

Abstract
Androgen receptor (AR) serves as a main therapeutic target for prostate cancer (PCa). However, resistance to anti-androgen therapy (SAT) inevitably occurs. Indomethacin is a nonsteroidal anti-inflammatory drug that exhibits activity against prostate cancer. Recently, we designed and synthesized a series of new indomethacin derivatives (CZ compounds) via Pd (II)-catalyzed synthesis of substituted N-benzoylindole. In this study, we evaluated the antitumor effect of these novel indomethacin derivatives in castration-resistant prostate cancer (CRPC). Upon employing CCK-8 cell viability assays and colony formation assays, we found that these derivatives had high efficacy against CRPC tumor growth in vitro. Among these derivatives, CZ-212-3 exhibited the most potent efficacy against CRPC cell survival and on apoptosis induction. Mechanistically, CZ-212-3 significantly suppressed the expression of AR target gene networks by degrading AR and its variants. Consistently, CZ-212-3 significantly inhibited tumor growth in CRPC cell line-based xenograft and PDX models in vivo. Taken together, the data show that the indomethacin derivative CZ-212-3 significantly inhibited CRPC tumor growth by degrading AR and its variants and could be a promising agent for CRPC therapy.
Keywords: castration-resistant prostate cancer (CRPC), indomethacin derivatives, CZ-212-3, androgen receptor (AR), patient-derived xenograft model (PDX)
Introduction
Prostate cancer (PCa) is the second leading cause of cancer-related death and the most common malignancy among men globally [1, 2]. The main clinically approved therapeutic options include surgery, radiation, androgen deprivation therapy (ADT), androgen receptor (AR)-targeted therapy and chemotherapy/immunotherapy [3]. After exposure to ADT, most PCa tumors inevitably develop into castrated-resistant prostate cancer (CRPC) [4–7], a deadly form of the disease. Two recently approved second-generation AR-targeting drugs, enzalutamide (ENZ) and abiraterone acetate (ABI), represent breakthroughs in CRPC treatment and show benefit in some patients [8]. Although initially effective, most tumors develop resistance to these drugs and progress to incurable diseases [9]. Therefore, there is an urgent need to explore new therapeutic strategies and develop specific agents for CRPC treatment.
Since AR plays a pivotal role in CRPC tumor growth and progression, current therapeutics mostly target the androgen-AR axis by either inhibiting AR activation (e.g., ENZ) or blocking androgen production (e.g., ABI). Numerous studies have demonstrated that de novo and acquired resistance are mainly caused by the generation of active AR mutations and variants [10]. Most AR variants are constitutively active and not responsive to anti-androgen therapies due to the lack of a functional ligand binding domain (LBD) [10, 11]. Therefore, agents against the AR LBD cannot fundamentally address the aforementioned therapeutic resistance, and developing agents to degrade AR and its variants may be an ideal strategy for CRPC therapy.
The FDA-approved nonsteroidal anti-inflammatory drug (NSAID) indomethacin, a cyclooxygenase inhibitor, is widely used to treat arthritis, gout, bursitis, and tendonitis to relieve pain, fever, joint stiffness, etc. [12]. Previous studies suggest that indomethacin exerts antitumor effects on multiple types of cancers [13, 14]. Indomethacin effectively inhibits growth and induces apoptosis in gastric cancer cells by impairing mitochondrial function [15]. Indomethacin also increases the response of colon cancer cells to the chemotherapy drug cisplatin and of melanoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) [16, 17]. Recent studies show that indomethacin suppresses the growth of prostate cancer cells both in vitro and in vivo by inhibiting AKR1C3 [18]. However, the efficacy of indomethacin against CRPC tumor growth appears limited [13, 18].
In this study, we observed that our designed and synthesized new indomethacin derivatives have high efficacy in reducing CRPC cell survival. CZ-212-3, the most potent derivative, significantly suppressed CRPC tumor cell growth both in vivo and in vitro by degrading AR and its variants. Thus, our findings suggest that the indomethacin derivative CZ-212-3 is as a promising agent for CRPC therapy.
Materials and methods
Synthesis and characterization of CZ compounds
CZ compounds were synthesized according to the reaction scheme shown in our previous research and were characterized by nuclear magnetic resonance (NMR) and mass spectrometry [19].
Reagents and antibodies
Enzalutamide was synthesized by WuXi AppTec. MG132, CHX and indomethacin were purchased from Selleck (Houston, Texas, USA). c-Myc (#18583), cleaved caspase-7 (#9491), GAPDH (#8884), PARP (#9532), p21 (#2947), ubiquitin (P4D1) (#3936) and AR-V7 (#68492) antibodies were purchased from Cell Signaling Technology (Boston, USA). Cyclin E (sc-247), p53 (sc-126), Ki67 (sc-23900) and AR (sc-7305) antibodies were purchased from Santa Cruz (CA, USA). AR (#06-680) antibody was purchased from Millipore (MMAS, USA). H3K27ac (ab4729) antibody was purchased from Abcam (Camb, UK).
Cell lines and cell culture
The human prostate cancer cell lines C42B, 22Rv1, C42B/ENZ, VCaP-CRPC, LNCaP, and HeLa were originally obtained from American Type Culture Collection (ATCC). All cells were cultured in RPMI-1640, except VCaP-CRPC, VCaP-CRPC, and HeLa cells, which were cultured in DMEM (Gibco). All media were supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin (10,000 U/mL, Life Technologies). All cells were cultured in a humidified atmosphere consisting of 5% CO2 at 37 °C.
