

Abstract
The combination of immunotherapy with other forms of treatment is an emerging strategy for boosting antitumor responses. By combining multiple modes of action, these combinatorial therapies can improve clinical outcomes through unique synergisms. Here, we show a microrobot-based strategy that integrates tumor tissue disruption with biological stimulation for cancer immunotherapy. The microrobot is fabricated by loading bacterial outer membrane vesicles onto a self-propelling micromotor, which can react with water to generate a propulsion force. When administered intratumorally to a solid tumor, the disruption of the local tumor tissue coupled with the delivery of an immunostimulatory payload led to complete tumor regression. Additionally, treatment of the primary tumor resulted in the simultaneous education of the host immune system, enabling it to control the growth of distant tumors. Overall, this work introduces a distinct application of microrobots in cancer immunotherapy and offers an attractive strategy for amplifying cancer treatment efficacy when combined with conventional therapies.
Keywords: micromotor, physical destruction, immunostimulant, in situ vaccination, cancer immunotherapy
Graphical Abstract
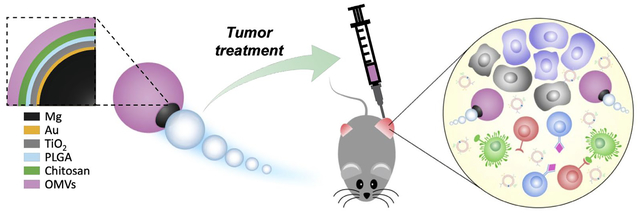
Micromotors coated with bacterial membrane are utilized for combinatorial cancer therapy. Upon intratumoral administration, the micromotors induce irreversible physical damage to the tumor tissue, while the immunostimulatory membrane recruits host immune cells to process tumor antigens in situ. Treatment of solid tumors using this platform results in robust antitumor immunity that can also control the growth of distant secondary tumors.
Immunotherapy has revolutionized how many types of cancer are managed in the clinic, oftentimes leading to drastically improved patient outcomes compared with traditional therapeutic modalities.[1, 2] Despite their many successes, most immunotherapies are only effective under specific conditions and work only for a subset of patients.[3, 4] To overcome these challenges, researchers have recently combined immunotherapeutic approaches with different types of conventional therapies, such as chemotherapy and radiotherapy.[5, 6] Combination immunotherapy is a powerful tactic that can synergistically amplify efficacy by addressing tumor immunosuppression from multiple angles.[7] Overall, combinatorial approaches hold great promise for expanding the utility of immunotherapies across a wider range of patients.
Microrobotic platforms, with their ability to actively deliver a diverse range of cargoes, have garnered significant attention over the past decade given their potential to be used for biomedical applications.[8–14] Compared with traditional delivery systems that rely on passive transport, microrobots utilize various actuation mechanisms to achieve active delivery that can more rapidly or more specifically localize a payload to a desired target.[15–17] These systems can also be designed with different biocompatible materials, such as magnesium (Mg) or zinc, to take advantage of the body’s natural fluids for propulsion.[18–20] Recently, it has been shown that surface functionalization of micromotors can be readily achieved using cell membrane coating technology,[21–23] enabling the resulting biomimetic construct to more effectively interface with its surroundings.[24, 25] Despite the numerous advantages of microrobots, their use for in vivo applications is still in its infancy.[8, 26] Thus far, the majority of studies have focused on the gastrointestinal tract, where microrobots have been used for detoxification, drug delivery, and immune modulation.[27] In order to push the microrobotics field forward and to expand their clinical utility, the identification of novel areas in which these platforms can excel is imperative.
Here, we report the development of a microrobot-based strategy that combines tissue disruption and biological stimulation at a tumor site towards enhanced cancer immunotherapy. Specifically, we utilized a Mg-based micromotor system that can interact with aqueous media to generate a reaction within a solid tumor, resulting in the disruption of the surrounding tumor tissue (Figure 1a). The compromised cancer cells could then act as a source of tumor antigens that were phagocytosed by local antigen-presenting cells to train the immune system.[28–30] To enhance their immune recruitment capability, the micromotors were loaded with bacteria-derived outer membrane vesicles (OMVs),[31] which contain a range of immunostimulatory molecules that have been leveraged for cancer treatment.[32–34] When administered intratumorally, the OMV-loaded micromotors (denoted ‘Motor-OMV’) were found to create large cavities and promote cell death. Concurrently, the immunogenic OMVs facilitated the effective recruitment and activation of immune cells, initiating a downstream cascade that ultimately suppressed the growth of both the treated tumor and a distal tumor. Through a localized administration, the Motor-OMV were found to be safe with no signs of short-term toxicity. Overall, this work successfully demonstrates that micromotor-assisted destruction can be combined with cancer immunotherapy to achieve potent antitumor effects and highlights the positive impact that continued development of microrobotics could have on the management of clinical diseases.
Figure 1.
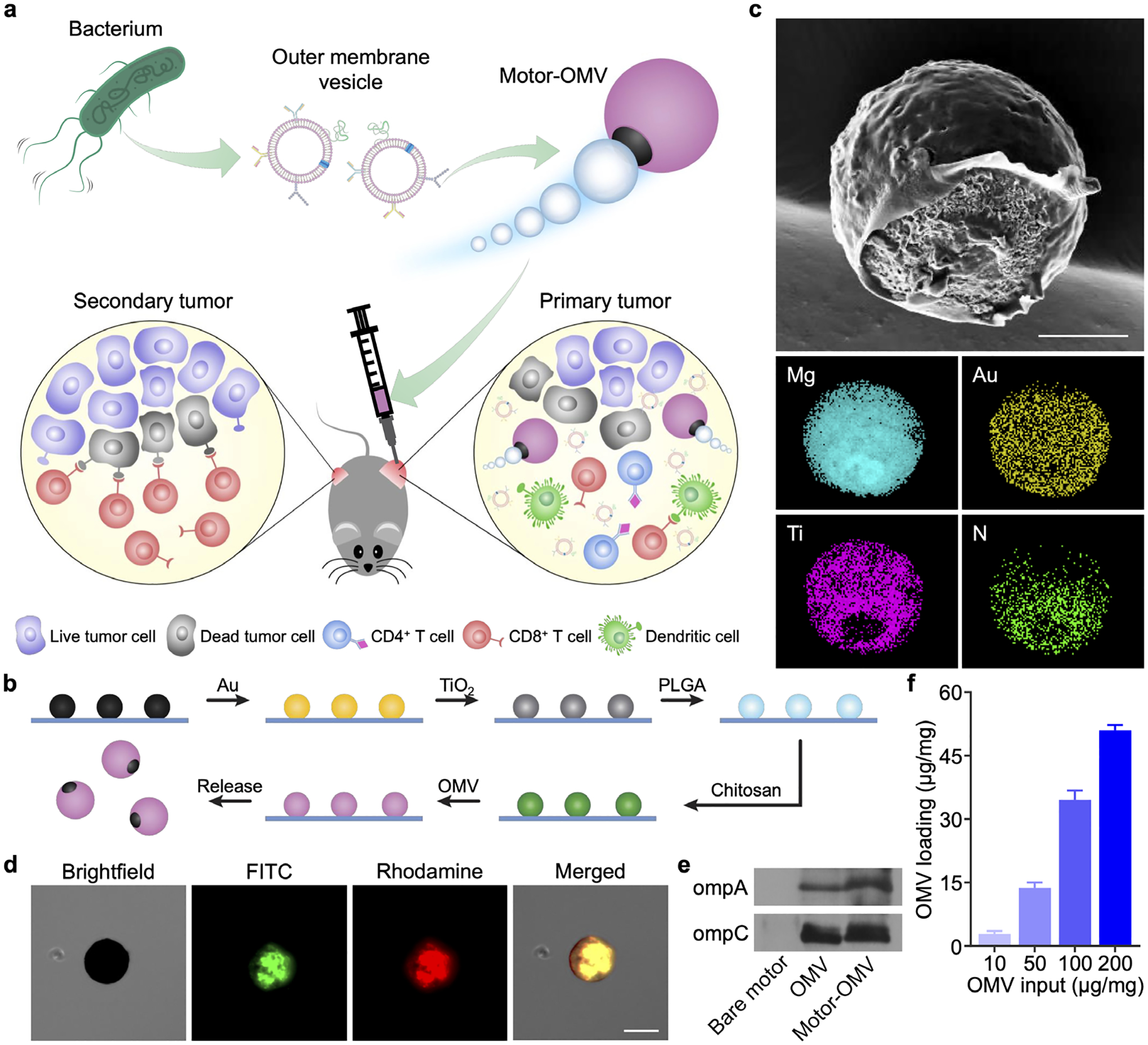
Fabrication and characterization of Motor-OMV. a) OMVs are derived from bacteria and loaded onto micromotors. The micromotors are intratumorally administered into mice to mechanically disrupt the tumor tissue while also promoting immune stimulation, leading to the generation of both local and systemic antitumor immunity. b) Mg microparticles on a glass slide are progressively coated with layers of Au, TiO2, PLGA, chitosan, and OMV to form Motor-OMV. c) Representative SEM image of a micromotor without the final OMV layer (scale bar = 10 μm). Corresponding EDX spectroscopy images of the micromotor show the distribution of Mg, Au, Ti, and nitrogen (N). d) Brightfield and fluorescence microscopy visualization of a representative Motor-OMV coated with FITC-labeled (green) chitosan and rhodamine-labeled (red) OMVs (scale bar = 25 μm). e) Western blot analysis for the presence of ompA and ompC on Motor-OMV. f) Quantification of total proteins on Motor-OMV per 1 mg of micromotor with varying initial OMV inputs (n = 3, mean + SD).
