

Abstract
Background:
Although the leucine-rich repeat kinase 2 p.G2019S mutation has been demonstrated to be a strong risk factor for PD, factors that contribute to penetrance among carriers, other than aging, have not been well identified.
Objectives:
To evaluate whether a cumulative genetic risk identified in the recent genome-wide study is associated with penetrance of PD among p.G2019S mutation carriers.
Methods:
We included p.G2019S heterozygote carriers with European ancestry in three genetic cohorts in which the mutation carriers with and without PD were selectively recruited. We also included the carriers from two data sets: one from a case-control setting without selection of mutation carriers and the other from a population sampling. Associations between polygenic risk score constructed from 89 variants reported recently and PD were tested and meta-analyzed. We also explored the interaction of age and PRS.
Results:
After excluding eight homozygotes, 833 p.G2019S heterozygote carriers (439 PD and 394 unaffected) were analyzed. Polygenic risk score was associated with a higher penetrance of PD (odds ratio: 1.34; 95% confidence interval: [1.09, 1.64] per +1 standard deviation; P = 0.005). In addition, associations with polygenic risk score and penetrance were stronger in the younger participants (main effect: odds ratio 1.28 [1.04, 1.58] per +1 standard deviation; P = 0.022; interaction effect: odds ratio 0.78 [0.64, 0.94] per +1 standard deviation and + 10 years of age; P = 0.008).
Conclusions:
Our results suggest that there is a genetic contribution for penetrance of PD among p.G2019S carriers. These results have important etiological consequences and potential impact on the selection of subjects for clinical trials.
Keywords: G2019S, genetics, LRRK2, Parkinson’s disease, polygenic risk score
Parkinson’s disease (PD) is a complex genetic disorder, where rare and highly damaging variants as well as common risk variants play a role in its etiology.1 The leucinc-rich repeat kinase 2 (LRRK2) p.G2019S (rs34637584) mutation is one of the major known contributors to PD.2,3 The p.G2019S mutation has an estimated prevalence of approximately 1% in the PD population of European ancestry, with much higher frequencies being reported for North African Berber Arab populations and European subpopulations with high Ashkenazi Jewish ancestry.4-7 The p.G2019S mutation is not fully penetrant, with risk of PD for carriers increasing with age. At age 80 years, 25% to 42.5% of carriers will have PD.7,8 A critical question that remains centers on what the determinants of p.G2019S penetrance are. Previously, it has been reported that a variant in DNM3, encoding the vesicular transport protein dynamin 3, is a potential LRRK2 p.G2019S age-at-onset modifier, lowering age at onset by approximately 8 years.9 However, other reports have not replicated this finding and instead nominated variants in SNCA as modifiers of LRRK2 mutation penetrance.10 The underlying pathogenic mechanism of LRRK2-linked PD is currently unknown, but a great deal of focus has been placed on the ability of the p.G2019S mutation to increase LRRK2 kinase activity, especially given that loss-of-function mutations do not seem to be contributing to disease.11,12
In the last several years, genome-wide association studies (GWAS) have identified an ever-increasing number of risk loci for PD. Individually, each of these loci confers modest effects on risk for disease. Polygenic risk score (PRS) represents known cumulative genetic risk across these loci in each assayed individual. PRS reveals that, collectively, these risk variants confer considerable risk for disease, with those in the top decile of genetic risk being 6-fold more likely to have PD than those in the lowest decile of genetic risk in the European population.1 PRS is also highly correlated with age at onset of PD13,14 and is associated with penetrance of damaging GBA variants.15 Here, we investigate the influence of the latest PD PRS on age at onset and penetrance of PD in p.G2019S carriers using several large cohorts.
