

Abstract
Target enrichment (such as Hyb-Seq) is a well-established high throughput sequencing method that has been increasingly used for phylogenomic studies. Unfortunately, current widely used pipelines for analysis of target enrichment data do not have a vigorous procedure to remove paralogs in target enrichment data. In this study, we develop a pipeline we call Putative Paralogs Detection (PPD) to better address putative paralogs from enrichment data. The new pipeline is an add-on to the existing HybPiper pipeline, and the entire pipeline applies criteria in both sequence similarity and heterozygous sites at each locus in the identification of paralogs. Users may adjust the thresholds of sequence identity and heterozygous sites to identify and remove paralogs according to the level of phylogenetic divergence of their group of interest. The new pipeline also removes highly polymorphic sites attributed to errors in sequence assembly and gappy regions in the alignment. We demonstrated the value of the new pipeline using empirical data generated from Hyb-Seq and the Angiosperms353 kit for two woody genera Castanea (Fagaceae, Fagales) and Hamamelis (Hamamelidaceae, Saxifragales). Comparisons of data sets showed that the PPD identified many more putative paralogs than the popular method HybPiper. Comparisons of tree topologies and divergence times showed evident differences between data from HybPiper and data from our new PPD pipeline. We further evaluated the accuracy and error rates of PPD by BLAST mapping of putative paralogous and orthologous sequences to a reference genome sequence of Castanea mollissima. Compared to HybPiper alone, PPD identified substantially more paralogous gene sequences that mapped to multiple regions of the reference genome (31 genes for PPD compared with 4 genes for HybPiper alone). In conjunction with HybPiper, paralogous genes identified by both pipelines can be removed resulting in the construction of more robust orthologous gene data sets for phylogenomic and divergence time analyses. Our study demonstrates the value of Hyb-Seq with data derived from the Angiosperms353 probe set for elucidating species relationships within a genus, and argues for the importance of additional steps to filter paralogous genes and poorly aligned regions (e.g., as occur through assembly errors), such as our new PPD pipeline described in this study. [Angiosperms353; Castanea; divergence time; Hamamelis; Hyb-Seq, paralogs, phylogenomics.]
High throughput sequencing (HTS) technologies, such as those associated with amplicon sequencing, restriction site digestion, target enrichment, and transcriptome sequencing, have empowered systematists and evolutionary biologists to infer phylogeny with genome-wide molecular markers for a better understanding of species relationships and to answer evolutionary questions with new perspectives that were not possible in the past (e.g., Pais et al. 2017. 2018; Dong et al. 2019; Fu et al. 2019; One Thousand Plant Transcriptomes Initiative 2019; Du et al. 2020; Gaynor et al. 2020; Zhou et al. 2020; Thomas et al. 2021; see reviews in Lemmon and Lemmon 2013; Dodsworth et al. 2019). Among these HTS technologies, target enrichment (Hyb-Seq in plants or sequence capture—Weitemier et al. 2014; and ultraconserved elements, UCEs, in animals—Faircloth et al. 2012) is highly promising and increasingly used for phylogenomic studies of lineages across different evolutionary timescales (e.g., Faircloth et al. 2013; McCormack et al. 2013; Leache et al. 2015; Léveillé-Bourret et al. 2018; Gaynor et al. 2020). The target enrichment method produces data from a targeted set of highly conserved genomic regions (and their flanking areas), often protein coding genes, using probes designed from prior knowledge of target sequences, either from the organism of interest, or a closely related species. The method is highly valued for its repeatability between experiments and between labs if the same probes are used (Harvey et al. 2016), and for generating a lasting and amplifiable resource for comparative studies at multiple taxonomic scales. Data from target enrichment have been shown to be suitable to phylogenomic studies of both deep and shallow phylogenetic divergence, depending on the probes used, because the data contain both conserved coding sequences and their flanking variable sequences (Lemmon et al. 2012; Faircloth et al. 2013; McCormack et al. 2013; Leache et al. 2015; Barrow et al. 2018; Léveillé-Bourret et al. 2018; Banker et al. 2020; Gaynor et al. 2020).
The development of the Angiosperms353 kit (Johnson et al. 2019), which captures 353 low copy nuclear genes across angiosperms, has enabled phylogenomic studies across angiosperm lineages from family to genus (e.g., Gaynor et al. 2020 for Diapensaceae; Larridon et al. 2020 for Cyperaceae; Murphy et al. 2020 for Nepenthes in Nepenthaceae; Shee et al. 2020 for Scheffera in Araliaceae). An explosion of phylogenomic studies using the Angiosperms353 probes is expected in the plant systematics community in the coming years. This endeavor will result in combinable data sets for building the “tree of life” of angiosperms through global-scale analysis (Dodsworth et al. 2019; Johnson et al. 2019). However, the universal probe kit has a disadvantage compared to taxon-specific kits in that the 353 target genes may or may not all be single copy across all species on which the kit is used, and probe binding affinity may cause probes to target unintended paralogous sequences (McCartney et al. 2016). In other words, the potential high divergence of some of the 353 target genes among the diverse angiosperm genomes poses a concern on possible prevalence of paralogs in the Hyb-Seq data. It is unknown if current bioinformatic pipelines developed for analyses of target enrichment data can reliably exclude paralogous gene copies in data derived from the Angiosperms353 probe kit.
Orthologs are genes related by descent from a common ancestor (due to a speciation event) and their evolutionary history tracks the phylogeny of species, while paralogs are products of gene duplication events. Theoretically, comparisons of paralogous copies of genes among species compromise phylogenetic inferences because the gene trees do not track speciation events, and hence, do not depict the true species relationships (Altenhoff et al. 2019; Fig. 1a). In the Hyb-Seq data or target enrichment data, in general, the paralogous genes might be “overlumped” by assembly methods which use sequence similarity thresholds to define homology. The overlumping of paralogs leads to inflation of sequence variation at those loci which may or may not affect the inference of species relationships but is expected to result in misestimation of branch lengths (and thus misestimates of divergence times). Therefore, excluding paralogs in phylogenetic studies using this type of data is pivotal, although paralogous gene sequences have value in other areas of comparative genomics (Madlung 2013; Limborg et al. 2016; McKinney et al. 2017). However, in Hyb-Seq data, orthologs and paralogs are often difficult to distinguish due to their high similarity in sequence identity (Altenhoff et al. 2019). All current pipelines for target enrichment data, including HybPiper (Johnson et al. 2016), PHYLUCE (Faircloth 2016), and SECAPR (Andermann et al. 2018), merely consider the sequence similarity in detecting paralogs.
Figure 1.

Concepts of ortholog-paralog and the key steps of workflow of HybPiper for target enrichment data analysis. a) Illustration of orthologs and paralogs. Orthologs are generated by speciation events, while paralogs are generated by gene duplication events. Speciation events are indicated by S1 and S2. b) The workflow of HybPiper, including reads mapping using BLASTx or BWA, de novo assembly of contigs using SPAdes, and paralogs detection.
A sequence similarity-based approach for calling paralogous genes may be sufficient for
phylogenetic studies using custom designed probes based on orthologous sequences encompassing
a closely related study group. However, for studies leveraging probes built from
evolutionarily distant taxa from the focal group of investigation, especially in groups where
gene and genome duplication are thought to be common such as plants, sequence similarity
between contig and target genes alone may not be sufficient for removing all paralogs.
Additional analyses of the sequence data may be needed to remove the potential paralogous
sequences before performing phylogenetic analyses. In this study, we propose supplementary
criteria to sequence similarity for detecting and removing problematic paralogous gene data
from Hyb-Seq by examining heterozygous sites within and among individuals in the aligned
sequences. Low rates of shared heterozygous sites across all samples in a species-level data
set is expected under the assumption that polymorphisms among species are more likely to be
fixed differences between paralogs over deep divergences (Eaton 2014; Eaton and Overcast 2020). Even at
the shallow level of phylogenetic divergence (e.g., population genetics), high shared
heterozygosity across all samples within a locus may also be attributed to paralogs (Hohenlohe et al. 2011; Harvey et al. 2016; McKinney et al. 2017).
Additionally, a high number of heterozygous sites of a locus within an individual may be
considered an indicator of gene duplication events or previously undetected polyploidy (Medina et al. 2019). Therefore, a high level of shared
heterozygosity at a site across individuals and high number of heterozygous sites within a
locus in an individual are both indicative of paralogy of the aligned gene sequences. In
pipelines developed for analyses of target enrichment data, usually an arbitrary cutoff of
sequence identity value between the contigs of a putative Hyb-Seq locus and the reference
target gene is used to determine if the locus contains paralogous sequences in an individual.
Currently, HybPiper (Johnson et al. 2016) uses BWA
(Li and Durbin 2009) or BLASTx (Altenhoff et al. 2019) to classify the raw reads into individual gene locus,
followed by SPAdes (Bankevich et al. 2012) to assemble
the reads in a given individual into contigs (Fig. 1b).
If multiple contigs with a  10
10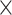 coverage depth in an
individual mapped to the same target gene with
coverage depth in an
individual mapped to the same target gene with 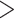 85% sequence identity, this
target gene is marked for presence of paralogs in the individual (Fig. 1b), which can be eliminated or addressed separately by investigators
to determine its orthology to sequences of the same locus of other individuals in subsequent
analyses.
85% sequence identity, this
target gene is marked for presence of paralogs in the individual (Fig. 1b), which can be eliminated or addressed separately by investigators
to determine its orthology to sequences of the same locus of other individuals in subsequent
analyses.
