

Abstract
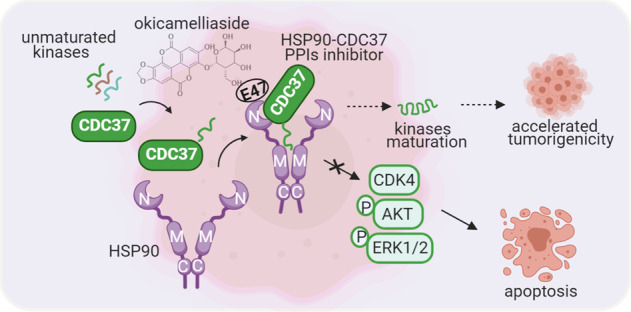
Heat shock protein 90 (HSP90) has been recognized as a crucial target in cancer cells. However, various toxic reactions targeting the ATP binding site of HSP90 may not be the best choice for HSP90 inhibitors. In this paper, an ellagic acid derivative, namely, okicamelliaside (OCS), with antitumor effects was found. To identify potential anti-cancer mechanisms, an OCS photosensitive probe was applied to target fishing and tracing. Chemical proteomics and protein-drug interaction experiments have shown that HSP90 is a key target for OCS, with a strong binding affinity (KD = 6.45 μM). Mutation analysis of the target protein and molecular dynamics simulation revealed that OCS could competitively act on the key Glu-47 site at the N-terminal chaperone pocket of HSP90, where the co-chaperone CDC37 binds to HSP90, affect its stability and reduce the ∆Gbind of HSP90-CDC37. It was demonstrated that OCS destroys the protein–protein interactions of HSP90-CDC37; selectively affects downstream kinase client proteins of HSP90, including CDK4, P-AKT473, and P-ERK1/2; and exerts antitumor effects on A549 cells. Furthermore, tumor xenograft experiments demonstrated high antitumor activity and low toxicity of OCS in the same way. Our findings identified a novel N-terminal chaperone pocket natural inhibitor of HSP90, that is, OCS, which selectively inhibits the formation of the HSP90-CDC37 protein complex, and provided further insight into HSP90 inhibitors for anti-cancer candidate drugs.
Keywords: HSP90, CDC37, okicamelliaside, antitumor, chaperone
Introduction
The successful execution of cellular processes depends on the coordinated interactions of proteins. A chaperone helps other proteins in the cell assemble, fold correctly, and stabilize, especially under conditions of cellular stress [1]. Heat shock protein 90 (HSP90) is a well-known 5′-adenosine triphosphate (ATP)-dependent protein chaperone that is essential for cell cycle regulation, proper protein assembly, kinase folding, cell differentiation, growth, and survival in many signaling pathways [2, 3]. Several HSP90 client proteins are closely related to the growth and proliferation of cancer cells. Targeting HSP90 to inhibit its client proteins and biological function is a therapeutic strategy for cancer [4–6].
Recently, protein–protein interactions (PPIs) have been shown to play a vital role in signal transduction pathways and networks in various physiological processes, particularly cancer-related processes. Disrupting protein complexes has become a new method of drug development. For example, there have been many studies on cancer treatment targeting the protein interactions of hypoxia-inducible factor-1 [7]. The novel potent TEAD inhibitor K-975 inhibits the YAP1/TAZ-TEAD PPI, demonstrating its suppressive effect on malignant pleural mesothelial tumors [8]. DDH-1, a new type of eIF4E/eIF4G PPI inhibitor, has shown anti-cancer activity in vivo and in vitro [9]. As a popular antitumor target protein, the development of HSP90 inhibitors for PPIs will also gain attention.
To date, more than 20 kinds of HSP90 inhibitors have entered clinical trials, and almost all of them have targeted the HSP90 ATPase pocket [10, 11]. However, the direct binding of the inhibitor to the ATPase pocket ultimately prevents the critical step of ATP hydrolysis and inevitably destroys all HSP90 clients. Due to various toxicities and heat shock reactions, none of them have been approved by the Food and Drug Administration [12, 13]. Therefore, new strategies should be developed to exert the antitumor effect of targeting HSP90.
Co-chaperones are also key regulators that drive multiple functions of the HSP90 chaperone mechanism. Multiple co-chaperones play an important role in achieving different functions and selectivity through direct PPIs with HSP90 [14, 15]. Protein kinases are HSP90 client proteins that play a crucial role in forming and developing tumors. Among them, more than half of the client proteins require co-chaperone cell division cycle 37 (CDC37) to regulate kinases such that they fold, transform, and mature appropriately [16]. As a co-chaperone protein that specifically targets kinases, before binding to open-state HSP90, CDC37 selectively recognizes and binds unfolded kinase clients and correctly folds overexpressed or mutated oncoproteins, thereby accelerating tumorigenicity [17]. These protein kinases contain receptor and non-receptor tyrosine kinases, lymphocyte-specific protein tyrosine kinases, and intracellular serine/threonine kinases, such as protein kinase B (AKT), extracellular regulated protein kinases (ERK)1/2, and cyclin-dependent kinase 4 (CDK4) [18]. In contrast to classic ATPase competitive HSP90 inhibitors, those that destroy HSP90-CDC37 PPIs selectively inhibit the kinase rather than inhibiting all HSP90 client proteins and have higher specificity and lower potential toxicity [19]. Hence, inhibitors that disrupt the interaction of HSP90-CDC37 will be a new strategy for the development of anti-cancer drugs.
In this study, we investigated a new natural inhibitor of HSP90, namely, okicamelliaside (OCS), derived from Camellia nitidissima Chi, that has antitumor activity. After target fishing and tracing, protein-drug interactions, and molecular dynamics simulations, the results demonstrated that OCS acts on the key Glu-47 site of HSP90. It disrupts the formation of the HSP90-CDC37 protein complex, inhibits the phosphorylation of HSP90 kinase client protein, and has an anti-cancer effect both in vitro and in vivo.