Materials and Methods
Whole-Genome Sequencing
Genetics and clinical data used in the preparation of this article were obtained from the Parkinson’s Progression Markers Initiative (PPMI) database (www.ppmi-info.org/data). (Eor up-to-date information on the PPMI study, visit www.ppmi-info.org.) In this study, we included three genetic cohorts: Parkinson’s Progression Markers Initiative Genetic Cohort (PPMI_GC); Parkinson’s Progression Markers Initiative Genetic Registry (PPMI_GR); and the LRRK2 Consortium Cohort (LCC), hereafter collectively named the Genetic Cohort Dataset. These cohorts selectively included people with specific high-risk genetic variants, such as carrying p.G2019S, for both cases and unaffected individuals. Whole-genome sequencing data were generated at the Laboratory of Neurogenetics at the National Institutes of Health, and detailed methods are available from the study website. After downloading, subsequent genotypes were filtered to exclude minor allele frequency of <0.01, missing rate > 5%, and the test for Hardy-Weinberg equilibrium threshold of 1.0E-4 in controls. Also, the participants with high missingness (sample call rate < 95%), sex discordance, extreme heterozygosity (F statistics <0.15), and excess relatedness (pair-wise kinship coefficients >0.125) were excluded from the analyses set. Following quality control, 185 carriers of p.G2019S (PD/non-PD = 88/97) in PPMI_GC, 75 (38/37) in PPMI_GR, and 175 (107/68) in LCC were included in the analysis. Among them, 7 individuals (3 in PPMI_GC, 3 in PPMI_GR, and 1 in LCC, all cases) were homozygotes.
Genotype Data From the International Parkinson Disease Genomics Consortium
Aggregated genotype data were obtained from the International Parkinson Disease Genomics Consortium (IPDGC), as previously described.1 In brief, genotypes were processed using standard pipelines and imputed using the HRC imputation panel r1.1 201616 through the Michigan imputation server under the default setting with phasing using the EAGLE option.17 Genotypes were filtered for imputation quality R2 > 0.8. Following quality-control criteria similar to that applied to the Genetic Cohort Dataset, a total of 227 carriers were included in the analysis, of which 208 were PD cases and 19 were controls. Among them, 1 PD patient was homozygous. For composing PRS, we used a threshold of R2 > 0.3 to obtain dosages of more risk-associated alleles because only 64 variants were passed with the cutoff of 0.8 in R2.
UK Biobank Data
The UK Biobank (UKBB) is a large, long-term biobank study in the UK that includes genetic data and a wide range of phenotypes of approximately 500,000 individuals.18 PD case-control status was based on multiple field codes, including 41202, 41204, 40002, and 20002. LRRK2 p.G2019S was directly genotyped in the UKBB cohort (rs34637584; n = 314), and the concordance rate in the whole-exome sequencing data19 was 100%. After applying the same quality-control steps as above, 179 carriers were identified, of which 6 were reported to be PD cases; all were heterozygotes.
PRS Calculation
PRS was calculated incorporating the risk loci previously associated with PD excluding the rs34637584, which is the mutation of LRRK2, P.G2019S.1 In the calculation of PRS, risk allele dosages were summed with weights as their published beta estimates, giving greater weight to alleles with higher estimates. Then, PRSs were standardized to each cohort level. For the UKBB and IPDGC imputed genotype data, not all 89 variants were available because of lower imputation scores (R2 cutoff of 0.3); therefore, 89 variants in the UKBB and a median of 89 variants (range, 82–89) in IPDGC were used. If a variant was missing, the weight of that variant was imputed with the cohort mean allele dosages.
Statistical Analysis
Our primary analysis was to test the association between PRS and PD odds using a fixed-effect meta-analysis model among the mutation carriers. First, a logistic regression model was applied for the main effect of PRS for the Genetic Cohort Dataset (PPMI_GC/PPMI_GR/LRRK2), IPDGC dataset, and UKBB dataset separately. Data were adjusted for age, sex, cohort, and PCs representing population structure. Across all datasets, linear age upon recruitment was used for carriers without PD and age at diagnosis for carriers with PD, whereas age at onset was used for the IPDGC dataset and age at recruitment for the UKBB dataset. Age data were centered—the original value subtracted by the mean age at each cohort—to accommodate square age variable in the later step. To adjust for population structure, PC1 to 10 were used for the Genetic Cohort Dataset and IPDGC dataset, and PC1 to 5 were used for the UKBB dataset because of its size. The associations between PD and PRS in three datasets were meta-analyzed using the fixed-effect model with the inverse variances as weights.