The immunostimulatory Motor-OMV were fabricated using a layer-by-layer process (Figure 1b). Mg microparticles, roughly 20 μm in size, were first dispersed on a glass slide and sputter-coated with gold (Au). The thin Au layer enables galvanic corrosion to speed up the reaction needed for propulsion and subsequently increases the overall speed of the micromotors.[35] Then, titanium dioxide (TiO2) was deposited by atomic layer deposition (ALD), covering the majority of the surface area while exposing a small opening at the contact point between the particle and the glass slide. The exposed Mg can react with the surrounding aqueous environment to produce the hydrogen bubbles needed for micromotor movement. On top of the TiO2 coating, layers of poly(lactic-co-glycolic acid) (PLGA) and chitosan were then added to reduce unwanted reaction of the Mg core during subsequent coating steps and to create a positively charged surface, respectively. Negatively charged Escherichia coli OMVs were loaded onto the surface by electrostatic interactions. Finally, free OMVs in the solution were washed out, and the OMV-loaded micromotors were dried under a vacuum. The final Motor-OMV formulation was stored at −20 °C for subsequent experiments.
Scanning electron microscopy (SEM) imaging was used to visualize the outer morphology of the micromotors without OMV loading, and energy-dispersive X-ray (EDX) spectroscopy with chemical mapping confirmed the presence of the Mg, Au, TiO2, and chitosan (Figure 1c). A small opening was clearly visualized in the Ti mapping where the micromotor had been in contact with the glass slide during fabrication; the same opening could not be visualized for the Au layer, as there was a lack of contrast due to the thinness of the coating. The presence of chitosan and OMVs on the final formulation was validated using Motor-OMV fabricated with both of the components fluorescently labeled (Figure 1d; Figure S1, Supporting Information). In this case, fluorescein isothiocyanate (FITC) was used in the chitosan layer while 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) was inserted into the OMVs. A clear overlap of both dyes with the micromotor was observed, indicating successful loading. An immunoblot for two abundant outer membrane markers, ompA and ompC,[36] provided further evidence for successful loading of the OMVs and confirmed the transfer of protein materials onto the micromotors during the fabrication process (Figure 1e). Loading optimization was performed by varying the OMV to micromotor ratio, and it was determined that an input of 100 μg of OMVs per 1 mg of micromotors yielded a good combination of loading yield and efficiency (Figure 1f).
After validating that the Motor-OMV were properly fabricated, we proceeded to evaluate their propulsion characteristics and their ability to facilitate disruption of tumor cells in vitro. Propulsion was analyzed in phosphate-buffered saline (PBS) at pH 5, where the induction of pitting corrosion leads to the reaction of Mg with water and generation of hydrogen bubbles (Figure 2a).[37, 38] Note that PBS at pH 5 was employed as the carrier solution in our subsequent in vivo studies. The propulsion of the micromotors was visualized under optical microscopy, and their average speed was determined to be 27 μm/s (Figure 2b,c; Video S1 and Video S2, Supporting Information). There were no differences in movement speed before and after the OMV loading. In addition, we assessed the propulsion of Motor-OMV at pH 6.5, which is more representative of the mildly acidic tumor microenvironment, and found that the average speed was slightly slower at 25 ± 2.6 μm/s (Figure S2; Video S3, Supporting Information). To analyze the destructive effects of Motor-OMV, a three-dimensional tumor spheroid model was developed using MC38 cells engineered to express green fluorescent protein (GFP). From fluorescent images of the spheroids, clear disruption to their structure could be seen after 4 h of incubation with Motor-OMV under normal physiological conditions (Figure 2d; Figure S3, Supporting Information). In the micromotor-treated samples, significant dissociation into individual tumor cells was observed. Comparatively, untreated spheroids displayed no noticeable change to their overall structure. An inert micromotor sample (denoted ‘static MP-OMV’), with no movement capability, was used as an additional control. Static MP-OMV were prepared and loaded with OMVs in the exact same fashion as Motor-OMV, except that the Mg cores were coated twice with TiO2 to prevent their reaction with the surrounding environment, thus enabling us to isolate active propulsion as a variable. When the spheroids were incubated with static MP-OMV, minimal changes to the macrostructure were observed, indicating clearly that the destruction caused by Motor-OMV resulted from their active propulsion.
Figure 2.
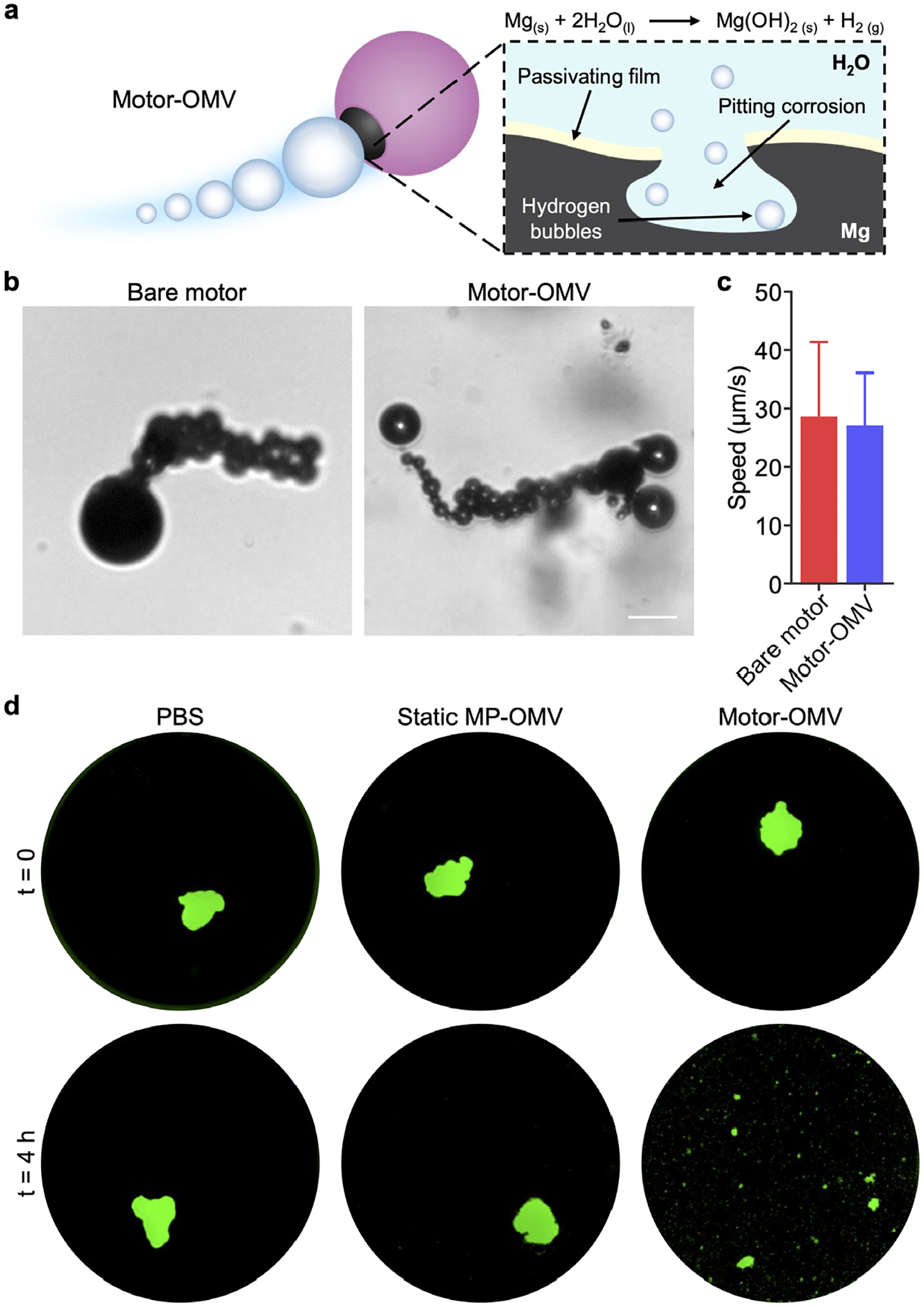
Micromotor propulsion and tumor spheroid destruction. a) Mg is left exposed in a small opening on Motor-OMV, allowing it to react with water in the surrounding environment to produce hydrogen gas bubbles for propulsion. The hydrogen production reaction is sustained by pitting corrosion. b) Optical images showing the trajectories of uncoated Mg micromotors and Motor-OMV in PBS at pH 5 (scale bar = 20 μm). c) Quantification of uncoated Mg micromotor and Motor-OMV speed (n = 5, mean + SD). d) Fluorescence visualization of representative GFP-expressing (green) MC38 spheroids before and after incubation with Motor-OMV or control samples at 37 °C for 4 h (well diameter = 6.4 mm).
To assess the disruption effects of Motor-OMV in a solid tumor environment, we utilized an MC38 colorectal cancer model. After subcutaneous inoculation into mice, the tumors were allowed to reach approximately 50 mm2 in size and were then intratumorally treated with Motor-OMV. One day after the treatment, the tumors were resected for histological sectioning and stained with hematoxylin and eosin (H&E) for visualization. It was observed that the Motor-OMV were able to rupture the solid tumor and generate cavities within the tissue (Figure 3a; Figure S4, Supporting Information). In contrast, tumors injected with either PBS or static MP-OMV displayed no change in their overall structure. For the tumors treated with Motor-OMV, brown particulates outlining portions of the cavities could be visualized at higher magnification (Figure 3b). The histological sections were also stained with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) to test for signs of cellular apoptosis. Strong signal was found at the disrupted interface of tumors that had received Motor-OMV treatment, even in areas with little micromotor accumulation (Figure 3c; Figure S5, Supporting Information). In contrast, baseline signal was observed in tumor sections from mice treated with PBS or static MP-OMV. To study whether large changes in temperature or pH may have contributed to the cavity formation, we allowed micromotors to fully react in vitro in carrier solution and measured each parameter (Figure S6, Supporting Information). There was a slight increase in temperature, and the pH increased from 5 to approximately physiological levels, thus suggesting that neither of these factors was likely to have caused significant physical destruction. We acknowledge that these preliminary tests cannot definitively rule out the contribution of either variable; however, the results imply that micromotor-assisted physical damage was likely to have played a role in the observed effects. In analyzing tumor cells isolated after Motor-OMV administration, we observed a significantly higher rate of necrosis compared with after static MP-OMV treatment (Figure S7, Supporting Information). It should be noted that necrotic cell death oftentimes leads to the release of various immunogenic damage-associated molecular patterns that can enhance immune responses.[39] Overall, these results confirmed that the reaction of the micromotors in the tumor microenvironment could rapidly induce irreversible damage to the local tumor structure and lead to enhanced tumor cell death.