Materials and methods
Reagents and materials
OCS was manufactured by us and tested using high-performance liquid chromatography (HPLC) with a purity of 98% (detailed data are shown in Figs. S1–S4). We designed and synthesized the OCS probe and OCS-modified functionalized magnetic microspheres, as shown in Figs. S5–S9. Tigecycline (TGC, T830543) and adriamycin (ADM, A899829) were purchased from Macklin (Shanghai, China). N3-Coumarin was synthesized by AppTec (Wuxi, China). DL-Dithiothreitol (DTT, A285) was purchased from Solarbio (Beijing, China). Geldanamycin (GDM, HY-15230) and D-luciferin potassium salt (HY-12591B) were obtained from MedChemExpress (Rocky Hill, USA). Primary antibodies against HSP90 (4877 S), CDC37 (4222 S), CDK4 (12790 S), AKT (4691 S), ERK1/2 (4695 S), GR (12041 T), P-AKT473 (4060 S), P-ERK1/2 (4370 S), P-GR (4161 S), β-actin (4970 S) and GAPDH (5174 S) were obtained from Cell Signaling Technology (Beverly, USA). Goat anti-rabbit IgG H&L (Alexa Fluor® 594) and goat anti-mouse IgG H&L (Alexa Fluor® 488) were obtained from Abcam (Cambridge, UK).
Cell culture and cell viability assessment
A549-LUC cells (CBP30021L) were purchased from Cobioer Biosciences Co., Ltd., and all other cells were provided by the American Type Culture Collection (Rockville, MD, USA). The HELA, HBSMC, CaCO-2, H9C2, N-HLF, A375, BEND.3, RAW264.7, HEPG2, H295R, and 293 T cell lines were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and 1% penicillin. H1299, H460, MCF-7, NIH/3T3, and BEAS-2B cells were cultured in RPMI-1640 medium, and A549, A549-LUC, and CHO cells were cultured in Ham’s F-12K medium. All cells were incubated in a humidified incubator at 37 °C and 5% CO2. According to the manufacturer’s instructions, cell viability was tested using the Cell Counting Kit-8 (CCK-8, Bioss, Beijing, China).
Transwell and wound-healing assays
According to a previous description [20], in the Transwell experiment, A549 cells were seeded in a Transwell cell culture chamber (3422, Corning) at a density of 5 × 104 cells per well. The cells were then incubated with OCS (10, 20, or 40 μM), the same amount of dimethyl sulfoxide (DMSO) (negative control), or TGC (10 μM, positive control) for 24 h. Paraformaldehyde 4% was used to fix the migrating cells, which were stained with 0.2% crystal violet and then observed under an optical microscope. In the wound-healing assay, scrapes were made with a 200-µL plastic pipette tip, and phosphate-buffered saline (PBS) was used to wash away the exfoliated cells. ADM was added as a positive control (1 μM) and incubated for 24 h. The images were analyzed using ImageJ software.
Annexin V-FITC/PI double-staining assays
To detect cell apoptosis, A549 cells were incubated with OCS (0, 10, 20, or 40 μM) or DMSO in the same amount for 24 h. After washing with PBS, the apoptotic cells were processed according to the Annexin V-FITC apoptosis detection kit instructions (A005-3, 7 Sea Pharmatech, Shanghai, China) and detected using a flow cytometer system (BDLSR Fortessa, Indianapolis, NJ, USA) within 30 min. Data were analyzed and processed using FlowJo 7.6.
Target fishing
A549 cells were incubated using a 10 μM OCS probe for 6 h. After washing three times with pre-cooled PBS, the cell lysate was extracted and quantified according to a previous method [21]. Functionalized magnetic microspheres and cell lysates were co-incubated at 4 °C to capture the target proteins. Then, the magnetic microspheres separated by magnetic properties and non-specific proteins were washed with pre-cooled PBS. The captured proteins were released by DTT (100 mM) at room temperature for 30 min. The supernatant was used for SDS-PAGE and Western blot analysis, and specific operations were performed according to a previously described method [22].
Co-localization
For the HSP90 and OCS probe co-localization imaging assays, A549 cells were seeded in 20-mm confocal dishes and divided into OCS (10 µM OCS), OCS probe (10 µM OCS probe), competition (10 µM OCS probe and 100 µM OCS), and control groups. At room temperature, the cells were fixed using 4% paraformaldehyde for 15 min and blocked with goat serum for 1 h. Then, the fixed cells were incubated with HSP90 antibody (1:500) overnight at 4 °C. Subsequently, goat anti-rabbit IgG H&L (Alexa Fluor® 594) secondary antibodies (1:1000) were added to the cells and incubated for 1 h. The click chemistry reaction was implemented in fixed cells with N3-coumarin (10 mM) to obtain a fluorescent probe and observed by confocal microscopy (Leica TCS SP8, Japan). Alexa Fluor 594 secondary antibodies (red): λEx: 594 nm and λEe: 617 nm; fluorescent probe (green): λEx: 488 nm and λEe: 520 nm.
For HSP90-CDC37 protein interaction positioning, the cells were divided into the control (Con) and OCS treatment groups (10 µM). After blocking, the cells were incubated using HSP90 antibody (rabbit mAb) and CDC37 (mouse monoclonal, 66420-1-Ig, Proteintech), followed by goat anti-rabbit IgG H&L (Alexa Fluor® 594) and goat anti-mouse IgG H&L (Alexa Fluor® 488) incubation separately. Tumor tissues were dewaxed, rehydrated, and boiled in sodium citrate to repair the antigen. After endogenous peroxidase was quenched, it was blocked with goat serum. The subsequent operations were consistent with the procedures performed on the cells. As mentioned above, fluorescence images of HSP90-CDC37 protein were obtained under a confocal microscope, where HSP90 fluorescence was red (λEx: 594 nm, λEe: 617 nm) and CDC37 fluorescence was green (λEx: 488 nm and λEe: 520 nm).
Molecular dynamics simulation and docking
The three-dimensional structures of HSP90 (PDB ID: 3O0I) and CDC37 (PDB ID: 1US7) can be downloaded from the RCSB protein database (www.rcsb.org). The Amber 14 and AmberTools 15 programs were used for molecular dynamics simulation of the selected docking positions. The molecular mechanics/generalized surface area method implemented in AmberTools 15 was used to calculate the binding free energy (∆Gbind, kcal/mol) [23–26]. All detailed operations are shown in the Supporting Information. Subsequently, based on the crystal structure of the HSP90-CDC37 protein complex, molecular docking was performed. The key residues on HSP90 (including E47 and Q133) were used to predict the potential binding sites of OCS and HSP90. AutoDock Vina 1.1.2 software was used to analyze the binding mode and sites between them.