In the secondary analysis, we analyzed the main effect of PRS as well as the interaction effect of age and PRS in the Genetic Cohort Dataset and IPDGC dataset. In this model, we adjusted for age, sex, cohort, top 10 PCs, and square age (model specifications are in the Supporting Information Appendix). We meta-analyzed the dataset level odds ratios (ORs) for the main effect and the interaction effect between age and PRS using the fixed-effect model with inverse-variance weighting. Square age was significantly associated with the PD odds in these two datasets; given that the UKBB dataset cannot accommodate the variable because of too few PD cases, we thus could not include square age in the primary analysis.
Also, the association of PRS on age at diagnosis (Genetic Cohort Dataset) or age at onset (IPDGC dataset) for the carriers with PD was analyzed in an exploratory manner. Adjusted covariates included age, square age, sex, cohorts, and PC1 to 10.
There were eight homozygotes of p.G2019S mutation in the analysis set, and we excluded them from all analyses. Analyses were conducted by using R software (version 3.5.1; https://cran.r-project.org/), with a significance level of 0.05 (two-sided). In the previous report of a case-control study of people with the European ancestry, the mean beta-coefficient estimation of +1 standard deviation (SD) in PRS was 0.709 (standard error [SE] = 0.072) with the sample size of 999 (case = 527, control = 472). If we would have had the same effect in the current data, we expect the effect size (beta estimate/standard deviation) of 0.312 requiring 216 participants per group for the power of 0.9 and a significance level of 0.05. With the sample size here, we had 90% power for the primary analysis. Similarly, if we expect the effect size of 0.074 derived from the previous report for the association between age at onset in European ancestry,13 at the significance level of 0.05 and the sample size of 433 (226 from the Genetic Cohort Dataset and 207 from the IPDGC dataset), the study has the power of 0.34.
Patients’ Consent
Participants’ information and genetic samples were obtained under appropriate written consent and with local institutional and ethical approvals for all cohorts and datasets in this study.
Results
Data Overview
In total, of three datasets there were 841 carriers, including 394 heterozygote carriers without PD, 439 heterozygote with PD, and 8 homozygotes with PD. Seven Homozygotes with PD in the Genetic Cohort Dataset had a higher PRS and older age at diagnosis compared to heterozygous PD cases (Table 1), although the differences were not statistically significant (P = 0.51 for PRS and P = 0.16 for age at diagnosis/onset). Because of the small number of homozygotes, we excluded them from downstream analyses.
TABLE 1.
Carriers of LRRK2 p.G2019S in three datasets
| Dataset |
Genetic Cohort Dataset |
IPDGC |
UKBB |
|||||
|---|---|---|---|---|---|---|---|---|
| LRRK2.pG2019S, allele count | 1 | 2 | 1 | 2 | 1 | |||
| PD status | Non-PD | PD | PD | Non-PD | PD | PD | Non-PD | PD |
| Total, n | 202 | 226 | 7 | 19 | 207 | 1 | 173 | 6 |
| Female, n | 102 | 120 | 4 | 10 | 102 | 0 | 90 | 1 |
| Age at study, year (SD) | 56.33 (13.59) | 67.00 (9.86) | 69.14 (2.06) | 56.58 (20.72) | — | — | 57.77 (7.76) | 66.17 (1.17) |
| Age at onset, year (SD) | — | — | — | — | 62.55 (11.58) | 61.00 (NA) | — | — |
| Age at diagnosis, year (SD) | — | 59.96 (10.51) | 65.68 (2.98) | — | — | — | — | — |
| PRS (SD) | −0.15 (1.03) | 0.12 (0.95) | 0.36 (0.17) | −0.25 (1.14) | 0.08 (0.87) | −1.17 (NA) | −0.11 (0.90) | 1.06 (0.52) |
Genetic Risk Score Modifies Penetrance of LRRK2 p.G2019S
The unadjusted plots of PRS among cases and unaffected individuals in the Genetic Cohort Dataset showed that the mean of the PRS was higher in cases among carriers (Fig. 1). The means of PRS in cases were also higher than those in controls among noncarriers in the IPDGC and UKBB datasets (Supporting Information Fig. S1). Meta-analysis results of the three datasets showed that a higher PRS was significantly associated with a higher OR of PD (OR, 1.34; 95% confidence interval [CI]: [1.09, 1.64] per +1 SD [per standard deviation of increase from the cohort mean; P = 0.005]; Fig. 2).