Cell viability, proliferation, and apoptosis assays
C42B (2×103 cells/well), C42B/ENZ (3×103 cells/well), 22Rv1 (2×103 cells/well), and VCaP-CRPC (5×103 cells/well) cells were plated in 96-well plates and treated with CZ compounds, indomethacin or vehicle control (dimethyl sulfoxide, DMSO, Sigma), and cell viability was determined at 96 h after plating with CCK-8 (Cell Counting Kit-8, Selleck, US). In addition, cell proliferation was measured by Cell-Titer GLO (Promega). For the apoptosis assay, caspase−3/7 activity was determined by using a luminescent caspase-Glo 3/7 assay kit (Promega Corporation, Madison, US) following the manufacturer’s instructions. All experimental points were set up in triplicate or sextuplicate as biological replicates, and the entire experiment was repeated three times.
Colony formation assay
The colony formation assay was performed as described previously [1, 20]. In detail, C42B (1×103 cells/well) and 22Rv1 (1×103 cells/well) cells were plated in 6-well plates, allowed to adhere for at least 24 h, and then treated with CZ compounds at different concentrations for an additional 14 days at 37 °C in 5% CO2 to allow colony formation. Colonies were washed in cold PBS, fixed in 4% paraformaldehyde for 20 min and then stained with 0.1% crystal violet solution. The number of colonies was counted after the cells were washed with PBS 3 times. The above assays were repeated three times.
Western blotting analysis
Western blot assays were performed as previously described [21]. After treatment with CZ compounds for the designated duration, cells were collected. The cells or tumor tissues were submerged in lysis buffer on ice for 30 min and then centrifuged at 4 °C for 15 min. Protein concentrations were quantified with the BCA protein assay (#23225, Thermo, US). Approximately 40 μg of protein in the whole-cell lysates was loaded in 8%-15% polyacrylamide gels, subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for 90 min, and transferred to a polyvinylidene fluoride (PVDF) membrane for 2 h. The membranes were blocked for 1 h at room temperature, incubated with appropriate primary antibody overnight, and probed with secondary antibody for 1 h at room temperature. Protein bands were detected by a ChemiDoc Molecular Imager system according to the operation protocol [22].
Flow cytometric analyses
C42B cells (1.5×105 cells/well) were seeded in 6-well plates, allowed to adhere for at least 24 h, and then treated with CZ compounds at different concentrations. The cells were digested by trypsin without EDTA and then collected after centrifugation at 4 °C. Cells were rinsed with prechilled PBS twice and then fixed in 70% ethanol overnight at 4 °C. The cells were washed with cold PBS two times and stained with RNase A solution (BD Bioscience) and propidium iodide (BD Bioscience) in 1× Annexin V binding buffer (BD Bioscience) for 30 min, following the manufacturer’s instructions. The above assays were repeated three times. Flow cytometric analysis was performed on a Beckman-Coulter Cytomics FC500 (Beckman).
Quantitative real-time PCR
Total RNA was collected from cells cultured in 6-well plates, and cDNA was prepared, amplified and measured in the presence of SYBR Green as described previously [23]. In detail, melting curve analysis was performed, and the fluorescence values were recorded. The experiments were repeated three times and the data are presented as values ± s.d. (SD). The sequences of the PCR analysis primers are listed in Supplementary Table 1.
RNA-sequence data analysis
C42B cells were treated with control or CZ-212-3 (0.5, 1 µM) for 24 h before RNA extraction. The concentration and quality of total RNA samples were first detected using an Agilent 2100 Bioanalyzer. A total amount of 1 μg of RNA was prepared for RNA-seq libraries using an Illumina Tru-Seq RNA Sample according to the manufacturer’s instructions. GSEA and Gene Ontology (GO) enrichment analysis were performed as described previously [24].
Luciferase reporter constructs and reporter-gene assay
Reporter-gene assays and transient transfection were performed as described previously [25]. A gene reporter assay was performed by transfecting HeLa cells with pcDNA-hAR, PGl3-5×ARE and TK-RL plasmids. HeLa cells were seeded in 96-well plates, allowed to adhere for at least 24 h, and then transfected with plasmids as indicated using Lipofectamine 2000 (Thermo Fisher, US). At 24 h after transfection, cells were treated with control or CZ compounds for another 24 h. Then, TK-RL and luciferase were detected with a luciferase assay substrate (Promega, US). All transfections were conducted in triplicate, and the experiments were repeated at least three times.
Immunohistochemistry (IHC)
Tumor tissue harvested from the PDX model mice was fixed in 4% paraformaldehyde and embedded in paraffin. Tissue slices (5-µm thickness) were incubated with Ki67 antibody at 4 °C overnight and then stained with the secondary antibody according to previous experiments [26].
ChIP-qPCR analysis
ChIP-qPCR analysis was performed as described previously [27]. The sequences of the PCR primers used in the ChIP assays are listed in Supplementary Table 1. ChIP-qPCR was performed with each experimental point in triplicate, and all experiments were repeated three times.