Interaction assay between OCS and HSP90
A series of classic techniques for verifying the interaction capacity of a small molecule ligand (OCS) and target protein HSP90 were applied, as described previously [27–31]. The binding between purified N-terminal HSP90 or its mutants and OCS was determined using the thermal shift assay, circular dichroism (CD) spectroscopy, surface plasmon resonance (SPR) analysis, microscale thermophoresis (MST), and fluorescence quenching assay. The detailed operation process is shown in the Supporting Information.
Co-immunoprecipitation assay
Briefly, A549 cell lysates were treated with 10 μM OCS or DMSO for 12 h and incubated with 5 μg of anti-HSP90 or anti-CDC37 at 4 °C overnight. Protein A/G magnetic agarose beads (HY-K0202, MedChemExpress) were pre-treated with 2% skimmed milk and added to lysates for another 2 h at 4 °C. Finally, the beads were washed five times using pre-cooled PBS and subjected to Western blot analysis.
Electron microscopy
According to previous reports [32], for staining of the protein complexes of HSP90 and CDC37, uranyl formate (pH 5.5–6.0) was used for negative staining on a 400-mesh copper grid (Pelco) with a thin carbon layer. The samples were imaged using TEM (FEI, Talos L120C G2) at 120 kV. A 4 k × 4 k CCD camera was used to record the micrographs at a nominal magnification of 30 k.
Animals and tumor xenograft experiments
Male BALB/c-nude mice (6 weeks old, ~20 g, specific pathogen-free) were purchased from Charles River Laboratories (SCXK2016-0006, Beijing, China). The mice were randomly divided into five groups (n = 6), and A549-LUC cells (1 × 107) were injected into the left armpit of nude mice. Treatment was initiated when the average tumor volume reached 100 mm3 [33]. Mice were intraperitoneally injected with OCS (3, 10 or 30 mg·kg−1·d−1), ADM (0.5 mg·kg−1·d−1) or 0.9% NaCl (as controls) for administration intervention. Tumor volume and body weight were measured every day. Four weeks after intraperitoneal administration, D-fluorescein potassium salt solution (15 mg/mL, 100 μL) was injected into the tail vein, and the fluorescence intensity of the tumor was monitored by bioluminescence measurement using a NightOWLIILB983 imaging system (Berthold Technologies, Germany). The mice were sacrificed, and the tumor tissue was excised and weighed. Then, the tumor was frozen in liquid nitrogen or fixed using paraformaldehyde for later use. All animal care and experimental procedures were approved by the Instructional Animal Care and Use Committee (DW20181223-35).
Immunohistochemistry
The tumor tissues were dewaxed, rehydrated, and boiled in sodium citrate for 10 min to retrieve the antigen. After endogenous peroxidase was quenched, the cells were incubated with CDK4, P-ERK1/2, and P-AKT473 antibodies (1:500) at 4 °C overnight. The diaminobenzidine (DAB) Substrate Kit (DA1015, Solarbio, Beijing, China) was used in the following operations. Biotin-conjugated secondary antibody (1:200) and horseradish peroxidase-conjugated streptavidin were sequentially incubated. DAB substrate was used to visualize the reaction, and the slides were washed in running tap water. The sections were counterstained with hematoxylin and observed under an optical microscope. In addition, data were analysed and processed using ImageJ.
Statistical analysis
The results are expressed as the mean ± standard error of the mean. Comparisons between two groups were made using Student’s t-test, and multiple data comparison was achieved using analysis of variance. All data were processed using GraphPad Prism 6.01 (La Jolla, CA, USA). P < 0.05 was considered a significant difference.
Results
OCS inhibited tumor cell growth and induced apoptosis
To verify the specificity of OCS for anti-cancer therapy, various types of cells (including tumor and normal cells) were tested. As shown in Fig. 1a, OCS showed a better inhibitory effect on HELA, HEPG2, and A549 tumor cells than most normal cells. A549 cells were selected for the following research. OCS inhibited viability in a time- and concentration-dependent manner in A549 cells (Fig. 1b). The IC50 value of OCS on A549 cells was ~22.7 μM at 72 h. In addition, in cell migration experiments, OCS reduced the number of migrating cells in a dose-dependent manner, similar to the positive control TGC (Fig. 1c). The wound-healing analysis showed that compared with the Con group, ADM almost completely inhibited cell migration, and OCS also inhibited the cell healing ability in a dose-dependent manner (Fig. 1d). To detect whether OCS induced apoptosis in A549 cells, double staining was performed using Annexin V-FITC/PI. The results revealed that the apoptosis rate increased from 8.47%±4.97% to 28.15%±6.86% when A549 cells were treated with 40 μM OCS for 24 h (Fig. 1e). These results demonstrate that OCS can inhibit the growth and proliferation of A549 cells and can induce apoptosis.
Fig. 1. OCS inhibited A549 cell growth and induced cell apoptosis.

a OCS administration was given to different tumor cell lines (purple) and normal cell lines (blue) and the IC50 value was measured. b The effect of OCS on the viability of A549 cells. c, d The effect of OCS on the migration and wound-healing assay of A549 cells. e Annexin V-FITC/PI double staining was used to detect the effect of OCS on A549 cell apoptosis. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Con, n = 3.
OCS targeted HSP90 and downregulated its kinase client proteins
To identify potential anti-cancer targets, an OCS photosensitive probe was synthesized. The structures of the OCS probe and its clicked product (a tracing probe synthesized by the cycloaddition reaction of OCS probe and coumarin azide), which were used for target tracing in A549 cells, are shown in Fig. 2a. The IC50 (29.03 μM) of the prepared OCS probe in A549 cells was consistent with the IC50 (22.7 μM) of OCS (Fig. S10), indicating that the OCS probe did not alter its antitumor activity and that it could be used in subsequent chemical biological tests.
Fig. 2. OCS targeted the HSP90 protein and affected downstream kinase client proteins.