FIG. 1.

Unadjusted PRS of cases and controls in genetic cohorts. The means of PRS in cases were higher than unaffected among LRRK2 p. G2019S mutation carriers. Age of recruitment for unaffected: LCC 51.5 (15.9), PPMI_GC 62.3 (7.5), and PPMI_GR 49.7 (15.0). Age at diagnosis for PD: LCC 58.1 (11.5), PPMI_GC 61.4 (8.9), and PPMI_GR 62.2 (10.1). All LRRK2 p.G2019S heterozygotes.
FIG. 2.

Meta-analysis for PRS and its interaction with age on penetrance. (A) Primary analysis of testing the association between PRS and PD showed that PRS was significantly associated with the PD odds. (B) Secondary analysis model with PRS and the interaction between PRS and age showed that the main effect of PRS as well as the interaction between PRS and age were significantly associated with the odds of PD, but in the opposite direction. Compared to the interaction effect among noncarriers in the IPDGC, the magnitude of the interaction among the mutation carriers was large. I_sq, I square (%); Q-test, P value for the test of heterogeneity. Odds ratios were adjusted for study, age, square age, sex, and PC1 to 10.
Stronger Association of Genetic Risk Score for PD Penetrance in Younger Carriers
In the secondary analysis examining the interaction between PRS and age, the main effect of PRS as well as the interaction between PRS and age were significantly associated with the odds of PD, but in the opposite direction (main effect: OR, 1.28 [1.04, 1.58] per +1 SD; P = 0.022; interaction effect: OR, 0.78 [0.64, 0.94] per +1 SD and +10 years of age; P = 0.008; Fig. 2). This suggests that in the studied cohorts, the magnitude of association between PRS and PD odds were stronger when the age of the participants was younger. We added post-hoc analyses stratified by age (<55, 55–65, and < 65 years) to illustrate the stronger association of PRS in younger age in carriers (Fig. 3). The analyses examining the main effect and interaction effect among nonmutation carriers in the IPDGC dataset showed that these effects were also significant in nonmutation carriers (the main term for +1 SD in PRS: OR, 1.6; P = 2.5E-245; the interaction term of +1 SD in PRS and + 10 years in age: 0.94; P = 1.4E-9). However, the magnitude of the interaction effect was smaller than that in LRRK2 p.G2019S carriers, and the magnitudes of associations were significantly different between carriers and noncarriers in the test of homogeneity (P = 0.044).
FIG. 3.
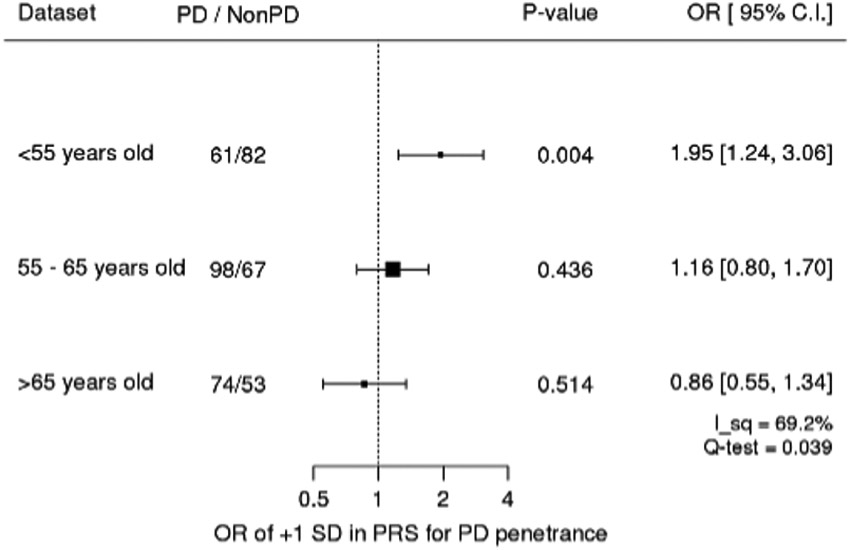
Age stratified associations of PRS on penetrance in Genetics Cohorts Dataset. Age-stratified analysis showed that PRS was significantly associated with youngerage. I_sq, I square (%); Q-test, P value for the test of heterogeneity. Odds ratios were adjusted for study, age, square age, sex, and PC1 to 10. All LRRK2 p.G2019S heterozygotes.