In addition, most pipelines for enrichment data implementing popular assemblers such as SPAdes (Bankevich et al. 2012) and Abyss (Simpson et al. 2009) for sequence assembly can only construct a single consensus sequence for a given locus in each individual (multiploidy) that represents the most frequent base of each site among read variants. This approach loses all information from heterozygous sites for identification of potential paralogs, which may result in data containing phylogenetic noises from paralogous genes that can mislead the inference of species relationship. Although SECAPR and scripts from Kates et al. (2018) can perform allele phasing, all of the presently widely used pipelines for handling target enrichment data do not make use of the information from heterozygous sites to detect paralogous sequences. To make use of heterozygous sites, such as to detect paralogs or for phylogenetic inference, modification of existing pipelines for enrichment data is needed. In this study, we developed a new pipeline that generates degenerate coded sequences (retaining information of heterozygous sites) from Hyb-Seq reads and uses criteria from both sequence similarity and quantity and distribution pattern of heterozygous sites for detection and cleaning of putative paralogs for downstream enrichment data analyses, which we call the Putative Paralogs Detection (PPD) pipeline (available on Github: https://github.com/Bean061/putative_paralog). We developed PPD by modifying HybPiper to code heterozygous sites in assemblies with IUPAC ambiguity codes, and to leverage these heterozygous sites for further filtering of putative paralogs (see details in Materials and Methods section). In order to demonstrate the value of the new pipeline, we compared the number of putative paralogous loci detected by PPD and HybPiper and evaluated the influence of paralogs on phylogenetic and divergence time dating analyses using Hyb-Seq data from the Angiosperms353 kit we generated for two diploid genera: Castanea (Chestnuts of Fagaceae) and Hamamelis (Witch-hazel of Hamamelidaceae). We further validated the paralogy of putative paralogous loci identified by PPD using a genome reference available for Castanea.
The chestnut genus Castanea Miller (Fagaceae) includes seven tree species, each restricted to eastern Asia (EA), eastern North America (ENA), or Europe. The species were divided into three sections (Dode 1908). Section Eucastanon Dode, includes the five species with three nuts per cupule: C. mollissima Blume and C. seguinii Dode from China and C. crenata Siebold & Zucc. from Japan, C. dentata (Marshall) Brokh. from North American, and C. sativa Mill. from Europe. Sections Balanocastanon Dode and Hypocastanon Dode each is monotypic including a single species and both make fruits containing one nut per cupule. Section Balanocastanon contains C. pumila (L.) Mill. from North America and Section Hypocastanon contains C. henryi (Skan) Rehder & Wilson from China. Within C. pumila, two varieties were recognized by Johnson (1988) and Nixon (1997), C. pumila var. pumila in the southeastern United States and C. pumila var. ozarkensis (Ashe) A.E. Murray limited to the Ozark mountains. Phylogenetic studies of Castanea were previously conducted using data from six chloroplast regions (Lang et al. 2006, 2007). The studies found that sect. Eucastanon is paraphyletic. The witch-hazel genus Hamamelis L. (Hamamelidaceae) is also a small woody genus consisting of six species of shrubs and small trees, isolated in EA and ENA. The EA species include H. mollis Oliv. from eastern and southern China (Chang 1979; Zhang and Lu 1995) and H. japonica Siebold & Zucc. from Japan (Sargent 1890; Ohwi 1978). The ENA species include H. virginiana L., that is widely distributed from Canada to the Gulf coast (Bradford and Marsh 1977), H. vernalis Sarg., a species endemic to the Ozark Mountains in Arkansas, Missouri, and eastern Oklahoma (Bradford and Marsh 1977), H. ovalis S.W. Leonard that is restricted to a small area of Mississippi (Leonard 2006), and H. mexicana Standl. endemic to northeastern Mexico (Standley 1937), which is also known as Hamamelis virginiana var. mexicana (Standl.) C.Lane. A few phylogenetic studies of Hamamelis were previously conducted using data from ITS, ETS, waxy gene, and several plastid genes (Wen and Shi 1999; Li et al. 2000; Xie et al. 2010). However, the species relationships within Hamamelis have remained uncertain due to low nodal support values and short internal branches, especially regarding the relationships within the ENA clade. Therefore, results from the study also allow us to evaluate the previous phylogenetic hypotheses and further resolve the species relationships within these two genera.
Materials and Methods
Data Generation
Preparation of DNA Samples
We generated data from 15 samples of Castanea, seven samples of
Hamamelis, and three samples of outgroups (Supplementary
Table S1 available on Dryad at https://doi.org/10.5061/dryad.ttdz08kwqhttps://doi.org/10.5061/dryad.ttdz08kwq), which covers all species of the
two genera. Outgroup species were chosen based on their phylogenetic positions in
Fagaceae and Hamamelidaceae, respectively inferred by Lang et al. (2006) and Xie et al.
(2010). Fothergilla and
Parrotiopsis were used as the outgroups of Hamamelis
while Quercus was used as the outgroup of Castanea.
Leaf samples were collected from the field or plants grown in arboreta or botanical
gardens (Supplementary Table S1
available on Dryad). Fresh leaves were stored in silica gel to dry. The dry leaves were
stored at  20
20 C until they were used for the DNA extraction.
C until they were used for the DNA extraction.
Total genomic DNAs were extracted from leaf samples using the CTAB protocol (Doyle 1991) with modification described in Cullings (1992) and Xiang et al. (1998). For leaf samples of Castanea that are
rich in secondary compounds, they were washed five times with 0.8 mL of a washing buffer
containing 10% polyethylene glycol, 0.35 M sorbitol, 50 mM Tris–HCl, 0.1% bovine serum
albumin, and 0.1% 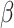 -mercaptoethanol (Sakaguchi et al. 2018; Zhou et al. 2020) prior to DNA extraction with the modified CTAB
method. The quality and quantity of DNA samples were first checked by 1% agarose gel
electrophoresis followed by measurement on a Nanodrop spectrophotometer (ThermoFisher)
and with a PicoGreen fluorescent dye assay (Life Technologies, ThermoFisher).
-mercaptoethanol (Sakaguchi et al. 2018; Zhou et al. 2020) prior to DNA extraction with the modified CTAB
method. The quality and quantity of DNA samples were first checked by 1% agarose gel
electrophoresis followed by measurement on a Nanodrop spectrophotometer (ThermoFisher)
and with a PicoGreen fluorescent dye assay (Life Technologies, ThermoFisher).
Library Preparation of Angiosperms353 Gene Enrichment and Sequencing
A total of 1000 ng DNA of each sample concentrated to 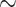 35
35
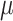 L was delivered to Rapid Genomics Lab
(Gainesville, Florida, USA) for Hyb-Seq library reconstruction and sequencing. The DNA
samples were pooled for hybridization to biotinylated probes using the Angiosperms353 v.
1 target capture kit (Johnson et al. 2019)
available from Arbor Biosciences (Arbor Biosciences, Ann Arbor, MI, USA). Sequencing of
DNAs pulled from the hybridization experiment was performed with Illumina MiSeq
(Illumina, San-Diego, CA, USA) to produce
L was delivered to Rapid Genomics Lab
(Gainesville, Florida, USA) for Hyb-Seq library reconstruction and sequencing. The DNA
samples were pooled for hybridization to biotinylated probes using the Angiosperms353 v.
1 target capture kit (Johnson et al. 2019)
available from Arbor Biosciences (Arbor Biosciences, Ann Arbor, MI, USA). Sequencing of
DNAs pulled from the hybridization experiment was performed with Illumina MiSeq
(Illumina, San-Diego, CA, USA) to produce 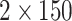 bp paired
end reads, as described in Gaynor et al.
(2020).
bp paired
end reads, as described in Gaynor et al.
(2020).
Locus Data Assembly and MSA Generation
All samples were demultiplexed using Illumina’s BCLtofastq by Arbor Biosciences. Raw
sequencing reads were then cleaned and trimmed by Trimmomatic v.0.38 (Bolger et al. 2014) using parameters MAXINFO:100:0.5 and
TRAILING:20. Subsequently, the HybPiper pipeline v. 3 (Johnson et al. 2016) was used to recover both coding sequences (CDS) and their
flanking intron/noncoding regions. The process includes three major steps: using the
nuclear sequences of Angiosperms353 genes (Johnson et al.
2019) as the references to capture all the reads from sequenced accessions via
the BWA option with default seed length k 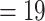 (Li and Durbin 2009), applying the SPAdes (Bankevich et al. 2012) to assemble reads into long
contigs, and implementing the intronerate.py module to recover “intron” and “supercontig”
(CDS
(Li and Durbin 2009), applying the SPAdes (Bankevich et al. 2012) to assemble reads into long
contigs, and implementing the intronerate.py module to recover “intron” and “supercontig”
(CDS 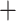 intron fragments) sequences. Then, we
used our PPD to generate multiple matrices to compare with those generated from HybPiper
(see details below). To assess the phylogenetic and divergence time dating effects of
paralogous genes we generated matrices consisting of supercontig sequences of three gene
groups trimmed with PPD: orthologous loci, paralogous loci, or all loci. The supercontig
matrices contained sequences of both coding and their flanking regions of the three
respective gene groups. The original supercontig matrices derived from HybPiper were
retained for comparison. To build the matrices of orthologous genes, the paralogs called
from HybPiper and PPD were manually removed from each all-gene matrix, while the matrices
of paralogous genes included the paralogs detected by HybPiper and paralogs detected by
PPD. Specifically, for the genes with paralog warning from HybPiper, we considered only
those loci with warnings for at least two individuals as paralogs. This conservative
approach followed Murphy et al. (2020) and was
based on 1) the fact that the reference sequences for the Angiosperms353 kit were putative
single copy genes from diverse, evolutionarily distant taxa, and 2) the observation of a
dissimilar sequence in one individual alone could be a random event or due to errors in
sequencing or sequence assembly in that individual, rather than true paralogy. To allow
different comparisons between the “consensus” matrices from HybPiper and
“degenerated” matrices from PPD, we generated
“consensus” matrices without (default of HybPiper), with gappy trimming
(s6 of part 2 of PPD), and with all PPD trimming steps (all steps of part 2 of PPD, see
details below). All data matrices and the relevant information are listed in Table 1.
intron fragments) sequences. Then, we
used our PPD to generate multiple matrices to compare with those generated from HybPiper
(see details below). To assess the phylogenetic and divergence time dating effects of
paralogous genes we generated matrices consisting of supercontig sequences of three gene
groups trimmed with PPD: orthologous loci, paralogous loci, or all loci. The supercontig
matrices contained sequences of both coding and their flanking regions of the three
respective gene groups. The original supercontig matrices derived from HybPiper were
retained for comparison. To build the matrices of orthologous genes, the paralogs called
from HybPiper and PPD were manually removed from each all-gene matrix, while the matrices
of paralogous genes included the paralogs detected by HybPiper and paralogs detected by
PPD. Specifically, for the genes with paralog warning from HybPiper, we considered only
those loci with warnings for at least two individuals as paralogs. This conservative
approach followed Murphy et al. (2020) and was
based on 1) the fact that the reference sequences for the Angiosperms353 kit were putative
single copy genes from diverse, evolutionarily distant taxa, and 2) the observation of a
dissimilar sequence in one individual alone could be a random event or due to errors in
sequencing or sequence assembly in that individual, rather than true paralogy. To allow
different comparisons between the “consensus” matrices from HybPiper and
“degenerated” matrices from PPD, we generated
“consensus” matrices without (default of HybPiper), with gappy trimming
(s6 of part 2 of PPD), and with all PPD trimming steps (all steps of part 2 of PPD, see
details below). All data matrices and the relevant information are listed in Table 1.