a The structures of OCS, OCS probe, and OCS click product. b SDS-PAGE and Western blot analysis of captured protein by OCS probe. Lane 1 shows the A549 cell lysate, Lane 2 shows the negative control without the OCS probe, and lane 3 shows the OCS probe-captured proteins. c Venn diagram analysis of OCS antitumor targets. The red intersection is the common target HSP90. d The co-localization of HSP90 (red) and OCS probe (green) in A549 cells (magnification 200×). e The effect of OCS and GDM on HSP90 representative client proteins. f The influence of OCS or GDM on the ATPase activity of HSP90. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Con, n = 3.
Next, the OCS probe was first used to capture the target protein from A549 cells using OCS-functionalized magnetic microspheres (the flow diagram is detailed in Fig. S9). The captured proteins were released from the magnetic beads by DTT reduction and analyzed using SDS-PAGE. As shown in Fig. 2b, compared with the control group (Lane 2), some potential target proteins were specifically enriched (Lane 3). The strips were subjected to enzymatic hydrolysis and identified using HPLC-MS/MS, and the identification results are shown in Table S1.
Furthermore, three groups of proteome data, including OCS probe captured protein, pharmMapper predicted OCS target proteins (http://www.lilab-ecust.cn/pharmmapper/), and cancer-related targets from the database of human genes (https://www.genecards.org/), were used for comprehensive analysis (Table S1). The Venn diagram is shown in Fig. 2c; only one likely target protein, HSP90, was recommended and then confirmed using Western blotting (Fig. 2b, right panel). In addition, co-localization of HSP90 and OCS probe was performed in A549 cells. As illustrated in Fig. 2d, almost no fluorescence was observed in the OCS control group; however, in the OCS probe group, the click product showed strong fluorescence (green) in the cytoplasm, which merged with that of the HSP90 antibody (red) very well, and the Pearson coefficient (PC) was approximately 0.79. In addition, the specific yellow merge was decreased (the PC value dropped to approximately 0.37) by 10-fold free OCS competition.
To further evaluate the impact of OCS on HSP90 downstream client proteins, Western blotting was used to detect representative kinase and non-kinase clients. Simultaneously, GDM, a classic ATP binding pocket inhibitor of the HSP90 N-terminus, was selected for comparative analysis. GDM directly binds to the ATPase pocket, which prevents ATP hydrolysis and inevitably destroys all HSP90 customer proteins [9]. The results are shown in Fig. 2e. In contrast to GDM, OCS reduced the phosphorylation status of the kinases AKT and ERK1/2 (P-AKT473 and P-ERK1/2, respectively) without changing the protein levels, and OCS treatment did not inhibit the expression of GR and P-GR, a non-kinase HSP90 client. Furthermore, we determined the effect of OCS on the ATPase of HSP90 according to a previously described method [34], and the detailed measurement process is shown in the Supporting Information. The activity of ATPase is reflected by detecting the decrease in NADPH at an absorbance of 340 nm. GDM (10 μM) exhibited a marked inhibitory effect on the ATPase activity of HSP90, while 100 μM GDM completely abolished the activity. However, 100 μM OCS had no effect on the ATPase of HSP90 protein (Fig. 2f). MST competition experiments also indicated that OCS had no effect on the ATP binding activity (shown in Figure S13). Based on the above results, we speculate that OCS may not be a classic HSP90 ATPase inhibitor but may be an emerging HSP90-CDC37 inhibitor that can selectively inhibit kinase clients.
OCS bound to the N-terminal CDC37 binding pocket of HSP90 and affected its stability
To confirm our hypothesis that OCS is an inhibitor of the HSP90-CDC37 complex, the Amber14 software package was used for molecular dynamics simulation. The root mean square deviation (RMSD) value of the protein skeleton is based on the starting structure along the simulation time, and it is plotted in Fig. S11. To obtain more information about the residues around the binding site and their contribution to the entire system, the electrostatic (∆Eele), van der Waals (∆Evdw), solvation (∆Esol), and total contribution (∆Etotal) of these residues were calculated using MMGBSA. In general, in the HSP90-CDC37 complex (Fig. 3a), most of the energy interactions for decomposition were derived from van der Waals interactions, mainly through hydrophobic interactions, such as Ala-117, Ala-121, Ala-124, Ala -126, Met-130, Phe-134, and Ile-214 of HSP90 and Met-164, Leu-165, Ala-204, and Leu-205 of CDC37 (Fig. S11). Detailed analysis showed that the residues Glu-47 and Gln-133 of HSP90 formed two strong hydrogen bond interactions, suggesting that the most critical sites for the binding between CDC37 and HSP90 are consistent with previous studies [26].
Fig. 3. OCS targeted the CDC37 binding pocket of HSP90 and affected protein stability.

The predicted binding modes were obtained from molecular dynamics simulation and decomposition of the binding energy on a per-residue basis of the (a) HSP90-CDC37 complex and (b) HSP90/OCS-CDC37 complex. c The RMSF of residues of the HSP90 protein in the HSP90-CDC37 complex and HSP90/OCS-CDC37 complex during the 40-ns simulation; the orange area is the ATPase binding pocket, and the blue area is the CDC37 binding site. d SPR analysis of interactions between OCS and HSP90. e The thermal shift assay to evaluate the thermal stability of HSP90 protein and OCS. f CD spectrum analysis of the influence of OCS on the HSP90 protein conformation.
In the HSP90/OCS-CDC37 complex (Fig. 3b), the main contribution originated from the interaction of van der Waals forces and hydrophobicity, such as Ala-117, Ala-121, Ala-126, and Phe-134 of HSP90 and Leu-165, Ala-204, and Leu-205 of CDC37 (Fig. S11). The results showed that although residue Glu-133 of HSP90 formed strong hydrogen bond interactions and electrostatic (∆Eele) contributions, it had the same ∆Eele value of −41.9 kcal/mol. However, the Glu-47 hydrogen bond interaction disappeared. In addition, the total free energy and estimated ∆Gbind of the CDC37-HSP90 and HSP90/OCS-CDC37 complexes calculated according to MMGBSA changed from −50.2 to −17.1 kcal/mol. It is speculated that OCS may be targeted to Glu-47 and weaken the interaction between CDC37 and HSP90.