Genetic Risk Score and Age at Onset/Diagnosis in LRRK2 p.G2019S Cases
Previously, we have shown that PRS is inversely correlated with age at onset in PD cases.13 In the current study, there was insufficient evidence that PRS was significantly associated with age at onset/diagnosis of PD in LRRK2 p.G2019S carriers. The directionality was the same as in the general population previously studied (−0.801 [−0.959, −0.643] per +1 SD in PRS), but the association did not reach significance level (Supporting Information Fig. S2). As mentioned in the power calculation analyses in Materials and Methods, this is an underpowered study, and therefore additional data are needed to confirm these results.
Exploratory Analysis for the Individual PD-Associated Variants
Although the power of the study is not sufficient, the motivation for this analysis was to qualitatively assess whether any of the risk variants could he strong penetrance modifiers. The Q-Q plot for the observed and expected P in -log10 scale showed an upper deviation from the expected line (lambda value of 1.34), indicating the contribution of these variants for the PD risk in p.G2019 LRRK2 heterozygous carriers. However, no single association stood out as a possible penetrance modifier after adjusting for multiple tests (Supporting Information Fig. S3). Furthermore, when the estimates for the associations of carriers and noncarriers of individual variants were compared, there were 10 variants whose tests of homogeneity were rejected at a significance level of 0.05 (Supporting Information Fig. S4; Supporting Information Table S2); however, none of these were significantly associated with age at onset/diagnosis among cases. Additionally, GBA mutations in LRRK2 p.G2019S carriers (Supporting Information Table S3) were analyzed, but there was no clear enrichment of GBA mutations in cases versus controls or for younger age at onset among cases.
Discussion
The aggregated genetic risks in the form of PRS that were constructed from the recent case-control GWAS of the European population was significantly associated with the risk of PD among heterozygous LRRK2 p.G2019S mutation carriers in our study. Although the estimated magnitude of association between PRS and PD in the UKBB seems unrealistically large (OR = 7.64 [1.34, 43.46]), the estimation has a large SE because of the small number of cases. In the meta-analysis, we used inverse-variance weighting, which took into account the certainty of the estimation when combining the results. The actual weights in the meta-analysis of the primary analysis were 88% from Genetic Cohort Dataset, 11% from IPDGC, and 1.3% from the UKBB. For the analysis including the interaction, we observed a negative association for interaction between PRS and age. This may he counterintuitive because the average age of PD diagnosis of the LRRK2 p.G2019S carriers is >55 years. The interpretation of the results is that the mutation plays a larger role as the carriers get older whereas the heavier genetic burden for PD in common European populations is important for carriers who develop PD at a younger age, although we did not have enough evidence for the association between PRS and age at onset/diagnosis likely because of limited power. Additionally, although age at onset and age at diagnosis is highly correlated, clinical site variation and differences in assessing this could affect the analysis. Therefore, more data are required to conclusively determine this point.
The risk of mutation carriers in the current study is seemingly representing the carriers with European ancestry (Supporting Information Table S1). In the IPDGG, 1.4% of the cases had the LRRK2 p.G2019S mutation, and 0.11% of the controls carried the mutation. The prevalence of PD in carriers is consistent with results from a previous report of 1% of PD patients carrying the mutation.7 Among the 362,884 participants passing filtering criteria in the UKBB, 0.049% were carriers of the LRRK2 p.G2019S mutation and approximately 10 times higher crude risk of PD associated with the mutation is agreeing with the IPDGC dataset. In addition, the meta-analysis does not show the heterogeneity of the estimation in three datasets.