Enzyme preparation and inhibition assays
Human recombinant AKR1C3 was prepared as described previously [28] and was used to reduce 9,10-phenanthrenequinone (PQ) in the presence of the coenzyme NADPH in vitro. Compounds were dissolved separately in DMSO and then diluted with 100 mmol/L potassium phosphate buffer (pH = 6.0) to final concentrations of 0–20 μM. Then, 93 μL of the compound solution containing 0.15 mM NADPH and 8.0 μM PQ was added to the 96-well plate. After preincubation for a specified time, 6 μL of AKR1C3 (0.33 μM) was added to each well, and the plates were shaken for a few seconds. The cells were incubated for another 10 min. The IC50 value of enzyme activity was detected as described previously [28].
Xenograft tumor models
Four-week-old male athymic nu/nu mice were purchased from GemPharmatech (Nanjing, China). To establish xenograft tumors, 22Rv1 cells (2×106) were suspended in 100 µL of PBS and Matrigel (1:1) and injected subcutaneously into the flank spine region on both sides of mice. When the volume of the tumor was ~100 mm3, the animals were randomly divided into two groups, and each group had seven or more animals according to the power calculation (http://www.biomath.info/power/) [29, 30]. The mice were intraperitoneally (i.p.) injected with control or CZ-212-3 5 times per week. The body weight of the mice and tumor volume were monitored every 2 days. The volume of the tumor was detected with calipers and calculated with the equation V = π × (length×width2)/6.
PDX model of prostate cancer
The patient-derived xenograft (PDX) model of prostate cancer (TM00298) was obtained from Hong-Wu Chen Laboratory. Four-week-old male NOD-SCID mice were subcutaneously injected with 10 mm3 minced tumor fragments. When the volume of the tumor was ~100 mm3, the animals were randomly divided into three groups: CZ-212-3 (50 mg/kg), ENZ (20 mg/kg), or vehicle; the mice were injected intraperitoneally (i.p.) 5 times per week (n = 6). At the end of the experiment, mice were sacrificed under euthanasia using a method approved by the animal ethics committee, and the tumors were collected, imaged, weighed and analyzed via hematoxylin and eosin (H&E) staining or Western blotting [31]. The tumor tissue, liver, lung and testes of mice were fixed in 4% paraformaldehyde before paraffin embedding. The samples were sliced into 5 µm sections and then stained with H&E. Histological features of the selected tissues and organs were observed under a microscope.
Statistical analysis
Cell culture-based experiments were conducted at least three times. As indicated, the experimental points were set up in triplicate or sextuplicate. All in vitro data are presented as the mean ± SD from three independent experiments, and in vivo data are presented as the mean ± SEM. Differences between groups were analyzed using two-tailed Student’s t test to compare the means. P < 0.05 was considered to be statistically significant.
Results
Design and optimization of indomethacin derivatives for advanced prostate cancer
We recently designed and synthesized a series of new indomethacin analogs (CZ compounds) [19]. Their molecular structures were shown in Fig. 1a. We used the prostate cancer cell line C42B to investigate the antitumor effects of different CZ compounds on prostate cancer. As shown in Fig. 1a, compounds CZ-199-1, CZ-212-2 and CZ-212-3 exhibited the most potent anti-CRPC activity. Structure-activity relationship (SAR) studies revealed that the activity of indomethacin and its derivatives is dependent on the core indole moiety, which exerts antitumor effects on multiple types of cancers [13]. Among the derivatives, those with the same substitution of R2 (CZ-217-2, 218-3, 221-3, 231-1, and 212-2) and replacement of R1 with OCH3 showed stronger activity than derivatives with other electron-donating groups, such as compound CZ-212-2 (Fig. 1). On the other hand, when the R1 group was OCH3, we changed the R2 of CZ-212-2 (IC50 = 1.254 μM) from a Cl group to an F group to obtain compound CZ-212-3 (IC50 = 1.088 μM), which shows potent cytotoxicity against advanced PCa cells and is very similar to CZ-212-2.
Fig. 1. Design and optimization of indomethacin derivatives for advanced prostate cancer.
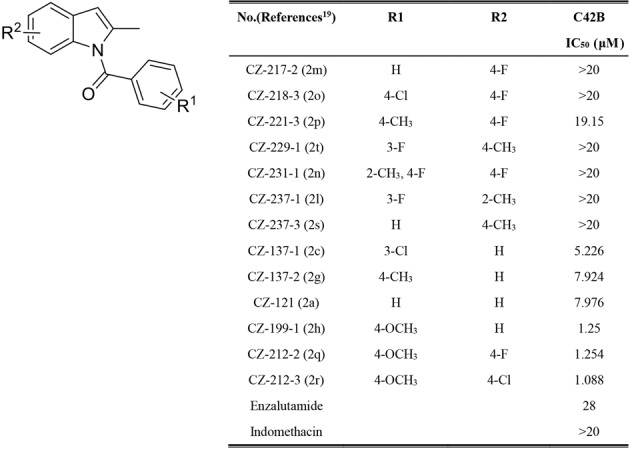
Structure of indomethacin derivatives and their half-maximum inhibitory concentration (IC50) values on CRPC C42B cell survival.