Figure 3.
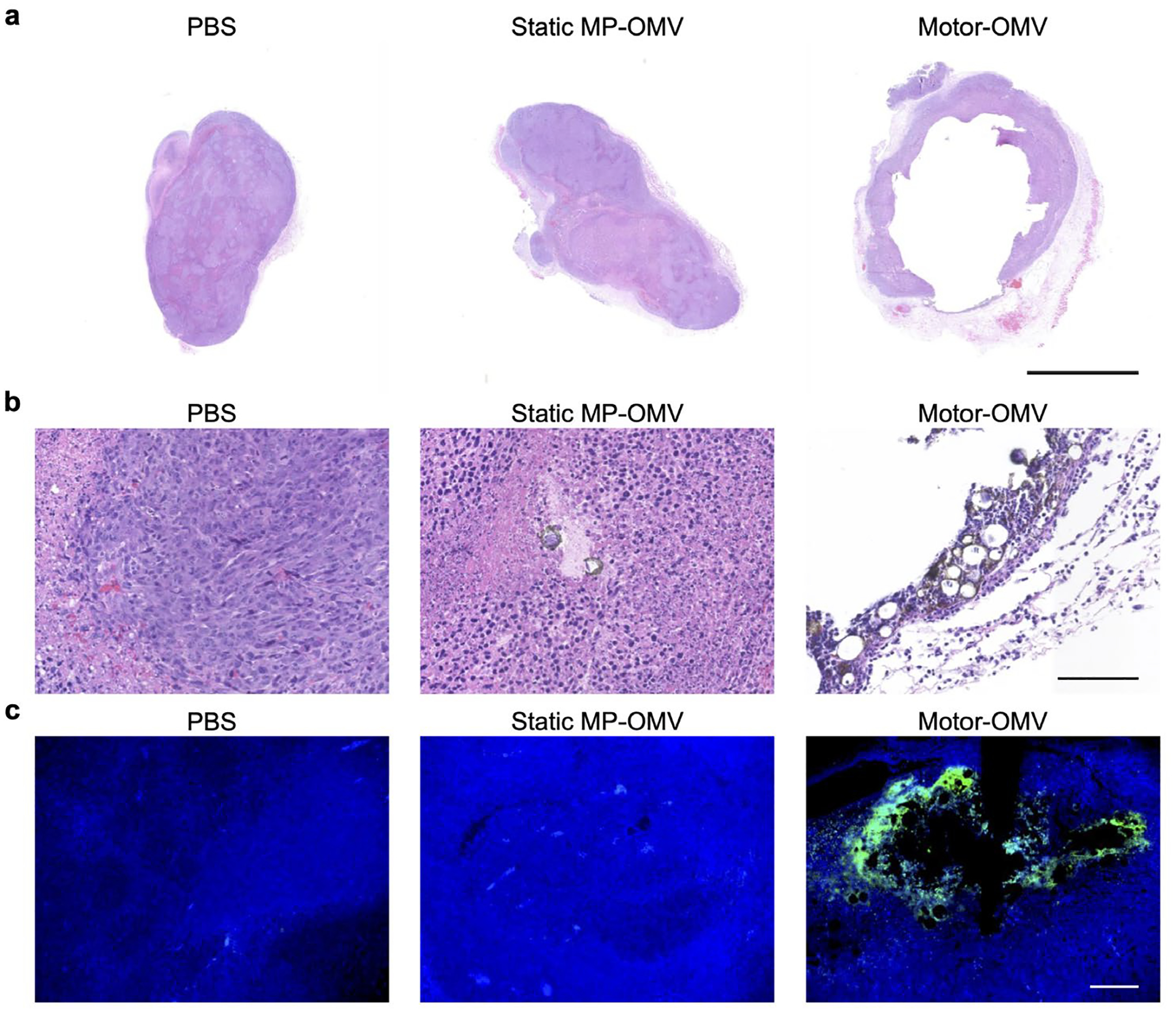
Mechanical destruction of solid tumor tissue in vivo by Motor-OMV. a) Representative H&E-stained whole tumor sections taken 1 day after treatment with Motor-OMV or control samples (scale bar = 2.5 mm). b) Representative H&E-stained tumor sections at higher magnification with micromotors visible in brown (scale bar = 100 μm). c) Fluorescence visualization of representative tumor sections stained with TUNEL (green) and DAPI (blue) (scale bar = 100 μm).
To evaluate the immunogenicity of Motor-OMV, we first studied their impact on dendritic cells using the murine DC2.4 cell line. The cells were pulsed with Motor-OMV or various control samples for 48 h and subsequently examined for signs of maturation. After treatment with Motor-OMV, clear upregulation of the maturation markers CD40, CD80, CD86, and major histocompatibility complex II (MHC-II) was observed (Figure 4a–h).[40] The expression levels were comparable to cells treated with OMVs alone at the same protein concentration, confirming that there was no loss of the immunostimulatory potential of OMVs after being incorporated onto the micromotors. On the other hand, cells treated with bare motors without OMVs retained basal levels of expression. Proinflammatory cytokine secretion into the cell culture supernatant was also assayed at different timepoints, and a clear upward trend over time was observed for the Motor-OMV and OMV groups (Figure 4i–l; Figure S8, Supporting Information). At 48 h, there were significant increases in tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), IL-12p40, and IL-1β. Consistent with the maturation studies, cytokine levels remained at basal levels for cells pulsed with bare motors. We also confirmed that there was no impact on DC2.4 cell viability when treated with Motor-OMV at the concentration used to study immune activation (Figure S9, Supporting Information). Overall, these results provided evidence that the Motor-OMV formulation could activate an innate immune response due to the presence of pathogen-associated molecular patterns present on the OMVs.[41]
Figure 4.
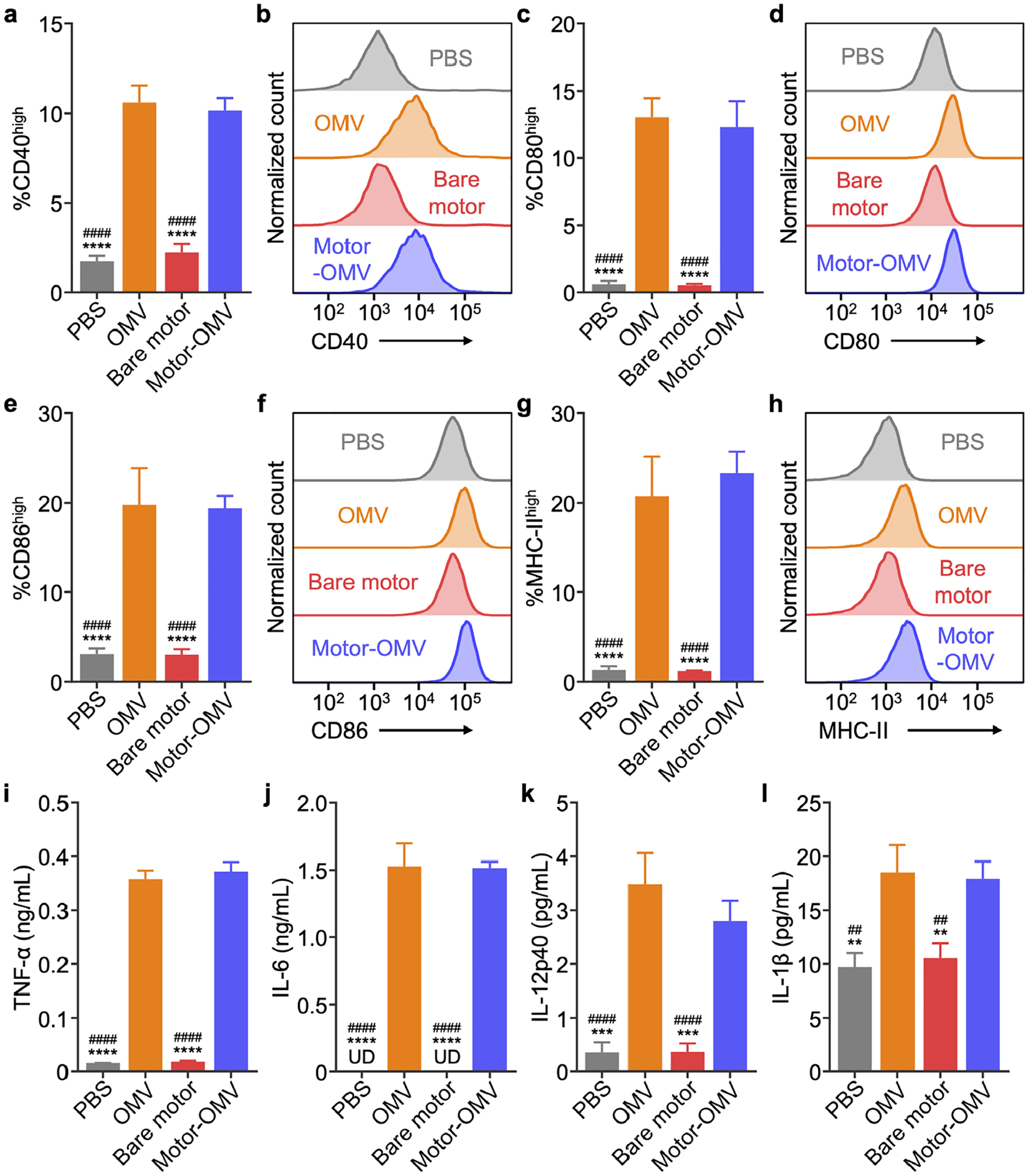
Activation of dendritic cells in vitro. a-h) Expression of maturation markers CD40 (a,b), CD80 (c,d), CD86 (e,f), and MHC-II (g,h) on DC2.4 after 2 days of incubation with Motor-OMV or control samples (n = 3, mean + SD). i-l) Concentration of TNF-α (i), IL-6 (j), IL-12p40 (k), and IL-1β (l) secreted by DC2.4 incubated with Motor-OMV or control samples for 2 days (n = 3, mean + SD; UD = undetectable). **p < 0.01, ***p < 0.001, and ****p < 0.0001 (compared to Motor-OMV); ##p < 0.01 and ####p < 0.0001 (compared to OMV); one-way ANOVA.