In addition, the root mean square fluctuations (RMSF) were calculated, which revealed residue flexibility. The RMSF values of HSP90 and CDC37 were less than 2 Å, and residues showed a small degree of flexibility, indicating that these residues appeared to be rigid during the simulation. The positions of the two binding pockets on the HSP90 N-terminus were compared and marked in Fig. 3c. Before and after OCS administration, the anterior segment sequence of the HSP90 N-terminus, which includes key Glu-47 residue sites, changed more than the posterior segment sequence that held residue Glu-133. The above molecular dynamics simulations provide a rational explanation of the interactions among OCS, HSP90, and CDC37, which provides valuable information on the interactions between HSP90 and CDC37.
To further evaluate the binding feasibility, the HSP90 N-terminal protein, including the chaperone pocket, was expressed and purified (Fig. S12) and used for interaction evaluation with OCS. According to the SPR results, OCS binds to the immobilized HSP90 protein, and the dissociation constant value (KD) was 6.45 μM (Fig. 3d). Thermal shift assays showed that OCS increased the melting temperature (Tm) of HSP90 protein and that 100 μM OCS increased the thermal stability of HSP90 by 1.76 °C (ΔTm) (Fig. 3e). Further CD chromatography was performed to test whether OCS affected HSP90 protein conformation. After treatment with OCS, the negative peak of the β-sheet conformation in the CD spectrum at approximately 230 nm showed a change (Fig. 3f). The above results indicated that OCS targeted the N-terminus of the HSP90 protein, changed the structural stability, and affected the interaction of HSP90 and CDC37.
OCS competes with Glu-47 of HSP90 with CDC37 and affects the interaction
To clarify the specific binding mode, a virtual dock was first performed. The docking results showed that OCS strongly interacted with Glu-47, Gly-132, and Phe-213 in HSP90 (Fig. 4a). In addition, the sequence alignment of HSP90 from different species (HUMAN, and MOUSE) showed that the HSP90 sequence is highly conserved (Fig. 4b). To verify the accuracy of the prediction, the above three key residues related to the binding of the HSP90-CDC37 pocket were mutated to alanine. The binding affinity (KD) between the mutants and OCS was measured using fluorescence quenching. As shown in Fig. 4c, compared to the KD value of WT HSP90 (19.71 μM), that of the E47A mutant (146.8 μM) was significantly lower, G132A (15.57 μM) and F213A (35.52 μM) mutants had weaker effects. In addition, in Fig. 4d, the MST assay also demonstrated the same result, and the KD value of the E47A mutant (67.0 μM) was much larger than that of WT HSP90 (8.6 μM). The results proved that OCS could bind to Glu-47 of HSP90 and compete for the key binding site of HSP90 and CDC37.
Fig. 4. The binding site assay between OCS and HSP90.

a Molecular docking analysis of OCS on the CDC37 binding pocket of HSP90; yellow circles indicate ATP binding sites, CDC37 binding pocket (black squares) is magnified in the right panel, and the key residues of Glu-47, Gly-132, and Phe-213, which interact with OCS, are highlighted. Hydrogen bonds are shown as yellow dotted lines. b Sequence comparison analysis of the N-terminal sequence of HUMAN and MOUSE species of HSP90. c Fluorescence quenching assay was used to test the combination of OCS and WT HSP90 or mutants (E47A, G132A, and F213A); **P < 0.01 vs. WT, n = 3. d The MST assay was used to confirm the binding capacity of OCS and the E47A mutant.
OCS disrupted the PPIs of HSP90-CDC37
To accurately evaluate the destruction efficiency of OCS on HSP90-CDC37 PPIs, co-immunoprecipitation between HSP90 and CDC37 was performed to detect the bait protein. As shown in Fig. 5a, whenever HSP90 or CDC37 was immunoprecipitated, OCS destroyed the HSP90-CDC37 PPIs in A549 cells. In addition, the co-localization test of HSP90 and CDC37 showed that the two proteins HSP90 (red) and CDC37 (green) could be mostly merged without OCS treatment in A549 cells, and the PC value was approximately 0.83 (Fig. 5b). When treated with 10 μM OCS, the yellow merge image was significantly less, and the PC value dropped to 0.56. The MST assay provided further detailed supporting evidence that OCS destroyed HSP90-CDC37 PPIs; the KD value of HSP90 and CDC37 protein was increased from 149 nM to 946 nM after OCS addition (Fig. 5c).
Fig. 5. OCS disrupted the HSP90-CDC37 interaction and selectively downregulated kinase clients of HSP90.

a Co-immunoprecipitation in A549 cells after treatment with OCS, **P < 0.01 and vs. Con; n = 3. b Co-localization of HSP90 (red) and CDC37 (green) in A549 cells, and Merge (yellow) indicated that the two proteins were located together. c MST was used to measure the effect of OCS on HSP90-CDC37 PPIs. d Negative staining electron microscopy analysis of the effect of 10 μM OCS on the HSP90-CDC37 protein complex. Scale bar: 150 nm. On the right side of the figure is an enlarged view of the white box shown. Scale bar: 50 nm. e Western blot analysis of the effect of different concentrations of OCS on HSP90, CDC37, CDK4, AKT, P-AKT473, ERK1/2, and P-ERK1/2, the representative kinase client proteins of HSP90. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Con, n = 3.
Furthermore, the phenomenon can be observed by a negative staining electron microscope. As shown in Fig. 5d, the wild-type HSP90 and CDC37 proteins were distributed in a relatively uniform form. However, the mixture of the two proteins showed obvious aggregates, and when OCS was incubated with the HSP90-CDC37 protein complex, the aggregates were dispersed. The results once again proved that OCS destroyed the PPIs of HSP90 and CDC37 proteins. OCS treatment induced the downregulation of representative kinase client proteins (CDK4, AKT, and ERK1/2) of HSP90-CDC37 in a dose-dependent manner (Fig. 5e). Therefore, OCS is a type of selective kinase client inhibitor that disrupts the HSP90-CDC37 interaction and will provide a lead compound for anti-cancer purposes.