The primary limitation of this study is the size, despite including several large cohorts. LRRK2 p.G2019S is rare in the general population, and therefore actively recruiting carriers would be a good strategy to increase power for studies such as ours. To investigate the penetrance of carriers, recruitment only focusing on carriers ia very effective, as shown in Table 1. More initiatives and future collaboration with clinical trials only targeting LRRK2 p.G2019S mutations are warranted. Currently, there are at least three clinical trials targeting LRRK2-linked PD: two use kinase inhibition, and the third focuses on reducing the protein load. As these trials move forward, important considerations are whether the disease mechanisms that cause LRRK2-linked PD are generalizable to typical PD, and whether it is possible to identify which LRRK2 mutation carriers will or will not express disease. Previously, we have shown that active randomization on genetic components of PD is important for successful clinical trials because unbalanced genetic risk may lead to the heterogeneity of intervention arms and lower the power of that trial.20 Our results suggest that actively balancing PRS is similarly important for a diseasc-modifying/-preventing trial targeting p.G2019S carriers.
Another limitation is that case/control status was determined at study enrollment. Some carriers who did not have PD at enrollment may develop the disease later. Although we tried to address this by adding age covariates in the analytical models, this type of mis-classification might change the results. Also, we used several types of ages for participants with PD in our models. We used age at diagnosis for the Genetic Cohort Dataset, but age at onset for the IPDGC dataset. These two measurements are expected to be highly correlated. A previous study reported a correlation coefficient of 0.917.13 We used age at recruitment for the UKBB because age at onset or age at diagnosis was unavailable in the dataset. Unless there was a systematic association between the duration of the disease at recruitment and variants, which we do not usually expect, the study estimations were not biased, although measurement variance might reduce the power of the study.
Taken together, we show that penetrance, and likely age at onset of LRRK2, is modified by PD PRS, implying a clear genetic influence.
Supplementary Material
Acknowledgments:
We thank all of the subjects who donated their time and biological samples to be a part of this study. We also thank all members of the International Parkinson Disease Genomics Consortium (IPDGC). See for a complete overview of members, acknowledgements, and funding: http://pdgenetics.org/partners. This work was supported, in part, by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services; project ZO1 AG000949.
Data used in preparation of this article were obtained from the Michael J Fox Foundation (MJFF)-sponsored LCC and PPMI database (www.ppmi-info.org/data). For up-to-date information on these studies, visit www.michaeljfox.org/lcc and www.ppmi-info.org. PPMI—a public-private partnership—is funded by The Michael J. Fox Foundation for Parkinson’s Research and funding partners, including AbbVie, Allergan, Avid Radiopharmaceuticals, Biogen, BioLegend, Bristol-Myers Squibb, (Celgene, Denali Incorporated, GE Healthcare, (Genentech, GlaxoSmithKline, Eli Lilly and Company, Lundbeck, Merck & Co., Meso Scale Discovery, Pfizer, Piramal, Prevail Therapeutics, Roche, Sanofi (Genzyme, Servier Laboratories, Takeda, Teva, UCB, Verily, Voyager Therapeutics, and Golub Capital (www.ppmi-info.org/fundingpartners). LCC is coordinated and funded by The Michael J. Fox Foundation for Parkinson’s research. The investigators within the LCC contributed to the design and implementation or the LCC and/or provided data and/or collected biospecimens, but did not necessarily participate in the analysis or writing of this report. The full list of LCC investigators can be found at: www.michaeljfox.org/lccinvestigators.
Footnotes
Relevant conflicts of interest/financial disclosures: Nothing to report.
Full financial disclosures and author roles may be found in the online version of this article.