Having confirmed the immune-priming capability of Motor-OMV in vitro, we proceeded to look at their effects in vivo using the same MC38 colorectal cancer model. Similar to the tumor destruction studies, mice were intratumorally treated with Motor-OMV once the tumor sizes reached approximately 50 mm2. On the following day, the tumors were dissected and processed into single-cell suspensions for immunophenotyping. Within the CD45+ leukocyte population, there was a significantly higher number of CD11c+F4/80− dendritic cells with strong expression of CD40, CD80, CD86, and MHC-II in the tumors treated with Motor-OMV as compared to the PBS-treated tumors (Figure 5a–h). There were no significant differences between the Motor-OMV and static MP-OMV groups, likely due to the stimulation of innate immune responses driven by the pathogen-associated molecular patterns present on the OMVs that were shared by both formulations.
Figure 5.
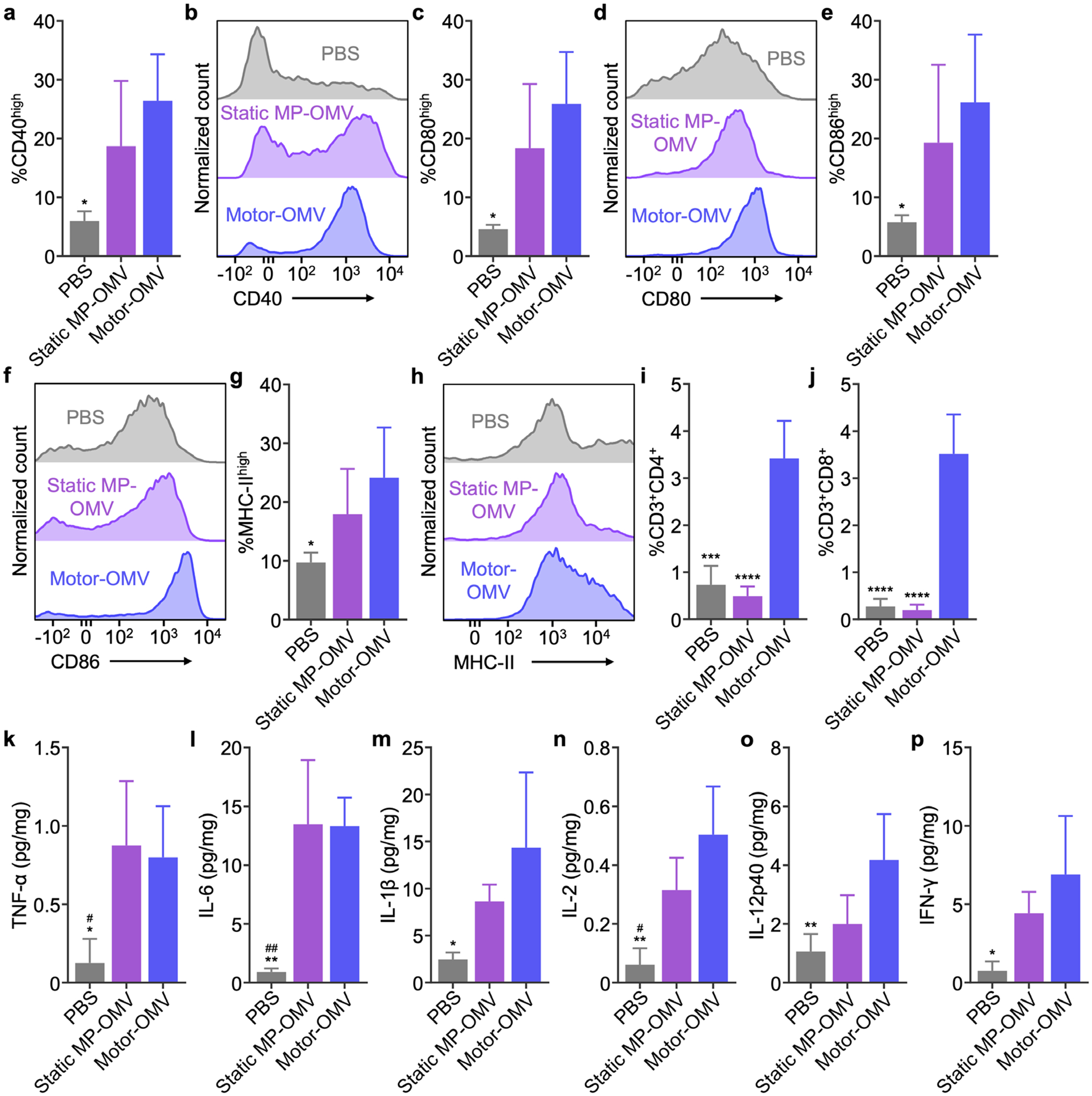
Immune recruitment and activation in vivo. a-h) Expression of maturation markers CD40 (a,b), CD80 (c,d), CD86 (e,f), and MHC-II (g,h) on CD11c+F4/80− dendritic cells within the CD45+ leukocyte population in tumors 1 day after intratumoral injection of Motor-OMV or control samples (n = 4, mean + SD). i,j) Percentage of infiltrating CD4+ (i) and CD8+ (j) T cells in tumors 4 days after Motor-OMV or control samples were administered intratumorally (n = 4, mean + SD). k-m) Levels of TNF-α (k), IL-6 (l), and IL-1β (m) in tumors 1 day after intratumoral treatment with Motor-OMV or control samples (n = 4, mean + SD). n-p) Levels of IL-2 (n), IL-12p40 (o), and IFN-γ (p) in tumors 4 days after Motor-OMV or control samples were injected intratumorally (n = 4, mean + SD). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 (compared to Motor-OMV); #p < 0.05 and ##p < 0.01 (compared to static MP-OMV); one-way ANOVA.
We next characterized infiltrating T cells within the tumor 4 days after treatment. Tumors treated with Motor-OMV had a significantly higher number of T cells with both the CD4+ and CD8+ phenotypes (Figure 5i,j). In contrast, treatment with static MP-OMV had negligible effects on T cell infiltration, indicating that, at the dosage we employed, the immunogenicity of the OMVs alone was not enough to activate strong cellular immunity. Upon further characterization, we observed a significantly higher number of activated CD8+ T cells after Motor-OMV treatment, as measured by CD25 and CD69 expression (Figure S10a,b, Supporting Information). The percentage of functional CD8+IFN-γ+ T cells was also significantly increased, while the percentage of CD4+ T cells with the Foxp3+ regulatory phenotype was significantly lower (Figure S10c,d, Supporting Information). To assess the antigen specificity of the infiltrating lymphocytes, we pulsed CD8+ cells isolated from treated tumors with MHC-I-restricted MC38 antigen epitopes, including peptides derived from Adpgk and p15E.[42, 43] After 7 days of restimulation, the number of CD3+CD8+ T cells was significantly higher in samples isolated from tumors treated with Motor-OMV (Figure S10e,f, Supporting Information). As cellular immunity mediated primarily by cytotoxic T cells is vital for tumor eradication, these results provided a promising indicator that antitumor immunity could be achieved using the Motor-OMV formulation.[44] Additionally, we assessed the cytokine profile inside the tumor after treatment with Motor-OMV. When examining the levels of TNF-α, IL-6, and IL-1β, which are frontline cytokines generally associated with innate immune activation,[45] all were significantly elevated after 1 day (Figure 5k–m). A similar result was observed on day 4 for cytokines involved in T cell activation, including IL-2, IL-12p40, and interferon-γ (IFN-γ) (Figure 5n–p).[46, 47] Treatment with static MP-OMV also resulted in elevated tumor cytokines, although the magnitude trended lower than what was observed for Motor-OMV. Taken together, the data here suggested that Motor-OMV could effectively recruit and activate immune cells towards an antitumor phenotype after in vivo administration.
The therapeutic efficacy of Motor-OMV was subsequently evaluated in a commonly employed bilateral tumor model. Mice were implanted with MC38 cells on the right flank to form a primary tumor and, at the same time, a secondary tumor with ten times fewer cells was inoculated into the left flank. Eight days later when tumor sizes reached approximately 30 mm2, mice were intratumorally treated every other day with 500 μg of Motor-OMV or various controls at the primary tumor site for a total of 4 treatments. Mice treated with Motor-OMV had a considerably smaller average sizes for both the treated primary tumor and the distant secondary tumor (Figure 6a–b; Figure S11, Supporting Information). Tumor growth slowed when treated with static MP-OMV, but the effects were not as profound as with active Motor-OMV, where 4 out of the 6 primary tumors were completely eradicated. Treatment with Motor-OMV was able to extend the median survival time to 38 days as compared to 18 days and 24 days for mice treated with PBS and static MP-OMV, respectively (Figure 6c). As Motor-OMV treatment utilizes antigenic material that is generated in situ by physical destruction, we elected to evaluate the broad applicability of the platform against additional solid tumor models, including CT26 colon adenocarcinoma (Figure 6d–f; Figure S12, Supporting Information) and the lowly immunogenic B16-F10 melanoma (Figure 6g–i; Figure S13, Supporting Information). In both cases, Motor-OMV outperformed the static MP-OMV control, significantly controlling primary and secondary tumor growth while extending median survival. Of note, Motor-OMV treatment resulted in complete primary tumor clearance in 67% and 33% of mice for the CT26 and B16-F10 models, respectively. In all three studies, the body weights of all mice remained similar throughout the course of the study, suggesting that there were no major adverse effects resulting from Motor-OMV treatment. To validate the importance of immune stimulation, we also tested the activity of bare micromotors without OMV coating against MC38 tumors, and no therapeutic benefit was observed (Figure S14, Supporting Information).
Figure 6.