OCS inhibited tumor growth of the A549-LUC xenograft model in vivo
To further evaluate the antitumor effect of OCS in vivo, different doses of OCS were used in nude mice bearing subcutaneous A549-LUC cell tumors. The experimental procedure is shown in Fig. 6a. The body weight and tumor growth conditions were measured and are shown in Fig. 6b, c. During OCS administration, compared with the Mod group, there was almost no effect on the weight of mice; however, OCS treatment decreased tumor growth in a dose-dependent manner. In the in vivo fluorescence imaging analysis, the total number of fluorescent photons was significantly reduced in a dose-dependent manner (Fig. 6d). The inhibitory effect of OCS on the growth of A549-LUC solid tumors was visible, as shown in Fig. 6e. These data suggest that OCS also has a better inhibitory effect on tumor growth in vivo.
Fig. 6. OCS exerted antitumor activity in vivo.

a Experimental flowchart. b The body weight and (c) the tumor growth curve. d Fluorescence imaging analysis of tumors in vivo. e Pictures and weight statistics of tumor tissues after treatment with different doses of OCS or ADM. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Mod, n = 6.
To uncover the mechanisms at the protein level in the resected tumor tissue, an immune co-localization assay between HSP90-CDC37 was performed on tumor tissues. As shown in Fig. 7a, the co-localization showed that the two proteins HSP90 (red) and CDC37 (green) could be mostly merged without OCS treatment in the Mod group (the PC value was ~0.91). In the OCS treatment group (30 mg/kg), there were fewer yellow merged images (the PC value was approximately 0.68). The results showed that OCS can destroy the PPIs of HSP90-CDC37 in tumor tissues. In addition, the downstream pathway of HSP90/CDC37 was investigated using Western blot analysis. As shown in Fig. 7b, OCS treatment selectively inhibited the levels of CDK4 and HSP90 kinase client protein (AKT and ERK1/2) phosphorylation, and the results were highly consistent with those of A549 cells. Immunohistochemistry also confirmed that compared with the Mod group, OCS (30 mg/kg) significantly reduced the expression of CDK4 and the phosphorylation of P-AKT473 and P-ERK1/2 in tumor tissues (Fig. 7c). These findings further confirm that OCS can inhibit HSP90 kinase client proteins and exert antitumor efficacy both in vivo and in vitro.
Fig. 7. The effect of OCS on kinase client proteins in tumor tissues.

a Co-localization of HSP90 (red) and CDC37 (green) in tumor tissues. Merged images (yellow) indicate that the two proteins were located together. b Western blot analysis of the representative client proteins of HSP90 in xenograft tumor tissues. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Mod, n = 3. c Immunohistochemical analysis of the effect of OCS on CDK4, P-AKT473 and P-ERK1/2 in tumor tissues. **P < 0.01, and ***P < 0.001 vs. Mod, n = 3. Scale bar: 100 μm.
Discussion
HSP90 protein is involved in the maturation and stabilization of many oncoproteins and telomerase [35]. The expression level of HSP90 in cancer cells is 2–10 times that of normal cells [36]. Therefore, in recent years, HSP90 has been considered one of the important targets for cancer treatment. HSP90 consists of four domains: (1) the highly conserved N-terminus, (2) the middle domain, and (3) the C-terminus. (4) The charged linker region connects the N-terminus to the middle domain [37]. By targeting different domains, HSP90 inhibitors are classified into different types [38]. GDM is the first HSP90 inhibitor based on benzoquinone ansamycins. It mimics ATP to competitively bind to the HSP90 N-terminal ADP/ATP pocket and blocks the natural substrate ATP [39]. GDM and its analogs have been shown to induce significant downregulation of multiple HSP90 client proteins and other downstream effectors and to reduce cell proliferation and motility. However, due to its low solubility and toxicity, its clinical potential is relatively poor [40]. To improve the pharmacological properties and increase the specificity, a series of purine analogs, including PU3 and resorcinol triazolone analogs, were designed and synthesized, such as ganetespib and other N-terminal ATP pocket inhibitors [41, 42]. In addition, due to the side effects of N-terminal inhibitors, HSP90 C-terminal binding inhibitors such as novobiocin and penicisulfuranol A inhibited the dimerization of HSP90 and the interaction of HSP90 with co-chaperones [43, 44]. With the emergence of small molecule inhibitors targeting PPIs, research has been conducted progressively on decoupling co-chaperone-HSP90 inhibitors.
However, due to the complexity of PPIs, few small molecule inhibitors can directly target HSP90-CDC37 PPIs. To date, only a few small molecules have been reported with the potential to destroy HSP90-CDC37 and exhibit anti-cancer activity. Synthetic compounds (DDO-5936 and VS-8) and some natural products (FW-04-806, celastrol withaferin A, and apigenin) merely show similar low activity or an unclear mechanism [15, 34, 45–48]. It was proposed that the plant extract celastrol could destroy HSP90-CDC37 PPIs. However, the direct binding site is still controversial and was identified using HSP90 N-terminus molecular docking or the HSP90 C-terminus fingerprint assay [49]. Others have reported natural inhibitors, including Qidong You’s group, which has conducted many studies on HSP90-CDC37 and uncovered VS-8, which targets HSP90-CDC37 PPIs and was discovered through virtual screening from 500 thousand compound databases (KD = 80.4 μM) [45], as well as DDO-5936 (KD = 3.86 μM) and 18 h (KD = 0.5 μM) obtained from compound structural optimization. The latter two compounds clarified the specific site of the inhibitor Glu-47 of HSP90, which provided important clues for the development of HSP90-CDC37 inhibitors [15, 50]. The study first reported that OCS, an ellagic acid glucoside, competes with Glu-47 of HSP90 for CDC37 with a high affinity (KD = 6.45 μM). Based on the previous molecular dynamics simulation, the results depict different flexibilities in the binding site of the HSP90-CDC37 complex in the presence and absence of OCS.
Currently, most reported HSP90 inhibitors prevent the catalytic cycle of HSP90 in the ADP-bound conformation by binding to the N-terminus of HSP90 in the ATPase pocket. They inactivate chaperone activity and lead to ubiquitination of client proteins and proteasome degradation. However, they cannot avoid damage to all HSP90 clients and cause off-target toxicity and heat stress [51]. Therefore, completely inhibiting HSP90 or directly targeting the ATPase binding site of HSP90 may not be the best choice for cancer treatment. Therefore, more high selectivity and low toxicity HSP90 inhibitors are needed.