Table 1.
Summary information of Angiosperms353 gene data obtained for Castanea and Hamamelis, including the loci number, total MSA length, average length per locus, segregating sites number and percentage, parsimony informative sites number and percentage, and concatenation-based tree and species tree topologies
| Genus | Matrix | Gene groups | Total number loci | Total MSA of (bp) | Average length per length (bp) | Total hypervariable locus (per gene) | Number of sites removed segregating sites (%) | Number of parsimony informative sites (%) | Concatenation-based tree/ASTRAL-III/SVDQuartets |
|---|---|---|---|---|---|---|---|---|---|
| Castanea | Untrimmed | Orthologs* | 333 | 1,277,569 | 3836.5 | — | 151,735 (11.9%) | 40,772 (3.2%) | Top1/Top1/Top1 |
| consensus | |||||||||
| Gappy trimmed | Orthologs | 333 | 878,334 | 2637.6 | — | 113,444 (12.9%) | 34,209 (3.9%) | Top1/Top1/Top1 | |
| consensus | |||||||||
| PPD Trimmed | Orthologs* | 333 | 823,951 | 2474.3 | 54,409 (163.4) | 54,410 (6.6%) | 15,007 (1.8%) | Top1/Top1/Top1 | |
| consensus | Paralogs | 11 | — | — | — | — | — | — | |
| Degenerated | All genes | 344 | 842,054 | 2447.8 | 55,220 (160.5) | 46,206 (5.5%) | 12,295 (1.5%) | Top1/Top1/Top1 | |
| Orthologs* | 296 | 718,887 | 2428.7 | 41,356 (139.7) | 40,148 (5.6%) | 10,638 (1.5%) | Top1/Top1/Top1 | ||
| Paralogs* | 48 | 123,167 | 2566.0 | 13,864 (288.8) | 6058 (4.9%) | 1657 (1.3%) | Top1/Top2/Top1 | ||
| Hamamelis | Untrimmed | Orthologs* | 344 | 760,767 | 2211.5 | — | 68,892 (8.9%) | 14,756 (2.3%) | Top5/Top4/Top4 |
| consensus | |||||||||
| Gappy trimed | Orthologs | 344 | 607,159 | 1765.0 | — | 54,136 (8.9%) | 14,110 (2.3%) | Top5/Top4/Top4 | |
| consensus | |||||||||
| PPD Trimmed | Orthologs* | 344 | 566,772 | 1647.6 | 38,050 (110.6) | 26,177 (4.6%) | 7043 (1.2%) | Top3/Top3/Top3 | |
| consensus | Paralogs | 2 | — | — | — | — | — | — | |
| Degenerated | All genes | 346 | 568,476 | 1643.0 | 33,743 (97.5) | 22,130 (3.9%) | 6292 (1.1%) | Top3/Top3/Top3 | |
| Orthologs* | 319 | 514,351 | 1612.4 | 29,628 (92.8) | 20,185 (3.9%) | 5693 (1.1%) | Top3/Top3/Top3 | ||
| Paralogs* | 27 | 54,125 | 2004.6 | 4115 (153.9) | 1945 (3.6%) | 599 (1.1%) | Top4/Top5/Top5 | ||
Notes: The “untrimmed consensus matrices” were the orthologs directly generated by HybPiper, the “gappy trimmed consensus matrices” were the orthologs generated by HybPiper trimmed by s6 of part 2 of PPD. the “consensus matrices” were generated with HybPiper with all the PPD trimming steps, and the “degenerated” data sets were all generated with all steps of PPD. The “consensus” matrix contains sequences with heterozygous sites represented by the most frequent base; “degenerated” matrix contains sequences with heterozygous sites represented by the IUPAC ambiguity codes. Top 1: Castanea, ((EA), (ENA, Europe)); Top 2: Castanea, ((EA, Europe), (ENA)); Top 3: Hamamelis, ((H. mollis, (H. japonica, ENA)); Top 4: Hamamelis, ((H. japonica, H. mollis), ENA); Top 5: Hamamelis, (H. japonica, (H. mollis, ENA)). MSA: Multiple Sequence Alignment. The matrices indicated by an asterisk were used for divergence time dating comparisons. Dash line indicates data not examined. Bolded font indicates the tree topologies are concordant in concatenation-based tree and species trees.
Pipeline Description
The putative paralogs pipeline (PPD) includes two major parts: first, generating “degenerated” matrices, and second, trimming highly heterozygous sites, misaligned regions, and particularly gappy columns and detection of paralogous genes (Fig. 2).
Figure 2.

Illustrated putative paralogs detection (PPD) pipeline analytical workflow. The
flowchart shows the basic PPD functions, including two major parts: 1) generating
“degenerated” sequences and 2) generating well-trimmed matrix and
detecting paralogous genes consisting of eight steps (s1–s8). See details in pipeline
description. MSA = multiple sequence alignment; OG  outgroup; Sp
outgroup; Sp  species.
species.
In the first half of the PPD pipeline, the “degenerated” sequences are built for HybPiper-derived supercontig or exon sequences (if the intron sequences were not captured or absent) of each locus using a bash script following Kates et al. (2018) (available on Github: https://github.com/Bean061/putative_paralog). This involves using the “consensus” sequences from HybPiper (Fig. 1b) as the references and mapping the raw reads back to the references in BWA with customized seed length according to the sequence length. As a higher seed length (BWA -k) value improves mapping quality (Robinson et al. 2017), we applied high seed length to ensure high quality mapping. Our sequencing method produced sequences of 150 bp for each read, we used a minimum seed length (-k) of 100 bp, instead of the default “-k” (19 bp). After mapping, the mapped duplicate reads are discarded using picard (https://broadinstitute.github.io/picard/). The program GATK (McKenna et al. 2010; DePristo et al. 2011) is then used to identify the variable sites using the HaplotypeCaller, with “-ploidy 2” parameter for diploid species, and SelectVariants functions. Finally, we use the FastaAlternateReferenceMaker function in GATK to convert the variable sites into the IUPAC coding to produce the “degenerated” (IUPAC) sequences for each gene.