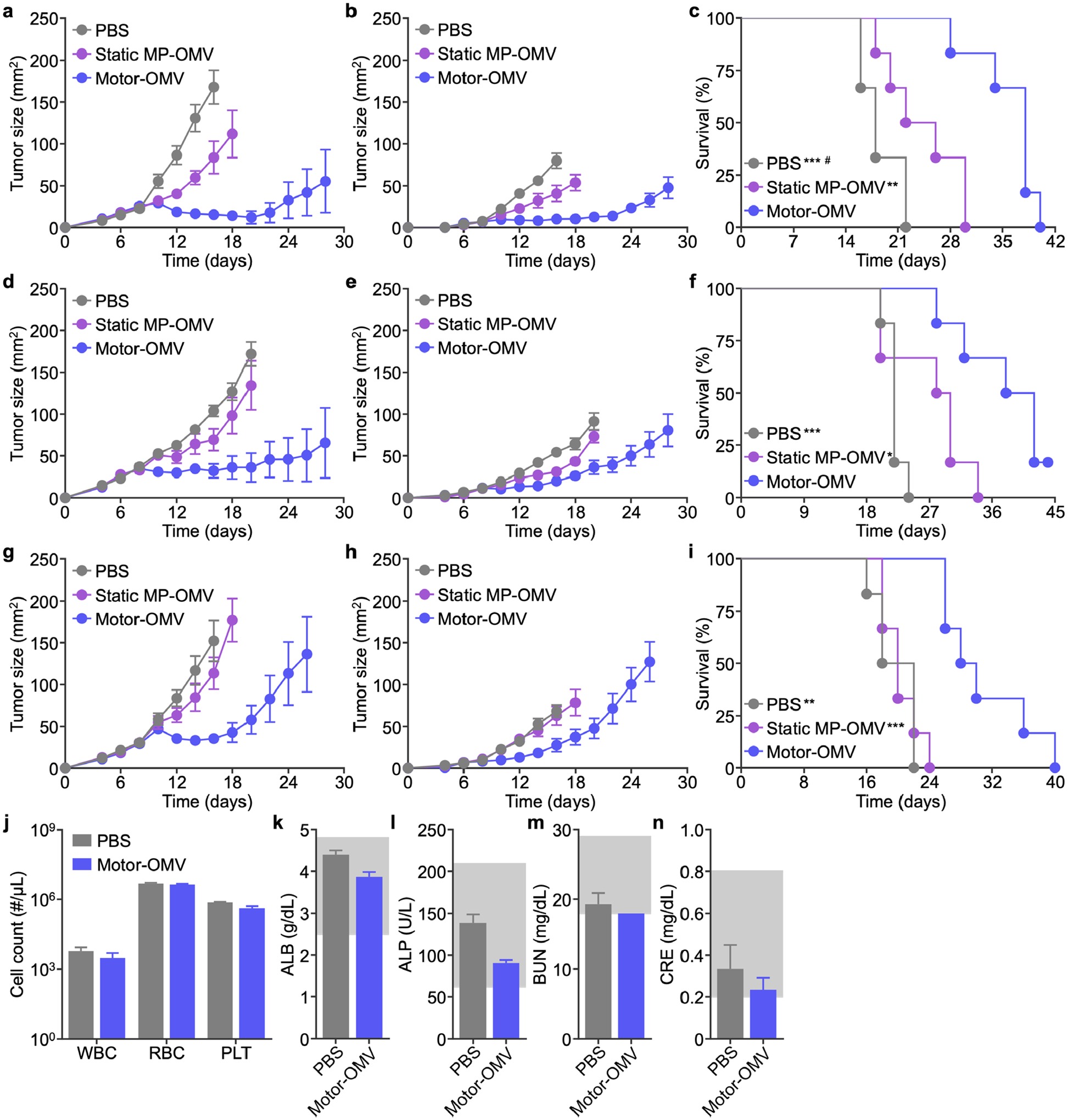
Therapeutic efficacy and safety in bilateral tumor models. a-i) MC38 (a-c), CT26 (d-f), or B16-F10 (g-i) cells were implanted subcutaneously into mice on the right flank to form a primary tumor and on the left flank at a lowered dosage to form a secondary tumor on day 0. Mice were intratumorally treated with Motor-OMV or control samples on days 8, 10, 12, and 14 at the primary tumor site. Average sizes of the primary (a, d, g) and secondary (b, e, h) tumors, as well as survival (c, f, i), were monitored over time (n = 6, mean ± SEM). j) Red blood cell (RBC), white blood cell (WBC), and platelet (PLT) counts 1 day after intratumoral treatment with Motor-OMV or PBS (n = 3, mean + SD). k-n) Levels of albumin (ALB, k), alkaline phosphatase (ALP, l), blood urea nitrogen (BUN, m), and creatinine (CRE, n) in the serum 1 day after intratumoral injection of Motor-OMV or PBS (n = 3, mean + SD). Gray boxes indicate the reference range for healthy mice. *p < 0.05, **p < 0.01, and ***p < 0.001 (compared to Motor-OMV); #p < 0.05 (compared to static MP-OMV); Mantel-Cox test.
To better understand the mechanism behind the improved control of secondary tumor growth, we evaluated the impact of Motor-OMV administration on the induction of systemic antitumor immunity. The splenocytes from treated tumor-bearing mice were isolated and then pulsed ex vivo with the Adpgk peptide, the p15E peptide, or MC38 whole cell lysate. After 7 days of restimulation, the number of CD3+CD8+ T cells was significantly higher in the Motor-OMV group compared with the PBS and static MP-OMV groups (Figure S15a–c, Supporting information). Furthermore, the concentrations of IL-2 (Figure S15d–f, Supporting Information) and IFN-γ (Figure S15g–i, Supporting Information) were significantly higher in the supernatant of the splenocyte cultures on day 4 and day 7, respectively. These results confirmed that Motor-OMV treatment was not only able to induce local immune cell recruitment and activation, but it also promoted the systemic generation of tumor-specific T cells that were able to control the growth of distant tumors through an abscopal effect.[48] The activation of systemic antitumor immunity can also help to mitigate potential concerns regarding the exacerbation of metastasis by physical tumor disruption.[49, 50]
To further evaluate safety, we quantified the number of red blood cells, white blood cells, and platelets in the blood of mice 1 day after treatment and observed no obvious differences (Figure 6j). Major serum biomarkers, including albumin, alkaline phosphatase, blood urea nitrogen, and creatinine, were also within normal ranges (Figure 6k–n). We also monitored serum cytokine concentrations over time and found that Motor-OMV elicited significantly lower levels of IL-6 and IL-12p40 compared with free OMVs, highlighting the advantage of coating the bacterial membrane material onto a micromotor substrate (Figure S16, Supporting Information). Overall, it was demonstrated that intratumoral administration of Motor-OMV could eradicate primary tumors and impair the growth of distant secondary tumors, while not exerting any noticeable toxicity.
In summary, we developed a micromotor formulation utilizing a combination of tumor tissue destruction and immune stimulation as a cancer therapy. The irreversible damage caused by micromotors significantly disrupted the tumor tissue and induced cell death. By incorporating highly immunogenic bacterial OMVs in the formulation, Motor-OMV were able to recruit immune cells into the tumor site to process tumor antigens from the dead cancer cells and elicit robust systemic anticancer immunity, which was essential for achieving tumor regression. In multiple cancer models, this approach was shown to completely eradicate a significant portion of the treated tumors while also controlling the growth of secondary tumors on the contralateral flank. In this case, we chose to employ bacterial OMVs due to their innate immunostimulatory properties and their safer nonreplicating nature. With further engineering, future platforms based on this combinatorial micromotor concept could utilize other types of immune agonists.[16] Additional payloads, including chemotherapeutics or other immunotherapeutics that increase tumor immunogenicity, could be loaded onto the micromotors in the PLGA or chitosan layer to amplify antitumor efficacy.[12, 15] The platform could also be combined with checkpoint blockade therapy to reduce global immune suppression, thus enhancing systemic antitumor immunity and allowing for improved control of distant tumors.[51] Motor-OMV propulsion can be fine-tuned by adjusting the size of the initial Mg microparticles, gold thickness, and carrier solution parameters such as ion concentration and pH. Additionally, pH-sensitive coatings could be employed to achieve better control over the timing of micromotor activation within the tumor. Because the Motor-OMV approach leverages the antigenic material within the tumor in situ, it will have broad applicability across various types of solid tumors.[52–54] The materials used in the micromotors are relatively safe, biodegradable, and inert, which may help to facilitate downstream clinical translation. Moreover, since the fabricated micromotors can be stored in dried form, we expect them to have a long shelf life. Overall, continued research along these lines is expected to advance the field of microrobotics and push it towards eventual clinical translation.
Experimental Section
Cell Lines.
DC2.4, an immortalized murine dendritic cell line isolated from a C57BL/6 mouse, was a gift from the Dong-Er Zhang laboratory. MC38, a murine colon adenocarcinoma cell line isolated from a C57BL/6 mouse, was a gift from the Jack Bui laboratory. CT26, a murine colon adenocarcinoma cell line derived from a BALB/c mouse, was a gift from the Silvio Gutkind laboratory. B16-F10, a murine melanoma derived from a C57BL/6 mouse, was obtained from the American Type Culture Collection (CRL-6475). Ampho-Phoenix, a retroviral transduction system derived from the HEK293T cell line, was obtained from the National Gene Vector Biorepository. All cell lines were cultured at 37 °C with a humidified atmosphere of 5% CO2 in T175 tissue culture flasks (Falcon) with Dulbecco’s modified Eagle medium (DMEM; Corning) supplemented with 10% (v/v) bovine growth serum (Hyclone) and 1% (v/v) penicillin-streptomycin (Gibco). CT26 cells were cultured using RPMI 1640 medium (Gibco) as a substitute for DMEM, and Ampho-Phoenix cells were maintained with an additional 200 μg/mL of hygromycin B (InvivoGen). All cell lines were tested for mycoplasma contamination monthly with a PCR detection kit (Applied Biological Materials).