CZ-212-3 inhibits CRPC cell survival by inducing apoptosis and G2 phase arrest of the cell cycle
We then examined the antitumor effect of CZ compounds in different prostate cancer cell lines. Our results showed that CZ compounds CZ-199-1, CZ-212-2, and CZ-212-3 strongly inhibited the viability of PCa cells in a dose-dependent manner, and the potency (indicated by the IC50 value, ~1 µM) was enhanced ~20-fold compared to that of indomethacin (IC50 > 20 µM). CZ-212-3 displayed the highest efficacy (Fig. 2a and Supplementary Fig. S1a-S1b). We also found that CZ compounds could effectively induce apoptosis of PCa cells, as indicated by upregulated Caspase-3/7 (Cas3/7) activities and higher protein levels of cleaved PARP1 and cleaved Caspase7 (Fig. 2b–c). Next, we sought to determine whether CZ compounds could inhibit PCa cell proliferation by arresting the cell cycle. Flow cytometry analysis demonstrated that CZ compounds resulted in cell cycle arrest at G2/M phase with a corresponding decrease in the percentages of cells in G1 and S phases. Consistently, Western blot analyses showed that CZ compounds increased the expression of the G2-related genes p53 and p21 (Fig. 2d–e). Furthermore, CZ compounds decreased the colony-forming ability of CRPC cells in a dose-dependent manner (Fig. 2f and Supplementary Fig. S1c). These data suggested that CZ compounds inhibit CRPC cell survival by inducing apoptosis and G2 cell cycle arrest in vitro.
Fig. 2. CZ-212-3 inhibits CRPC cell survival by inducing apoptosis and cell cycle arrest at G2 phase.

a Cell viability as measured by Cell-Titer GLO (Promega) of CRPC cells treated with the indicated concentrations of indomethacin, CZ-199-1, CZ-212-2, or CZ-212-3 for 4 days. b Apoptosis as detected by Caspase-3/7 (Cas3/7) activities in C42B cells treated with vehicle or the indicated CZ compounds for 24 h. c Immunoblotting analysis of the indicated proteins in C42B cells treated with vehicle or the indicated CZ compounds for 24 h. d C42B cells were exposed to vehicle or the indicated concentrations of CZ-212-3 for 24 h. Then, the cells were harvested, and cell cycle arrest was analyzed by flow cytometry. e Immunoblotting analysis of the indicated proteins in C42B cells treated with vehicle or the indicated CZ compounds for 24 h. f C42B and 22Rv1 cells were treated with vehicle or the indicated concentrations of CZ-199-1, CZ-212-2, or CZ-212-3 for 14 days, after which colony formation was assessed. **P < 0.01, ***P < 0.001; Student’s t test, n = 3. All data above are shown as the mean ± SD.
CZ-212-3 controls the AR-dependent gene programs
To investigate the underlying mechanism by which CZ compounds inhibit CRPC cell survival, we performed RNA-seq on the transcriptomes of C42B cells treated with the most potent compound, CZ-212-3. Since AR plays a pivotal role in CRPC tumor cell growth, we first examined the effect of CZ-212-3 on the AR pathway and found that CZ-212-3 strongly inhibited AR-induced gene expression and increased AR-suppressed gene expression (Fig. 3a). Gene set enrichment analysis (GSEA) also showed that CZ-212-3 robustly disrupted AR target gene programs (Supplementary Fig. S2a). In addition, our qRT-PCR results verified that CZ-212-3 strongly inhibited the expression of AR target genes such as KLK2, KLK3, ANCCA, FKBP5 and NKX3.1 but had a minimal effect on AR mRNA expression (Fig. 3b). We then determined whether CZ-212-3 suppressed gene expression by directly affecting AR binding to the regulatory sites of target genes. The results showed that CZ-212-3 strongly reduced the occupancy of AR and transcriptional activation-linked histone marker H3K27ac on KLK3 enhancer regions (Fig. 3c). In line with its effects on AR programs, CZ-212-3 also effectively suppressed the expression of androgen-induced genes related to cell proliferation and survival (Fig. 3d).
Fig. 3. CZ-212-3 controls AR-dependent gene activity.

a Heat map displaying the fold changes in gene expression in C42B cells treated with CZ-212-3 (0.5, 1 µM) for 24 h, compared to vehicle (DMSO) as detected by RNA-seq. Androgen-responsive AR activity signature genes that showed altered expression are displayed. b qRT-PCR analysis of the indicated genes in C42B cells treated with vehicle or CZ-212-3 (0.5 µM) for 24 h. c Genome browser display of AR-binding events on the promoter and enhancer of the KLK3 gene based on data from a previous ChIP-seq analysis (GSE72714) (upper). ChIP-qPCR analysis of relative AR or histone modifications at the KLK3 gene promoter and enhancer in C42B cells treated with vehicle, 1 µM CZ-212-3 or 20 µM ENZ for 24 h (bottom). d Heat map of differences in the expression of genes involved in cell proliferation and survival in C42B cells treated with the indicated concentrations of CZ compounds for 24 h. Gene expression profiling was performed with RNA sequencing. Data are shown as the mean ± SD, **P < 0.01, ***P < 0.001; Student’s t test, n = 3.