The second half of the PPD pipeline trims alignments and detects paralogs, and includes 8
steps: s1) Resort gene files: Use all “degenerated” sequence files from
every individual as the input, and then sort the “degenerated” sequences
orthologous to the 353 reference genes in each sample into individual locus files
according to gene names. s2) Sequence filtering: Filter the sequences with more than 5%
(default) heterozygosity according to the percentage information of heterozygous sites in
every sequence because a sequence with a high percentage of heterozygous sites may
indicate sequencing or assembly errors of the particular locus. This setting can be
changed by users with “-he” parameter. s3–s5) MSA generating: To obtain a better alignment
result, the reference sequence of each locus is added for alignment using MAFFT
(–adjustdirection –maxiterate 1000 –globalpair) (Katoh and
Standley 2013). The reference sequences are removed before trimming of the
aligned sequences in s6 and s7. s6) MSA trimming: Remove the gappy sites (i.e., sites
missing in 50% or more individuals) using TrimAl (default “-gt 0.51”) (Carretero-Paulet and Fares 2012), a threshold based on
the simulation study by Wiens and Morrill (2011)
which showed that adding a set of characters with data for 50% of the species is either
beneficial or harmless for phylogenetic study. We found the gappy regions were extensive
and mostly at two ends spanning the intron/flanking regions of the gene sequence alignment
in a locus, which might be attributed to erroneous assembly with a small number of raw
reads in a few individuals. Therefore, we excluded these regions from the alignment to
remove the influence of the gappy sites in phylogenetic analyses. The “-gt” parameter can
also be customized (this parameter is identical to the “-gt” in TrimAl). s7) MSA further
trimming: Detect and trim the hypervariable sites or regions using a sliding window
method. The polymorphic sites in the ingroups meeting the requirement in each window were
marked and then removed from all individuals (including the sites in outgroup species) by
TrimAl. The maximum number of sites in a sliding window can be modified by the “-mi”
parameter and sliding window length can be modified by the “-w” parameter in PPD. The
default values for “-mi” and “-w” are 4 and 20, respectively, which represent if there are
more than 4 polymorphic sites (not counting sites with heterozygous bases/degenerate
sites) in a 20 bp sliding window (representing 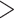 25% variable sites) all
of the polymorphic sites will be marked and removed by TrimAl. For polymorphic sites
attributed solely to differences in sequences of the outgroups and meeting the requirement
of more than eight polymorphic sites (changeable via “-mo” parameter in PPD, default is
eight) present in each 20 bp window, they are marked and replaced by a dash “-” in the
sequence of the outgroup and the sites are not removed from any individuals to retain
information likely phylogenetically informative among ingroup taxa. These criteria should
be adjusted according to observation of the nontrimmed taxa MSA. We used the
25% variable sites) all
of the polymorphic sites will be marked and removed by TrimAl. For polymorphic sites
attributed solely to differences in sequences of the outgroups and meeting the requirement
of more than eight polymorphic sites (changeable via “-mo” parameter in PPD, default is
eight) present in each 20 bp window, they are marked and replaced by a dash “-” in the
sequence of the outgroup and the sites are not removed from any individuals to retain
information likely phylogenetically informative among ingroup taxa. These criteria should
be adjusted according to observation of the nontrimmed taxa MSA. We used the
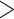 25% cutoff for our data based on the
assumption that such high rates of sequence variation in the 353 genes and their flanking
regions among our study ingroup species is unlikely true and may represent alignment
ambiguity due to errors from sequence assembly. Our visual inspection of the BWA mapping
result found the hypervariable sites had extremely low mapping quality, for example, low
depth of mapped reads (less than 5 reads) and many wrongly mapped reads. Including these
sites would inflate sequence variation, thus, the branch length in phylogenetic
inferences. s8) Paralog identification: Consider a locus as a paralog if it contains one
or more heterozygous site(s) that are shared by 50% (default) or more individuals. The
threshold of shared percentage and the number of heterozygous sites can be adjusted by the
user using the “-hs” parameter and “-nh” parameter, respectively. For example, in Figure 2, a hypothetical MSA of a locus/gene (on the left
side) shows sequence with high heterozygous sites (Sp1), a polymorphic site that is
heterozygous in
25% cutoff for our data based on the
assumption that such high rates of sequence variation in the 353 genes and their flanking
regions among our study ingroup species is unlikely true and may represent alignment
ambiguity due to errors from sequence assembly. Our visual inspection of the BWA mapping
result found the hypervariable sites had extremely low mapping quality, for example, low
depth of mapped reads (less than 5 reads) and many wrongly mapped reads. Including these
sites would inflate sequence variation, thus, the branch length in phylogenetic
inferences. s8) Paralog identification: Consider a locus as a paralog if it contains one
or more heterozygous site(s) that are shared by 50% (default) or more individuals. The
threshold of shared percentage and the number of heterozygous sites can be adjusted by the
user using the “-hs” parameter and “-nh” parameter, respectively. For example, in Figure 2, a hypothetical MSA of a locus/gene (on the left
side) shows sequence with high heterozygous sites (Sp1), a polymorphic site that is
heterozygous in 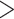 50% samples/individuals of a diploid
organism (labeled as polymorphic site 2), and a sequence containing a region with apparent
alignment ambiguity due to error in contig assembly (shown as hypervariable sites compared
to the rest). Identical heterozygous site(s) shared by over 50% individuals (Polymorphic
site 2) in the MSA is used as the indication of presence of paralogs in the locus and is
the criterion for calling putative paralogs in the PPD.
50% samples/individuals of a diploid
organism (labeled as polymorphic site 2), and a sequence containing a region with apparent
alignment ambiguity due to error in contig assembly (shown as hypervariable sites compared
to the rest). Identical heterozygous site(s) shared by over 50% individuals (Polymorphic
site 2) in the MSA is used as the indication of presence of paralogs in the locus and is
the criterion for calling putative paralogs in the PPD.
Phylogenetic Analyses
Concatenation-based tree
Phylogenetic analyses of the concatenated Hyb-Seq data were performed for the supercontig data matrices of the three gene groups generated from PPD as well as supercontig data matrices of the orthologs derived from HybPiper listed in Table 1 using a maximum likelihood method implemented in IQ-TREE v. 1.6.12 (Nguyen et al. 2015) partitioned by genes. All analyses used the TESTNEW option to obtain the best molecular model per partition. UF bootstrap was applied to evaluate the topology (Hoang et al. 2018). To test the congruence among different partition methods and phylogenetic methods, we also ran a phylogenetic analysis with the best merged partitions suggested by ModelFinder using MFP-MERGE in IQ-TREE (Lanfear et al. 2012) and conducted analyses without any partition using RAxML (Stamatakis 2014) and MrBayes (Ronquist and Huelsenbeck 2003) for the “degenerated” orthologous data matrices derived from PPD pipeline (for details, see Supplementary Information available on Dryad). The RAxML and MrBayes analyses above were all conducted on the CIPRES Science Gateway Portal (Miller et al. 2010).
Coalescent-based species tree
We used both ASTRAL-III (Zhang et al. 2018) and SVDQuartets (Chifman and Kubatko 2014) to generate the coalescent-based species trees. For the analyses with ASTRAL-III, we used gene trees from IQ-TREE for both genera as the input and ran ASTRAL-III with the default parameters. For the analyses with SVDQuartets, we used concatenated multilocus data as the input. Then, PAUP* v4.0a166 (Swofford 2003) was used to generate a total of 100,000 quartets with 100 bootstrap replicates and then the quartet assembly method QFM was used to produce a summary tree (Reaz et al. 2014), following Zhou et al. (2020).
All concatenation-based trees and coalescent-based species trees were visualized and edited in FigTree v.1.4.4 (Rambaut 2012) and edited with ggtree [R] (Yu et al. 2018) and Adobe Illustrator 2020 (Adobe Systems, San Francisco, CA, USA).
Divergence Time Analyses
We employed BEAST2 2.6.2 (Bouckaert et al. 2014) to estimate the divergence times of lineages within each genus. BEAST2 can consider information at heterozygous sites in divergence time estimation. The divergence time analyses were conducted for orthologous and paralogous matrices generated from the PPD (marked with asterisk in Table 1) to allow comparisons and assess the effect of paralogous genes. The divergence time analysis was also conducted for “consensus” supercontig matrices of orthologs with and without PPD trimming to allow comparison between “degenerated” orthologous gene data derived from PPD and the “consensus” orthologous gene data from the HybPiper alone. The stem ages of Castanea and Hamamelis were constrained based on fossil evidence, as 66 to 72 Ma (lognormal) and 50 to 56 Ma (lognormal), respectively (for details, see Supplementary Information available on Dryad).
Divergence time analyses were run under the GTR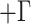 molecular model
for all orthologous gene matrices and HKY
molecular model
for all orthologous gene matrices and HKY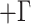 molecular model
for paralogous gene matrices for both Castanea and
Hamamelis, the best models for each on the BIC values from jModelTest
(Darriba et al. 2012). An uncorrelated lognormal
relaxed clock (Drummond et al. 2006) and the
birth–death process model (Stadler 2010) were
implemented in the analyses. To account for the fact that our sampling in
Castanea contained two samples per species, which violates assumptions
of the BD model, we performed an additional analysis of the orthologous gene data by using
a single sample per species to evaluate the impact of this violation. To facilitate
comparisons among data sets and between undated and dated phylogenies, we included the
original sampling of Castanea in divergence time analyses of all data
sets. We run our analyses as a single concatenated supermatrix, as divergence time
analyses using concatenated unpartitioned supermatrices compared with gene partitioned
matrices of genomic data results in similar divergence times, but the concatenated data
sets were more efficient than the partitioned data sets in attaining suitable effective
sample sizes (Voloch and Schrago 2012). We set the
mean GrowthRate (net diversification rate) to have a uniform distribution with a range of
0–100, with an initial value of 0.0, and the relative death rate (extinction
rate/speciation rate) to have a 0–1 range, with an initial value of 0.5. These values were
chosen based on the estimated average net diversification rate and extinction rate in
plants (De Vos et al. 2015). Because constraints on
node times can interact with constraints on other nodes and can also impact the divergence
times of nodes that are elsewhere on the tree, we ran “empty” Markov Chain Monte Carlo
analyses by adopting the prior settings but without using the sequence data to determine
if the marginal densities of calibrated nodes matched the calibration densities, a desired
property of a calibrated tree prior (Heled and Drummond
2012). These analyses yielded approximations to the prior distributions. To
ensure that the prior distributions were well approximated, these “empty” MCMC runs all
had effective sample sizes that exceeded 200. We found congruence between the priors and
their approximations. Then, we ran the analyses with data for 200 million generations,
with sampling of trees every 10,000 generations. Quality of the runs and parameter
convergence were assessed using Tracer v.1.6.0 (Rambaut et
al. 2018). The maximum credibility tree of median heights was then constructed
using TreeAnnotator after discarding 20% trees as burn-in.
molecular model
for paralogous gene matrices for both Castanea and
Hamamelis, the best models for each on the BIC values from jModelTest
(Darriba et al. 2012). An uncorrelated lognormal
relaxed clock (Drummond et al. 2006) and the
birth–death process model (Stadler 2010) were
implemented in the analyses. To account for the fact that our sampling in
Castanea contained two samples per species, which violates assumptions
of the BD model, we performed an additional analysis of the orthologous gene data by using
a single sample per species to evaluate the impact of this violation. To facilitate
comparisons among data sets and between undated and dated phylogenies, we included the
original sampling of Castanea in divergence time analyses of all data
sets. We run our analyses as a single concatenated supermatrix, as divergence time
analyses using concatenated unpartitioned supermatrices compared with gene partitioned
matrices of genomic data results in similar divergence times, but the concatenated data
sets were more efficient than the partitioned data sets in attaining suitable effective
sample sizes (Voloch and Schrago 2012). We set the
mean GrowthRate (net diversification rate) to have a uniform distribution with a range of
0–100, with an initial value of 0.0, and the relative death rate (extinction
rate/speciation rate) to have a 0–1 range, with an initial value of 0.5. These values were
chosen based on the estimated average net diversification rate and extinction rate in
plants (De Vos et al. 2015). Because constraints on
node times can interact with constraints on other nodes and can also impact the divergence
times of nodes that are elsewhere on the tree, we ran “empty” Markov Chain Monte Carlo
analyses by adopting the prior settings but without using the sequence data to determine
if the marginal densities of calibrated nodes matched the calibration densities, a desired
property of a calibrated tree prior (Heled and Drummond
2012). These analyses yielded approximations to the prior distributions. To
ensure that the prior distributions were well approximated, these “empty” MCMC runs all
had effective sample sizes that exceeded 200. We found congruence between the priors and
their approximations. Then, we ran the analyses with data for 200 million generations,
with sampling of trees every 10,000 generations. Quality of the runs and parameter
convergence were assessed using Tracer v.1.6.0 (Rambaut et
al. 2018). The maximum credibility tree of median heights was then constructed
using TreeAnnotator after discarding 20% trees as burn-in.