In contrast to classic HSP90 inhibitors, as a new type of natural inhibitor, OCS competes with CDC37 at the Glu-47 residue of HSP90, disrupts the formation of the HSP90-CDC37 complex, impedes the dimerization of HSP90, selectively inhibits the phosphorylation level of downstream protein kinases, and exerts antitumor activity. The benefits provide further insights into the regulation of the interaction between HSP90-CDC37 and other chaperone molecules in cancer treatment and provide a basis for developing alternative drugs against the CDC37 pocket of HSP90.
Supplementary information
Acknowledgements
This study was supported by Guangxi Innovation-driven Development 20 Special Foundation Project (Guike No. AA18118049). We would like to thank Nature Research Editing (https://authorservices.springernature.com/) for English language editing.
Author contributions
Conceptualization, GB and YYH; Data curation and formal analysis, CJC; Funding acquisition, XTH; EWH and GB; Investigation, CJC and YYH; Methodology, CJC; KXL; MZ; LLY; FKS and WBW; Supervision, GB; Validation, CJC and YYH; Writing-original draft, CJC; Writing-review & editing, GB and YYH.
Competing interests
The authors declare no competing interests.
Contributor Information
Yuan-yuan Hou, Email: houyy@nankai.edu.cn.
Gang Bai, Email: gangbai@nankai.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00737-x.
References
- 1.Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–55. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- 2.Mahalingam D, Swords R, Carew JS, Nawrocki ST, Bhalla K, Giles FJ. Targeting HSP90 for cancer therapy. Br J Cancer. 2009;100:1523–9. doi: 10.1038/sj.bjc.6605066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiosis G, Dickey CA, Johnson JL. A global view of Hsp90 functions. Nat Struct Mol Biol. 2013;20:1–4. doi: 10.1038/nsmb.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buc Calderon P, Beck R, Glorieux C. Targeting hsp90 family members: a strategy to improve cancer cell death. Biochem Pharmacol. 2019;164:177–87. doi: 10.1016/j.bcp.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 5.Huang X, Yan X, Zhang Q, Hong P, Zhang W, Liu Y, et al. Direct targeting of HSP90 with daurisoline destabilizes β-catenin to suppress lung cancer tumorigenesis. Cancer Lett. 2020;489:66–78. doi: 10.1016/j.canlet.2020.05.024. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed A, Tait SWG. Targeting immunogenic cell death in cancer. Mol Oncol. 2020;14:2994–3006. doi: 10.1002/1878-0261.12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Xi W, Li X, Sun H, Li Y. Advances in inhibition of protein-protein interactions targeting hypoxia-inducible factor-1 for cancer therapy. Bioorg Med Chem. 2019;27:1145–58. doi: 10.1016/j.bmc.2019.01.042. [DOI] [PubMed] [Google Scholar]
- 8.Kaneda A, Seike T, Danjo T, Nakajima T, Otsubo N, Yamaguchi D, et al. The novel potent TEAD inhibitor, K-975, inhibits YAP1/TAZ-TEAD protein-protein interactions and exerts an anti-tumor effect on malignant pleural mesothelioma. Am J Cancer Res. 2020;10:4399–415. [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Wang L, Zhang S, Fan J, Yang H, Li Q, et al. Novel eIF4E/eIF4G protein-protein interaction inhibitors DDH-1 exhibits anti-cancer activity in vivo and in vitro. Int J Biol Macromol. 2020;160:496–505. doi: 10.1016/j.ijbiomac.2020.05.233. [DOI] [PubMed] [Google Scholar]
- 10.Bhat R, Tummalapalli SR, Rotella DP. Progress in the discovery and development of heat shock protein 90 (Hsp90) inhibitors. J Med Chem. 2014;57:8718–28. doi: 10.1021/jm500823a. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Carbonero R, Carnero A, Paz-Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol. 2013;14:e358–69. doi: 10.1016/S1470-2045(13)70169-4. [DOI] [PubMed] [Google Scholar]
- 12.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823:742–55. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajan A, Kelly RJ, Trepel JB, Kim YS, Alarcon SV, Kummar S, et al. A phase I study of PF-04929113 (SNX-5422), an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumor malignancies and lymphomas. Clin Cancer Res. 2011;17:6831–9. doi: 10.1158/1078-0432.CCR-11-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calderwood SK. Molecular cochaperones: tumor growth and cancer treatment. Science. 2013;2013:217513. doi: 10.1155/2013/217513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Retzlaff M, Hagn F, Mitschke L, Hessling M, Gugel F, Kessler H, et al. Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol Cell. 2010;37:344–54. doi: 10.1016/j.molcel.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Pearl LH. Hsp90 and Cdc37—a chaperone cancer conspiracy. Curr Opin Genet Dev. 2005;15:55–61. doi: 10.1016/j.gde.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Stepanova L, Yang G, DeMayo F, Wheeler TM, Finegold M, Thompson TC, et al. Induction of human Cdc37 in prostate cancer correlates with the ability of targeted Cdc37 expression to promote prostatic hyperplasia. Oncogene. 2000;19:2186–93. doi: 10.1038/sj.onc.1203561. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Zhang L, Li L, Jiang J, Zheng Z, Shang J, et al. Small-molecule inhibitor targeting the Hsp90-Cdc37 protein-protein interaction in colorectal cancer. Sci Adv. 2019;5:eaax2277. doi: 10.1126/sciadv.aax2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith JR, Workman P. Targeting CDC37: an alternative, kinase-directed strategy for disruption of oncogenic chaperoning. Cell Cycle. 2009;8:362–72. doi: 10.4161/cc.8.3.7531. [DOI] [PubMed] [Google Scholar]
- 20.Zhang W, Gao J, Cheng C, Zhang M, Liu W, Ma X, et al. Cinnamaldehyde enhances antimelanoma activity through covalently binding ENO1 and exhibits a promoting effect with dacarbazine. Cancers. 2020;12:311. doi: 10.3390/cancers12020311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao J, He X, Ma Y, Zhao X, Hou X, Hao E, et al. Chlorogenic acid targeting of the AKT PH domain activates AKT/GSK3β/FOXO1 signaling and improves glucose metabolism. Nutrients. 2018;10:1366. doi: 10.3390/nu10101366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng C, Shen F, Ding G, Liu A, Chu S, Ma Y, et al. Lepidiline a improves the balance of endogenous sex hormones and increases fecundity by targeting HSD17B1. Mol Nutr Food Res. 2020;64:e1900706. doi: 10.1002/mnfr.201900706. [DOI] [PubMed] [Google Scholar]