To provide further evidence that CZ-212-3 reduced CRPC cell viability by inhibiting AR activity, we also investigated the effect of compound CZ-212-3 on cell proliferation in C42B cells transfected with AR siRNA and showed that AR silencing blocked the inhibitory effect of CZ-212-3 on cell viability (Supplementary Fig. S2b). Furthermore, qRT-PCR experiments showed that AR silencing could block the inhibitory effect of CZ-212-3 on the expression of AR target genes (Supplementary Fig. S2c). Taken together, our results strongly suggested that CZ-212-3 inhibits CRPC cell survival by downregulating the AR-dependent gene program.
CZ-212-3 promotes the degradation of AR and its variants through the proteasome pathway
Since the above results showed that CZ compounds could effectively suppress AR function, we next examined whether CZ-212-3 controlled AR activity and/or expression. Employing an AR-driven KLK3 enhancer luciferase reporter system, we found that CZ-212-3 strongly suppressed the activity of endogenous AR (Fig. 4a and b, Supplementary Fig. S3a), and the results of the immunoblotting analysis showed that CZ compounds strongly suppressed the expression of AR, its variants and its target gene c-Myc in C42B and 22Rv1 cells (Fig. 4c and d). In contrast, indomethacin did not affect the expression of AR or c-Myc (Supplementary Fig. S3b). Since CZ-212-3 did not significantly change the mRNA level of AR (Fig. 3b), we investigated whether CZ compounds affected AR expression at the posttranslational level. C42B cells were treated with the protein synthesis inhibitor cycloheximide (CHX) in the presence or absence of CZ-212-3. Immunoblotting results showed that AR protein levels were significantly decreased in cells treated with CHX and CZ-212-3 compared with cells treated with CHX alone (Fig. 4e), which indicated that CZ-212-3 reduced AR expression through degradation. We then tested whether CZ compounds inhibited the expression of AR and its variants through the proteasome pathway. Our results showed that treatment with the proteasome inhibitor MG132 could effectively rescue the downregulation of AR, AR variants and c-Myc induced by CZ-212-3 in C42B and 22Rv1 cells (Fig. 4e). We then performed molecular docking to demonstrate whether CZ-212-3 could bind to the AR ligand binding domain to induce degradation of AR protein. As shown in Supplementary Fig. S3c, CZ-212-3 can tightly bind to AR via strong hydrogen bonds with residues ARG752 and ASN705 and π-π interactions with PHE764 in the active site. Our immunoblotting assay demonstrated that CZ-212-3 did not induce AR degradation via ubiquitination (Supplementary Fig. S3d), which suggested that CZ-212-3 induced AR degradation via other degradation pathways, such as the lysosomal and caspase pathways. These data collectively supported the notion that CZ compounds promote the degradation of AR and its variants through the proteasome pathway.
Fig. 4. CZ-212-3 promotes the degradation of AR protein through the proteasome pathway.

a ARE-guided gene reporter assays with HeLa cells showing the effects of pcDNA-hAR on PGl3-5×ARE-luc activation. b ARE-guided gene reporter assays with 22Rv1 cells showing the effects of CZ-212-3 on AR activation. The fold change indicated the activities of AR under the influence of CZ-212-3 compared to those in response to control treatment. c Immunoblotting analysis of AR (full length) and c-Myc in C42B cells treated with vehicle, CZ-212-2, or CZ-212-3 at the indicated concentrations for 24 h. d Immunoblotting analysis of AR (full length), AR-V7 (variants) and c-Myc in 22Rv1 cells treated with vehicle, CZ-212-2, or CZ-212-3 at the indicated concentrations for 24 h. e Immunoblotting analysis of AR and c-Myc in C42B cells treated with cycloheximide (CHX) and the indicated concentrations of CZ-212-3 for various lengths of time (left). Immunoblotting analysis of AR and c-Myc in C42B and 22Rv1 cells treated with various concentrations of CZ-212-3 with or without MG132 (10 μM) for 24 h (right). The data are shown as the mean ± SD. *P < 0.05, **P < 0.01, n = 3.
CZ-212-3 inhibits prostate cancer tumor growth in vivo
We next examined the antitumor effect of CZ compounds on prostate cancer in vivo. Mice bearing 22Rv1 xenograft tumors were treated with CZ-212-3 or vehicle via intraperitoneal injection 5 times per week. Our results showed that CZ-212-3 significantly inhibited tumor growth and decreased tumor weight without affecting mouse body weight (Fig. 5a, b and Supplementary Fig. S4a-S4b). We then employed a patient-derived prostate cancer xenograft model (PDX) to examine the efficacy of CZ-212-3 in a more clinical-like setting. The results showed that CZ-212-3 significantly reduced tumor volume and tumor weight compared with those in the vehicle control group in the PDX model (Fig. 5c, f). Tumor sections analyzed by immunohistochemistry with a Ki67 antibody showed that CZ-212-3 strongly inhibited PCa cell proliferation (Fig. 5g). H&E staining showed no obvious histologic change in testes or livers of mice treated with CZ-212-3 (Supplementary Fig. S4c). In xenograft tumors, in line with our above in vitro data, we observed the same reduction in the expression of AR, AR variants and c-Myc and the induction of the apoptosis markers cleaved PARP1 and cleaved caspase-7 upon CZ-212-3 treatment (Fig. 5h). In addition, ChIP analysis revealed that CZ-212-3 significantly reduced AR binding at the enhancer region of the AR target gene PSA (Fig. 5i). Taken together, these data show that CZ-212-3 inhibits prostate cancer tumor growth in vivo by suppressing the AR signaling pathway.