Assessment of PPD Success Rate on Paralogs Identification
To test whether the putative paralogs detected by PPD were true paralogs and assess the false positive and false negative rates of PPD in identifying paralogs, we conducted nucleotide BLAST (Altschul et al. 1990) search to determine if the putative orthologs and paralogs would map to one or more regions of reference genome sequences. One Castanea species has a published genome (C. mollissima ASM1418300v1 from Wang et al. 2020) but no species of Hamamelis has genome sequences available. Because considering BLAST results using distant genome references may not reflect gene paralogy correctly, we assessed the success rate of PPD in identification of paralogous genes only in Castanea samples using the Castanea reference genome. We considered a locus to be confirmed as paralogous when its sequence from any Castanea sample had two or more BLAST hits on the reference genome and/or had a BLAST hit to a genome location different from that of other samples with 90 percent of identity with at least 500 bp mapping length in the separate regions of the reference genome. We calculated the success rate of PPD in paralogous gene identification as the number paralogous loci confirmed by the BLAST mapping analyses divided by the total number of paralogous loci identified by PPD. We also assessed the failure rate of PPD in calling paralogous genes by mapping the pooled sequences of all species of a putative orthologous locus to the reference genome. If sequences of a gene locus are mapped to more than one region in the reference genome, we recorded it as a case of false orthology. We also evaluated if false orthology and false paralogs influenced our phylogenetic analyses by repeating IQ-TREE analyses described above on a matrix that contained PPD orthologs and putative false paralogs but excluding any putative false orthology from BLAST results.
Results
The number of loci, alignment length, average length per locus, total hypervariable sites removed, number of segregating sites, and number of parsimony informative sites varied among the three gene groups and between genera (Table 1). We found no sequences with excess heterozygosity and thus no sequences were removed from our data due to the presence of excess within-individual heterozygosity (5% or more). In the “consensus” matrices generated from HybPiper, approximately an average of 1120 bp in Castanea and 446 bp in Hamamelis were removed from each locus through the gap-trimming step in PPD. Through the PPD sliding window trimming process, approximately an average of 163 bp and 110 bp hypervariable sites from each locus were detected and removed from Castanea and Hamamelis, respectively.
Paralog Detection in Hyb-Seq Data
The gene matrices generated by HybPiper had paralog warning for 11 loci shared in two or more individuals (Gene 6048, 6954, 4951, 4724, 5940, 6387, 6570, 7583, 7324, 5138, 5941) out of a total of 344 genes sequenced in Castanea, but only two putative paralogs (Gene 5463, 5347) out of 346 genes in Hamamelis were identified based on the same criteria. In contrast, our PPD pipeline (in conjunction with HybPiper) detected 48 and 27 paralogs in Castanea and Hamamelis, respectively (Table 1). We found 31 (77.5%) out of 40 paralogs from PPD had multiple hits to the Castanea reference genome, while 9 (22.5%) paralogs had one hit based on the BLAST results (Table 2; Supplementary Tables S2 and S3 available on Dryad). In orthologous genes detected by PPD, we found 255 (83.9%) out of 304 orthologs had only a single hit (i.e., all samples mapped only to a single region of the genome), while 46 (15.1%) putative orthologs had multiple hits (Table 2; Supplementary Tables S4 and S5 available on Dryad). Phylogenetic analyses that also excluded orthologs with multiple BLAST hits and included paralogs with single BLAST hits were qualitatively the same as all other PPD analyses described below (Supplementary Fig. S1 available on Dryad). As a comparison, we found 11 paralogs by HybPiper, eight of which differed from paralogs from PPD. Four (36.4%) out of 11 paralogs from HybPiper had multiple hits to the Castanea reference genome, while six (54.5%) paralogs had one hit and one putative paralog had no hits (Table 2). Among the 333 orthologous genes from HybPiper, 258 (77.5%) had single hit, 73 (21.9%) had multiple hits, and 2 (0.6%) had no hits (Table 2).
Table 2.
The number of genes with different hits using BLAST against Castanea genome
| PPD | HybPiper | |||||
|---|---|---|---|---|---|---|
| Identified genes | Multiple hits | Single hit | No hit | Multiple hits | Single hit | No hit |
| Paralogs | 31 (77.5%) | 9 (22.5%) | 0 (0.0%) | 4 (36.4%) | 6 (54.5%) | 1 (9.1%) |
| Orthologs | 46 (15.1%) | 255 (83.9%) | 3 (1.0%) | 73 (21.9%) | 258 (77.5%) | 2 (0.6%) |
Phylogenetic Analyses of Orthologous Gene Data
The phylogenetic analyses of the orthologous gene data from PPD using IQ-TREE (with gene partition and best merged partition), RAxML, and MrBayes resulted in the same tree topologies with strong nodal support in both Castanea (Fig. 3a; Supplementary Figs. S2–S4 available in Dryad) and Hamamelis (Fig. 3b and Supplementary Figs. S5–S7 available on Dryad). The coalescent-based species trees reconstructed from ASTRAL-III and SVDQuartets for each genus also had the same topology identical to the concatenation-based tree (Fig. 4). In Castanea, the reciprocal monophyly of species from EA and ENA were recovered for each region, and the European species C. sativa was placed as the sister to the American clade (Fig. 4a). In Hamamelis, species from ENA form a monophyletic group sister to H. japonica with H. mollis diverging out first, sister to the remaining species. However, the node connecting the ENA clade and H. japonica was not well supported in ASTRAL-III (0.59) but well supported in SVDQuartets (90) (Fig. 4b).
Figure 3.

Comparison of concatenation-based trees of Castanea and Hamamelis resulting from phylogenetic analyses of PPD (“degenerated”) and HybPiper (“consensus”) data of orthologs. a) “degenerated”, orthologs in Castanea from PPD. b) “degenerated”, orthologs in Hamamelis from PPD. c) PPD-trimmed “consensus,” orthologs in Castanea from HybPiper. d) PPD-trimmed “consensus,” orthologs in Hamamelis from HybPiper. e) untrimmed “consensus,” orthologs in Castanea from HybPiper. f) untrimmed “consensus,” orthologs in Hamamelis from HybPiper. All analyses were performed using the IQ-TREE partitioned by genes. The topologies of a) and b) are identical to the best merged partitioned concatenation-based trees from IQ-TREE and unpartitioned concatenation-based trees from RAxML and MrBayes (Supplementary Figs. S2–S7 available on Dryad). Quercus castaneifolia was used as the outgroup for Castanea while Parrotiopsis jacquemontiana and Fothergilla gardenii were used as outgroups of Hamamelis. EA = eastern Asia; ENA = eastern North America.}
Figure 4.

Coalescent-based species trees of Castanea and Hamamelis reconstructed using the orthologous gene data from PPD with ASTRAL-III and SVDQuartets. Numbers on the branches are support values from ASTRAL-III (the fractions of quartet trees supporting the node) and SVDQuartets (bootstrap support), respectively. Quercus castaneifolia was used as the outgroup of Castanea while Parrotiopsis jacquemontiana and Fothergilla gardenii were used as outgroups of Hamamelis. EA = eastern Asia; ENA = eastern North America.
Phylogenetic analysis of the orthologous gene data from HybPiper alone with and without PPD trimming steps resulted in different results in the two genera considered. In Castanea, the same topology was recovered from orthologous gene data for HybPiper matrices with and without PPD trimming, and this topology was the same as the topology recovered from the full PPD pipeline (compare Figs. 3a,c,e). In Hamamelis, the analysis of the untrimmed matrix resulted in a tree with a topology different from the tree from the PPD and trimmed HybPiper data (compare Figs. 3b,d,f). In both genera, the branch lengths in HybPiper data-based trees were substantially longer than trees based on the PPD data, especially in the trees from the untrimmed HybPiper consensus data.
Divergence Time Analyses of Orthologous Genes
Castanea
Divergence time analyses of the PPD-derived data including all samples (i.e.,
“degenerated” supercontigs of orthologous genes) estimated the crown
age of the genus (splitting of the EA and ENA clades) as the early Miocene (17.9 Ma, 95%
HPD: 14.3–21.8 Ma). Within the genus, other divergence occurred in the mid-Miocene and
late Miocene (Fig. 5a and Supplementary
Table S6 available on Dryad). The European chestnut (C.
sativa) diverged from the two ENA species in the mid-Miocene (13.6 Ma, 95%
HPD: 10.9–16.7 Ma). The divergence times estimated from analysis with one sample per
species were highly similar to those based on full sampling (two samples per species)
for Castanea, with differences of median values
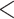 1 million years (Supplementary
Fig. S8 available on Dryad).
1 million years (Supplementary
Fig. S8 available on Dryad).
Figure 5.

Results of divergence times (median) of Castanea (left column) and Hamamelis (right column) estimated using different Hyb-Seq data. a) and b), the results are from PPD-generated orthologous data. c and d), results are from “consensus” data through HybPiper and PPD trimming steps. e and f), the results are from PPD-generated paralogous data. g and h) results are from “consensus” data without any trimming steps. The number at each node represents the median divergence time, while the node bars represent its 95% HPD. Circle 1 represents the fossil calibration of Castanea, while circle 2 represents the fossil calibration of Hamamelis. All topologies were drawn by ggtree in R. EA = eastern Asia; ENA = eastern North America.
Divergence times (median values) estimated from the HybPiper-derived data were approximately 11 million years (untrimmed) and two million years (trimmed) older, respectively, for all the nodes (Fig. 5a,c,g and Supplementary Table S6 available on Dryad). The divergence times estimated from the paralogous genes were two to a few million years older than the estimates based on the orthologous gene data (Fig. 5e and Supplementary Table S6 available on Dryad).