To produce the GFP-expressing MC38 cell line, Ampho-Phoenix cells were transfected using Lipofectamine 2000 (Life Technologies) with 10 μg of the pQCXIH retroviral expression vector (Clontech) containing the GFP gene.[55] Two days later, Ampho-Phoenix cells were removed from the virus-containing media by centrifugation. The supernatant was mixed with polybrene (Millipore Sigma) to a concentration of 4 μg/mL and used to transduce MC38 by spinfection at 800 g for 90 min. The next day, the transduced MC38 cells were selected with 500 μg/mL of hygromycin B to enrich the GFP-positive population. Finally, MC38-GFP cells were sorted for the top 5% of positive cells with a Becton Dickinson Influx sorter and maintained in 200 μg/mL of hygromycin B for future experiments. GFP expression was verified with a Becton Dickinson Accuri C6 flow cytometer and analyzed using FlowJo software.
Animal Care.
Mice (6 to 8 weeks old) were housed in an animal facility at the University of California San Diego (UCSD) under federal, state, local, and National Institutes of Health (NIH) guidelines. All animal experiments were performed in accordance with NIH guidelines and approved by the Institutional Animal Care and Use Committee of UCSD under protocol number S09388.
Bacterial OMV Derivation.
OMV collection was done following a modified version of previously published protocols.[56, 57] Briefly, DH5α E. coli (New England Biolabs) was cultured overnight in Luria-Bertani broth (Becton Dickinson) in a rotatory shaker and refreshed with the medium at a 1:100 dilution. After 24 h, the E. coli bacteria were pelleted at 4,500 g for 10 min, and the supernatant was filtered with 0.45-μm PES vacuum filters (Nalgene). Then, 4 L of filtrate was concentrated nearly 100 times using a KrosFlo KR2i tangential flow filtration system equipped with a hollow fiber mPES membrane column with a molecular weight cutoff of 30 kDa (Spectrum). The OMVs in the concentrate were pelleted out at 150,000 g for 2 h at 4 °C with a Beckmann Optima XPN-80 ultracentrifuge and resuspended with DNase/RNase-free water (Invitrogen). Protein content was determined with a BCA protein assay kit (Pierce) according to the manufacturer’s instructions, and the OMVs were stored at −80 °C for future use.
Fabrication of Motor-OMV.
Mg microparticles (TangShan WeiHao Magnesium Powder Co.) with an average size of 20 ± 5 μm were utilized as the core for Motor-OMV fabrication. To remove any impurities, the Mg microparticles were washed with acetone and dried under a nitrogen stream. Washed particles were then dispersed onto a glass slide and sputtered with a thin Au layer using a Denton Discovery 18 instrument for 3 s under a vacuum of 5 × 10−6 Torr, DC power of 200 W, argon flow of 2.4 mT, and a rotation speed of 13 rpm. Then, the microparticles were coated with TiO2 by ALD for 1000 cycles at 100 °C with a Beneq TFS 200 system. ALD is a chemical vapor deposition technique which uses gas phase reactants, leading to uniform coatings over the Mg microparticles while leaving a small opening of approximately 2 μm at the contact point of the particle with the glass slide. Subsequently, the particles were coated in 100 μl of 1% (w/v) PLGA (Millipore Sigma) dissolved in ethyl acetate (Millipore Sigma), followed by coating in 100 μl of 0.05% (w/v) chitosan (Millipore Sigma), 0.1% (w/v) sodium dodecyl sulfate (SDS; Millipore Sigma), and 0.02% (v/v) acetic acid (Millipore Sigma) in water. The coated motors were collected off the glass slide and aliquoted into microcentrifuge tubes for OMV coating.
To fabricate Motor-OMV, OMVs in water were added into tubes containing dry micromotors at a 1:5, 1:10, 1:20, or 1:100 weight ratio and sonicated in a Fisher CPXH 2800 ultrasonic bath for 15 min. Then, micromotors were washed twice with ethanol by centrifuging at 1,500 g for 5 min. After removing the supernatant, any residual ethanol was evaporated in a fume hood to form dry Motor-OMV pellets. The static MP-OMV control was fabricated using Mg microparticles that were coated with two layers of TiO2 via ALD and loaded with OMVs following the same procedures. The double coating fully covered the surface of the Mg, avoiding reaction with the surrounding environment. Besides the loading optimization study, all other experiments employed Motor-OMV fabricated at the 1:10 OMV to micromotor weight ratio. All dry micromotors were stored at −20 °C if not used immediately.
Characterization of Motor-OMV.
SEM images of micromotors without OMV coating were acquired with an FEI Quanta 250 ESEM microscope, using an acceleration voltage of 10 kV. EDX spectroscopy mapping analysis was performed using an Oxford EDS detector coupled to the SEM microscope and operated by Pathfinder software. To verify successful loading of the OMVs, fluorescent Motor-OMV were prepared following the same protocol as above with the chitosan layer labeled using FITC (λex/λem = 490/525 nm; Fluka) and OMVs labeled with 1% (wt/wt) 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (λex/λem = 560/583 nm; Avanti Polar Lipids). Fluorescence images were captured using an EVOS FL microscope coupled with either a 10× or 40× objective with the GFP and RFP filters.
To determine the OMV loading, dry Motor-OMV were resuspended in 2% SDS and vortexed briefly. The samples were then incubated at 37 °C for 30 min and sonicated for 2 min in a Fisher FS30D bath sonicator. Micromotors were spun down at 10,000 g for 5 min, and the protein content in the supernatant was analyzed with a BCA protein assay kit. The background from an equivalent amount of bare motors was subtracted from each measurement to obtain the final loading value.
To confirm the OMVs were properly transferred onto the micromotor surface, dry Motor-OMV were resuspended with DNase/RNase-free water at a final protein concentration of 500 μg/mL. Samples were then diluted with NuPAGE lithium dodecyl sulfate sample buffer (Novex) at a 3:1 ratio and denatured at 70 °C for 30 min in a Fisher Isotemp digital dry bath. To ensure the proteins were properly dislodged from the micromotors, each sample was sonicated for 1 min before loading them into a 12-well Bolt 4–12% bis-tris gel (Novex). Protein separation was resolved in a mini blot module using Bolt MOPS SDS running buffer (Novex) at 165 V for 45 min. Proteins from the gel were then transferred onto a 0.45-μm nitrocellulose membrane (Pierce) at 15 V for 30 min in Bolt transfer buffer (Novex) supplemented with 10% (v/v) methanol (Fisher Scientific). To identify specific outer membrane proteins, the membrane was first blocked in 5% (w/v) milk (Apex Bioresearch Products) in PBS (Teknova) with 0.05% (v/v) Tween 20 (National Scientific) for 1 h and then stained with either an anti-ompA rabbit antibody (Biorbyt) or anti-ompC rabbit antibody (MyBioSource) at 1 μg/mL overnight at 4 °C. Excessive antibodies were washed off with 0.05% Tween 20 in PBS 3 times for 5 min each before incubating with a horseradish peroxidase-conjugated anti-rabbit secondary antibody (Biolegend) for 2 h at room temperature. The membrane was washed again as before and further cleaned with distilled water before development with ECL western blotting substrate (Pierce) using autoradiography films (Genesee Scientific) in an ImageWorks Mini-Medical/90 Developer.
Propulsion Activity of Motor-OMV.
Autonomous Motor-OMV propulsion was observed by placing the different micromotor formulations in PBS (pH 5 or pH 6.5). Images and videos were acquired with a Nikon Eclipse 80i upright microscope coupled with a 10× or 40× objective lens and a QuantEM 512/SC camera. The speed of Motor-OMV was characterized using MetaMorph software, which calculates the distance traveled during a given time frame. To visualize the destruction of tumor spheroids, MC38-GFP cells were seeded into 96-well assay plates (Corning) that were precoated with 1% (w/v) agarose (Apex Bioresearch Products) at 2 × 104 cells per well. Six days later, spheroids were isolated from each well, washed 3 times with PBS, and then suspended in PBS (pH 5). The individual spheroids were mixed with solutions containing either Motor-OMV (1 mg), static MP-OMV, or PBS (pH 5) at a 1:1 volume ratio. The integrity of the spheroids was monitored immediately before mixing with the samples and after incubating for 4 h at 37 °C. Prior to imaging, bubbles that had formed as a result of the micromotor reaction were removed. Images were obtained with a Keyence BZ-X710 fluorescence microscope using a 4× objective lens equipped with the GFP filter (UCSD Specialized Cancer Center Support P30 Grant 2P30CA023100) and analyzed with BZ-X Analyzer software. To assess the potential impact of Motor-OMV on local temperature and pH, each parameter was measured before and 10 min after the addition of dry micromotors into 2 mL of PBS (pH 5) at a final concentration of 20 mg/mL.
In Vivo Destruction Activity.
To study the destruction capability of the micromotors in a solid tumor environment, 1 × 106 MC38 cells were inoculated into syngeneic C57BL/6NHsd mice (Envigo) on both flanks. Once the tumors reached approximately 50 mm2 on day 10, mice were randomly distributed and injected intratumorally with either Motor-OMV (1 mg), static MP-OMV, or PBS (pH 5). On the following day, the mice were euthanized and the tumors were collected in their entirety. The tissues were fixed in 10% (v/v) formalin (Fisher Scientific) for 24 h, dehydrated with ethanol, and sectioned for either H&E staining or fluorescent TUNEL. Histological preparation was performed by the Moores Cancer Center Tissue Technology Shared Resource (Cancer Center Support Grant P30CA23100). H&E images were acquired with a Hamamatsu NanoZoomer 2.0-HT Slide Scanner (UCSD School of Medicine Microscopy Core Grant NS047101) and analyzed with the NanoZoomer Digital Pathology software. TUNEL slides were further stained with VECTASHIELD mounting medium with DAPI (Vector Laboratories), and images were acquired with a Keyence BZ-X710 fluorescence microscope using a 20× objective lens with the DAPI and GFP filters.