Fig. 5. CZ-212-3 inhibits prostate cancer tumor growth in vivo.

a Effects of the indicated treatments (CZ-212-3, 50 mg/kg, i.p. (n = 10)) or vehicle (n = 8) five times per week on the growth of 22Rv1 xenografts. Treatment started when the 22Rv1 tumor volume reached ~50 mm3; the mean tumor volume ± SEM is shown. Significance was calculated using Student’s t test, **P < 0.01. b The weights of 22Rv1 tumors from mice treated with CZ-212-3 (50 mg/kg, i.p., 5 times a week, n = 10) or vehicle (n = 8 mice per group) for 16 days. The mean tumor weight ± SEM is shown. Student’s t test, **P < 0.01. c Effects of CZ-212-3 (50 mg/kg, i.p., 5 times a week), ENZ (20 mg/kg, i.p., 5 times a week), and vehicle on the growth of PCa TM00298 PDX xenografts (n = 6 mice per group). The mean tumor volume ± SEM is shown. Significance was calculated using Student’s t test, ***P < 0.001. d Effects of CZ-212-3 (50 mg/kg, i.p., 5 times a week), ENZ (20 mg/kg, i.p., 5 times a week), and vehicle on the growth of PCa TM00298 PDX xenografts (n = 6 mice per group). Representative tumor images are shown. e The weights of PCa TM00298 PDX tumors from mice treated with CZ-212-3 (50 mg/kg, i.p., 5 times a week), ENZ (20 mg/kg, i.p., 5 times a week) or vehicle (n = 6 mice per group). The mean tumor weight ± SEM is shown. Student’s t test, ***P < 0.001. f Body weight of mice from different treatment groups described in Fig. 5c is shown during the 36-day treatment period (presented as the mean ± SEM, n = 6). g Representative photographs of 22Rv1 xenografts from mice treated i.p. with vehicle or 50 mg/kg CZ-212-3 with corresponding IHC for Ki67. Scale bar was 100 μm (left). Representative immunohistochemistry (IHC) quantification of Ki67 positivity in PCa TM00298 PDX tumors (right). Data represent the mean ± SEM. Significance was calculated using Student’s t test, where ***P < 0.001. h Western blot of 22Rv1 xenograft tumors after 16-day of treatment with vehicle or CZ-212-3 as described in Fig. 5c. Cell lysate for each lane was obtained from homogenized tumor tissues randomly combined from three mice in the vehicle, CZ-212-3 and ENZ groups. i ChIP-qPCR analysis of relative AR bound at the KLK2/KLK3 promoter and enhancer regions in PCa TM00298 PDX tumors described in Fig. 5c. Cell lysate for each lane was obtained from homogenized tumor tissues randomly combined from three mice from the vehicle and CZ-212-3 groups. Student’s t test, *P < 0.05, n = 3.
Discussion
Currently approved therapeutics that target the anti-androgen-AR axis, such as enzalutamide and abiraterone, represent a breakthrough for CRPC therapy. However, this class of drugs could not overcome resistance induced by AR mutations and variants. The majority of clinical trials have indicated that AR variants (AR-V7) confer resistance to and result in poorer outcomes in CRPC patients with AR-targeting therapies [32]. Developing agents that promote the protein degradation of AR and its variants or block their expression were suggested as two ideal CRPC therapeutic strategies. Here, we discovered and characterized a series of novel indomethacin-derived compounds that degrade AR, which could efficiently inhibit CRPC tumor cell growth both in vivo and in vitro.
AR and AR variants, such as AR gene amplification, AR mutation, changes in AR auxiliary regulatory factors, activation of other nonsteroidal substances and other signaling pathways for AR, play critical roles in CRPC, and the generation of AR variants is involved in the occurrence of CRPC [33]. Inhibiting AR decreases cell proliferation and ultimately inhibits cancer growth. Currently, targeted therapies only focuses on AR activity, and there is no drug therapy for AR variations. For example, clinical studies of patients with advanced PCa demonstrated that AR-V7 expression is one of the most common variants in PCa samples [34]. Higher expression of AR-V7 was reported to be associated with resistance to enzalutamide and abiraterone and with progression of CRPC in primary tumors [35]. Therefore, it remains a great challenge to overcome drug resistance in advanced PCa. USP14 stabilizes MDM2 via direct degradation in PCa after treatment with small-molecule inhibitors [36]. Recently, with the discovery of AR-V7 and agents targeting the AR N-terminal domain (NTD), DNA binding of AR and degradation of AR protein have increased [37, 38]. Thus, protein degradation may be a potential therapeutic strategy [39].
Indomethacin is widely used to relieve pain, fever and inflammation with good tolerance and safety [12]. Indomethacin could effectively inhibit gastric cancer cell growth and induce apoptosis through impaired mitochondrial function[15]. Indomethacin also increased the colon cancer cell response to the chemotherapy drug cisplatin and the melanoma cancer cell response to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis [16, 17]. Recent studies suggested that indomethacin, acting as an AKR1C3 inhibitor, exerted an anti-CRPC effect by suppressing androgen synthesis and AR activation [18]. Indomethacin and enzalutamide for CRPC treatment have been investigated in ongoing clinical trials. However, the efficacy of indomethacin on CRPC cell survival is limited, and the development of novel and more potent indomethacin derivatives is needed.