Hamamelis
Divergence time analyses of the PPD-derived data showed the crown node of Hamamelis (splitting of H. mollis from the remaining species) was dated back to the late Oligocene (e.g., 27.6 Ma with the 95% HPD as 24.0–31.6 Ma; Fig. 5b and Supplementary Table S6 available on Dryad). The divergence of H. japonica from the ENA clade was dated to the early Miocene (e.g., 23.3 Ma, with the 95% HPD as 20.2-26.7 Ma; Fig. 5b and Supplementary Table S6 available on Dryad). Divergence events within the American clade were dated to the late Miocene for H. virginiana and the Pliocene for the other species (Fig. 5b and Supplementary Table S6 available on Dryad). Similarly, the divergence times estimated from HybPiper-derived data were approximately 6–10 million years (untrimmed) and up to three million years (trimmed) older, respectively, for all nodes (Fig. 5d,h; Supplementary Table S6 available on Dryad).
Divergence time analyses of the paralogous gene data detected by PPD showed the median were highly similar at some nodes but younger or older at other nodes with differences within four or five million years, compared to the estimates from the “degenerated” orthologous gene data (Fig. 5f and Supplementary Table S6 available on Dryad). However, the 95% HPD were much higher at all nodes, indicating greater uncertainty.
Discussion
Impacts of Paralogs and the Value of the PPD
Our results showed that our new pipeline (PPD) identified many more putative paralogs than HybPiper. Although the “consensus” sequence data generated from HybPiper may produce the phylogenetic tree with the same topology as the tree from the “degenerated” sequence data derived from PPD, the HybPiper data contained many more “false” phylogenetic informative sites (due to the presence of paralogous genes and consensus coding of the sequences), resulting in longer branches affecting divergence time estimation (Figs. 3 and 5; Supplementary Table S6 available on Dryad). The sequence data with better cleaning of paralogs and coded with the “degenerated” method are advantageous for phylogenomic studies, as they contain more accurate information for phylogenetic and divergence time estimations. Comparisons of the PPD data with “consensus” data with and without PPD trimming steps (Figs. 3 and 5) indicated that the observed differences in branch lengths and divergence times cannot be explained by differences in trimming alone and sequence coding and paralogs also affected branch lengths and divergence time estimation. Furthermore, our phylogenetic analyses of the loci containing paralogous genes often resulted in phylogenies different from those inferred from data of the orthologous genes in Hamamelis (Supplementary Fig. S9 available on Dryad). The divergence times estimated from data including potential paralogous genes (i.e., the “consensus” data matrices from HybPiper) or from the paralogous genes identified from PPD are older and have larger HPDs, likely due to the additional variable sites introduced by gene paralogy (Fig. 5 and Supplementary Table S6 available on Dryad). Our results clearly highlighted the negative impacts of paralogous gene content in phylogenetic analyses and that paralogous gene content either inflates estimates of divergence time or increases uncertainty of divergence time estimation in Castanea and Hamamelis (Fig. 5 and Supplementary Table S6 available on Dryad). Comparisons of the PPD data with the “consensus” and untrimmed “consensus” data from HybPiper further indicated that the effects of sequence trimming on branch lengths and divergence time estimation were major, greater than the influences of sequence coding and paralogs in our case (Figs. 3 and 5). These results together strongly support that additional steps following HybPiper to “polish” data from Hyb-Seq of Angiosperms353 probe kit are necessary before phylogenetic and downstream analyses. Moreover, we show that the PPD pipeline can effectively clean alignments with user-defined trimming and identify paralogs in these alignments to produce higher quality data for phylogenetic and divergence time dating analyses. The “degenerated” matrix generated from the PPD using the IUPAC ambiguity codes are suitable for a wide range of modern phylogenetic tools for phylogeny and divergence time estimation, including RAxML (Stamatakis 2014), IQ-TREE (Nguyen et al. 2015), SVDQuartet (Chifman and Kubatko 2014), BEAST2 (Bouckaert et al. 2014) that has an option to treat ambiguity-coded positions as informative.
Accuracy Rate and Caveats of PPD in Paralogs Identification
Through BLAST mapping analysis with the Castanea mollissima genome, the
paralogy of most of the PPD identified paralogous loci (31 out of 40 at a rate of 77.5%)
were confirmed by two or more hits. The remaining nine paralogs each had a single hit in
the genome (i.e., all samples mapped only to a single region), which represented
false-positive paralogs, may be explained by loss of the duplicated paralogous loci in the
reference genome and/or incompleteness of the reference genome. Additional
Castanea genomes that may become available in the future will help
further test this hypothesis. Alternatively, small-scale duplication events (e.g., Hudson et al. 2011; Carretero-Paulet and Fares 2012; Rensing
2014) that are prevalent in Castanea plants may be missed based
on the settings we used for BLAST (such as a 500 bp length), leading to the false
classification of a putative paralog as having only a single hit. We found that five out
of these nine loci have only one heterozygous site shared by 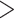 50%
individuals. The single shared heterozygous site in these five paralogs could be a result
of occurrence by chance or sequencing errors. If users want to minimize such potentially
false identification of paralogs and they can use a more conservative approach by
increasing the number of heterozygous sites shared by
50%
individuals. The single shared heterozygous site in these five paralogs could be a result
of occurrence by chance or sequencing errors. If users want to minimize such potentially
false identification of paralogs and they can use a more conservative approach by
increasing the number of heterozygous sites shared by 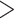 50%
individuals. However, this may result in the potential of missing true paralogous loci. If
no reference genome is available for verification of paralogy of loci, and given that
sequences for numerous loci are available from Hyb-Seq for phylogenetic analyses, we
recommend a more aggressive approach to removing paralogs, such as the one adopted in our
study.
50%
individuals. However, this may result in the potential of missing true paralogous loci. If
no reference genome is available for verification of paralogy of loci, and given that
sequences for numerous loci are available from Hyb-Seq for phylogenetic analyses, we
recommend a more aggressive approach to removing paralogs, such as the one adopted in our
study.
Our mapping analysis also indicated that PPD outperformed HybPiper alone at identifying true orthologs. We found 255 out 303 (83.9%) orthologous genes identified by PPD were true orthologs (evidenced by a single hit in the BLAST analysis), compared with only 77.5% from HybPiper alone. Additionally, 46 of the orthologous genes from PPD had two hits (15.1%), indicating paralogy of these loci according to our mapping criterion, while 73 (21.9%) of putative orthologs from HybPiper alone had multiple hits. This may indicate that both HybPiper and PPD do not remove all potential paralogs, but with only a single reference genome available, it is also possible these putative orthologs mapping multiple times could reflect errors in reference genome assemblies. Regardless of the origin of these putative paralogs missed by PPD, excluding them from phylogenetic analyses did not result in substantial differences in phylogenetic results between the original orthologous PPD matrix and one without these genes, indicating that a small percentage of “false” orthologs is tolerable. However, researchers may choose to validate the PPD identified paralogs and orthologs for their taxa with reference genome available and further refine the data, as done with Castanea in our study Overall, compared to HybPiper, PPD generated more accurate orthologous gene data for phylogenetic and downstream analyses (Table 1 and Supplementary Table S7 available on Dryad).
Taxonomy and Relationships within Castanea and Hamamelis
Our phylogenetic data do not agree with the morphology-based classification scheme of three sections in Castanea (Sect. Eucastanon, Sect. Balanocastanon, and Sect. Hypocastanon) (Dode 1908; Johnson 1988). Our result indicated that Sect. Eucastanon that included C. dentata, C. sativa, C. mollissima, C. seguinii, and C. crenata is paraphyletic and the character of one nut per cupule in C. pumila (ENA) and C. henryi (EA) is homoplasy. Our results also support that ENA clade is sister to European C. sativa with high support value (Figs. 3a and 4a). The taxonomic status of the Allegheny chinkapin (C. pumila) and the Ozark chinkapin has been disputed (Johnson 1988; Nixon 1997). Johnson (1988) considered the Ozark chinkapin as a variety of C. pumila, while Nixon (1997) regarded it as a separate species C. ozarkensis. In our study, all individuals representing C. pumila including the Ozark chinkapins formed a monophyletic group sister to C. dentata with strong support. Therefore, our phylogenomic study does not support the recognition of C. ozarkensis as a distinct species. However, the hypothesis should be further tested with population level sampling of related taxa.
In Hamamelis, our result suggested a similar topology with previous phylogenies using data from ITS, ETS, waxy gene, and several plastid genes (Wen and Shi 1999; Li et al. 2000; Xie et al. 2010), which showed H. mollis diverged first, followed by the divergence between H. japonica and ENA clade. The ENA clade was a well-supported monophyletic clade. Different from previous studies, our concatenation-based tree showed a well resolved relationship among ENA clade using nuclear gene data, indicating H. virginiana is the first diverged species, followed by the divergence of H. vernalis, and H. mexicana is sister to H. ovalis (Fig. 3b). However, our coalescent-based species tree showed a different topology within the ENA clade, uniting H. ovalis and H. vernalis as the sister group but with low support values (Fig. 4b). This conflict suggests there might be incomplete lineage sorting or gene flow among these three taxa in North America. The node connecting H. japonica and ENA clade is also relatively low in the species tree reconstructed with ASTRAL-III (0.59; Fig. 4b), indicating another phylogenetic conflict among gene trees and the possibility of ancient gene flow or incomplete lineage sorting.
In conclusion, PPD, the pipeline we have described here, improves the quality of data obtained from Hyb-Seq for phylogenomic analyses through detection of additional paralogous genes and removal of hypervariable regions. Through empirical studies in Castanea and Hamamleis, our study demonstrated that data derived from HybPiper without the filtering steps implemented in PPD biased phylogenetic and divergence time estimation. Although our results focused on expanding HybPiper to improve detection of paralogs, our study also highlights the importance of accounting for potential paralogous genes in phylogenomic studies. As such, we recommend that phylogenomic analyses account for paralogs, such as through our PPD tool, particularly when the study group of interest belongs to lineages where gene duplication could be a concern.
