

T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematologic neoplasm derived from transformed T-cell progenitors. Even though intensive chemotherapy results in long-term survival in ∼80% of children and the majority of adults, this treatment is associated with severe side effects and relapsed disease is refractory to treatment.1 Better understanding of the molecular pathogenesis of T-ALL to develop targeted therapies with greater efficacy and lower toxicities is needed. Aberrant cell growth and proliferation of T-ALL lymphoblasts are sustained by activating strong oncogenic drivers, such as NOTCH1, which is activated in >60% of T-ALL cases by genetic mutations in the NOTCH1 gene itself and regulators of NOTCH1 signaling (eg, FBXW7).2 Multiple elegant studies demonstrate that NOTCH1 directly activates MYC transcription and that MYC is an essential downstream effector contributing to multiple aspects of leukemogenesis.3–5 Dysregulated MYC signaling drives T-ALL proliferation and maintains the activity of leukemia stem cells.6 The essential role of MYC in T-ALL development is solidly validated in multiple leukemia models where MYC inhibition blocks tumor growth.7 Together, these data suggest that targeting MYC may be efficacious in T-ALL therapy. However, direct inhibition of MYC has proven difficult to date due to the lack of functional MYC domains for small molecule binding.
Alternative efforts have focused on indirectly inhibiting MYC by targeting MYC posttranslational regulation, such as accelerating MYC degradation.8 In a recent study published in Cancer Cell, Jiang and colleagues uncover a new molecular mechanism underlying MYC protein stabilization and the molecular pathogenesis of T-ALL that provides a novel potential strategy for therapeutically targeting MYC.9 The authors performed co-immunoprecipitation to search for MYC interacting proteins in T-ALL and identified Aurora B kinase (AURKB) as a novel MYC binding partner. Using in vitro kinase assays and mass spectrometry analysis, they found that AURKB catalyzed a phosphorylation reaction on MYC at the serine 67 residue (S67). The authors then assessed MYC protein stability using phosphor-mimetic or deficient mutants, and demonstrated that S67 phosphorylation stabilized MYC protein and enhanced its transcriptional activity.
It is well established that MYC protein stability is regulated by phosphorylation. In particular, enhanced phosphorylation at serine 62 residue (S62) prolongs MYC protein half-life.8 Previous reports have revealed protein kinases that catalyze S62 phosphorylation in a variety of tumor contexts, such as breast cancer and colorectal cancer,10 leading to stabilized MYC and enhanced oncogenic potency. Interestingly, S62 phosphorylation is required for GSK3β to recognize MYC and phosphorylate the threonine 58 residue (T58), resulting in the FBXW7-mediated polyubiquitination and 26S proteasomal degradation of MYC.11 In contrast to S62 phosphorylation, Jiang et al show that S67 phosphorylation blocks the interaction between MYC and GSK3β, thereby inhibiting the T58 phosphorylation and MYC degradation. Both S62 and S67 reside in the N-terminal MYC Box I region that constitutes a phosphor-degron with several conserved serine and threonine residues and controls MYC protein degradation. It is possible that phosphor-S67 prevents MYC from binding to GSK3β and acts in concert with S62 phosphorylation to promote MYC accumulation. Yet, the sequential order of these phosphorylation events in vivo and how they affect each other remains to be elucidated.
Jiang et al further showed that stabilized MYC directly bound to the AURKB locus and activates its transcription, resulting in a positive feedback loop that amplifies MYC oncogenic activity (Fig. 1). The authors went on to show that the AURKB-MYC axis is essential for T-ALL progression using a MYC transgenic T-ALL zebrafish model. They showed that co-expressing MYC and AURKB transgenes significantly accelerated leukemogenesis in comparison to expressing the MYC transgene alone. To address the functional importance of the MYC S67 phosphorylation event, they generated phosphor-mimetic and -deficient mutants. By expressing these mutant alleles, Jiang and colleagues revealed that constitutive phosphorylation at the S67 residue enhanced T-cell leukemogenesis, resembling the co-expression of MYC and AURKB transgenes, whereas loss of phosphorylation failed to do so. This study thus presents compelling in vivo evidence to support that this new mechanism for MYC activation contributes to leukemogenesis.
Figure 1.