Supporting Data
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
References
- 1.Nalls MA, Blauwendraat C, Vallerga CL, et al. Expanding Parkinson’s disease genetics: novel risk loci, genomic context, causal insights and heritable risk. bioRxiv. 2019. Available at: https://www.biorxiv.org/content/10.1101/388165v3. Accessed January 9, 2020. [Google Scholar]
- 2.Nichols WC, Pankratz N, Hernandez D, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 2005;365:410–412. [DOI] [PubMed] [Google Scholar]
- 3.Gilks WP, Abou-Sleiman PM, Gandhi S, et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 2005;365:415–416. [DOI] [PubMed] [Google Scholar]
- 4.Bras JM, Guerreiro RJ, Ribeiro MH, et al. G2019S dardarin substitution is a common cause of Parkinson’s disease in a Portuguese cohort. Mov Disord 2005;20:1653–1655. [DOI] [PubMed] [Google Scholar]
- 5.Ozelius LJ, Senthil G, Saunders-Pullman R, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med 2006;354:424–425. [DOI] [PubMed] [Google Scholar]
- 6.Lesage S, Dürr A, Tazir M, et al. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med 2006;354:422–423. [DOI] [PubMed] [Google Scholar]
- 7.Healy DG, Falchi M, O’sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol 2008;7:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee AJ, Wang Y, Alcalay RN, et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov Disord 2017;32:1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trinh J, Gustavsson EK, Vilariño-Güell C, et al. DNM3 and genetic modifiers of age of onset in LRRK2 Gly2019Ser parkinsonism: a genome-wide linkage and association study. Lancet Neurol 2016;15:1248–1256. [DOI] [PubMed] [Google Scholar]
- 10.Fernández-Santiago R, Garrido A, Infante J, et al. α-synuclein (SNCA) but not dynamin 3 (DNM3) influences age at onset of leucine-rich repeat kinase 2 (LRRK2) Parkinson’s disease in Spain. Mov Disord 2018;33:637–641. [DOI] [PubMed] [Google Scholar]
- 11.Blauwendraat C, Reed X, Kia DA, et al. ; COURAGE-PD (Comprehensive Unbiased Risk Factor Assessment for Genetics and Environment in Parkinson’s Disease) Consortium, the French Parkinson’s Disease Consortium, and the International Parkinson’s Disease Genomics Consortium (IPDGC). Frequency of loss of function variants in LRRK2 in Parkinson disease. JAMA Neurol 2018;75:1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whiffin N, Armean MI, Kleinman A, et al. Human loss-of-function variants suggest that partial LRRK2 inhibition is a safe therapeutic strategy for Parkinson’s disease. bioRxiv. 2019. Available at: https://www.biorxiv.org/content/10.1101/561472v1. Accessed January 9, 2020. [Google Scholar]
- 13.Blauwendraat C, Heilbron K, Vallerga CL, et al. ; International Parkinson’s Disease Genomics Consortium (IPDGC). Parkinson’s disease age at onset genome-wide association study: defining heritability, genetic loci, and α-synuclein mechanisms. Mov Disord 2019;34:866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nalls MA, Escott-Price V, Williams NM, et al. Genetic risk and age in Parkinson’s disease: continuum not stratum. Mov Disord 2015;30:850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blauwendraat C, Reed X, Nalls M, Krohn L, Heilbron K. Genetic modifiers of risk and age at onset in GBA positive Parkinson’s disease and Lewy body dementia. Brain 2020;143:234–248, 10.1093/brain/awz350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016;48:1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das S, Forer L, Schönherr S, et al. Next-generation genotype imputation service and methods. Nat Genet 2016;48:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Hour VC, Ioanna T, Beckman DJ, et al. Whole exome sequencing and characterization of coding variation in 49,960 individuals in the UK Biobank. bioRxiv. 2019. Available at: https://www.biorxiv.org/content/10.1101/572347v1. Accessed January 9, 2020. [Google Scholar]
- 20.Leonard H, Blauwendraat C, Krohn L, et al. Genetic variability and potential effects on clinical trial outcomes: perspectives in Parkinson’s disease. bioRxiv. 2018. Available at: https://www.biorxiv.org/content/10.1101/427385v2.abstract. Accessed November 29, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.