Acknowledgments
We thank the Soltis lab at Florida Museum of Natural History, CX Fu lab at Zhejiang University, Gao lab at Kunming Institute of Botany, Chinese Academy of Sciences, JC Raulston Arboretum, Arnold Arboretum, University of Washington Botanic Gardens, P Jones at Sarah Duke Garden, UNC herbarium, J Lee from Korea research institute of bioscience and biotechnology and Y Ru from Heidelberg University for providing some leaf tissues, J.L. Thorne from North Carolina State University and M Johnson from Texas Tech University for discussion of the PPD pipeline. Special thanks to three anonymous reviewers and editors for precious suggestions. We also thank SR Manchester and K Pigg for discussion of Castanea and Hamamelis fossils. Finally, we thank Z Du from Wuhan Botanical Garden to run pilot tests for the PPD with data from Cornus and Aesculus.
Supplementary Material
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.ttdz08kwq.
Demultiplexed sequence data are available for download from the NCBI Sequence Read Archive (SRA) (BioProject PRJNA670453).
Funding
The work was supported by an NSF grant of the United States [DEB – 1442161 to Q.Y.(J.) Xiang]. This work was also benefited from the USDA National Institute of Food and Agriculture, Hatch project 02718; [NSF DEB – 1754376 supported J.S.].
References
- Altenhoff A.M., Glover N.M., Dessimoz C.. 2019. Inferring orthology and paralogy. In: Anisimova M., editor. Evolutionary genomics. Methods in Molecular Biology, vol. 1910. New York: Humana. p. 149–175. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Andermann T., Cano Á., Zizka A., Bacon C., Antonelli A.. 2018. SECAPR—a bioinformatics pipeline for the rapid and user-friendly processing of targeted enriched Illumina sequences, from raw reads to alignments. PeerJ. 6:e5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker S.E., Lemmon A.R., Hassinger A.B., Dye M., Holland S.D., Kortyna M.L., Ospina O.E., Ralicki H., Lemmon E.M.. 2020. Hierarchical hybrid enrichment: multitiered genomic data collection across evolutionary scales, with application to chorus frogs (Pseudacris). Syst. Biol. 69:756–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., Pyshkin A.V., Sirotkin A.V., Vyahhi N., Tesler G., Alekseyev M.A., Pevzner P.A.. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow L.N., Lemmon A.R., Lemmon E.M.. 2018. Targeted sampling and target capture: assessing phylogeographic concordance with genome-wide data. Syst. Biol. 67:979–996. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R., Heled J., Kuhnert D., Vaughan T., Wu C.H., Xie D., Suchard M.A., Rambaut A., Drummond A.J.. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford J.L., Marsh D.L.. 1977. Comparative Studies of the Witch Hazels Hamamelis virginiana and H. vernalis. J. Ark. Acad. Sci. 31(1):29-31. [Google Scholar]
- Capella-Gutierrez S., Silla-Martinez J.M., Gabaldon T.. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero-Paulet L., Fares M.A.. 2012. Evolutionary dynamics and functional specialization of plant paralogs formed by whole and small-scale genome duplications. Mol. Biol. Evol. 29:3541–3551. [DOI] [PubMed] [Google Scholar]
- Chang H.T. 1979. Hamamelidaceae. In: Florae Reipublicae Popularis Sinicae, vol. 35. Beijing: Science Press. p. 36-116. [Google Scholar]
- Chifman J., Kubatko L.. 2014. Quartet inference from SNP data under the coalescent model. Bioinformatics 30:3317–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullings K.W. 1992. Design and testing of a plant-specific PCR primer for ecological and evolutionary studies. Mol. Ecol. 1:233–240. [Google Scholar]
- Darriba D., Taboada G.L., Doallo R., Posada D.. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods. 9:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., McKenna A., Fennell T.J., Kernytsky A.M., Sivachenko A.Y., Cibulskis K., Gabriel S.B., Altshuler D., Daly M.J.. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos J.M., Joppa L.N., Gittleman J.L., Stephens P.R., Pimm S.L.. 2015. Estimating the normal background rate of species extinction. Conserv. Biol. 29:452–462. [DOI] [PubMed] [Google Scholar]
- Dode L.A. 1908. Notes dendrologiques. Paris: Au Siège de la Société.p. 1-166. [Google Scholar]
- Dodsworth S., Pokorny L., Johnson M.G., Kim J.T., Maurin O., Wickett N.J., Forest F., Baker W.J.. 2019. Hyb-Seq for flowering plant systematics. Trends Plant Sci. 24:887–891. [DOI] [PubMed] [Google Scholar]
- Dong Y., Chen S., Cheng S., Zhou W., Ma Q., Chen Z., Fu C.-X., Liu X., Zhao Y., Soltis P.S., Wong G.K.-S., Soltis D.E., Xiang Q.-Y.. 2019. Natural selection and repeated patterns of molecular evolution following allopatric divergence. eLife 8:e45199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle J. 1991. DNA protocols for plants. In: Hewitt G.M., Johnston A.W.B., Young J.P.W., editors. Molecular Techniques in Taxonomy. NATO ASI Series (Series H: Cell Biology), vol. 57. Berlin, Heidelberg: Springer. p. 283–293. [Google Scholar]
- Du Z.-Y., Harris A., Xiang Q.-Y. (Jenny). 2020. Phylogenomics, co-evolution of ecological niche and morphology, and historical biogeography of buckeyes, horsechestnuts, and their relatives (Hippocastaneae, Sapindaceae) and the value of RAD-Seq for deep evolutionary inferences back to the Late Cretaceous. Mol. Phylogenet. Evol. 145:106726. [DOI] [PubMed] [Google Scholar]
- Drummond A.J., Ho S.Y., Phillips M.J., Rambaut A.. 2006. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton D.A.R. 2014. PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics 30:1844–1849. [DOI] [PubMed] [Google Scholar]
- Eaton D.A.R., Overcast I.. 2020. ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics 36:2592–2594. [DOI] [PubMed] [Google Scholar]
- Faircloth B.C. 2016. PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics 32:786–788. [DOI] [PubMed] [Google Scholar]
- Faircloth B.C., McCormack J.E., Crawford N.G., Harvey M.G., Brumfield R.T., Glenn T.C.. 2012. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst. Biol. 61:717–726. [DOI] [PubMed] [Google Scholar]
- Faircloth B.C., Sorenson L., Santini F., Alfaro M.E.. 2013. A phylogenomic perspective on the radiation of ray-finned fishes based upon targeted sequencing of ultraconserved elements (UCEs). PLoS One 8:e65923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C.N., Mo Z.Q., Yang J.B., Ge X.J., Li D.Z., Xiang Q.J., Gao L.M.. 2019. Plastid phylogenomics and biogeographic analysis support a trans-Tethyan origin and rapid early radiation of Cornales in the Mid-Cretaceous. Mol. Phylogenet. Evol. 140:106601. [DOI] [PubMed] [Google Scholar]
- Gaynor M.L., Fu C., Gao L., Lu L., Soltis D.E., Soltis P.S.. 2020. Biogeography and ecological niche evolution in Diapensiaceae inferred from phylogenetic analysis. J. Syst. Evol. 58(5):646–662. [Google Scholar]
- Harvey M.G., Smith B.T., Glenn T.C., Faircloth B.C., Brumfield R.T.. 2016. Sequence capture versus restriction site associated DNA sequencing for shallow systematics. Syst. Biol. 65:910–924. [DOI] [PubMed] [Google Scholar]
- Heled J., Drummond A.J.. 2012. Calibrated tree priors for relaxed phylogenetics and divergence time estimation. Syst. Biol. 61:138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang D.T., Chernomor O., Von Haeseler A., Minh B.Q., Vinh L.S.. 2018. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35:518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenlohe P.A., Amish S.J., Catchen J.M., Allendorf F.W., Luikart G.. 2011. Next-generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and westslope cutthroat trout: SNP discovery: next generation sequencing. Mol. Ecol. Resour. 11:117–122. [DOI] [PubMed] [Google Scholar]
- Hudson C.M., Puckett E.E., Bekaert M., Pires J.C., Conant G.C.. 2011. Selection for higher gene copy number after different types of plant gene duplications. Genome Biol. Evol. 3:1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson G.P. 1988. Revision of Castanea sect Balanocastanon (Fagaceae). J. Arnold Arbor. 25–49. [Google Scholar]
- Johnson M.G., Gardner E.M., Liu Y., Medina R., Goffinet B., Shaw A.J., Zerega N.J.C., Wickett N.J.. 2016. HybPiper: extracting coding sequence and introns for phylogenetics from high-throughput sequencing reads using target enrichment. Appl. Plant Sci. 4:1600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.G., Pokorny L., Dodsworth S., Botigué L.R., Cowan R.S., Devault A., Eiserhardt W.L., Epitawalage N., Forest F., Kim J.T., Leebens-Mack J.H., Leitch I.J., Maurin O., Soltis D.E., Soltis P.S., Wong G.K., Baker W.J., Wickett N.J.. 2019. A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering. Syst. Biol. 68:594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kates H.R., Johnson M.G., Gardner E.M., Zerega N.J.C., Wickett N.J.. 2018. Allele phasing has minimal impact on phylogenetic reconstruction from targeted nuclear gene sequences in a case study of Artocarpus. Am. J. Bot. 105:404–416. [DOI] [PubMed] [Google Scholar]
- Katoh K., Standley D.M.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30:772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R., Calcott B., Ho S.Y.W., Guindon S.. 2012. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29:1695–1701. [DOI] [PubMed] [Google Scholar]
- Lang P., Dane F., Kubisiak T.L.. 2006. Phylogeny of Castanea (Fagaceae) based on chloroplast trnT-L-F sequence data. Tree Genet. Genomes. 2:132–139. [Google Scholar]
