Potential Clinical Applications. Oxid Med Cell Longev. 2021;2021:6687386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jia J, Jin H, Nan D, et al. New insights into targeting mitochondria in ischemic injury. Apoptosis. 2021;26(3–4):163–183. [DOI] [PubMed] [Google Scholar]
- [6].Fan JL, Nogueira RC, Brassard P, et al. Integrative physiological assessment of cerebral hemodynamics and metabolism in acute ischemic stroke. J Cereb Blood Flow Metab 2021. 271678X211033732 10.1177/0271678X211033732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liu X, Wang X, Zhang L, et al. By targeting apoptosis facilitator BCL2L13, microRNA miR-484 alleviates cerebral ischemia/reperfusion injury-induced neuronal apoptosis in mice. Bioengineered. 2021;12(1):948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhou X, Wang Z, Xu B, et al. Long non-coding RNA NORAD protects against cerebral ischemia/reperfusion injury induced brain damage, cell apoptosis, oxidative stress and inflammation by regulating miR-30a-5p/YWHAG. Bioengineered. 2021;12(2):9174–9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wu M, Gu X, Ma Z.. Mitochondrial Quality Control in Cerebral Ischemia-Reperfusion Injury. Mol Neurobiol. 2021;58(10):5253–5271. [DOI] [PubMed] [Google Scholar]
- [10].You H, Jin Y, Kang J, et al. Mitochondrial serine protease Omi/HtrA2 accentuates brain ischemia/reperfusion injury in rats and oxidative stress injury in vitro by modulating mitochondrial stress proteins CHOP and ClpP and physically interacting with mitochondrial fusion protein OPA1. Bioengineered. 2020;11(1):1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yapijakis C. Regulatory Role of MicroRNAs in Brain Development and Function. Adv Exp Med Biol. 2020;1195:237–247. [DOI] [PubMed] [Google Scholar]
- [12].Wang J, Wang A, He H, et al. Trametenolic acid B protects against cerebral ischemia and reperfusion injury through modulation of microRNA-10a and PI3K/Akt/mTOR signaling pathways. Biomed Pharmacother. 2019;112:108692. [DOI] [PubMed] [Google Scholar]
- [13].Liang Y, Xu J, Wang Y, et al. Inhibition of MiRNA-125b Decreases Cerebral Ischemia/Reperfusion Injury by Targeting CK2alpha/NADPH Oxidase Signaling. Cell Physiol Biochem. 2018;45(5):1818–1826. [DOI] [PubMed] [Google Scholar]
- [14].Chen Z, Yang J, Zhong J, et al. MicroRNA-193b-3p alleviates focal cerebral ischemia and reperfusion-induced injury in rats by inhibiting 5-lipoxygenase expression. Exp Neurol. 2020;327:113223. [DOI] [PubMed] [Google Scholar]
- [15].Lai N, Wu D, Liang T, et al. Systemic exosomal miR-193b-3p delivery attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage in mice. J Neuroinflammation. 2020;17(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xiao Z, Wei Z, Deng D, et al. Downregulation of Siah1 promotes colorectal cancer cell proliferation and migration by regulating AKT and YAP ubiquitylation and proteasome degradation. Cancer Cell Int. 2020;20(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Franck T, Krueger R, Woitalla D, et al. Mutation analysis of the seven in absentia homolog 1 (SIAH1) gene in Parkinson’s disease. J Neural Transm (Vienna). 2006;113(12):1903–1908. [DOI] [PubMed] [Google Scholar]
- [18].Tian X, Gong L, Jin A, et al. E3 ubiquitin ligase siah1 nuclear accumulation is critical for homocysteine-induced impairment of C6 astroglioma cells. Mol Med Rep. 2019;20(3):2227–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yao RQ, Ren C, Xia ZF, et al. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. 2021;17(2):385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xu Z, Sproul A, Wang W, et al. Siah1 interacts with the scaffold protein POSH to promote JNK activation and apoptosis. J Biol Chem. 2006;281(1):303–312. [DOI] [PubMed] [Google Scholar]
- [21].Teng L, Chen W, Yin C, et al. Dexmedetomidine Improves Cerebral Ischemia-Reperfusion Injury in Rats via Extracellular Signal-Regulated Kinase/Cyclic Adenosine Monophosphate Response Element Binding Protein Signaling Pathway. World Neurosurg. 2019;127:e624–e30. [DOI] [PubMed] [Google Scholar]