- 23.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J Chem Theory Comput. 2013;9:3878–88. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 24.Sousa da Silva AW, Vranken WF. ACPYPE—antechamber python parser interface. BMC Res Notes. 2012;5:367. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2005;26:114. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25:247–60. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Wang Z, Ma X, Yang S, Hu X, Tao J, et al. Glycyrrhetinic acid binds to the conserved P-loop region and interferes with the interaction of RAS-effector proteins. Acta Pharm Sin B. 2019;9:294–303. doi: 10.1016/j.apsb.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med. 2009;15:750–6. doi: 10.1038/nm.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751:119–39. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Manvar D, Mishra M, Kumar S, Pandey VN. Identification and evaluation of anti hepatitis C virus phytochemicals from Eclipta alba. J Ethnopharmacol. 2012;144:545–54. doi: 10.1016/j.jep.2012.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schweiger R, Soll J, Jung K, Heermann R, Schwenkert S. Quantification of interaction strengths between chaperones and tetratricopeptide repeat domain-containing membrane proteins. J Biol Chem. 2013;288:30614–25. doi: 10.1074/jbc.M113.493015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen PH, Unger V, He X. Structure of full-length human PDGFRβ bound to its activating ligand PDGF-B as determined by negative-stain electron microscopy. J Mol Biol. 2015;427:3921–34. doi: 10.1016/j.jmb.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei Y, Yang Q, Zhang Y, Zhao T, Liu X, Zhong J, et al. Plumbagin restrains hepatocellular carcinoma angiogenesis by suppressing the migration and invasion of tumor-derived vascular endothelial cells. Oncotarget. 2017;8:15230–41. doi: 10.18632/oncotarget.14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem. 2009;284:35381–9. doi: 10.1074/jbc.M109.051532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banerji U. Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res. 2009;15:9–14. doi: 10.1158/1078-0432.CCR-08-0132. [DOI] [PubMed] [Google Scholar]
- 36.Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213–7. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 37.Krishna P, Gloor G. The Hsp90 family of proteins in Arabidopsis thaliana. Cell Stress Chaperones. 2001;6:238–46. doi: 10.1379/1466-1268(2001)006<0238:thfopi>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 39.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA. 1994;91:8324–8. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cervantes-Gomez F, Nimmanapalli R, Gandhi V. Transcription inhibition of heat shock proteins: a strategy for combination of 17-allylamino-17-demethoxygeldanamycin and actinomycin D. Cancer Res. 2009;69:3947–54. doi: 10.1158/0008-5472.CAN-08-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taldone T, Chiosis G. Purine-scaffold Hsp90 inhibitors. Curr Top Med Chem. 2009;9:1436–46. doi: 10.2174/156802609789895737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jhaveri K, Ochiana SO, Dunphy MP, Gerecitano JF, Corben AD, Peter RI, et al. Heat shock protein 90 inhibitors in the treatment of cancer: current status and future directions. Expert Opin Investig Drugs. 2014;23:611–28. doi: 10.1517/13543784.2014.902442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donnelly AC, Mays JR, Burlison JA, Nelson JT, Vielhauer G, Holzbeierlein J, et al. The design, synthesis, and evaluation of coumarin ring derivatives of the novobiocin scaffold that exhibit antiproliferative activity. J Org Chem. 2008;73:8901–20. doi: 10.1021/jo801312r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai J, Chen A, Zhu M, Qi X, Tang W, Liu M, et al. Penicisulfuranol A, a novel C-terminal inhibitor disrupting molecular chaperone function of Hsp90 independent of ATP binding domain. Biochem Pharmacol. 2019;163:404–15. doi: 10.1016/j.bcp.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 45.Wang L, Li L, Zhou ZH, Jiang ZY, You QD, Xu XL. Structure-based virtual screening and optimization of modulators targeting Hsp90-Cdc37 interaction. Eur J Med Chem. 2017;136:63–73. doi: 10.1016/j.ejmech.2017.04.074. [DOI] [PubMed] [Google Scholar]
- 46.Huang W, Ye M, Zhang LR, Wu QD, Zhang M, Xu JH, et al. FW-04-806 inhibits proliferation and induces apoptosis in human breast cancer cells by binding to N-terminus of Hsp90 and disrupting Hsp90-Cdc37 complex formation. Mol Cancer. 2014;13:150. doi: 10.1186/1476-4598-13-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grover A, Shandilya A, Agrawal V, Pratik P, Bhasme D, Bisaria VS, et al. Hsp90/Cdc37 chaperone/co-chaperone complex, a novel junction anticancer target elucidated by the mode of action of herbal drug Withaferin A. BMC Bioinform. 2011;12:S30. doi: 10.1186/1471-2105-12-S1-S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao M, Ma J, Zhu HY, Zhang XH, Du ZY, Xu YJ, et al. Apigenin inhibits proliferation and induces apoptosis in human multiple myeloma cells through targeting the trinity of CK2, Cdc37 and Hsp90. Mol Cancer. 2011;10:104. doi: 10.1186/1476-4598-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG, et al. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther. 2008;7:162–70. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 50.Wang L, Jiang J, Zhang L, Zhang Q, Zhou J, Li L, et al. Discovery and optimization of small molecules targeting the protein-protein interaction of heat shock protein 90 (Hsp90) and cell division cycle 37 as orally active inhibitors for the treatment of colorectal cancer. J Med Chem. 2020;63:1281–97. doi: 10.1021/acs.jmedchem.9b01659. [DOI] [PubMed] [Google Scholar]
- 51.Luo M, Meng Z, Moroishi T, Lin KC, Shen G, Mo F, et al. Heat stress activates YAP/TAZ to induce the heat shock transcriptome. Nat Cell Biol. 2020;22:1447–59. doi: 10.1038/s41556-020-00602-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.