To study necrotic cell death, tumor cells were first dissociated by cutting them into small 1–3 mm pieces, followed by digestion with occasional shaking at 37 °C for 30 min in the presence of 1 mg/mL collagenase D (Roche) and 100 μg/mL DNase I (Roche) in DMEM. The tissue was then passed through a 70-μm mesh cell strainer (Fisher Scientific) and spun down at 700 g for 5 min. Red blood cells (RBCs) in the mixture were lysed with a commercial lysis buffer (Biolegend) according to the manufacturer’s instructions. Cells were incubated with 1% (w/v) bovine serum albumin (BSA; Millipore Sigma) in PBS on ice for 15 min to block nonspecific binding and then further incubated with TruStain FcX PLUS anti-mouse CD16/32 (Biolegend) for 15 min on ice to block Fc receptor binding. Samples were first stained with Pacific Blue-conjugated anti-mouse CD45 (30-F11, Biolegend) and washed with 1% BSA in PBS before further staining with APC-labeled annexin V (Life Technologies) and propidium iodide (Biolegend) in annexin V binding buffer (Biolegend) according to manufacturer’s instructions. Single-stained controls were prepared for gating purposes. Data were acquired with a Becton Dickinson FACSCanto II flow cytometer and analyzed using FlowJo software.
Dendritic Cell Activation.
DC2.4 cells were seeded into 12-well tissue culture plates (Genesee Scientific) at 1 × 105 cells per well and allowed to adhere overnight. Equivalent amounts of Motor-OMV (500 ng/mL OMVs & 15.5 μg/mL micromotors), bare motors, free OMVs, and PBS (pH 5) were added into different wells and incubated with the cells. At different timepoints (4 h, 8 h, 12 h, 24 h, and 48 h), the supernatant was collected from each well for analysis by an enzyme-linked immunosorbent assay (ELISA), and the media was replenished. Cytokine content in the supernatant was analyzed using the appropriate ELISA reagents (Table S1, Supporting Information) according to the manufacturer’s instructions. At the final timepoint, the cells were collected with 1 mM ethylenediaminetetraacetic acid (EDTA) in PBS (Corning) and centrifuged at 700 g for 5 min. Cells were then blocked with 1% BSA in PBS on ice to remove nonspecific binding. Each sample was split into four for staining (Table S1, Supporting Information). After 30 min of incubation at 4 °C, the samples were washed three times with 1% BSA in PBS, and data were acquired with a Becton Dickinson Accuri C6 flow cytometer. Appropriate dye-labeled isotype antibody controls were used to assist with gating. To determine viability, DC2.4 cells were treated for 2 days and collected as described above. The cells were then blocked with 1% BSA in PBS and stained with FITC-labeled annexin V (Biolegend) and propidium iodide in annexin V binding buffer according to manufacturer’s instructions. Single-stained controls were prepared for gating purposes. Data were acquired with a Becton Dickinson FACSCanto II flow cytometer. All flow cytometry data were analyzed using FlowJo software.
In Vivo Immune Stimulation.
For tumor implantation, 1 × 106 MC38 cells were subcutaneously inoculated into C57BL/6NHsd mice on both flanks. Once tumor sizes reached approximately 50 mm2, mice were intratumorally injected with the different formulations as before. The mice were euthanized, and the tumors were collected either 1 day or 4 days post-administration. To determine the cytokine content, tumors were weighed and homogenized with a Biospec Mini-Beadbeater-16 in 1 mL of PBS. Tissue homogenates were spun down at 10,000 g for 5 min, and the supernatant was clarified through 0.22-μm PVDF syringe filters (CellTreat). Samples were stored at −20 °C until ELISA analysis (Table S1, Supporting Information).
For immunophenotyping, tumors were dissociated as described above, followed by RBC lysis. Cells were then stained with the Zombie Aqua fixable viability kit (Biolegend) or LIVE/DEAD fixable aqua dead cell stain kit (Life Technologies) for 30 min at room temperature and washed once with 1% BSA in PBS. Afterwards, the cells were blocked with 1% BSA on ice for 15 min and then with TruStain FcX PLUS anti-mouse CD16/32 antibody for another 15 min on ice. Cells were stained with the appropriate antibodies (Table S1, Supporting Information) for 30 min at 4 °C before the unbound antibodies were washed out as before. For intracellular staining, cells were prepared using a True-Nuclear transcription factor buffer set (Biolegend) according to the manufacturer’s instructions after surface staining. Afterwards, the cells were stained with the appropriate intracellular antibodies (Table S1, Supporting Information) for 30 min at room temperature in the permeabilization buffer. Unbound antibodies were then washed away once with the permeabilization buffer and once with 1% BSA in PBS. Single-stained and fluorescence minus one control samples were prepared similarly for gating and compensation purposes.
Antigen Specificity.
To confirm the antigen specificity of the T cells generated after treatment, C57BL/6NHsd mice were inoculated subcutaneously with MC38 cells. Once the tumors reached approximately 50 mm2 in size on day 10, they were treated with the different formulations. Four days later, the mice were euthanized, and the tumor tissue and spleen were collected. Tumor dissociation was performed as previously described. After RBC lysis, CD8+ cells were isolated by magnetic separation using CD8a MicroBeads (Ly-2, Miltenyi Biotec) on MACS LS separation columns (Miltenyi Biotec) per the manufacturer’s instructions. Purified CD8+ cells were seeded into 12-well suspension plates (Genesee Scientific) at 1 × 105 cells per well. To derive splenocytes, the spleen tissue was physically sheared and extruded through a 70-μm mesh cell strainer. The cells were centrifuged at 700 g for 5 min, and RBCs were lysed as before. Splenocytes were seeded into 6-well suspension plates (Genesee Scientific) at 5 × 106 cells per well.
CD8+ cells were cultured in Iscove’s modified Dulbecco’s medium (Gibco) supplemented with 10% (v/v) USDA-certified fetal bovine serum (Gibco), 1% (v/v) GlutaMAX (Gibco), 1% penicillin-streptomycin, and 55 μM 2-mercaptoethanol (Gibco). Splenocytes were cultured in the same medium but with an additional 10 ng/mL of granulocyte/macrophage-colony stimulating factor (Biolegend). For restimulation, cells were pulsed with 1 μg/mL of an Adpgk peptide (ASMTNMELM; Biomatik), 1 μg/mL of a p15E peptide (KSPWFTTL; Biomatik), or 20 μg/mL of MC38 whole cell lysate. To prepare the whole cell lysate, MC38 cells at full confluency were detached with 1 mM EDTA in PBS, spun down at 700 g for 5 mins, and the pellet was resuspended in DNase/RNase-free water. The solution was then frozen at −80 °C and thawed in a 37 °C water bath for a total of three cycles. Protein content was determined with a BCA protein assay kit. Supernatant from the splenocyte cultures was collected on day 4 and day 7 for ELISA analysis (Table S1, Supporting Information). After 7 days of restimulation, cells were collected, blocked with 1% BSA in PBS on ice for 15 min, blocked with TruStain FcX PLUS anti-mouse CD16/32 for another 15 min, and then stained with the appropriate antibodies (Table S1, Supporting Information). Unbound antibodies were washed out with 1% BSA in PBS, and the cells were stained with 7-AAD viability staining solution (Biolegend) for a minimum of 10 min. Cell concentration was determined by premixing the cells with a known amount of CountBright absolute counting beads (Life Technologies), followed by data acquisition with a Becton Dickinson FACSCanto II flow cytometer and analysis using FlowJo software.
Therapeutic Efficacy.
Bilateral tumor models were employed to study the therapeutic potential of Motor-OMV. For the MC38 model, 1 × 106 cells were subcutaneously inoculated into the right flank of C57BL/6NHsd mice for the primary tumor and 1 × 105 cells were inoculated into the contralateral flank for the secondary tumor. The same inoculation dosages were utilized to generate the primary and secondary tumors for the CT26 model in BALB/c mice (Charles River Laboratory). For the B16-F10 model, 5 × 105 cells were subcutaneously inoculated into the right flank of C57BL/6NHsd mice for the primary tumor and 1 × 105 cells were inoculated into the contralateral flank for the secondary tumor. Once the primary tumor reached approximately 30 mm2 on day 8, mice were intratumorally treated with either Motor-OMV (500 μg), static MP-OMV, bare motors, or PBS (pH 5) every other day for a total of 4 treatments. Both tumors were monitored with calipers every other day starting on day 4, and the body weight of each mouse was monitored every week starting on day 0. The tumor sizes were calculated as the length × width and the experimental endpoint was predefined as either the primary or secondary tumor exceeding 200 mm2.
In Vivo Safety Studies.
To evaluate the safety of the micromotor formulation, MC38 tumor-bearing C57BL/6NHsd mice were intratumorally treated with 1 mg of Motor-OMV or PBS (pH 5) when tumor sizes reach approximately 50 mm2 on day 10 post-inoculation. After 24 h, blood was collected via the submandibular vein and allowed to coagulate. The serum for chemistry analysis was then collected by centrifuging the coagulated blood at 3,000 g for 5 min. A separate small aliquot of blood was collected into Microvette 100 potassium-EDTA blood collection tubes (Sarstedt) for hematological analysis. All tests were performed by the Animal Care Program Diagnostic Services Laboratory at UCSD. To monitor serum cytokine levels, MC38 tumor-bearing mice were intratumorally administered with 1 mg of Motor-OMV or an equivalent amount of free OMVs once the tumors reached approximately 50 mm2 on day 10. Blood was collected via the submandibular vein at 6 h, 24 h, and 48 h after treatment, and the serum was derived as before. Serum from unmanipulated tumor-bearing mice was used to establish a baseline. The samples were stored at −20 °C until ELISA analysis (Table S1, Supporting Information).
Statistical Analysis.
All data were analyzed without any preprocessing. For tumor growth kinetics, data are presented as mean ± SEM, while all other data are presented as mean ± SD. A minimum sample size of 3 was used for all studies in which statistical analysis was performed. Comparisons between two groups were done with an unpaired two-tailed Student’s t-test. For studies with three or more groups, one-way ANOVA with Turkey’s post-hoc analysis was used to determine significance. Survival curves were compared using the Mantel-Cox test. All statistical analysis was performed using GraphPad Prism 8.