- Lang P., Dane F., Kubisiak T.L., Huang H.. 2007. Molecular evidence for an Asian origin and a unique westward migration of species in the genus Castanea via Europe to North America. Mol. Phylogenet. Evol. 43:49–59. [DOI] [PubMed] [Google Scholar]
- Larridon I., Villaverde T., Zuntini A.R., Pokorny L., Brewer G.E., Epitawalage N., Fairlie I., Hahn M., Kim J., Maguilla E., Maurin O., Xanthos M., Hipp A.L., Forest F., Baker W.J.. 2020. Tackling rapid radiations with targeted sequencing. Front. Plant Sci. 10:1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leache A.D., Chavez A.S., Jones L.N., Grummer J.A., Gottscho A.D., Linkem C.W.. 2015. Phylogenomics of phrynosomatid lizards: conflicting signals from sequence capture versus restriction site associated DNA sequencing. Genome Biol. Evol. 7:706–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon A.R., Emme S.A., Lemmon E.M.. 2012. Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst. Biol. 61:727–744. [DOI] [PubMed] [Google Scholar]
- Lemmon E.M., Lemmon A.R.. 2013. High-throughput genomic data in systematics and phylogenetics. Annu. Rev. Ecol. Evol. Syst. 44:99–121. [Google Scholar]
- Leonard S. 2006. A new species of witch-hazel (Hamamelis: Hamamelidaceae) apparently endemic to Southern Mississippi. SIDA, Contributions to Botany 22(2):849–856. [Google Scholar]
- Léveillé-Bourret É., Starr J.R., Ford B.A., Moriarty Lemmon E., Lemmon A.R.. 2018. Resolving rapid radiations within angiosperm families using anchored phylogenomics. Syst. Biol. 67:94–112. [DOI] [PubMed] [Google Scholar]
- Li H., Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Bogle A.L., Klein A.S., Donoghue M.J.. 2000. Phylogeny and biogeography of Hamamelis (Hamamelidaceae). Harv. Pap. Bot. 5:171–178. [Google Scholar]
- Limborg M.T., Seeb L.W., Seeb J.E.. 2016. Sorting duplicated loci disentangles complexities of polyploid genomes masked by genotyping by sequencing. Mol. Ecol. 25:2117–2129. [DOI] [PubMed] [Google Scholar]
- Madlung A. 2013. Polyploidy and its effect on evolutionary success: old questions revisited with new tools. Heredity 110:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney D.L., Walker R.M., Morris S.W., McIntosh A.M., Porteous D.J., Evans K.L.. 2016. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genom. Data. 9:22–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack J.E., Hird S.M., Zellmer A.J., Carstens B.C., Brumfield R.T.. 2013. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol. Phylogenet. Evol. 66:526–538. [DOI] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A.. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney G.J., Waples R.K., Seeb L.W., Seeb J.E.. 2017. Paralogs are revealed by proportion of heterozygotes and deviations in read ratios in genotyping-by-sequencing data from natural populations. Mol. Ecol. Resour. 17:656–669. [DOI] [PubMed] [Google Scholar]
- Medina R., Johnson M.G., Liu Y., Wickett N.J., Shaw A.J., Goffinet B.. 2019. Phylogenomic delineation of Physcomitrium (Bryophyta: Funariaceae) based on targeted sequencing of nuclear exons and their flanking regions rejects the retention of Physcomitrella, Physcomitridium and Aphanorrhegma. J. Syst. Evol. 57:404–417. [Google Scholar]
- Miller M.A., Pfeiffer W., Schwartz T.. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE), 14Nov. 2020; New Orleans, LA. p. 1–8. [Google Scholar]
- Murphy B., Forest F., Barraclough T., Rosindell J., Bellot S., Cowan R., Golos M., Jebb M., Cheek M.. 2020. A phylogenomic analysis of Nepenthes (Nepenthaceae). Mol. Phylogenet. Evol. 144:106668. [DOI] [PubMed] [Google Scholar]
- Nguyen L.T., Schmidt H.A., Von Haeseler A., Minh B.Q.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon K. 1997. Castanea. In: Flora of North America Editorial Committee, editors. Flora of North America North of Mexico, Vol. 3. New York: Oxford University Press. p. 439–442. [Google Scholar]
- Ohwi J. 1978. Hamamelis. In: Flora of Japan. Tokyo: Shibundo Co. Ltd. Publishers. p. 1–724. [Google Scholar]
- One Thousand Plant Transcriptomes Initiative. 2019. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574:679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pais A.L., Li X., Jenny Xiang Q.-Y.. 2018. Discovering variation of secondary metabolite diversity and its relationship with disease resistance in Cornus florida L. Ecol. Evol. 8:5619–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pais A.L., Whetten R.W., Xiang Q.-Y.J.. 2017. Ecological genomics of local adaptation in Cornus florida L. by genotyping by sequencing. Ecol. Evol. 7:441–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. 2012. FigTree v1. 4. Available from: https://github.com/rambaut/figtree/releases/tag/v1.4.4. [Google Scholar]
- Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A.. 2018. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67:901–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaz R., Bayzid M.S., Rahman M.S.. 2014. Accurate phylogenetic tree reconstruction from quartets: a heuristic approach. PLoS One 9:e104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensing S.A. 2014. Gene duplication as a driver of plant morphogenetic evolution. Curr. Opin. Plant Biol. 17:43–48. [DOI] [PubMed] [Google Scholar]
- Robinson K.M., Hawkins A.S., Santana-Cruz I., Adkins R.S., Shetty A.C., Nagaraj S., Sadzewicz L., Tallon L.J., Rasko D.A., Fraser C.M., Mahurkar A., Silva J.C., Dunning Hotopp J.C.. 2017. Aligner optimization increases accuracy and decreases compute times in multi-species sequence data. Microb. Genom. 3:e000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F., Huelsenbeck J.P.. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S., Takahashi D., Setoguchi H., Isagi Y.. 2018. Genetic structure of the clonal herb Tanakaea radicans (Saxifragaceae) at multiple spatial scales, revealed by nuclear and mitochondrial microsatellite markers. Plant Species Biol. 33:81–87. [Google Scholar]
- Sargent C.S., editor. 1890. Hamamelideae-Sapotaceae. In: The silva of North America, vol. 5. Boston and New York: Houghton, Mifflin. [Google Scholar]
- Shee Z.Q., Frodin D.G., Cámara-Leret R., Pokorny L.. 2020. Reconstructing the complex evolutionary history of the Papuasian Schefflera radiation through herbariomics. Front. Plant Sci. 11:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J.T., Wong K., Jackman S.D., Schein J.E., Jones S.J.M., Birol I.. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res. 19:1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler T. 2010. Sampling-through-time in birth-death trees. J Theor Biol. 267:396–404. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley P.C. 1937. Studies of American plants, VII. Field Mus. Nat. Hist., Bot. ser. 17:155–224. [Google Scholar]
- Swofford D.L. 2003. PAUP*: phylogenetic analysis using parsimony (*and other methods) Version 4. Sinauer Associates, Sunderland, Massachusetts. Available from: https://paup.phylosolutions.com/. [Google Scholar]
- Thomas S.K., Liu X., Du Z., Dong Y., Cummings A., Pokorny L., Xiang Q.-Y., Leebens-Mack J.. 2021. Comprehending the cornales: phylogenetic reconstruction of the order using the angiosperms 353 probe set. Am. J. Bot. 108. doi: 10.1002/ajb2.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloch C.M., Schrago C.G.. 2012. Impact of the partitioning scheme on divergence times inferred from mammalian genomic data sets. Evol. Bioinform. 8:EBO.S9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Tian S., Sun X., Cheng X., Duan N., Tao J., Shen G.. 2020. Construction of pseudomolecules for the Chinese Chestnut (Castanea mollissima) genome. G3-GENES GENOM. GENET. 10:3565–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitemier K., Straub S.C.K., Cronn R.C., Fishbein M., Schmickl R., McDonnell A., Liston A.. 2014. Hyb-Seq: combining target enrichment and genome skimming for plant phylogenomics. Appl. Plant Sci. 2:1400042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J., Shi S.. 1999. A phylogenetic and biogeographic study of Hamamelis (Hamamelidaceae), an eastern Asian and eastern North American disjunct genus. Biochem. Syst. Ecol. 27:55–66. [Google Scholar]
- Wiens J.J., Morrill M.C.. 2011. Missing data in phylogenetic analysis: reconciling results from simulations and empirical data. Syst. Biol. 60:719–731. [DOI] [PubMed] [Google Scholar]
- Xiang Q.-Y., Crawford D.J., Wolfe A.D., Tang Y.-C., DePamphilis C.W.. 1998. Origin and biogeography of Aesculus L. (Hippocastanaceae): a molecular phylogenetic perspective. Evolution 52:988–997. [DOI] [PubMed] [Google Scholar]
- Xie L., Yi T.-S., Li R., Li D.-Z., Wen J.. 2010. Evolution and biogeographic diversification of the witch-hazel genus (Hamamelis L., Hamamelidaceae) in the Northern Hemisphere. Mol. Phylogenet. Evol. 56:675–689. [DOI] [PubMed] [Google Scholar]
- Yu G., Lam T.T.-Y., Zhu H., Guan Y.. 2018. Two methods for mapping and visualizing associated data on phylogeny using Ggtree. Mol. Biol. Evol. 35:3041–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Rabiee M., Sayyari E., Mirarab S.. 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 19:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Lu A.. 1995. Hamamelidaceae: geographic distribution, fossil history and origin. Chin. Sci. Abstracts Ser. B. 6:37. [Google Scholar]
- Zhou W., Xiang Q.-Y., Wen J.. 2020. Phylogenomics, biogeography, and evolution of morphology and ecological niche of the eastern Asian–eastern North American Nyssa (Nyssaceae). J. Syst. Evol. 58:571–603. [Google Scholar]