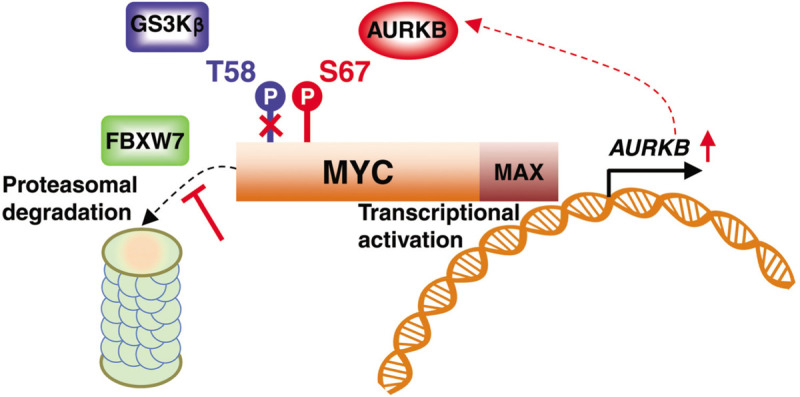
AURKB directly phosphorylates MYC, counteracting GSK3β-mediated T58 phosphorylation, FBXW7-mediated polyubiquitination, and proteasomal degradation. Stabilized MYC directly activates AURKB transcription, constituting a reciprocal activation loop between MYC and AURKB.
For therapeutic purposes, the authors evaluated the efficacy of an AURKB inhibitor-based targeted strategy in T-ALL. They found that the AURKB inhibitor AZD1152, either alone or in combination with vincristine (a first-line drug used in treating T-ALL), exhibited potent antileukemia effect. Notably, these treatments selectively repressed FBXW7 wild-type T-ALL and had minimal effect on T-ALL with FBXW7 inactivating mutations, suggesting that FBXW7 gene mutation status may serve as a biomarker predicting drug sensitivity. AURKB is a well-characterized cell-cycle regulator, which is overexpressed in both ALL and acute myeloid leukemia (AML), and essential for leukemic cell growth, proliferation, and survival. It is very likely that AZD1152 inactivates both cell cycle targets and MYC to exert its antileukemic function. To determine the essential role of MYC down-regulation in AZD1152-induced cell death, the authors introduced a phosphor-mimetic MYC S67 allele in AZD1152-sensitive KOPTK1 cells and showed that this overexpression significantly rescued cell death and reverted MYC target gene down-regulation. Moreover, depleting MYC in AZD1152-resistant Jurkat cells sensitized these cells to drug treatment, suggesting that decreased MYC expression is required for T-ALL cells to respond to AZD1152.
In summary, this work from Jiang and colleagues proposes a mechanism-based MYC-targeted approach that has the potential to improve T-ALL therapy. Oncoproteins of the MYC family are major drivers of human cancers and are overexpressed in about 50% of cancers.12 Whether MYC S67 phosphorylation is involved in the development of other tumors remains elusive. It is also unknown whether this phosphorylation promotes MYC stabilization and whether it is regulated by AURKB in other tumor contexts. If so, the implications of this study may pave a new avenue for treatment of other MYC-dependent tumors. This elegant work provides convincing evidence that S67 phosphorylation is a novel modification event to trigger MYC activation and is important for oncogenic functions, at least in T-ALL, thereby presenting an essential stepping stone to fully understand MYC functions in cancer.
REFERENCES
- [1].Raetz EA, Teachey DT. T-cell acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 2016;2016(1):580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer 2016;16(8):494–507. [DOI] [PubMed] [Google Scholar]
- [3].Weng AP, Millholland JM, Yashiro-Ohtani Y, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev 2006;20(15):2096–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu H, Chi AW, Arnett KL, et al. Notch dimerization is required for leukemogenesis and T-cell development. Genes Dev 2010;24(21):2395–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yashiro-Ohtani Y, Wang H, Zang C, et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc Natl Acad Sci U S A 2014;111(46):E4946–E4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].King B, Trimarchi T, Reavie L, et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 2013;153(7):1552–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 1999;4(2):199–207. [DOI] [PubMed] [Google Scholar]
- [8].Farrell AS, Sears RC. MYC degradation. Cold Spring Harb Perspect Med 2014;4(3): [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jiang J, Wang JC, Yue M, et al. Direct phosphorylation and stabilization of MYC by Aurora B kinase promote T-cell leukemogenesis. Cancer Cell 2020;37(2):200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tan J, Li Z, Lee PL, et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov 2013;3(10):1156–1171. [DOI] [PubMed] [Google Scholar]
- [11].Welcker M, Orian A, Jin J, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A 2004;101(24):9085–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dang CV. MYC on the path to cancer. Cell 2012;149(1):22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]