- [22].Li W, Zhu Q, Xu X, et al. MiR-27a-3p suppresses cerebral ischemia-reperfusion injury by targeting FOXO1. Aging (Albany NY). 2021;13(8):11727–11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cui L, Cao W, Xia Y, et al. Ulinastatin alleviates cerebral ischemia-reperfusion injury in rats by activating the Nrf-2/HO-1 signaling pathway. Ann Transl Med. 2020;8(18):1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang J, Zhang W, Ma B, et al. A novel biscoumarin derivative dephosphorylates ERK and alleviates apoptosis induced by mitochondrial oxidative damage in ischemic stroke mice. Life Sci. 2021;264:118499. [DOI] [PubMed] [Google Scholar]
- [25].Chen X, Li C, Li J, et al. Upregulation of miR-1306-5p decreases cerebral ischemia/reperfusion injury in vitro by targeting BIK. Biosci Biotechnol Biochem. 2019;83(12):2230–2237. [DOI] [PubMed] [Google Scholar]
- [26].Shen L, Gan Q, Yang Y, et al. Mitophagy in Cerebral Ischemia and Ischemia/Reperfusion Injury. Front Aging Neurosci. 2021;13:687246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Su XT, Wang L, Ma SM, et al. Mechanisms of Acupuncture in the Regulation of Oxidative Stress in Treating Ischemic Stroke. Oxid Med Cell Longev. 2020;2020:7875396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gonzalez MC, Romero JM, Ingaramo MC, et al. Enhancement by Gospel protein of GAPDH aggregation induced by nitric oxide donor and its inhibition by NAD+. FEBS Lett. 2016;590(14):2210–2220. [DOI] [PubMed] [Google Scholar]
- [29].Jin Y, Ratnam K, Chuang PY, et al. A systems approach identifies HIPK2 as a key regulator of kidney fibrosis. Nat Med. 2012;18(4):580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Manuel GE, Johnson T, Liu D. Therapeutic angiogenesis of exosomes for ischemic stroke. Int J Physiol Pathophysiol Pharmacol. 2017;9(6):188–191. [PMC free article] [PubMed] [Google Scholar]
- [31].Liang Q, Yang J, and He J, et al. Stigmasterol alleviates cerebral ischemia/reperfusion injury by attenuating inflammation and improving antioxidant defenses in rats. Biosci Rep. 2020;40(4):BSR20192133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang BY, Wang GR, Ning WH, et al. Electroacupuncture Pretreatment Elicits Tolerance to Cerebral Ischemia/Reperfusion through Inhibition of the GluN2B/m-Calpain/p38 MAPK Proapoptotic Pathway. Neural Plast 2020. 8840675 10.1155/2020/8840675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang D, Pan N, Jiang C, et al. LncRNA SNHG8 sponges miR-449c-5p and regulates the SIRT1/FoxO1 pathway to affect microglia activation and blood-brain barrier permeability in ischemic stroke. J Leukoc Biol. 2021. DOI: 10.1002/JLB.1A0421-217RR [DOI] [PubMed] [Google Scholar]
- [34].Rasheed M, Liang J, Wang C, et al. Epigenetic Regulation of Neuroinflammation in Parkinson’s Disease. Int J Mol Sci. 2021;23(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kim T, Mehta SL, and Morris-Blanco KC, et al. The microRNA miR-7a-5p ameliorates ischemic brain damage by repressing alpha-synuclein. Sci Signal. 2018;11(560):eaat4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen Y, Lian YJ, Ma YQ, et al. LncRNA SNHG1 promotes alpha-synuclein aggregation and toxicity by targeting miR-15b-5p to activate SIAH1 in human neuroblastoma SH-SY5Y cells. Neurotoxicology. 2018;68:212–221. [DOI] [PubMed] [Google Scholar]
- [37].Lei Y, Jin X, Sun M, et al. miR-129-5p Ameliorates Ischemic Brain Injury by Binding to SIAH1 and Activating the mTOR Signaling Pathway. J Mol Neurosci. 2021;71(9):1761–1771. [DOI] [PubMed] [Google Scholar]
- [38].Zhao B, Li D, Zhang S, et al. Dexmedetomidine attenuates cerebral ischemia-reperfusion injury in rats by inhibiting the JNK pathway. Ann Palliat Med. 2021;10(6):6768–6778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data generated or analyzed during this study are included in this published article.