In the present study, we created a series of new indomethacin derivatives that exhibit stronger inhibition of CRPC cell survival than indomethacin does. CZ-212-3 is one of the most effective anti-CRPC agents. Meanwhile, indomethacin potently inhibited the activity of AKR1C3, while CZ-212-3 had a weak inhibitory effect on AKR1C3 activity (Supplementary Fig. S5a), which suggested that indomethacin and CZ-212-3 exhibit anti-CRPC activity via different mechanisms. Our further studies demonstrated that CZ-212-3 could bind to and degrade AR and AR variant proteins in CRPC cells. We also demonstrated that CZ-212-3 reduced AR binding to its target loci and the abundance of the local activating histone mark H3K27ac. CZ-212-3 potently repressed the growth of CRPC cells in vitro and in vivo via inhibition of AR signaling. Since our studies showed that CZ-212-3 degrades the protein of both AR and AR variants in CRPC cells, this compound may provide therapeutic advantages over antiandrogen- and AR-FL-targeted treatments, particularly in CRPC tumors with high levels of AR splice variants. In conclusion, our results collectively indicate that the indomethacin derivative CZ-212-3 might significantly inhibit CRPC tumor growth by degrading AR and its variants and serve as a promising agent for CRPC therapy.
Supplementary information
Acknowledgements
This research was supported by the National Natural Science Foundation of China (81872891, 21801260, 82003651 and 21971267), the Guangdong Natural Science Funds for Distinguished Young Scholar (No. 2019B151502016), the Guangzhou Science and Technology Basic Research Program (No. 202002020082), the National Major Special Projects for the Creation and Manufacture of New Drugs (2019ZX09301104), and the Fundamental Research Funds for the Central Universities (No. 19ykzd23 and 2021qntd44).
Author contributions
JJW and DPZ conceived and designed the experiments. HW, ZC, GDC, JHC, XHZ, and SSY performed the experiments. YFG, MYW, and JPL carried out the molecular docking procedure. JJW, PY, PQL, and DPZ wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Hong Wang, Zhe Chang, Guo-di Cai.
Contributor Information
De-peng Zhao, Email: zhaodp5@mail.sysu.edu.cn.
Jun-jian Wang, Email: wangjj87@mail.sysu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00738-w.
References
- 1.Wang J, Zou JX, Xue X, Cai D, Zhang Y, Duan Z, et al. ROR-gamma drives androgen receptor expression and represents a therapeutic target in castration-resistant prostate cancer. Nat Med. 2016;22:488–96. doi: 10.1038/nm.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen HG, Conn CS, Kye Y, Xue L, Forester CM, Cowan JE, et al. Development of a stress response therapy targeting aggressive prostate cancer. Sci Transl Med. 2018;10:1–11. [DOI] [PMC free article] [PubMed]
- 4.Chang AJ, Autio KA, Roach M, 3rd, Scher HI. High-risk prostate cancer-classification and therapy. Nat Rev Clin Oncol. 2014;11:308–23. doi: 10.1038/nrclinonc.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heidenreich A, Pfister D, Merseburger A, Bartsch G. Castration-resistant prostate cancer: where we stand in 2013 and what urologists should know. Eur Urol. 2013;64:260–5. doi: 10.1016/j.eururo.2013.05.021. [DOI] [PubMed] [Google Scholar]
- 6.Ferraldeschi R, Welti J, Luo J, Attard G, de Bono JS. Targeting the androgen receptor pathway in castration-resistant prostate cancer: progresses and prospects. Oncogene. 2015;34:1745–57. doi: 10.1038/onc.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ervik B, Nordoy T, Asplund K. Hit by waves-living with local advanced or localized prostate cancer treated with endocrine therapy or under active surveillance. Cancer Nurs. 2010;33:382–9. doi: 10.1097/NCC.0b013e3181d1c8ea. [DOI] [PubMed] [Google Scholar]
- 8.Gustafson JL, Neklesa TK, Cox CS, Roth AG, Buckley DL, Tae HS, et al. Small-molecule-mediated degradation of the androgen receptor through hydrophobic tagging. Angew Chem Int Ed Engl. 2015;54:9659–62. doi: 10.1002/anie.201503720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol. 2015;67:470–9. doi: 10.1016/j.eururo.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–38. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu J, Van der Steen T, Tindall DJ. Are androgen receptor variants a substitute for the full-length receptor? Nat Rev Urol. 2015;12:137–44. doi: 10.1038/nrurol.2015.13. [DOI] [PubMed] [Google Scholar]
- 12.Liedtke AJ, Adeniji AO, Chen M, Byrns MC, Jin Y, Christianson DW, et al. Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (Type 5 17beta-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J Med Chem. 2013;56:2429–46. doi: 10.1021/jm3017656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol Cancer Ther. 2017;16:35–44. doi: 10.1158/1535-7163.MCT-16-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bredt AB, Girey GJ. Antipyretic effects of indomethacin in liver metastases of solid tumors. Cancer. 1982;50:1430–3. doi: 10.1002/1097-0142(19821001)50:7<1430::AID-CNCR2820500732>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 15.Mazumder S, De R, Debsharma S, Bindu S, Maity P, Sarkar S, et al. Indomethacin impairs mitochondrial dynamics by activating the PKCζ–p38–DRP1 pathway and inducing apoptosis in gastric cancer and normal mucosal cells. J Biol Chem 2019;294:8238–58. [DOI] [PMC free article] [PubMed]