Supplementary Material
Acknowledgements
J.Z. and E. K. contributed equally to this work. This work is supported by the National Institutes of Health under Award Number R01CA200574 and the Defense Threat Reduction Agency Joint Science and Technology Office for Chemical and Biological Defense under Grant Number HDTRA1-18-1-0014. R.M-U. acknowledges the support from the Fulbright grants and the Conicyt PFCHA/Doctorado becas Chile/2015-56150011.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Mellman I, Coukos G, Dranoff G, Nature 2011, 480, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].McNutt M, Science 2013, 342, 1417. [DOI] [PubMed] [Google Scholar]
- [3].Martin JD, Cabral H, Stylianopoulos T, Jain RK, Nat. Rev. Clin. Oncol 2020, 17, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kroll AV, Fang RH, Jiang Y, Zhou J, Wei X, Yu CL, Gao J, Luk BT, Dehaini D, Gao W, Zhang L, Adv. Mater 2017, 29, 1703969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sang W, Zhang Z, Dai Y, Chen X, Chem. Soc. Rev 2019, 48, 3771. [DOI] [PubMed] [Google Scholar]
- [6].Liang JL, Luo GF, Chen WH, Zhang XZ, Adv. Mater 2021, DOI: 10.1002/adma.202007630. [DOI] [PubMed] [Google Scholar]
- [7].Khalil DN, Smith EL, Brentjens RJ, Wolchok JD, Nat. Rev. Clin. Oncol 2016, 13, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li J, Esteban-Fernandez de Avila B, Gao W, Zhang L, Wang J, Sci. Robot 2017, 2, eaam6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Medina-Sanchez M, Xu H, Schmidt OG, Ther. Deliv 2018, 9, 303. [DOI] [PubMed] [Google Scholar]
- [10].Liu K, Ou J, Wang S, Gao J, Liu L, Ye Y, Wilson DA, Hu Y, Peng F, Tu Y, Appl. Mater. Today 2020, 20, 100694. [Google Scholar]
- [11].Hortelao AC, Carrascosa R, Murillo-Cremaes N, Patino T, Sanchez S, ACS Nano 2019, 13, 429. [DOI] [PubMed] [Google Scholar]
- [12].Karshalev E, Zhang Y, Esteban-Fernandez de Avila B, Beltran-Gastelum M, Chen Y, Mundaca-Uribe R, Zhang F, Nguyen B, Tong Y, Fang RH, Zhang L, Wang J, Nano Lett 2019, 19, 7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wan M, Li T, Chen H, Mao C, Shen J, Angew. Chem. Int. Ed. Engl 2021, 60, 13158. [DOI] [PubMed] [Google Scholar]
- [14].Ou J, Liu K, Jiang J, Wilson DA, Liu L, Wang F, Wang S, Tu Y, Peng F, Small 2020, 16, 1906184. [DOI] [PubMed] [Google Scholar]
- [15].Esteban-Fernandez de Avila B, Angsantikul P, Li J, Angel Lopez-Ramirez M, Ramirez-Herrera DE, Thamphiwatana S, Chen C, Delezuk J, Samakapiruk R, Ramez V, Obonyo M, Zhang L, Wang J, Nat. Commun 2017, 8, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang C, Fernandez de Avila BE, Mundaca-Uribe R, Lopez-Ramirez MA, Ramirez-Herrera DE, Shukla S, Steinmetz NF, Wang J, Small 2020, 16, 1907150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schmidt CK, Medina-Sanchez M, Edmondson RJ, Schmidt OG, Nat. Commun 2020, 11, 5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mundaca-Uribe R, Esteban-Fernandez de Avila B, Holay M, Lekshmy Venugopalan P, Nguyen B, Zhou J, Abbas A, Fang RH, Zhang L, Wang J, Adv. Healthc. Mater 2020, 9, 2000900. [DOI] [PubMed] [Google Scholar]
- [19].Xu C, Wang S, Wang H, Liu K, Zhang S, Chen B, Liu H, Tong F, Peng F, Tu Y, Li Y, Nano Lett 2021, 21, 1982. [DOI] [PubMed] [Google Scholar]
- [20].Xu X, Huo ZY, Guo JM, Liu H, Qi XL, Wu ZH, Bio-Des. Manuf 2020, 3, 133. [Google Scholar]
- [21].Fang RH, Kroll AV, Gao W, Zhang L, Adv. Mater 2018, 30, 1706759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang HY, Li ZS, Wu ZG, He Q, Adv. Ther 2019, 2, 1900096. [Google Scholar]
- [23].Gao CY, Lin ZH, Lin XK, He Q, Adv. Ther 2018, 1, 1800056. [Google Scholar]
- [24].Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L, Nature 2015, 526, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M, Nat. Mater 2009, 8, 543. [DOI] [PubMed] [Google Scholar]
- [26].Wang W, Zhou C, Adv. Healthc. Mater 2020, 10, 2001236. [DOI] [PubMed] [Google Scholar]
- [27].Soto F, Chrostowski R, Front. Bioeng. Biotechnol 2018, 6, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Min Y, Roche KC, Tian S, Eblan MJ, McKinnon KP, Caster JM, Chai S, Herring LE, Zhang L, Zhang T, DeSimone JM, Tepper JE, Vincent BG, Serody JS, Wang AZ, Nat. Nanotechnol 2017, 12, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering S, Nat. Nanotechnol 2016, 11, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Newman JH, Chesson CB, Herzog NL, Bommareddy PK, Aspromonte SM, Pepe R, Estupinian R, Aboelatta MM, Buddhadev S, Tarabichi S, Lee M, Li S, Medina DJ, Giurini EF, Gupta KH, Guevara-Aleman G, Rossi M, Nowicki C, Abed A, Goldufsky JW, Broucek JR, Redondo RE, Rotter D, Jhawar SR, Wang SJ, Kohlhapp FJ, Kaufman HL, Thomas PG, Gupta V, Kuzel TM, Reiser J, Paras J, Kane MP, Singer EA, Malhotra J, Denzin LK, Sant’Angelo DB, Rabson AB, Lee LY, Lasfar A, Langenfeld J, Schenkel JM, Fidler MJ, Ruiz ES, Marzo AL, Rudra JS, Silk AW, Zloza A, Proc. Natl. Acad. Sci. U. S. A 2020, 117, 1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kaparakis-Liaskos M, Ferrero RL, Nat. Rev. Immunol 2015, 15, 375. [DOI] [PubMed] [Google Scholar]
- [32].Holay M, Guo Z, Pihl J, Heo J, Park JH, Fang RH, Zhang L, ACS Appl. Bio Mater 2020, 4, 3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kim OY, Park HT, Dinh NTH, Choi SJ, Lee J, Kim JH, Lee SW, Gho YS, Nat. Commun 2017, 8, 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Qing S, Lyu C, Zhu L, Pan C, Wang S, Li F, Wang J, Yue H, Gao X, Jia R, Wei W, Ma G, Adv. Mater 2020, 32, 2002085. [DOI] [PubMed] [Google Scholar]
- [35].Gao W, Feng X, Pei A, Gu Y, Li J, Wang J, Nanoscale 2013, 5, 4696. [DOI] [PubMed] [Google Scholar]
- [36].Weiner JH, Li L, Biochim. Biophys. Acta 2008, 1778, 1698. [DOI] [PubMed] [Google Scholar]
- [37].Lee G, Park J, Geochim. Cosmochim. Acta 2013, 102, 162. [Google Scholar]
- [38].Makar GL, Kruger J, Int. Mater. Rev 1993, 38, 138. [Google Scholar]
- [39].Messmer MN, Snyder AG, Oberst A, Cell Death Differ 2019, 26, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mbongue JC, Nieves HA, Torrez TW, Langridge WH, Front. Immunol 2017, 8, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ellis TN, Kuehn MJ, Microbiol. Mol. Biol. Rev 2010, 74, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, Modrusan Z, Mellman I, Lill JR, Delamarre L, Nature 2014, 515, 572. [DOI] [PubMed] [Google Scholar]
- [43].Melero I, Quetglas JI, Reboredo M, Dubrot J, Rodriguez-Madoz JR, Mancheno U, Casales E, Riezu-Boj JI, Ruiz-Guillen M, Ochoa MC, Sanmamed MF, Thieblemont N, Smerdou C, Hervas-Stubbs S, Cancer Res 2015, 75, 497. [DOI] [PubMed] [Google Scholar]
- [44].Grivennikov SI, Greten FR, Karin M, Cell 2010, 140, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tosi MF, Allergy Clin J. Immunol 2005, 116, 241. [DOI] [PubMed] [Google Scholar]
- [46].Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C, Immunity 2009, 31, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kennedy R, Celis E, Immunol. Rev 2008, 222, 129. [DOI] [PubMed] [Google Scholar]
- [48].Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC, Nat. Rev. Cancer 2018, 18, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Alieva M, van Rheenen J, Broekman MLD, Clin. Exp. Metastasis 2018, 35, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tohme S, Simmons RL, Tsung A, Cancer Res 2017, 77, 1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Drake CG, Lipson EJ, Brahmer JR, Nat. Rev. Clin. Oncol 2014, 11, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G, Nat. Rev. Immunol 2017, 17, 97. [DOI] [PubMed] [Google Scholar]
- [53].Shen Z, Song J, Yung BC, Zhou Z, Wu A, Chen X, Adv. Mater 2018, 30, 1704007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhou J, Kroll AV, Holay M, Fang RH, Zhang L, Adv. Mater 2020, 32, 1901255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jiang Y, Krishnan N, Zhou J, Chekuri S, Wei X, Kroll AV, Yu CL, Duan Y, Gao W, Fang RH, Zhang L, Adv. Mater 2020, 32, 2001808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gao W, Fang RH, Thamphiwatana S, Luk BT, Li J, Angsantikul P, Zhang Q, Hu CM, Zhang L, Nano Lett 2015, 15, 1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhang Y, Chen Y, Lo C, Zhuang J, Angsantikul P, Zhang Q, Wei X, Zhou Z, Obonyo M, Fang RH, Gao W, Zhang L, Angew. Chem. Int. Ed. Engl 2019, 58, 11404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.