- 16.Brunelli C, Amici C, Angelini M, Fracassi C, Belardo G, Santoro MG. The non-steroidal anti-inflammatory drug indomethacin activates the eIF2alpha kinase PKR, causing a translational block in human colorectal cancer cells. Biochem J. 2012;443:379–86. doi: 10.1042/BJ20111236. [DOI] [PubMed] [Google Scholar]
- 17.Tse AK, Cao HH, Cheng CY, Kwan HY, Yu H, Fong WF, et al. Indomethacin sensitizes TRAIL-resistant melanoma cells to TRAIL-induced apoptosis through ROS-mediated upregulation of death receptor 5 and downregulation of survivin. J Invest Dermatol. 2014;134:1397–407. doi: 10.1038/jid.2013.471. [DOI] [PubMed] [Google Scholar]
- 18.Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, et al. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015;75:1413–22. doi: 10.1158/0008-5472.CAN-14-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang Z, Ma T, Zhang Y, Dong Z, Zhao H, Zhao D. Synthesis of functionalized indoles via palladium-catalyzed cyclization of N-(2-allylphenyl) benzamide: a method for synthesis of indomethacin precursor. Molecules. 2020;25:1233. [DOI] [PMC free article] [PubMed]
- 20.Rada M, Vasileva E, Lezina L, Marouco D, Antonov AV, Macip S, et al. Human EHMT2/G9a activates p53 through methylation-independent mechanism. Oncogene. 2017;36:922–32. doi: 10.1038/onc.2016.258. [DOI] [PubMed] [Google Scholar]
- 21.Ponnusamy S, Coss CC, Thiyagarajan T, Watts K, Hwang DJ, He Y, et al. Novel selective agents for the degradation of androgen receptor variants to treat castration-resistant prostate cancer. Cancer Res. 2017;77:6282–98. doi: 10.1158/0008-5472.CAN-17-0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saito Y, Li L, Coyaud E, Luna A, Sander C, Raught B, et al. LLGL2 rescues nutrient stress by promoting leucine uptake in ER+ breast cancer. Nature. 2019;569:275–9. doi: 10.1038/s41586-019-1126-2. [DOI] [PubMed] [Google Scholar]
- 23.Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, et al. Small-molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell. 2019;36:483–97. doi: 10.1016/j.ccell.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell. 2017;32:474–89. doi: 10.1016/j.ccell.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asim M, Tarish F, Zecchini HI, Sanjiv K, Gelali E, Massie CE, et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun. 2017;8:374. doi: 10.1038/s41467-017-00393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen W, Zhang W, Ye W, Wang H, Zhang Q, Shen J, et al. SR9009 induces a REV-ERB dependent anti-small-cell lung cancer effect through inhibition of autophagy. Theranostics. 2020;10:4466–80. doi: 10.7150/thno.42478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han W, Gao S, Barrett D, Ahmed M, Han D, Macoska JA, et al. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene. 2018;37:710–21. doi: 10.1038/onc.2017.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng X, Jiang Z, Li X, Zhang C, Li Z, Wu Y, et al. Screening, synthesis, crystal structure, and molecular basis of 6-amino-4-phenyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitriles as novel AKR1C3 inhibitors. Bioorg Med Chem. 2018;26:5934–43. doi: 10.1016/j.bmc.2018.10.044. [DOI] [PubMed] [Google Scholar]
- 29.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–82. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen HG, Yang JC, Kung HJ, Shi XB, Tilki D, Lara PN, Jr., et al. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene. 2014;33:4521–30. doi: 10.1038/onc.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng G, Zhang Q, Pan J, Lee Y, Ouari O, Hardy M, et al. Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis. Nat Commun. 2019;10:2205. doi: 10.1038/s41467-019-10042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharp A, Coleman I, Yuan W, Sprenger C, Dolling D, Rodrigues DN, et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest. 2019;129:192–208. doi: 10.1172/JCI122819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shankar E, Franco D, Iqbal O, El-Hayek V, Gupta S. Novel approach to therapeutic targeting of castration-resistant prostate cancer. Med Hypotheses. 2020;140:109639. doi: 10.1016/j.mehy.2020.109639. [DOI] [PubMed] [Google Scholar]
- 34.Lee CH, Ku JY, Ha JM, Bae SS, Lee JZ, Kim CS, et al. Transcript levels of androgen receptor variant 7 and ubiquitin-conjugating enzyme 2C in hormone sensitive prostate cancer and castration-resistant prostate cancer. Prostate. 2017;77:60–71. doi: 10.1002/pros.23248. [DOI] [PubMed] [Google Scholar]
- 35.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–13. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian Z, D’Arcy P, Wang X, Ray A, Tai YT, Hu Y, et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood. 2014;123:706–16. doi: 10.1182/blood-2013-05-500033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–8. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–56. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Islam MT, Zhou X, Chen F, Khan MA, Fu J, Chen H. Targeting the signalling pathways regulated by deubiquitinases for prostate cancer therapeutics. Cell Biochem Funct. 2019;37:304–19. doi: 10.1002/cbf.3401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.